
INTRODUCTION
Breast cancer is the most frequently diagnosed cancer among women in the U.S. and is the second leading cause of cancer-related mortality in this population [1]. Metastatic disease is the cause of the majority of cancer-related death [1]. Elucidating and targeting the processes that lead to metastasis could prevent mortality due to breast and other cancers. Breast cancer is a heterogeneous disease that is typically classified according to the expression of three cell surface receptors, i.e. estrogen receptor, progesterone receptor, and HER2 receptor [2]. Estrogen receptor and HER2 can be targeted therapeutically which leads to a more favorable prognosis for women with tumors expressing these receptors. Alternatively, 10-15% of all breast cancers do not express any of these receptors and are referred to as Triple Negative Breast Cancer (TNBC) [3]. Lacking tumor-specific molecular targets, TNBCs are treated via surgery, chemotherapy, and radiation but nevertheless have a worse prognosis compared to other subtypes of breast cancer [4].
Cyclooxygenase enzymes (COX-1 and COX-2) catalyze the rate-limiting step in the production of eicosanoids from arachidonic acid, an omega-6 fatty acid found in the cell membrane. The main eicosanoid product found in tumors is the inflammatory mediator prostaglandin E2 (PGE2) [5, 6]. Like other cancers of epithelial origin, aberrant expression of COX-2 is found in approximately half of all breast cancers, and elevated COX-2 and PGE2 levels are associated with a poor prognosis [5–10]. PGE2 is actively exported by multiple drug resistance-associated protein 4 (MRP4) into the extracellular space where it can bind one of four cognate, G-protein coupled receptors (EP1-4) and induce several signaling pathways [11–23]. Activation of EP2 and EP4 receptors has been associated with several contributing factors in tumor progression including angiogenesis, cellular proliferation, migration, invasion, metastasis, immune evasion, and support of a cancer stem-like cell phenotype [14–23]. MRP4 has been associated with a poor prognosis in tumors of the blood, brain, colon, liver, lung, pancreas, and prostate, but has only been briefly described in breast cancer [24–31]. Extracellular PGE2 can be imported through the prostaglandin transporter (PGT) via exchange with intracellular lactate and metabolized by 15-prostaglandin dehydrogenase (15-PGDH) [32–36]. 15-PGDH is a tumor suppressor gene in breast cancer as lack of sufficient expression of this enzyme can result in the accumulation of PGE2 thus leading to sustained PGE2 signaling [35, 37]. Metabolized PGE2 cannot bind EP receptors; therefore, PGT and 15-PGDH are both required to terminate PGE2-activated signaling [38].
Using large publicly available data sets, we recently reported that MRP4, PGT and 15-PGDH were differentially expressed among distinct breast cancer molecular subtypes [39]. In basal type breast cancer or TNBC, high COX-2, high MRP4, low PGT, and low 15-PGDH mRNA expression levels were observed. The current study examines the functional significance of this observation.
RESULTS
Based on our previous observations that primary breast cancers of molecularly defined subtypes differentially expressed members of the COX-2/PGE2 pathway at the mRNA level, we characterized a panel of breast cancer cell lines reflecting breast cancer subtypes to further test the hypothesis that aggressive, metastatic, and basal type breast cancers would display a pattern of COX-2 pathway expression leading to high PGE2 in the tumor microenvironment. MRP4 protein expression was elevated in the MCF10A (normal mammary epithelium), MDA-MB-231 (basal), MDA-MB-436 (basal), and BT549 (basal) cell lines compared to MCF7 (luminal) and MDA-MB-468 (basal) cells (Figure 1A). MRP4 was not detected in T47D (luminal) cells. Overall, elevated MRP4 expression was somewhat correlated with cell lines of aggressive molecular subtype (basal) compared to luminal malignant cell lines.
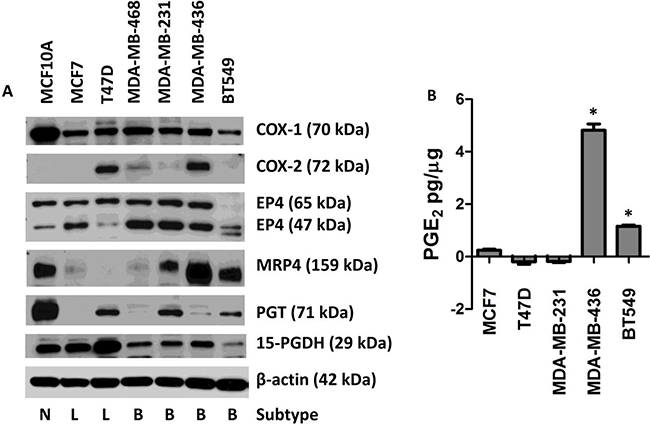
Figure 1: Expression of the COX-2/PGE2 pathway proteins in seven human breast cell lines leading to extracellular accumulation of PGE2. MCF10A cells were compared to six human breast cancer cell lines. A. Protein lysates from MCF10A, MCF7, T47D, MDA-MB-468, MDA-MB-231, MDA-MB-436, and BT549 cells were blotted for the following proteins: COX-1, COX-2, EP4, MRP4, PGT, and 15-PGDH. β-actin was used as a loading control. Breast cancer subtypes are indicated below the blot by Normal (N), Luminal (L), and Basal (B). B. Conditioned media was collected from sub-confluent cell culture and assayed for PGE2. Total PGE2 (pg) in the conditioned media was normalized to total cellular protein (μg). * p < 0.01 relative to the other four cell lines. PGE2 content is expressed as mean ± SEM of triplicate determinations.
MCF10A cells had the highest PGT protein expression (Figure 1A). T47D, MDA-MB-231, and BT549 cell lines displayed moderate levels of PGT while MCF7, MDA-MB-468, and MDA-MB-436 cell lines expressed either low or no detectable PGT. Expression of the PGE2-metabolizing enzyme, 15-PGDH, in MCF10A, MCF7, and T47D cells was higher than in the four basal cell lines (Figure 1A). The observed protein expression pattern for 15-PGDH is characteristic of a tumor suppressor gene [35, 37]. Consistent with a house-keeping role, COX-1 protein expression was detected in all cell lines (Figure 1A). COX-2 protein was detected in four of seven cell lines (Figure 1A).
The EP4 receptor has been detected at multiple sizes due to the extent of glycosylation of the protein; the apparent size ranging between 47 and 65 kDa. EP4 expression at the larger (65 kDa) isoform was detected at similar levels in all cell lines except BT549 which had barely detectable EP4 expression at this size. Higher expression of the 47 kDa isoform was detected in the basal cell lines compared to normal (MCF10A) and luminal cell lines (Figure 1A).
Taken together these studies reveal complex expression patterns of PGE2 family members in individual cell lines reflecting the marked heterogeneity of human breast cancer. In each cell line, a different combination of these proteins could lead to dysregulated levels of PGE2 in the tumor microenvironment; however, some general patterns have emerged. We saw that more aggressive cell lines generally express higher levels of MRP4 and reduced levels of PGT and 15-PGDH. The net result would be higher levels of PGE2 in the tumor microenvironment due to increased PGE2 export by MRP4 and decreased PGE2 import and metabolism via PGT and 15-PGDH, respectively. Conversely, less aggressive, luminal cell lines tended to have lower levels of MRP4, higher levels of PGT, and higher levels of 15-PGDH expression. These patterns are consistent with our previous report that primary basal type or TNBC breast cancers typically express high COX-2, high MRP4, low PGT, and low 15-PGDH mRNA, and also with our mRNA expression analysis of these cell lines (data not shown) [39].
Given these differential expression patterns of the PGE2 pathway members, we asked what impact these differences have on net PGE2 production by breast cancer cells. We analyzed PGE2 production in conditioned media via ELISA and saw that MDA-MB-436 cells produce high levels of PGE2 (4.82 pg/μg cellular protein) (Figure 1B). These cells express both COX-1 and COX-2 proteins along with elevated MRP4 and reduced PGT (Figure 1A). BT549 cells accumulate moderate levels of PGE2 (1.16 pg/μg cellular protein) in conditioned media (Figure 1B). These cells do not express high levels of COX-2; however, they express elevated levels of MRP4 while not expressing 15-PGDH that would metabolize any PGE2 produced from COX-1 (Figure 1A). PGT expression is also low in these cells so that extracellular PGE2 would not be imported efficiently and could accumulate in the conditioned media. PGE2 levels were low in the conditioned media of MCF7 cells (Figure 1B) consistent with the negligible expression of COX-2 and MRP4. Thus, PGE2 detected in the conditioned media would be dependent on COX-1 and export would be dependent on the low level of MRP4 or passive diffusion (Figure 1A). In addition, MCF7 cells express 15-PGDH which would suppress the amount of PGE2 detected in the conditioned media from these cells. T47D cells favor PGE2 metabolism as there was less PGE2 detected in the conditioned media compared to the unconditioned media (Figure 1B). The PGE2 detected in the unconditioned media likely came from the serum component of the growth media. Since T47D cells express PGT and high levels of 15-PGDH, they are poised to robustly metabolize PGE2, even though COX-2 is expressed (Figure 1A). These cells also lack MRP4 expression so PGE2 is not actively exported (Figure 1A). Likewise, a net decrease in PGE2 was also detected from the growth media of MDA-MB-231 cells (Figure 1B). This is consistent with the low endogenous COX-2 levels in these cells and moderate expression of MRP4, PGT, and 15-PGDH (Figure 1A). 15-PGDH expression in MDA-MB-436 cells is equal to other cells that do not accumulate PGE2 in the conditioned media, but since the ratio of MRP4-to-PGT is high, any PGE2 in the cells would be exported instead of being metabolized.
Since MRP4 exports PGE2 from a variety of cell types, elevated MRP4 expression in the tumor could be a mechanism by which malignant cells maintain elevated PGE2 in the tumor microenvironment [13, 25, 29–31, 40, 41]. In order to determine the role of MRP4 in PGE2 export from tumor cells, we used both genetic and pharmacologic approaches to perturb MRP4 activity and measured PGE2 accumulation in the conditioned media of two basal type cell lines. The MDA-MB-231 cell line expresses elevated levels of MRP4, and PGE2 production is inducible by inflammatory stimuli. The migratory and metastatic behaviors of MDA-MB-231 cells are partially dependent on PGE2 signaling [42]. The MDA-MB-436 cell line expresses high MRP4 and produces PGE2 without exogenous stimulation. MDA-MB-231 cells were treated with PMA (phorbol ester) to induce COX-2 and accumulation of PGE2 in the conditioned media [43]. We employed siRNA targeting of ABCC4 to reduce expression of MRP4 and examined the level of PGE2 in the conditioned media. Using two siRNA constructs, we achieved significant (35-75%) reduction in MRP4 expression relative to scramble control cells (Figure 2B). In MDA-MB-231 cells, stimulation with PMA (80 nM, 1 hr) resulted in a 25-fold increase in PGE2 accumulation in the conditioned media compared to unstimulated cells (Figure 2A). Fifty-four to sixty-six percent less PGE2 was exported from MDA-MB-231 cells when they were transfected with siRNA targeting ABCC4 and briefly stimulated with PMA compared to PMA-treated scramble siRNA control (p < 0.05). The PGE2 export role of MRP4 was confirmed in a second basal type cell line. Like MDA-MB-231 cells, MDA-MB-436 cells accumulated significantly less PGE2 in the conditioned media when MRP4 expression was reduced with siRNA (Figure 2C, 2D).
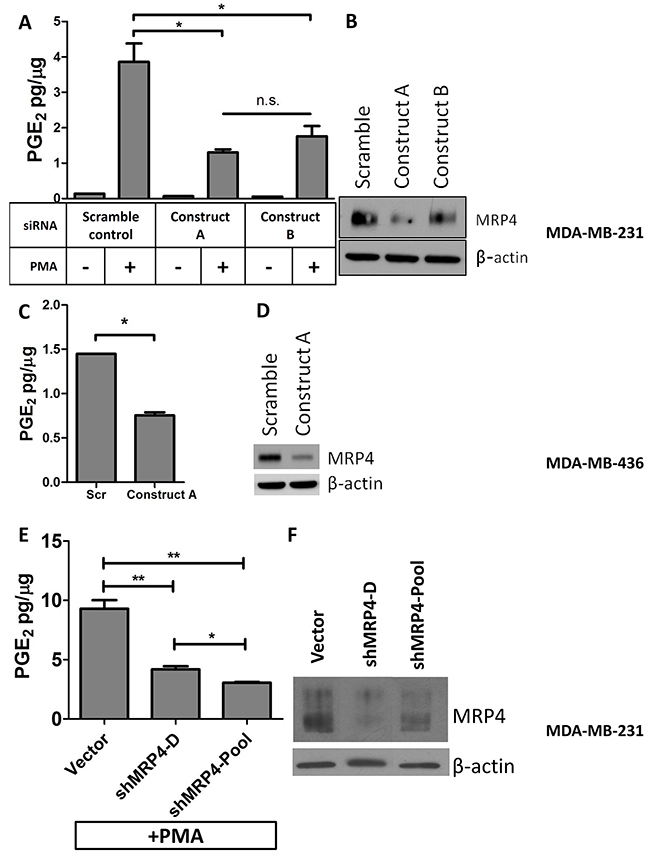
Figure 2: Knockdown of MRP4 in MDA-MB-231 and MDA-MB-436 cells suppresses the export of PGE2. MDA-MB-231 cells were transfected with MRP4 siRNA (3 nmol/L) for 24 hours before being stimulated with PMA (80 nmol/L, 1 hr) and replacing growth media. After 16 hours, conditioned media and total protein lysate was collected. A. Conditioned media from MRP4-silenced cells (MDA-MB-231) stimulated with PMA was assayed for PGE2 and total protein. B. A representative western blot of MDA-MB-231 cells transfected with siRNA (3 nmol/L) and stimulated with PMA shows decreased expression of MRP4. MDA-MB-436 cells were transfected with MRP4 siRNA (10 nmol/L). C. Conditioned media from MDA-MB-436 cells was collected after overnight incubation and assayed for PGE2 content. D. A representative western blot showing 68% decreased MRP4 expression following siRNA transfection of MDA-MB-436 cells. E. MDA-MB-231 vector control, clone (shMRP4-D), and pool (shMRP4-Pool) populations of stable MRP4 knockdown cells were briefly stimulated with 80 nM PMA and incubated overnight in fresh growth medium. Conditioned media was collected and assayed for PGE2. F. A representative western blot showing decreased MRP4 expression of MDA-MB-231 clone shMRP4-D and shMRP4-Pool cell lines compared to vector control cells. PGE2 is expressed as mean ± SEM pg/μg protein from triplicate determinations. β-actin was used as a loading control.* p < 0.05, ** p < 0.01, n.s. = not significant.
Stable MRP4 knockdown clones from MDA-MB-231 cells were also generated and characterized for decreased MRP4 expression. MRP4 protein expression was reduced by 62-67% relative to vector control (Figure 2E). MDA-MB-231 cells (one clone and one pooled population) with decreased MRP4 expression were briefly stimulated with PMA to induce COX-2 and to produce PGE2. As observed in transient MRP4 knockdown, cells with decreased MRP4 expression exported less PGE2 compared to vector control confirming the results obtained using transient gene silencing (Figure 2F).
Two pharmacologic inhibitors of MRP4 (Tyrphostin AG1478, MK571) were also employed to evaluate the role of MRP4 in PGE2 export. PMA-stimulated MDA-MB-231 cells or MDA-MB-436 cells were treated with inhibitor and evaluated for PGE2 accumulation in the conditioned media. Tyrphostin AG1478, a tyrosine kinase inhibitor with recently identified inhibitory activity on MRP4 [44], inhibited PGE2 accumulation in both cell lines in a dose-dependent manner (Figure 3A, 3C). In MDA-MB-231 cells, PGE2 export was decreased by 92%, 65%, and 36% in the presence of Tyrphostin AG1478 at 10, 5, and 2.5 μmol/L, respectively (p < 0.001). Likewise, PGE2 export by MDA-MB-436 cells was significantly decreased by 52%, 46%, and 14% in the presence of Tyrphostin AG1478 at 10, 5, and 2.5 μmol/L, respectively (p < 0.01). Treatment with 50 μM, but not 25 μM, MK571 resulted in a reduction in the level of PGE2 in the conditioned media of both MDA-MB-231 and MDA-MB-436 cells when compared to vehicle control (Figure 3B, 3D).
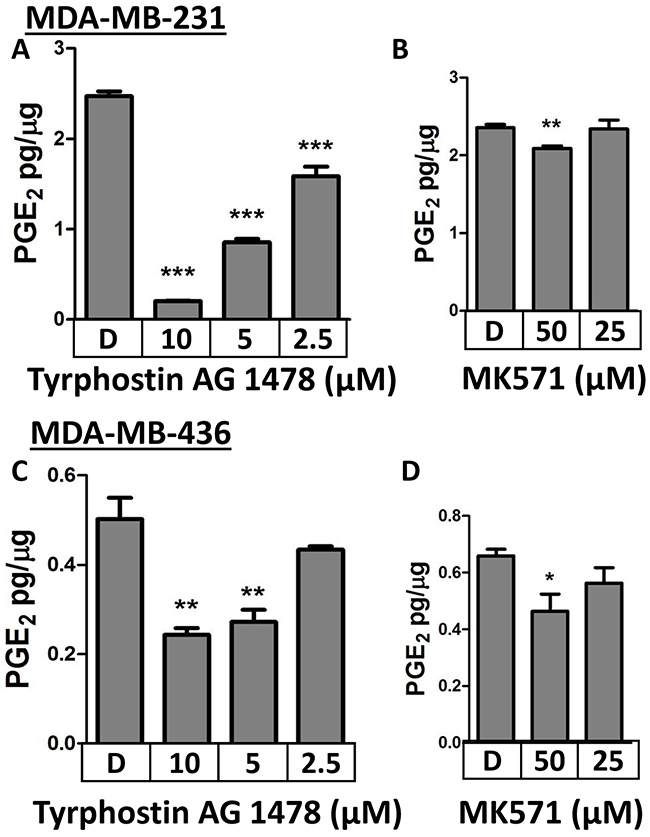
Figure 3: Pharmacologic inhibition of MRP4 with Tyrphostin AG1478 or MK571 suppresses PGE2 export. A. MDA-MB-231 cells were stimulated briefly with 80 nM PMA before the media was replaced with the indicated concentrations of A. Tyrphostin, B. MK571, or DMSO. MDA-MB-436 cells were treated with the indicated concentrations of C. Tyrphostin D. MK571, or DMSO. PGE2 accumulation in the conditioned media after 18 hours was quantified by enzyme immunoassay. PGE2 is expressed as mean ± SEM pg/μg protein from triplicate determinations. * p < 0.05, ** p < 0.01*** p < 0.001 relative to DMSO.
Suppression or inhibition of MRP4 decreases the export of PGE2 from basal or TNBC breast cancer cells. Conversely, MCF7 cells were used for MRP4 over-expression experiments due to the low level of endogenous MRP4 expression as well as being representative of an estrogen and progesterone receptor positive, luminal subtype cell line. MCF7 cells were transfected to express either empty vector control or an MRP4 expression plasmid (pcDNA3.1(-)-MRP4-Zeo) and two lines were selected (MCF7-MRP4-2 and MCF7-MRP4-3) along with a control line stably expressing empty vector (MCF7-Vec). MRP4 expression in MCF7-MRP4-2 and MCF7-MRP4-3 cells was 70-100-fold higher than in MCF7-Vec cells (Figure 4A). The ectopically expressed MRP4 protein was determined to be the correct size via western blot suggesting that post-translational modifications, such as glycosylation, were properly applied. Compared to MDA-MB-436 cells, MCF7-Vec cells accumulate less PGE2 overall, reflecting the lower endogenous COX-2 levels observed in these cells (Figure 1 and Figure 4). PGE2 release is modestly increased by stimulation with lipopolysaccharide (LPS, 10 ug/mL, 24 hours), but this increase was not statistically significant and was not further enhanced by over-expression of MRP4 in these cells (Figure 4B). This suggests that the combination of COX-2 expression and enforced MRP4 expression in MCF7 cells is not sufficient to accumulate PGE2 in the conditioned media.
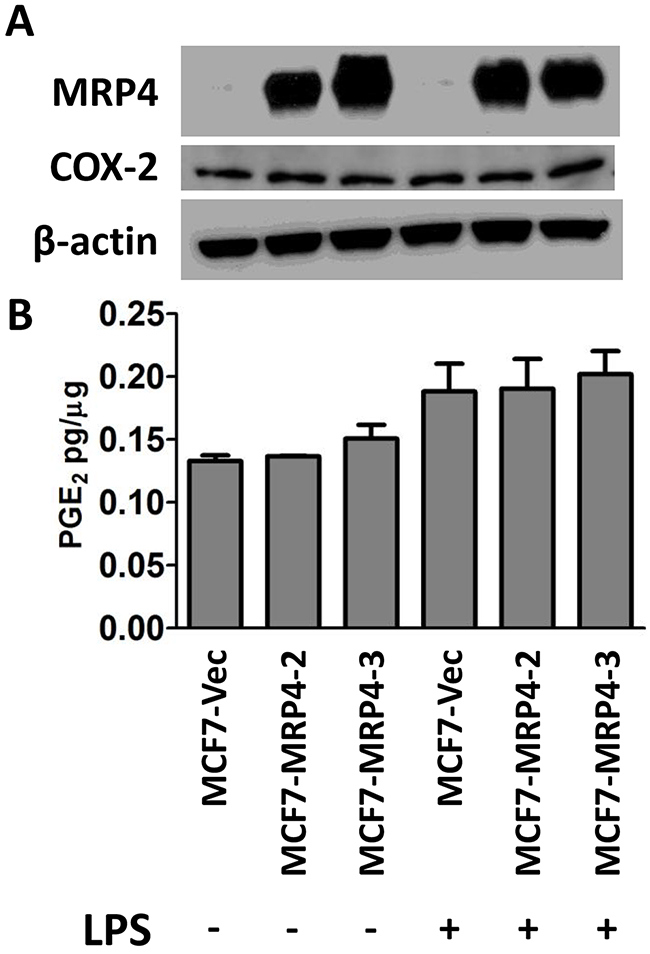
Figure 4: Stable over-expression of MRP4 does not enhance PGE2 export from MCF7 cells. A. Vector-expressing control cells (MCF7-Vec) or MRP4-expressing cells (MCF7-MRP4-2, MCF7-MRP4-3) were stimulated with 10 μg/mL LPS for 24 hours. Total protein lysate was collected for western blot to determine relative levels of MRP4 and COX-2 expression. β-actin was used as a loading control. B. Conditioned media was collected and assayed for PGE2. PGE2 is expressed as mean ± SEM pg/μg protein from triplicate determinations. Western blot lanes are vertically aligned with corresponding PGE2 levels.
In order to confirm that these genetic (RNA-interference and over-expression) and pharmacologic approaches were specifically affecting MRP4 functions, we employed a drug resistance assay to determine the effect of these perturbations on the sensitivity of these cells to the cytotoxic drug 6-mercaptopurine (6-MP), a known substrate for MRP4 [45]. In the instance of high MRP4 expression and activity, 6-MP does not accumulate in the cell to the degree that causes cytotoxicity and apoptosis; this reduced cytotoxicity is expressed as an increase in IC50.
MRP4 expression was suppressed via siRNA or shRNA in MDA-MB-231, MDA-MB-436, and BT549 cells, and sensitivity to 6-MP was determined and expressed as the IC50. The ratio of the IC50 of cells transfected with siRNA against ABCC4 compared to the IC50 of control transfected cells was calculated and this ratio is expressed as “fold sensitization.” In MDA-MB-231 cells (moderate basal MRP4 expression), reducing the level of MRP4 by either siRNA or shRNA resulted in an approximate 2-fold increased sensitivity to 6-MP (Table 1). Clones MDA-MB-231 shMRP4-B and MDA-MB-231 shMRP4-C had an approximate 80% decrease in MRP4 expression relative to vector control (data not shown). In MDA-MB-436 and BT549 cells (high basal MRP4 expression), reducing the expression of MRP4 using siRNA resulted in a 1.43–1.74-fold increase in cytotoxicity mediated by 6-MP. These data are consistent with a mechanism by which reduced MRP4 expression leads to reduced export of 6-MP resulting in higher accumulation of 6-MP and enhanced cell killing. Conversely, MCF7 cells that stably over-express MRP4 (MCF7-MRP4-2, MCF7-MRP4-3) were approximately 2-fold more resistant to 6-MP (higher IC50) when compared to MCF7 cells expressing empty vector, consistent with increased export of 6-MP (Table 1). Consistent with the genetic data, MRP4 inhibition with MK571 or Tyrphostin AG 1478 also increased the sensitivity to 6-MP in the three MRP4-expressing basal cell lines. Table 2 summarizes the fold sensitization as a result of treatment with MK571 or Tyrphostin AG 1478. Inhibitor concentrations were selected such that inhibitor treated cells had equivalent viability to vehicle-treated cells after 3 days. Pharmacologic inhibition of MRP4 by MK571 resulted in a 1.47, 1.76, and 2.94-fold increase in sensitivity to 6-MP in MDA-MB-436, BT549, and MDA-MB-231 cells, respectively. Tyrphostin AG 1478 (10 μM) induced a 2.86- and 4.83-fold increase in sensitivity to 6-MP in MDA-MB-436 and BT549 cells, respectively. The modulation of resistance to 6-MP by targeting MRP4 shows that these perturbations specifically affect MRP4 and not the entire PGE2 pathway.
Table 1: Genetic suppression of MRP4 increases sensitivity to 6-MP while ectopic over-expression of MRP4 increases resistance to 6-MP
Genetic Treatment |
Fold Sensitizationa |
||
|---|---|---|---|
MDA-MB-231 |
|||
siRNA |
Construct A |
2.25 |
** |
Construct B |
2.22 |
* |
|
shRNA |
Clone 2 |
2.22 |
* |
Clone 4 |
1.97 |
* |
|
MDA-MB-436 |
|||
siRNA |
Construct A |
1.43 |
* |
Construct C |
1.63 |
* |
|
BT549 |
|||
siRNA |
Construct A |
1.60 |
** |
Construct C |
1.74 |
* |
|
MCF7 |
Fold Resistanceb |
||
MRP4 stable overexpression |
Sub-line M2 |
1.93 |
** |
Sub-line M3 |
2.12 |
* |
|
aSensitization by RNA-interference relative to control transfection
bResistance by MCF7 cells expressing MRP4 relative to vector control
* p < 0.05, ** p < 0.01
Table 2: Pharmacologic inhibition of MRP4 increases sensitivity to 6-MP
Pharmacologic Treatment |
μM |
Fold Sensitizationa |
|
|---|---|---|---|
MDA-MB-231 |
|||
MK571 |
50 |
2.94 |
* |
MDA-MB-436 |
|||
MK571 |
25 |
1.47 |
* |
Tyrphostin AG 1478 |
10 |
2.86 |
** |
BT549 |
|||
MK571 |
50 |
1.76 |
** |
Tyrphostin AG 1478 |
10 |
4.83 |
** |
aSensitization by MRP4 inhibitors relative to vehicle treatment
* p < 0.05, ** p < 0.01
Independent of MRP4 expression level, treatment of MDA-MB-231 cells with a range of exogenous PGE2 did not significantly affect proliferation of these cells (data not shown). Additionally, we saw no significant change in proliferation when MRP4 protein levels were altered genetically (data not shown).
Since elevated COX-2 and PGE2 levels are indicators of poor prognosis in several types of cancer including breast, and we have shown that MRP4 contributes to the amount of PGE2 produced by breast cancer cells, we investigated the role of MRP4 on both primary tumor growth and metastatic potential. MDA-MB-231 cells (5×105) stably expressing vector control or shRNA targeting MRP4 along with stable expression of the luciferase enzyme (MDA-MB-231 shRNA/Luc, Figure 5A) were implanted subcutaneously proximal to the mammary fat pad of female BALB/c SCID mice; tumor growth was monitored by caliper, and metastatic spread was estimated by bioluminescent imaging [46]. At day +90, 9 of 10 mice injected with either MDA-MB-231 shVec/Luc or MDA-MB-231 shMRP4-C/Luc had developed subcutaneous tumors. All ten mice injected with MDA-MB-231 shMRP4-D/Luc cells had established subcutaneous tumors. Mean tumor volumes were not different among mice injected with control versus either MRP4 knockdown cell line (Figure 5B). Upon reaching 18 mm in longest tumor diameter, mice were euthanized; blood, lung, and tumor tissues were collected for analysis. Tumor weight upon necropsy was used as a confirmatory measure of tumor growth. Consistent with the estimated tumor volumes, excised tumor weight was not significantly different among the three cell lines expressing different levels of MRP4 (Figure 5C). We observed a similar expression pattern of ABCC4 mRNA among the tumor tissues generated from the knockdown cell lines (Figure 5D) compared to the protein expression of the primary cell lines used for injection (Figure 5A). Tumors derived from MDA-MB-231 shVec/Luc cells had higher ABCC4 mRNA expression compared to tumors derived from MDA-MB-231 shMRP4-C/Luc or MDA-MB-231 shMRP4-D/Luc MRP4 knockdown clones indicating that the tumor MRP4 phenotype at the end of the observation period reflected the phenotype of the initial implanted tumor cells. Based on these observations, we conclude that the absence of any effect of MRP4 downregulation on primary tumor growth is not due to upregulation of MRP4 in vivo.
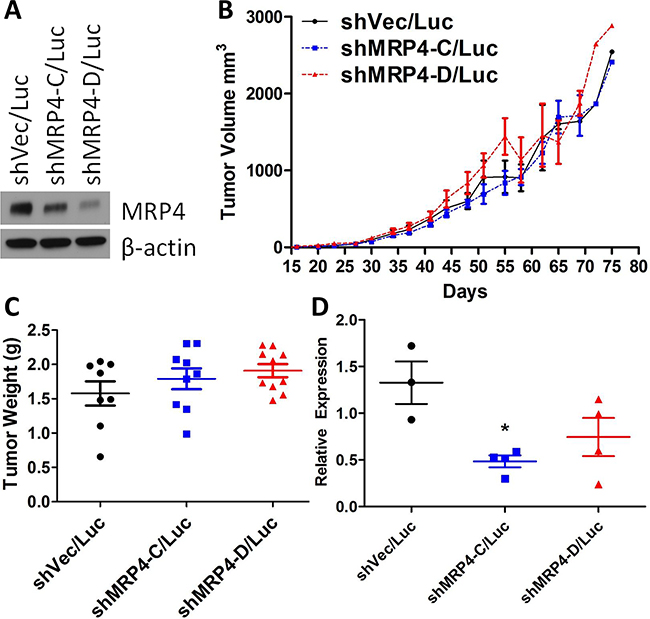
Figure 5: Subcutaneous xenografts of MDA-MB-231 shMRP4/Luc cells showed no difference in growth despite differences in MRP4 expression. A. Western blot showing relative MRP4 expression levels of vector control (shVec/Luc) and shRNA knockdown clones (shMRP4-C/Luc and shMRP4-D/Luc) prior to injection in mice. β-actin was used as a loading control. B. Tumor volume (mean ± SEM) estimated from diameter measurements of subcutaneous tumors. Volume = (long diam.)(perpendicular diam.)2(π/6). C. Weight of excised subcutaneous MDA-MB-231 shRNA/Luc tumors. D. Relative expression of ABCC4 mRNA from excised MDA-MB-231 shMRP4/Luc xenograft tumors. Each point represents one tumor sample. * p = 0.0093 compared to MDA-MB-231 shVec/Luc tumor samples.
Whole-animal bioluminescence imaging was monitored over the course of tumor growth in order to detect spontaneous metastases from the mammary gland-implanted tumor. We detected markedly more bioluminescence in the lungs of mice injected with vector control MDA-MB-231 shVec/Luc cells compared to the mice injected with either of two MRP4 knockdown clones (MDA-MB-231 shMRP4-C/Luc and MDA-MB-231 shMRP4-D/Luc) (Figure 6A). Thus, differences in MRP4 expression did not impact the growth of the primary tumor, but had a profound effect on the ability of tumor cells to establish spontaneous pulmonary metastases. Bioluminescence in the lungs of mice injected with shVec/Luc cells increased over time, consistent with the continuing expansion of metastatic lesions whereas very little, if any, growth of either cell line expressing shMRP4 occurred during the observation period. The reduced metastatic potential in MRP4 knockdown cells was confirmed in a second independent experiment (data not shown). In order to confirm the presence of human breast cancer cells in the lung tissue of these mice, total RNA was isolated from lung tissue and evaluated by qPCR for the presence of human GAPDH (using human-specific GAPDH primer set) compared to mouse Gapdh mRNA. When the average relative expression of GAPDH/Gapdh detected in the MDA-MB-231 shVec/Luc samples was set equal to 1, lower levels of GAPDH/Gapdh were observed in the lungs of mice bearing tumors with suppressed MRP4 expression (MDA-MB-231 shMRP4-C/Luc, MDA-MB-231 shMRP4-D/Luc) (Figure 6B).
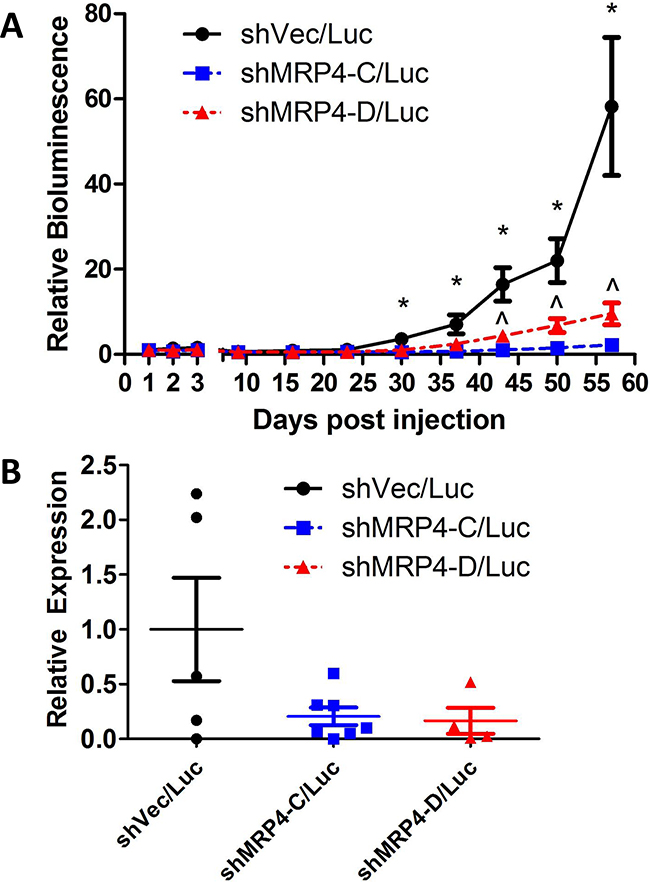
Figure 6: Suppressed expression of MRP4 decreased metastatic load in mice bearing subcutaneous MDA-MB-231 shMRP4/Luc tumors. Weekly pulmonary region bioluminescence readings were taken from 9-10 mice/group with established subcutaneous MDA-MB-231 shMRP4/Luc tumors. A. Bioluminescence signal was corrected for background luminescence and mean ± SEM of 9-10 mice per group is reported relative to 1 day post-injection pulmonary bioluminescence. The following symbols represent the results from t-tests performed at each timepoint. *: p < 0.05 between vector and knockdown clones (shVec/Luc vs. shMRP4-C/Luc, shVec/Luc vs. shMRP4-D/Luc), ^: p < 0.05 between knockdown clones (shMRP4-C/Luc vs. shMRP4-D/Luc). B. Relative expression of human GAPDH relative to mouse Gapdh in lung samples from mice that bore the indicated tumor xenografts.
DISCUSSION
We recently reported the analysis of gene expression of COX-2 pathway members in a large publicly available breast cancer database [39]. That study identified a pattern of expression in TNBC and basal type breast cancer which should result in high PGE2 in the tumor microenvironment i.e., high COX-2, high MRP4, low PGT, and low 15-PGDH. No biochemical data was available in that dataset which prompted the current study to characterize the functional importance of this pattern. The current study further supports our central hypothesis that the COX-2/PGE2 pathway is dysregulated in aggressive cancers [7, 8, 47]. We have now shown that breast cancer subtypes have different expression profiles of the PGE2 pathway members and that MRP4 expression was strongly elevated in aggressive (IDC, TNBC, basal type) breast cancer cell lines relative to less aggressive, luminal cell lines. Given that MRP4 is also expressed in normal epithelium, it was not surprising that we detected this protein in MCF10A cells; however, in malignant cell lines, MRP4 expression correlated with an aggressive phenotype [13, 40]. We also saw a trend of decreased 15-PGDH protein expression in aggressive breast cancer cell lines relative to normal mammary epithelium consistent with the reported tumor suppressor role of 15-PGDH [35, 37]. These data support the hypothesis that other members of the PGE2 pathway besides COX-2, in particular MRP4, PGT, and 15-PGDH, may have an impact on the intratumoral levels of PGE2 [5, 7, 9, 34, 38, 48].
Previously unknown, we have now shown that breast cancer cells with elevated MRP4 expression would be able to more efficiently export PGE2 into the microenvironment. A tumor microenvironment with elevated PGE2 could be established and sustained when MRP4 expression is elevated even in tumors in which COX-2 expression is not elevated, and this identifies a second potential mechanism, beyond COX-2, to achieve high PGE2 in the tumor microenvironment. This model of high-MRP4 expression also assumes low expression of PGT and 15-PGDH which would otherwise metabolize intratumoral PGE2. We demonstrate that, in TNBC particularly, MRP4 could also play a role in maintaining a tumor microenvironment with elevated PGE2 levels, a clinical parameter that has been associated with poor outcome [5, 15, 49, 50]. Notably, MRP4 but not MRP1-3 or MRP5 transports PGE2 (and PGE1) [51]. In addition to PGE2, MRP4 exports nucleoside analogs, e.g. cAMP and cGMP, which could contribute to malignant behavior. While previous findings from our group and others suggest that metastatic potential downstream of PGE2/EP4 signaling is not strictly dependent on cAMP [16, 19, 52], we cannot rule out a role of nucleoside transport by MRP4. Future studies will examine these additional potential mechanisms by which MRP4 promotes metastasis.
We have now shown that suppressing MRP4 expression leads to decreased metastatic potential. We showed previously that PGE2 activates EP4 to support breast cancer metastasis [16, 53]. Our current studies support the hypothesis that elevated MRP4 could be a mechanism underlying high PGE2/EP4 signaling and this mechanism may be particularly important in basal and TNBC. There is extensive literature demonstrating that high PGE2 is associated with enhanced metastatic potential [5, 47, 54]. Consistent with this mechanism, we observed reduced metastatic capacity from mammary gland implanted tumors with reduced MRP4 expression. Growth of primary tumors was not apparently affected by changes in MRP4 levels.
PGE2 elicits diverse biological responses in tumor cells and heterogeneous tumor tissue in a cell type-specific manner [42]. PGE2 signaling is not directly associated with proliferation of MDA-MB-231 breast tumor cells. Increased MRP4 expression has been correlated with increased proliferation in other tumor types but this was not linked mechanistically to PGE2 production [24, 27, 28, 31]. Reducing expression of MRP4 in either MDA-MB-436 or MDA-MB-231 cells did not affect cell proliferation in vitro. We have shown that MRP4 contributes to PGE2 accumulation in vitro and metastatic progression in vivo, but this does not definitively exclude the possibility that another substrate of MRP4 besides PGE2 is also contributing to these effects. MDA-MB-436 cells express both COX-1 and COX-2, and PGE2 content in the conditioned media of these cells is influenced by the activity of MRP4. Consistent with the proliferation data, we did not see differences in primary tumor growth with respect to MRP4 expression level in MDA-MB-231 xenograft experiments; we further conclude that MRP4 does not directly affect cell proliferation. These data are consistent with the reports that PGE2 is not directly associated with proliferation in all breast cancer [42].
Taken together, we show that suppression of MRP4 expression in a metastatic, basal type breast cancer cell line (MDA-MB-231) decreased the ability of a subcutaneous primary tumor to develop spontaneous metastases when compared to MDA-MB-231 cells with high endogenous levels of MRP4. Our findings are consistent with our central hypothesis that MRP4 is functioning in the tumor microenvironment of the established primary tumor by increasing the level of PGE2 which can act in an autocrine or paracrine manner and is available to diverse cells in the heterogeneous tumor, enhancing metastatic potential and progression of the tumor. MRP4 could be a contributing factor in the accumulation of PGE2 in the tumor microenvironment preferentially in TNBC and basal subtype tumors compared to luminal breast tumors and should be considered as a potential molecular target in this subtype with overall poor outcomes.
MATERIALS AND METHODS
Human cell lines originally obtained from ATCC (Manassas, VA) were cultured in a 5% CO2 atmosphere at 37°C. MCF10A cells were maintained in DMEM/F12 (Corning, Corning, NY) supplemented with 5% horse serum, 20 ng/ml epidermal growth factor, 10 μg/ml insulin, 1 ng/ml cholera toxin, 100 μg/ml hydrocortisone, 1% penicillin and 1% streptomycin. The following breast cancer cell line media (Corning) was supplemented with 10% fetal bovine serum (FBS) (Gemini Bio-products, Atlanta Biologicals), 1% penicillin/streptomycin (Gemini Bio-products), and 2 mM L-glutamine (Gemini Bio-products). MCF7, MDA-MB-231, and MDA-MB-468 cells were grown in Dulbecco’s Modified Eagle Medium (DMEM) with 1 g/L glucose. T47D cells were grown in RPMI 1640 medium. MDA-MB-436 cells were grown in DMEM/F12 medium. BT549 cells were grown in DMEM with 4.5 g/L glucose. Cell lines were recently (June, 2016) authenticated by STR typing using the Promega Geneprint 10 system in comparison to ATCC STR databases. At the same time, the GenePrint 5x mouse primer pair mix was used to rule out contamination with mouse DNA.
Control and shRNA plasmids (Origene) targeting ABCC4 were transfected into retroviral packaging phoenix cells (Allele Biotechnology, San Diego, CA) with Lipofectamine 2000 (Thermo Fisher). Forty-eight hours after transfection, retrovirus-containing medium was collected and centrifuged to pellet any cell debris. The supernatant containing the viral particles was transferred to new tubes and stored at -80°C. Viral medium was added with 4 μg/mL polybrene (American Bioanalytical) to MDA-MB-231 cells for 2 days before being passaged into 1.0 μg/mL puromycin (Invitrogen) selection. After 1 week of selection, surviving cells were characterized by quantitative PCR and western blot for ABCC4/MRP4 knockdown compared to vector control cells, and the pooled populations with the greatest knockdown were subcloned for further characterization. Clones derived from puromycin-resistant single-cell cultures with stable MRP4 knockdown were cultured with viral media containing a luciferase expression plasmid for 3 days before being passaged into hygromycin B selection (Corning). After 2 weeks, resistant cells were characterized for luciferase expression by adding luciferin (PerkinElmer) (250 μg/mL) to the cells and evaluating bioluminescent intensity via luminometer (Berthold Technologies, U.S.A., Oak Ridge, TN). Signal 200-fold over background was considered sufficient for use in in vivo experiments.
MRP4 over-expression was performed both transiently and stably. Transient transfection of MCF7 cells with the MRP4 expression plasmid pcDNA3.1(-)MRP4-Zeo (a generous gift from H. Hayashi, University of Tokyo) or pcDNA3.1(+)-Zeo (a generous gift from I. Lindberg, University of Maryland) empty vector was conducted using Lipofectamine 3000 (Thermo Fisher) at a ratio of 2 μg DNA to 3 μL Lipofectamine 3000. Stable MRP4 expressing MCF7 sub-lines were generated similar to the transient cells, but with the addition of Zeocin (100 μg/mL) (Thermo Fisher) to growth media. Cells were passaged every 3-4 days with fresh Zeocin. After 3 weeks, surviving cells were characterized by western blot for relative MRP4 expression. Two cell lines expressing MRP4 (MCF7-MRP4-2 and MCF7-MRP4-3) and one cell line expressing vector (MCF7-Vec) were used to evaluate PGE2 accumulation and 6-MP resistance.
RNA, cDNA, qPCR
Total RNA was isolated from cultured cells using the NucleoSpin RNA kit (Machery-Nagel) according to the manufacturer’s instructions. Isolation of RNA for siRNA screening was performed using the DirectZol RNA isolation kit (Zymo). Total RNA from mouse tissue was isolated using TRIzol following the manufacturer’s protocol (Thermo Fisher Scientific). cDNA was synthesized from 500-1000 ng total RNA using the qScript cDNA SuperMix (Quanta) according to the manufacturer’s instructions. ABCC4 and GAPDH expression were performed in triplicate using probe-based primer sets and iQ Supermix (Bio-Rad) with approximately 100 ng cDNA per reaction. Relative gene expression was determined using the 2-ΔΔCt method with GAPDH as the reference gene. Results are representative of replicate experiments and expressed as relative expression ± standard deviation [55].
Protein isolation
Total cellular protein was collected from cultured cells following a wash with cold phosphate buffered saline (PBS). Lysis buffer was comprised of RIPA buffer (Sigma-Aldrich) supplemented with 1% protease inhibitor (Sigma-Aldrich), 1% phenylmethylsulfonyl fluoride (PMSF, Sigma-Aldrich), sodium orthovanadate (2 mM, Sigma-Aldrich), and sodium fluoride (5 mM, New England Biolabs). Lysis buffer was added to adherent cells and incubated on ice for 10 minutes. Alternatively, cells were detached using trypsin, resuspended in growth media, and centrifuged. The cell pellet was resuspended in lysis buffer. Lysates were vortexed 2-3 times over 20 minutes and otherwise kept on ice. Lysates were clarified by centrifugation at 8000 x g for 10 minutes at 4°C. Clarified, soluble protein was transferred to a new tube and stored at -80°C. Protein concentration of these clarified lysates was determined by the Bradford protein quantification assay (Thermo Fisher Scientific).
Western immunoblotting
Equal amounts of protein (20-50 μg) were combined with 4x Laemmli sample buffer and β-mercaptoethanol (2.5% final) (Bio-Rad) and incubated at 95°C for 5 minutes before being loaded into SDS-PAGE gels for electrophoresis. Separated proteins were transferred to PVDF membrane using the Trans-Blot Turbo system (Bio-Rad) and blocked in 5% milk in wash buffer (phosphate buffered saline plus 0.1% Tween-20, PBS-T). Membranes were incubated overnight at 4°C with primary antibodies under gentle rocking. Membranes were washed and incubated for 1 hour with secondary antibodies at room temperature under gentle rocking. Membranes were incubated in an ECL (Pierce, Bio-Rad, or GE) reagent for 5 minutes and exposed to x-ray film to obtain the relative protein expression. Primary antibodies against COX-2, EP4, 15-PGDH, and PGT were from Cayman Chemical (Ann Arbor, MI). Primary antibodies against COX-2 and COX-1 were from Cell Signaling Technologies (CST). Primary antibody against MRP4 was from Enzo Life Sciences (M4I-10). The primary antibody against beta-actin (AC-15) was from Sigma-Aldrich. Milk (5%) in PBS-T was used for diluting primary and secondary antibodies. Horseradish Peroxidase (HRP)-conjugated secondary antibodies were used in the following concentration ranges: anti-rabbit (Bio-Rad) 1:5,000, anti-mouse (KPL) 1:5,000 – 1:10,000, and anti-rat (CST) 1:3,000 – 1:5,000.
PGE2 determination assay
PGE2 levels in conditioned media were determined using the Prostaglandin E2 EIA kit (Cayman Chemical) according to the manufacturer’s protocol. For normalization, PGE2 content is expressed in pg/μg cellular protein. When using MDA-MB-231 cells for PGE2 export experiments, cells were stimulated with 80 nM PMA (Sigma-Aldrich) in fresh growth medium for 1 hour at 37°C. Stimulation media was removed and replaced with fresh growth media for 16 hours at 37°C. The conditioned medium and whole cell lysate were collected and stored at -80°C. MRP4 inhibitors were added to both stimulation and replacement media when indicated. MCF7 cells were stimulated with 10 μg/mL lipopolysaccharide (LPS, Sigma-Aldrich) for 24 hours and conditioned medium was collected along with total cellular protein.
Chemicals
MK571 (Cayman, Calbiochem) was dissolved to 15 mM in DMSO, aliquoted, and stored at -20°C. Tyrphostin AG 1478 (Sigma-Aldrich) was dissolved to 10 mM in DMSO and stored at 4°C. 6-Mercaptopurine (6-MP, Sigma-Aldrich) was dissolved to 250 mM in 1 M NaOH (Sigma-Aldrich) and stored at -20°C for up to 2 weeks. A 4-fold dilution series of 6-MP was prepared in 1 M NaOH and added to growth media so that the maximal final NaOH concentration was 4 mM. There were no proliferative changes between cells cultured in growth media alone or in growth media with 4 mM NaOH. Luciferin (PerkinElmer) was dissolved in sterile PBS to 40 mg/mL and stored, protected from light, at -20°C.
Human breast cancer xenograft
MDA-MB-231 cells (5×105) stably expressing both shRNA targeting ABCC4 and luciferase were injected subcutaneously into female BALB/c-SCID mice (Jackson). The MDA-MB-231 cell line was selected for these xenograft experiments as it reliably established tumors and spontaneous metastatic lesions. Tumor growth was measured twice weekly and approximate tumor volume was calculated by “Volume = (A)(B2)(π/6),” where A = long diameter and B = perpendicular diameter. All animal experiments were conducted in compliance with University of Maryland Institutional Animal Care and Use Committee.
Statistical analysis
The Student’s t-test was used to compare two experimental groups. A p-value < 0.05 was considered to be significant (*). 1-way ANOVA was used to analyze multiple experimental conditions, and a Bonferroni post-test was used to determine the significance of each pair of conditions tested. Results are representative of at least 3 experiments.
ACKNOWLEDGMENTS
We thank Dr. Hisamitsu Hayashi (University of Tokyo) for generously providing the MRP4 expression plasmid.
CONFLICTS OF INTEREST
Authors have no conflicts of interest to disclose.
GRANT SUPPORT
This work was supported by a Veterans Affairs Merit Award (AMF).
REFERENCES
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015; 65: 5–29. doi: 10.3322/caac.21254.
2. Onitilo AA, Engel JM, Greenlee RT, Mukesh BN. Breast Cancer Subtypes Based on ER/PR and Her2 Expression: Comparison of Clinicopathologic Features and Survival. Clin Med Res. 2009; 7: 4–13. doi: 10.3121/cmr.2009.825.
3. Bauer KR, Brown M, Cress RD, Parise CA, Caggiano V. Descriptive analysis of estrogen receptor (ER)-negative, progesterone receptor (PR)-negative, and HER2-negative invasive breast cancer, the so-called triple-negative phenotype. Cancer. 2007; 109: 1721–8. doi: 10.1002/cncr.22618.
4. Rakha EA, Ellis IO. Triple-negative/basal-like breast cancer: review. Pathology (Phila). 2009; 41: 40–7. doi: 10.1080/00313020802563510.
5. Ristimäki A, Sivula A, Lundin J, Lundin M, Salminen T, Haglund C, Joensuu H, Isola J. Prognostic significance of elevated cyclooxygenase-2 expression in breast cancer. Cancer Res. 2002; 62: 632–5.
6. Howe LR. Inflammation and breast cancer. Cyclooxygenase/prostaglandin signaling and breast cancer. Breast Cancer Res. 2007; 9: 210. doi: 10.1186/bcr1678.
7. Parrett M, Harris R, Joarder F, Ross M, Clausen K, Robertson F. Cyclooxygenase-2 gene expression in human breast cancer. Int J Oncol. 1997; 10: 503–7.
8. Soslow RA, Dannenberg AJ, Rush D, Woerner BM, Khan KN, Masferrer J, Koki AT. COX-2 is expressed in human pulmonary, colonic, and mammary tumors. Cancer. 2000; 89: 2637–45.
9. Denkert C, Winzer K-J, Müller B-M, Weichert W, Pest S, Köbel M, Kristiansen G, Reles A, Siegert A, Guski H, Hauptmann S. Elevated expression of cyclooxygenase-2 is a negative prognostic factor for disease free survival and overall survival in patients with breast carcinoma. Cancer. 2003; 97: 2978–87. doi: 10.1002/cncr.11437.
10. Shim JY, An HJ, Lee YH, Kim SK, Lee KP, Lee KS. Overexpression of cyclooxygenase-2 is associated with breast carcinoma and its poor prognostic factors. Mod Pathol Off J U S Can Acad Pathol Inc. 2003; 16: 1199–204. doi: 10.1097/01.MP.0000097372.73582.CB.
11. Furugen A, Yamaguchi H, Tanaka N, Shiida N, Ogura J, Kobayashi M, Iseki K. Contribution of multidrug resistance-associated proteins (MRPs) to the release of prostanoids from A549 cells. Prostaglandins Other Lipid Mediat. 2013; 106: 37–44. doi: 10.1016/j.prostaglandins.2013.08.002.
12. Tanaka N, Yamaguchi H, Mano N. Transport of eicosapentaenoic acid-derived PGE3, PGF(3α), and TXB3 by ABCC4. PloS One. 2014; 9: e109270. doi: 10.1371/journal.pone.0109270.
13. Russel FGM, Koenderink JB, Masereeuw R. Multidrug resistance protein 4 (MRP4/ABCC4): a versatile efflux transporter for drugs and signalling molecules. Trends Pharmacol Sci. 2008; 29: 200–7. doi: 10.1016/j.tips.2008.01.006.
14. Reader J, Holt D, Fulton A. Prostaglandin E2 EP receptors as therapeutic targets in breast cancer. Cancer Metastasis Rev. 2011; 30: 449–63. doi: 10.1007/s10555-011-9303-2.
15. Fulton AM, Ma X, Kundu N. Targeting prostaglandin E EP receptors to inhibit metastasis. Cancer Res. 2006; 66: 9794–7. doi: 10.1158/0008-5472.CAN-06-2067.
16. Kundu N, Ma X, Holt D, Goloubeva O, Ostrand-Rosenberg S, Fulton AM. Antagonism of the prostaglandin E receptor EP4 inhibits metastasis and enhances NK function. Breast Cancer Res Treat. 2009; 117: 235–42. doi: 10.1007/s10549-008-0180-5.
17. Kundu N, Ma X, Kochel T, Goloubeva O, Staats P, Thompson K, Martin S, Reader J, Take Y, Collin P, Fulton A. Prostaglandin E receptor EP4 is a therapeutic target in breast cancer cells with stem-like properties. Breast Cancer Res Treat. 2013; 143: 19–31. doi: 10.1007/s10549-013-2779-4.
18. Wang X, Klein RD. Prostaglandin E2 induces vascular endothelial growth factor secretion in prostate cancer cells through EP2 receptor-mediated cAMP pathway. Mol Carcinog. 2007; 46: 912–23. doi: 10.1002/mc.20320.
19. Kim JI, Lakshmikanthan V, Frilot N, Daaka Y. Prostaglandin E2 promotes lung cancer cell migration via EP4-betaArrestin1-c-Src signalsome. Mol Cancer Res MCR. 2010; 8: 569–77. doi: 10.1158/1541-7786.MCR-09-0511.
20. Zhang Y, Daaka Y. PGE2 promotes angiogenesis through EP4 and PKA Cγ pathway. Blood. 2011; 118: 5355–64. doi: 10.1182/blood-2011-04-350587.
21. Yang L, Huang Y, Porta R, Yanagisawa K, Gonzalez A, Segi E, Johnson DH, Narumiya S, Carbone DP. Host and direct antitumor effects and profound reduction in tumor metastasis with selective EP4 receptor antagonism. Cancer Res. 2006; 66: 9665–72. doi: 10.1158/0008-5472.CAN-06-1271.
22. Xin X, Majumder M, Girish GV, Mohindra V, Maruyama T, Lala PK. Targeting COX-2 and EP4 to control tumor growth, angiogenesis, lymphangiogenesis and metastasis to the lungs and lymph nodes in a breast cancer model. Lab Investig J Tech Methods Pathol. 2012; 92: 1115–28. doi: 10.1038/labinvest.2012.90.
23. Konya V, Marsche G, Schuligoi R, Heinemann A. E-type prostanoid receptor 4 (EP4) in disease and therapy. Pharmacol Ther. 2013; 138: 485–502. doi: 10.1016/j.pharmthera.2013.03.006.
24. Zhao X, Guo Y, Yue W, Zhang L, Gu M, Wang Y. ABCC4 is required for cell proliferation and tumorigenesis in non-small cell lung cancer. OncoTargets Ther. 2014; 7: 343–51. doi: 10.2147/OTT.S56029.
25. Noori-Daloii MR, Saffari M, Raoofian R, Yekaninejad M, Dinehkabodi OS, Noori-Daloii AR. The multidrug resistance pumps are inhibited by silibinin and apoptosis induced in K562 and KCL22 leukemia cell lines. Leuk Res. 2014; 38: 575–80. doi: 10.1016/j.leukres.2013.10.028.
26. Holla VR, Backlund MG, Yang P, Newman RA, DuBois RN. Regulation of prostaglandin transporters in colorectal neoplasia. Cancer Prev Res (Phila Pa). 2008; 1: 93–9. doi: 10.1158/1940-6207.CAPR-07-0009.
27. Borel F, Han R, Visser A, Petry H, van Deventer SJH, Jansen PLM, Konstantinova P, with collaboration of the Réseau Centre de Ressources Biologiques Foie (French Liver Biobanks Network) F. Adenosine triphosphate-binding cassette transporter genes up-regulation in untreated hepatocellular carcinoma is mediated by cellular microRNAs. Hepatology. 2012; 55: 821–832. doi: 10.1002/hep.24682.
28. Huynh T, Norris MD, Haber M, Henderson MJ. ABCC4/MRP4: a MYCN-regulated transporter and potential therapeutic target in neuroblastoma. Front Oncol. 2012; 2: 178. doi: 10.3389/fonc.2012.00178.
29. Montani M, Hermanns T, Herrmanns T, Müntener M, Wild P, Sulser T, Kristiansen G. Multidrug resistance protein 4 (MRP4) expression in prostate cancer is associated with androgen signaling and decreases with tumor progression. Virchows Arch Int J Pathol. 2013; 462: 437–43. doi: 10.1007/s00428-013-1390-8.
30. Henderson MJ, Haber M, Porro A, Munoz MA, Iraci N, Xue C, Murray J, Flemming CL, Smith J, Fletcher JI, Gherardi S, Kwek C-K, Russell AJ, et al. ABCC multidrug transporters in childhood neuroblastoma: clinical and biological effects independent of cytotoxic drug efflux. J Natl Cancer Inst. 2011; 103: 1236–51. doi: 10.1093/jnci/djr256.
31. Copsel S, Garcia C, Diez F, Vermeulem M, Baldi A, Bianciotti LG, Russel FGM, Shayo C, Davio C. Multidrug resistance protein 4 (MRP4/ABCC4) regulates cAMP cellular levels and controls human leukemia cell proliferation and differentiation. J Biol Chem. 2011; 286: 6979–88. doi: 10.1074/jbc.M110.166868.
32. Kochel TJ, Fulton AM. Multiple Drug Resistance-Associated Protein 4 (MRP4), Prostaglandin Transporter (PGT), and 15-Hydroxyprostaglandin Dehydrogenase (15-PGDH) as Determinants of PGE2 Levels in Cancer. Prostaglandins Other Lipid Mediat. 2015; 116–117: 99–103. doi: 10.1016/j.prostaglandins.2014.11.003.
33. Chi Y, Suadicani SO, Schuster VL. Regulation of prostaglandin EP1 and EP4 receptor signaling by carrier-mediated ligand reuptake. Pharmacol Res Perspect. 2014; 2: e00051. doi: 10.1002/prp2.51.
34. Eruslanov E, Daurkin I, Ortiz J, Vieweg J, Kusmartsev S. Pivotal Advance: Tumor-mediated induction of myeloid-derived suppressor cells and M2-polarized macrophages by altering intracellular PGE2 catabolism in myeloid cells. J Leukoc Biol. 2010; 88: 839–48. doi: 10.1189/jlb.1209821.
35. Tai H-H. Prostaglandin catabolic enzymes as tumor suppressors. Cancer Metastasis Rev. 2011; 30: 409–17. doi: 10.1007/s10555-011-9314-z.
36. Chan BS, Endo S, Kanai N, Schuster VL. Identification of lactate as a driving force for prostanoid transport by prostaglandin transporter PGT. Am J Physiol Ren Physiol. 2002; 282: F1097-1102. doi: 10.1152/ajprenal.00151.2001.
37. Wolf I, O’Kelly J, Rubinek T, Tong M, Nguyen A, Lin BT, Tai H-H, Karlan BY, Koeffler HP. 15-hydroxyprostaglandin dehydrogenase is a tumor suppressor of human breast cancer. Cancer Res. 2006; 66: 7818–23. doi: 10.1158/0008-5472.CAN-05-4368.
38. Nomura T, Lu R, Pucci ML, Schuster VL. The Two-Step Model of Prostaglandin Signal Termination: In Vitro Reconstitution with the Prostaglandin Transporter and Prostaglandin 15 Dehydrogenase. Mol Pharmacol. 2004; 65: 973–8. doi: 10.1124/mol.65.4.973.
39. Kochel TJ, Goloubeva OG, Fulton AM. Upregulation of Cyclooxygenase-2/Prostaglandin E2 (COX-2/PGE2) Pathway Member Multiple Drug Resistance-Associated Protein 4 (MRP4) and Downregulation of Prostaglandin Transporter (PGT) and 15-Prostaglandin Dehydrogenase (15-PGDH) in Triple-Negative Breast Cancer. Breast Cancer Basic Clin Res. 2016; 10: 61–70. doi: 10.4137/BCBCR.S38529.
40. Masereeuw R, Russel FGM. Regulatory pathways for ATP-binding cassette transport proteins in kidney proximal tubules. AAPS J. 2012; 14: 883–94. doi: 10.1208/s12248-012-9404-z.
41. Sinha C, Ren A, Arora K, Moon C-S, Yarlagadda S, Zhang W, Cheepala SB, Schuetz JD, Naren AP. Multi-drug resistance protein 4 (MRP4)-mediated regulation of fibroblast cell migration reflects a dichotomous role of intracellular cyclic nucleotides. J Biol Chem. 2013; 288: 3786–94. doi: 10.1074/jbc.M112.435925.
42. Robertson FM, Simeone A-M, Mazumdar A, Shah AH, McMurray JS, Ghosh S, Cristofanilli M. Molecular and pharmacological blockade of the EP4 receptor selectively inhibits both proliferation and invasion of human inflammatory breast cancer cells. J Exp Ther Oncol. 2008; 7: 299–312.
43. Tong M, Ding Y, Tai H-H. Reciprocal regulation of cyclooxygenase-2 and 15-hydroxyprostaglandin dehydrogenase expression in A549 human lung adenocarcinoma cells. Carcinogenesis. 2006; 27: 2170–9. doi: 10.1093/carcin/bgl053.
44. Cheung L, Yu DMT, Neiron Z, Failes TW, Arndt GM, Fletcher JI. Identification of new MRP4 inhibitors from a library of FDA approved drugs using a high-throughput bioluminescence screen. Biochem Pharmacol. 2015; 93: 380–8. doi: 10.1016/j.bcp.2014.11.006.
45. Hayashi H, Naoi S, Nakagawa T, Nishikawa T, Fukuda H, Imajoh-Ohmi S, Kondo A, Kubo K, Yabuki T, Hattori A, Hirouchi M, Sugiyama Y. Sorting nexin 27 interacts with multidrug resistance-associated protein 4 (MRP4) and mediates internalization of MRP4. J Biol Chem. 2012; 287: 15054–65. doi: 10.1074/jbc.M111.337931.
46. Li Y, Reader JC, Ma X, Kundu N, Kochel T, Fulton AM. Divergent roles of CXCR3 isoforms in promoting cancer stem-like cell survival and metastasis. Breast Cancer Res Treat. 2015; 149: 403–15. doi: 10.1007/s10549-014-3229-7.
47. Greenhough A, Smartt HJM, Moore AE, Roberts HR, Williams AC, Paraskeva C, Kaidi A. The COX-2/PGE pathway: key roles in the hallmarks of cancer and adaptation to the tumor microenvironment. Carcinogenesis. 2009; 30: 377–86. doi: 10.1093/carcin/bgp014.
48. Hwang D, Scollard D, Byrne J, Levine E. Expression of cyclooxygenase-1 and cyclooxygenase-2 in human breast cancer. J Natl Cancer Inst. 1998; 90: 455–60.
49. Half E, Tang XM, Gwyn K, Sahin A, Wathen K, Sinicrope FA. Cyclooxygenase-2 expression in human breast cancers and adjacent ductal carcinoma in situ. Cancer Res. 2002; 62: 1676–81.
50. Sivula A, Talvensaari-Mattila A, Lundin J, Joensuu H, Haglund C, Ristimäki A, Turpeenniemi-Hujanen T. Association of cyclooxygenase-2 and matrix metalloproteinase-2 expression in human breast cancer. Breast Cancer Res Treat. 2005; 89: 215–20. doi: 10.1007/s10549-004-0714-4.
51. Reid G, Wielinga P, Zelcer N, van der Heijden I, Kuil A, de Haas M, Wijnholds J, Borst P. The human multidrug resistance protein MRP4 functions as a prostaglandin efflux transporter and is inhibited by nonsteroidal antiinflammatory drugs. Proc Natl Acad Sci U S A. 2003; 100: 9244–9. doi: 10.1073/pnas.1033060100.
52. Ritter CA, Jedlitschky G, Meyer zu Schwabedissen H, Grube M, Köck K, Kroemer HK. Cellular export of drugs and signaling molecules by the ATP-binding cassette transporters MRP4 (ABCC4) and MRP5 (ABCC5). Drug Metab Rev. 2005; 37: 253–78. doi: 10.1081/DMR-200047984.
53. Ma X, Kundu N, Rifat S, Walser T, Fulton AM. Prostaglandin E Receptor EP4 Antagonism Inhibits Breast Cancer Metastasis. Cancer Res. 2006; 66: 2923–7. doi: 10.1158/0008-5472.CAN-05-4348.
54. Menter DG, Dubois RN. Prostaglandins in cancer cell adhesion, migration, and invasion. Int J Cell Biol. 2012; 2012: 723419. doi: 10.1155/2012/723419.
55. Bookout AL, Mangelsdorf DJ. Quantitative real-time PCR protocol for analysis of nuclear receptor signaling pathways. Nucl Recept Signal. 2003; 1: e012. doi: 10.1621/nrs.01012.