
INTRODUCTION
Nasopharyngeal carcinoma (NPC) is a head and neck cancer arising from the epithelial cells in the lining of nasopharynx [1]. The prevalence is distinctively manifested geographically and ethnically, with a high incidence in some parts of the world, including southern China, Southeast Asia, northern Africa, Alaska, and Greenland [2]. In the endemic regions, the incidence of 25 cases per 100,000 population has been recorded annually, which is about 25-fold higher than the rest of the world [3–5]. The differences in the geographical and racial distribution indicate the multifactorial etiology of the particular tumor; these include environmental factors, food consumption, the infection of Epstein-Barr virus (EBV), a lymphotropic human herpesvirus, and genetic susceptibility. This remarkable distribution of NPC indicates that the development of nasopharyngeal cancer may be associated with the genetic and environmental factors [3].
Hitherto, radiotherapy and concurrent chemoradiotherapy are the primary treatments of primary NPC [6]. The tumor cells of NPC are sensitive to radiotherapy; however, the use of radiotherapy, chemoradiotherapy, or targeted radiotherapy has not significantly improved the survival rate of the patients. An overall locoregional failure after the initial treatment is approximately 10% [7]. The regional lymph node, distant metastasis, and loco-regional recurrence are the major indications of the therapeutic failure of NPC [8–10]. Elucidation of the underlying mechanism of NPC genesis is necessitated in order to improve the efficacy of the therapy.
Apoptosis is a host mechanism to eliminate the unwanted cell clones and serve as the end effect of antitumor immune surveillance. The extrinsic or the death receptor pathway contributes to the removal of the mutant or transformed cells; the pathway is activated by death ligands elaborated by the effector cells in antitumor immunity, cytotoxic T lymphocytes (CTL), and natural killer (NK) cells.
NF-κB is a transcription factor, a heterodimer of different subunits, and is implicated in immune response, inflammation, and carcinogenesis [11]. The subunits belong to a protein family comprising of RelA (p65), RelB, c-Rel, p50 (p105 precursor), and p52 (p100 precursor) [12]. The subunits form homo- or heterodimeric complexes with each other and also with their inhibitory proteins (I-κB proteins) [11]. In the quiescent physiologic state, NF-κB is sequestered in the cytoplasm in an inactive form and associated with I-κB proteins like, IkI- κB a or the precursor proteins, p100 and p105. The I-κBs molecules are phosphorylated by I-κB B kinases (IKKs) before they are dissociated from the complex. They are ubiquitinated after the dissociation, followed by degradation. The degradation of I-κB from the NF-κB : I-κB complex is an essential step for activating the released NF-κB [13–15]. It binds to specific DNA sequences on the regulatory elements upstream of the coding region of a gene, through the Rel homology region.
NF-κB transcriptionally activates anti-apoptotic genes that function in the death-inducing signaling complex (DISC) as well as in the mitochondria. These anti-apoptotic genes, such as IAP-1, IAP-2, c-FLIP, TRAF1, and TRAF2, coordinate their inhibitory effects to interfere with the receptor-mediated death at the DISC. IAP-1 and IAP-2 can interact directly with caspases to block their activation, thereby facilitating their degradation [16].
As a cytotoxic cytokine in this category, TRAIL can kill tumor cells in vivo without harming the normal tissues [17]. TRAIL binds to death receptor 4 (DR4)/TRAIL-R1 and DR5/TRAIL-R2, which are mainly expressed on transformed cells and triggers apoptotic signal initiated by the cleavage of caspase-8. In contrast to tumor cells, non-neoplastic cells tend to express the TRAIL decoy receptors DR3/TRAIL-R3 and DR6/TRAIL-R4; the host cells are rendered less sensitive to TRAIL-mediated death. [18, 19]. The finding has initiated the development of TRAIL receptor (TRAIL-R) agonists as an anticancer drug in clinical therapy. This hypothesis is further supported by the finding that malignant cells could resist TRAIL-mediated cytotoxicity, mainly because of the silencing of tumor suppressor genes that downregulate anti-apoptotic signaling pathways [20].
Tumor suppressor genes (TSGs) are frequently lost in malignancies. With the loss-of-function of these TSGs, the cells are no longer harnessed in growth and proliferation by regulation of the cell cycle entry, thus, triggering apoptosis [21, 22].
BLU encodes a protein of the family that possesses a motif of zinc finger myelogenous Nervy domain (ZMYND). Some of the proteins, like ETO/ZMYND3, are implicated in the genesis of malignancies through protein-protein interactions, as repressors of transcription. A fused protein, AML, comprising of ETO and RUNX, is formed due to the chromosomal translocation t(8:21) in the genesis of acute myelogenous leukemia. ETO of the ZMYND family associates with histone deacetylase (HDAC) to suppress the transformation potential of oncogene RUNX2 [23].
BLU is highly expressed in many normal tissue types including lung, testes, and the uterine cervix. However, it is downregulated in lung, breast, kidney, neuroblastoma, and esophageal squamous cell carcinoma [24–27]. It is also frequently observed to be hypermethylated in primary tumors [28–31]. The structural features of the BLU protein involve the C-terminal region encompassing the MYND domain. The domain consists of several cysteine and histidine amino acid residues, which may be involved in protein–protein interactions to repress gene transcription
The tumor suppression of BLU has been indicated by its frequent loss in some human malignancies, but the underlying mechanisms are largely unknown. Previously, we have shown that similar to the tumor suppressors RASSF1A [32] and NGDR 2 [33], BLU down-regulates JNK signaling and inhibits the promoter activity of cyclin D1, thereby reducing its expression level to arrest the cell cycle in G1 phase [34]. The inhibition of cell cycle entry contributes to the proliferation inhibition of BLU. Park et al. have shown that BLU potentiated apoptosis induced by the chemotherapeutic agent, paclitaxel to exert tumor suppression [35]. Promotion of paclitaxel induced by BLU correlated with enhanced activities of pro-apoptotic molecules Bax and p21, and inactivation of BclxL and NF-κB inducing Bcl-2, both are anti-apoptotic.
In the present study, we explored whether BLU inhibited the NF-κB pathway, and whether the NF-κB-dependent factors cFLIP and cIAP2 promoted death receptor-induced apoptosis.
RESULTS
Growth inhibitory effects on HNE1 cells exerted in vitro and in vivo by BLU
The expression of EGFP-tagged BLU in HNE1 cells infected with pCD316-Ad5, and 20 and 40 MOI BLU-Ad5 was confirmed by green fluorescent signals (Figure 1A). After infection with 0, 5, 10, 20, and 40 MOI BLU-Ad5, the viability of the cells assayed using the CCK8 kit was found to be decreased in a dose-dependent manner, with a significant difference at each dose (Figure 1B), suggesting that BLU possessed an inhibitory effect on the cell proliferation. The viability assay revealed that the recombinant pCD316 adenovirus also presented cytotoxicity albeit much weaker (data not shown). When intratumorally injected into human NPC xenografts in nude mice, the growth of the tumors was remarkably inhibited by recombinant BLU-Ad5 (Figure 1C and 1D).
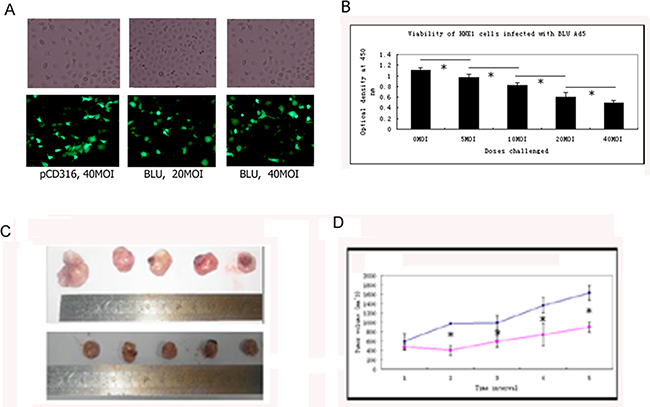
Figure 1: Growth inhibitory effects on HNE1 cells exerted in vitro and in vivo by BLU. A. Re-expression of BLU was confirmed the presence of green fluorescence viewed with microscopy. B. Growth inhibitory effect of BLU on HNE1 cells was measured with CCK8 kit. Significant differences was suggested by P<0.05 depicted with an asterisk (*). C. Growth of xenografted HNE1 cells was inhibited by BLU in Balb/C mice. Two weeks after subcutaneously inoculation, palpable tumors were identified. Recombinant BLU Ad5 and the control adenovirus with pCD316 were injected intratumorally. The long and short radiuses were measured with caliber every other day. The mice were sacrificed after two-week observation. The tumors were harvested by surgical removal and photographed (C); Upper panel: control group with injection of pCD316 Ad5; lower panel: experimental group with injection of BLU Ad5; D. The tumor volumes were calculated according to the formula v=ab2/2; a and b were the long and short radiuses mentioned above. The growth curve was plotted according to the tumor size registered every other day. Data derived from tests with three groups each containing 6 mice and presented as mean ± SD. The asterisk (*) in D depicted p< 0.05 in statistics.
TRAIL induced apoptosis in HNE1 cells was potentiated by expression of BLU
The microscopic assessment of the PI-stained HNE1 cells’ nucleus, post stimulation with 200 ng/ml TRAIL with 10 μg/mL CHX, revealed substantial apoptosis. The cell number decreased significantly, and the fragmented nuclei were visible (Figure 2A). A death ligand of TNF superfamily, TRAIL, activated two arms of reaction: pro-apoptotic and activation of anti-apoptotic survival supporting the NF-κB pathway. Apoptosis is induced only when the de novo protein synthesis mediated by NF-κB activation is inhibited by CHX. The turnover of the phosphatidylserine to the external membrane is a characteristic of apoptosis. When co-incubated with the designated dose of TRAIL and CHX, the annexin V positive and low PI cell populations increased (in Q4 quadrant); the change was pronounced in BLU-expressing cells (Figure 2B and 2C). The cells are classified into two types based on the mitochondrial involvement to activate the downstream caspases in death receptor-induced apoptosis. The DISC formed on the membrane of type II cells recruits less caspase-8 and weakly triggers the caspase cascade than type I cells. Both the permeabilization of the mitochondrial membrane and the release of a pro-apoptotic factor to cytoplasm occurred in the cells in the current study, indicating a type II phenotype. Disruption of the mitochondrial membrane potential (MMP) is preceded by the alteration of the transition pore of the membrane. As assayed by FACS-based TMRE, the change was rather pronounced in BLU-expressing HNE-1 cells challenged by TRAIL (Figure 2D and 2E).
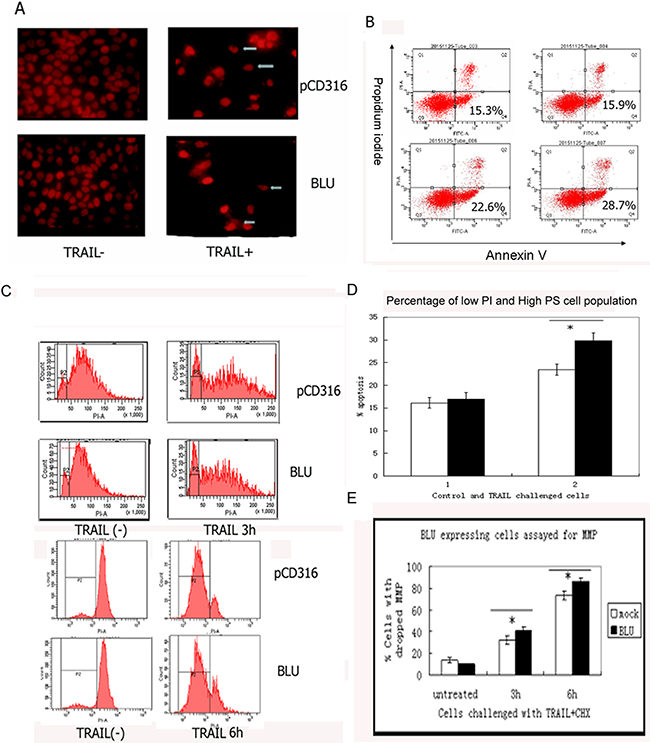
Figure 2: TRAIL induced apoptosis in HNE1 cells was potentiated by expression of BLU. A. The morphology of TRAIL treated HNE1 cells. The cells were co-incubated with 200 ng/ml TRAIL, then were collected and stained with PI, and viewed with fluorescence microscopy. Apoptosis was confirmed by the fragmented nuclei and reduced cell numbers. B. Turnover of surface phosphatidylserine (PS) on TRAIL treated HNE1 cells. The cells grown in 6 well culture plates were treated as above. They were harvested, stained with annexin V kit and analyzed with a FACScan. The quadrant 4 (Q4) with low PI and high PS represented apoptosis. The size of Q4 population of HNE1 infected with BLU Ad5, and pCD316Ad5, untreated and treated with TRAIL were calculated. The data was presented in C. as means ±SD from at least three independent tests. D. Drop of mitochondrial membrane potential in TRAIL challenged HNE1 cells. The cells were treated with TRAIL, and stained with TMRE. The drop of mitochondrial membrane potential (MMP) on apoptosis contributed to a depolarized peak left to the main peak of normal MMP. The data from at least three independent tests was summarized in E. and presented as mean ± SD.
TRAIL-induced apoptosis was potentiated in BLU-expressing cells, as suggested by promoted cleavage of caspase-8 and PARP by BLU
The expression of receptors, DR4 or TRAIL R1 and DR5 or TRAIL R2 on HNE1 cells, was present on pCD316-Ad5 or BLU-Ad5 infected HNE1 cells’ surface as detected by immunoblotting (Figure 3A). This expression confirmed that TRAIL possessed the ability to trigger apoptosis in this cell line. During death receptor-induced apoptosis, the aggregated death receptor formed a DISC on the inner surface of the membrane, and the complex recruited initiators such as caspases 8 and 10, catalyzed by autohydrolysis. The zymogen form of caspase-8 was presented as a dimer of isoforms a and b, which were manifested as dimers of 55 and 58kDa, respectively. As a result of co-incubation with TRAIL, 41kDa and 43kDa dimers were generated; the cleavage was remarkable in TRAIL-treated BLU-expressing cells, and additional procaspase-8 was cleaved (Figure 3B). When challenged with TRAIL, PARP was cleaved as shown by the generation of a 95kDa band. Similar to the cleavage of caspase-8, more PARP was degraded in BLU-expressing cells, as indicated by the low amount of the upper band. The data also suggested that cleavage of the apoptotic substrate was promoted by BLU. Although the expression of BLU normally did not trigger remarkable morphological apoptosis, the immunoblot of the BLU-expression showed PARP degradation, a characteristic of apoptosis (Figure 3C). The lanes 1, 2, 3, 4 represented pCD316-Ad5 and BLU-Ad5, untreated with TRAIL; pCD316-Ad5 and BLU-Ad5 treated with TRAIL, respectively.
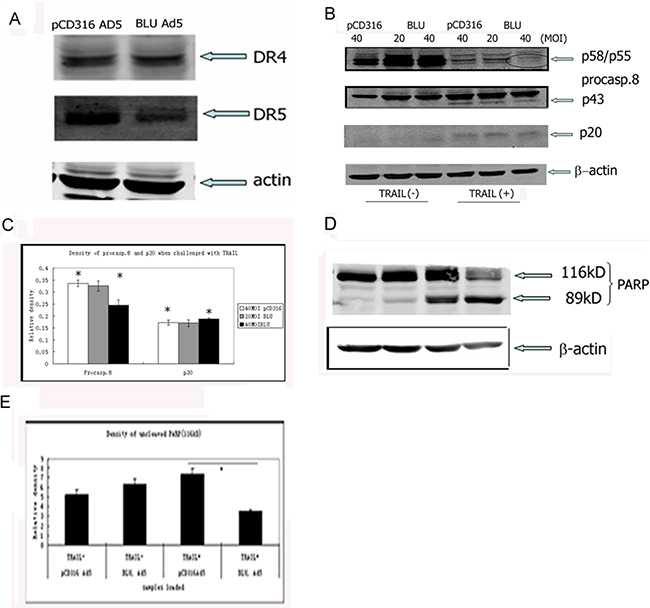
Figure 3: BLU promoted TRAIL-induced caspase-8 and PARP degradation. A. The apoptosis triggering ability of TRAIL in HNE1 cells was confirmed by the expression of death receptor 4 (DR4) and DR5. Total proteins of HNE1 cells infected with pCD316 Ad5 and BLU Ad5 were separated by SDS-PAGE, immunoblotted and probed with antibodies, H-130 (against DR4) and D-6 (against DR5). The loading of proteins was calibrated by β-actin (lowest panel). B. Cleavage of an initiator caspase, caspase-8, was assayed with total proteins extracted from HNE1 cells infected with pCD316 Ad5 and BLU Ad5, then challenged with TRAIL. They were immunoblotted and probed with specific antibody C15. The cleavage was pronounced in TRAIL treated BLU expressing cells. C. The relative expression of procaspase-8 to that of β-actin in cells was quantified by a scanning densitometer. The control expression of β-actin was set as 1. D. Under similar conditions described in (B), PARP was cleaved. The amount of cleavage of PARP was compared in TRAIL-treated BLU-Ad5 and pCD316 Ad5infected HNE1 cells. The weak degradation of PARP in TRAIL-treated BLU-Ad5-infected cells suggested that BLU was pro-apoptotic. E. The relative levels of 116kDa PARP expression to that of β-actin in cells were quantified by a scanning densitometer. The control expression of β-actin was set as 1. The asterisk (*) depicted p<0.05.
Expression of BLU downregulated NF-κB signaling by inhibiting reporter activity and reducing the expression IkapakB kinase alpha (IKKα)
Death receptor-induced apoptosis is regulated by a set of molecules structurally similar to caspase-8, caspase-8-like apoptosis regulator (CLAR), and other anti-apoptotic factors. A well-known member of this family, cFLIPL (cellular FLICE inhibitory protein, long form) is transcription factor NF-κB-dependent. cFLIPL negatively regulates the activity of caspase-8 as a dominant negative binding partner. With the expression of cFLIPL, the caspase-8 activity is decreased. Therefore, we examined the level of cFLIPL. The data showed that in BLU-expressing HNE1 cells, the expression of cFLIPL was downregulated; also cIAP2 was downregulated in BLU Ad5 in comparison with pCD316 Ad5 infected HNE1 cells (Figure 4A). The quantified data in Figure 3A were presented in Figure 4C. Since the induction of CLAR and cIAP2 is NF-κB-dependent, we tested whether the expression of BLU blocked the NF-κB signaling. Additionally, the level of IKKalpha, the component of the complex which catalyzes phosphorylation of I–κ kB and the transcription factor p65 NF-κB, was reduced in BLU-expressing HNE-1 cells (Figure 4B, 4D). The pConA- κB-luc reporter containing three κB binding sites revealed low activity when co-transfected to HNE1 cells with BLU, or to cells infected with BLU-Ad5 (Figure 4E).
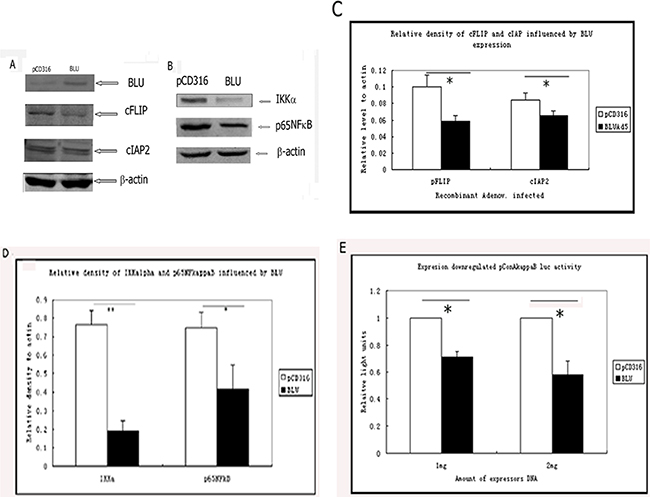
Figure 4: Expression of BLU-inhibited signaling of NF-κB and downregulated NF-κB-induced anti-apoptotic factor cFLIP and cIAP2. A. Expression of cFLIPL and cIAP2 by HNE1 cells infected with pCD316-AD5 or BLU-Ad5. The total proteins from the lysates of cells were immunoblotted and probed with anti-cFLIPL mAb (clone NF6), and anti cIAP2 rabbit polyclonal antibody. The difference of cFLIP and cIAP2 expression level in BLU-expressing and control cells was suggested. B. The levels of IKKα and p65NF-κB, regulator and an active subunit of NF-κB pathway was evaluated by immunoblotting in BLU-expressing and control cells. The representative data were derived from at least two independent tests. C. The relative levels of cFLIP and cIAP2 expression to that of β-actin in cells were quantified by a scanning densitometer. The control expression of β-actin was set as 1. p-values between 40 MOI pCD316 Ad5 and BLU Ad5 in comparison to cFLIP and cIAP2 were less than 0.05. D. The relative levels of IKKα and p65NF-κB expression to that of actin in cells were quantified by a scanning densitometer. The control expression of β-actin was set as 1. *: p<0.05; **: p<0.01. E. Reporter assay of NF-κB binding activity of BLU. HNE1 cells were co-transfected with 2μg pCD316 or 2 μg BLU pCD316 +0.1 μg pConA-κB-luc, and 1 μg pCD316 or 1 μg BLU pCD316 +0.1 μg pConA-κB-luc, together with renilla luciferase expressor plasmid to calibrate transfection efficiency. The lysate of the cells was mixed with luciferrin and the luciferase activity was recorded by a luminometer. The ratio of the values of firefly (from pConA κB luc.) and renilla luciferase activity was calculated. The values were presented as mean ± SD, derived from at least three independent experiments.
DISCUSSION
The epigenetic and/or genetic inactivation of TSGs contributes towards the occurrence of malignancy, because of the loss of functions such as inhibition of cell cycle entry and triggering of apoptosis in the host [21, 22]. Such anomaly is implicated in the proliferation of cancer. It has been reported that the re-expression of the frequently lost TSGs restores the normal modulation of cell cycle entry and apoptosis by inhibition of some signaling pathway [32–35]. In the present study, we have shown that re-expression of BLU, a TSG mapped on the frequently lost chromosomal region, 3p21, potentiated TRAIL-induced apoptosis.
Antitumor immune surveillance is exerted by two sets of effector cells, CTLs and NK cells [38]. The cells function to eradicate the tumor cells by releasing cytotoxic molecules such as TRAIL and Fas ligand, to stimulate DR-induced or extrinsic apoptosis. The data in the present study suggested that TRAIL possessed the potent ability to kill NPC-derived NHE1 cells. However, various strategies have been shown to be utilized by cancer cells, to evade host immune surveillance and facilitate outgrowth of the malignant clones [39]. Wennerberg et al. reported that an anticancer chemotherapeutic agent, doxorubicin, augmented the activity of NK cells by modulating the signaling TRAIL receptor [40]; the activity of TRAIL is regulated by cancer-related genes and miRNAs [20]. The findings that BLU-enhanced apoptosis induced by a chemotherapeutic agent, paclitaxel, prompted to investigate whether BLU potentiated DR-induced apoptosis mediated by TRAIL, in order to promote antitumor immune surveillance.
We have shown that infection with BLU-expressing recombinant BLU-Ad5 remarkably reduced the viability of NPC-derived HNE1 cells, and intratumoral injection of the same recombinant adenovirus inhibited the growth of human NPC xenografts in nude mice. The data further validated the tumor suppressive potential of the BLU gene. Functionally, BLU may suppress the tumor formation and progression via intracellular pathway targeting. Our previous study indicated that BLU downregulated the JNK pathway and inhibited the promoter activity of CCND1, coding for cyclin D1, hence arresting the cell cycle in G1 phase [34]. Other studies have shown that BLU also downregulated proangiogenic cytokine to inhibit angiogenesis [41]; it also inactivated anti-apoptotic members of the Bcl-2 family to promote paclitaxel-induced apoptosis [35]. In contrast to the findings that forced expression mediated by adenoviral vector was unable to trigger apoptosis in lung cancer-derived cells [42], our study suggested that BLU was pro-apoptotic in HNE1 cells as it induced the cleavage of 116kDa PARP.
Potentiation of TRAIL-induced apoptosis has been observed in our study. Flow cytometry based staining with annexin V, which detects phosphatidylserine (PS) on the outer membrane, revealed only minor difference in apoptotic populations in control cells when infected with BLU-Ad5 vs. pCD316 Ad5. However, the apoptotic population was significantly increased in TRAIL-treated BLU-expressing cells. Stimulation by TRAIL triggered degradation of an initiator caspase, caspase-8. In HNE1 cells infected with BLU-Ad5 and pCD316 Ad5, the procaspase-8 isoforms degraded when challenged with TRAIL. The TRAIL induced cleavage of procaspase-8 was promoted when the cells were infected with 40 MOI BLU-Ad5. At this dose, the apoptosis potentiation by BLU was suggested.
Caspase-8 presents a differential ability to activate within DISC in order to stimulate the downstream executive caspases like caspase-3, 6, and 7 [43]. Therefore, the cells are classified into two types, depending on whether caspase-8 cleaves a BH3-like Bcl-2 family protein, BID [44, 45] to permeabilize the outer membrane of mitochondria and trigger the release of pro-apoptotic factors, cytochrome C and apoptosis inducing factor (AIF) [46, 47]. In Type II cells, the mitochondria are perturbed, and apoptosomes are formed to activate the caspase cascade, leading to morphological apoptosis [48, 49]. The drop of MMP was observed in HNE1 cells co-incubated with TRAIL, characteristic of the type II cells. The change in the mitochondrial membrane was also pronounced in BLU-expressing cells.
DR induced-apoptosis is regulated at the level of DISC, mediated by the ratio of caspase-8 and a structurally similar molecule cFLIPL [50]. cFLIPL, of long isoform but not the short isoform c-FLIPS, has been shown to be required for caspase-8 activation [51], although the role of the short isoform of FLIP S in inhibition of extrinsic apoptosis could not be entirely excluded [52]. The current study revealed that the expression of BLU reduced the level of cFLIPL, stimulated the initiator caspase-8, thereby enhanced death receptor-induced apoptosis. Clinicopathologically, the c-FLIP overexpression was correlated with the poor prognosis of NPC [53].
The present study demonstrated that cIAP2, another NF-κB-dependent anti-apoptotic factor [54], which is overexpressed in EBV-associated NPC [55] was downregulated in BLU-expressing HNE1 cells. The reduced expression of cFLIPL and cIAP2 by BLU led to the hypothesis that tumor suppression by BLU was exerted by blocking the NF-κB signaling.
A recently identified tumor suppressor in NPC [56] and esophageal carcinoma, cysteine-rich intestinal protein 2 (CRIP2) encoded by a gene on chromosome 14 has been shown to inhibit NF-κB-dependent proangiogenic cytokine transcription in NPC cells, and its ectopic expression triggers apoptosis in esophageal carcinoma [57].
A tumor suppressor, Von Hippel-Lindau (VHL) on chromosomal 3p25 region, has been shown to suppress NF-κB pathway and enhance TNF-induced apoptosis in renal cell carcinoma (RCC) [58, 59]. The regional deletions on the short arm of chromosome 3 (3p) in RCC is the prototypic alteration in the malignancy-related karyotype of chromosome 3. Additionally, the anomaly of the putative tumor suppressor genes on 3p14, 3p21, and 3p25 is the crucial event in the carcinogenesis of nasopharynx [60, 61]. High expression of inhibitor of differentiation 1 (Id1) and p65 NF-κB have been reported to be correlated with poor prognosis of NPC patients [62].
NF-κB is an anti-apoptotic factor that is implicated in immune response, inflammation, and occurrence of malignancies [11, 15]. During quiescent physiological condition, NF-κB remains inactive because of heterodimerization with an inhibitor subunit I-κB. When I-κB is phosphorylated by upstream kinases, it dissociated from the complex, followed by ubiquitination and degradation. Then, the subunits, such as p65NF-κB, are activated to function as transcription factors [12–14].
The downregulation of the subunit p65 of NF-κB and an upstream kinase IKKalpha caused by the expression of BLU were examined in our study. IKK alpha catalyzes the phosphorylation of I-κB, leading to the activation p65 NF-κB [12]. Though mechanisms remain to be elucidated, the expression of BLU reduced the level of IKKα and p65NF-κB and inhibited the activity of NF–κB reporter pConA-κB-luc.
In immune-deficient individuals, EBV is activated in B lymphocytes to express genomic products with potential of NF-κB induction to transform host cells; the resultant proliferative lymphoid clones progress to lymphoma. Blocking the activated NF-κB with chemical inhibitors such as BAY11-7082 has been promising in treating such disorders [63]. The present study together with other reports [58], suggested NF-κB signaling to be suppressed by a TSG, and it is warranted further validation as a target for anticancer therapy. In summary, promotion of death receptor induced apoptosis by BLU through inhibition of NFκB pathway was depicted in Figure 5.
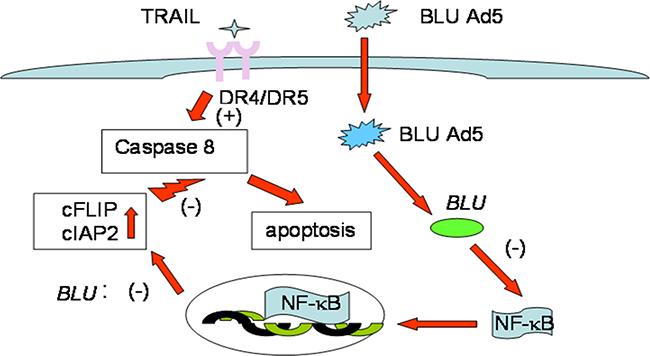
Figure 5: Schematic pattern of Promotion of death receptor induced apoptosis by BLU. In the present experiment, tumor suppressor BLU which is frequently lost in NPC was ectopically expressed by transfer of adenoviral vector transfer. The expression of BLU inhibited signaling of NF-κB hence reduced the expression of anti-apoptotic factors cFLIP and cIAP2. Caspase 8 cleavage triggered by ligation of DR4 and DR5 by TRAIL was enhanced, and apoptosis induced by the two death receptors was promoted and this could contribute to strengthen antitumor immunity.
MATERIALS AND METHODS
Cells, viral stock, and plasmids
HNE-1 cells derived from undifferentiated nasopharyngeal carcinoma in southern China were a kind gift from Prof. Qian Tao (Chinese University of Hong Kong). They were cultured in DMEM (Gibco Biotechnology, Guangzhou, China) supplemented with 10% fetal calf serum (FCS; Gibco Biotechnology), with or without penicillin + streptomycin (PEST; Gibco Biotechnology), incubated at 37°C with 5% carbon dioxide. Recombinant BLU pCD316 plasmid was constructed by cloning a cDNA fragment of BLU in the SacI-SalI site of CMV-derived shuttle vector pCD316. The vector with disrupted E1 and E3 sites was constructed by Dr. Frank Graham [34, 36]. TRAIL with EGFP expression was a kind gift from Dr. Bingliang Fang (MD Anderson Cancer Center) [37]. NF-κB reporter plasmid, pConA-κB-luc, contains three κB binding sites upstream of the coding cassette of luciferase, a kind gift from Prof. Martin Rowe, University of Birmingham, UK.
Transfection and viral infection
The NPC cells HNE1 were seeded in 6-well plates at a density of 1-2×105cells/ml. Recombinant BLU-Ad5 was generated by co-transfection of BLU-pCD316 and at least two coding plasmids of the adenoviral capsid protein into packaging cells, with the total amount of 8.5×1011 PFU. The cells were then infected with various doses of recombinant BLU-Ad5 adenovirus and pCD316-Ad5 for at least 24h after replacing the previous medium. The incorporation of the adenoviral vector was confirmed by fluorescence microscopy and Western blotting. The cells infected with BLU and empty vector were challenged with recombinant TRAIL (Pepro Tech, Guangzhou, China). Sterile coverslips were placed on the bottom of the plates. Alternatively, the cells were transfected with TRAIL-EGFP or the empty vector EGFP-N1 utilizing the K2 kit containing Multifier and transfection reagent (Biontex, Martinsried, Germany; a kind gift from Dr. Roland Klösel), according to the manufacturer’s protocol. Briefly, the cells were co-incubated with Multiplier for 2h at 37°C, before the DNA and the lipid transfection reagent mixture was gently added to the cells. The expression of the introduced tagged gene was confirmed by fluorescence microscopy. Then, the cells were harvest and fixation with 1:1 acetone-methanol. The cells on the coverslips were stained with 1:100 propidium iodide (PI, Sigma) and screened for apoptotic nuclei induced by TRAIL.
Cell viability
1×105 cells were seeded in 96-well plates and infected with 0, 5, 10, 20, 40 MOI/cell BLU Ad5 adenovirus for 48h, in triplicate wells. The cells were then washed with sterile PBS, CCK8 reagents were added, and co-incubated for 12h, followed by an assessment on a Biotek, plate reader. The mean values and standard deviation (SD) were calculated and represented as mean ± SD derived from at least three independent experiments.
Luciferase assay of reporter genes
The cells were co-transfected with BLU pCD316 or empty vector pCD316, pConA-κB-luc and Renilla Luciferase pGL2 (Promega) with K2 kit according to the manufacturer’s instructions. The cells were harvested by lysis with 1x passive lysis buffer (PLB), the lysates collected into microcentrifuge tubes, and the supernatant saved after ultracentrifugation. The luciferase activity was incorporated with a Turner luminometer by mixing the supernatant with luciferin. The relative light activity was presented as the ratio of firefly and renilla luciferase activities. The data was presented as mean ± SD derived from at least three independent tests.
Western blotting
The cells grown on 6-well plates or Petri dishes were detached by lysis with RIPA buffer containing Triton X-100. The lysates were mixed with 2x Laemmli buffer and DNA strands sheared by sonication. Subsequently, the protein lysates were denatured at 95°C for 5min. The total protein was resolved by polyacrylamide gel electrophoresis (PAGE) and transferred to nitrocellulose membrane. The membrane was blocked with 5% non-fat milk in PBS and then incubated with primary antibodies either at RT for 1h or 4°C overnight. The primary antibodies used in the present study were: rabbit anti-human BLU (1:1000, AbCam, Guangzhou, China), murine anti-human actin (1:2000, Sigma, Guangzhou, China), murine anti-human TRAILR1/DR4 (H-130) and TRAILR2/DR5 (D-6) (1:1000, from Santa Cruz, Guangzhou, China). The following antibodies were purchased from Cell Signal Transduction (CST), Guangzhou, China: murine anti-caspase 8, clone C15 (1:500), murine anti-human poly (ADP-ribose) polymerase (PARP) clone C-20 (1:1000), murine anti-IKKα and anti-p65 NF–kapaB (1:1000). The specific murine anti-cFLIP monoclonal antibody, NF6, was a kind gift from Professor Peter Krammer (Program on Tumor Immunogenetics, Deutsches Krebsforschungszentrum, Heidelberg, Germany). After three washes with 0.1% Tween 20 in PBS, the membrane was probed with ultra red labeled anti-murine or rabbit immunoglobulin G (IgG) (Invitrogen, Guangzhou), and the reaction was developed with Odyssey, as previously described [34].
Apoptosis induction and flow cytometry
The cells were infected with BLU-Ad5 or pCD316-Ad5, and challenged by co-incubation with TRAIL+CHX or transfected with TRAIL vector. The cells were harvested, pooled in complete medium, and fixed, followed by sterile PBS washes. Subsequently, the cells were resuspended in PBS, stained using annexin V detection kit, and assessed by FACScan. The cell numbers on quadrant 4, with low PI and high annexin V staining, were considered as apoptotic. Concurrently, the cells with similar parameters were stained with tetramethylrhodamine ethyl ester (TMRE, Immunohistochemistry) and the populations with disrupted mitochondrial membrane potential (MMP), characteristic of apoptosis, were calculated. The data were presented as means ± standard deviation (SD) derived from at least three independent experiments
Effect of recombinant BLU Ad5 on xenograft in nude mice
The animal breeding conditions and experimental protocols were approved by the medical ethics committee of Guangdong Medical University. Balb/c nude mice of both sexes, aged 4-6 weeks, were purchased from Kunming Institute of Zoology, Chinese Academy of Sciences (CAS). The mice were divided into two groups: BLU Ad5 injection and control with pCD316 Ad5. For each group, at least three batches of mice were used, and each batch contained ten mice. The data from at least six mice were used in case of the death of mice during the experiments. 2×106 HNE-1 cells were resuspended in 0.2mL serum-free RPMI, and the suspensions were injected subcutaneously. The palpable tumors were identified in two weeks after inoculation. At this time, 3.5×106 PFU BLU-Ad5 was injected intratumorally. Before and after injection, the lengths of the long (a) and short (b) axes were measured with a caliber. The size of the xenograft tumors was measured every two days and allowed to grow for three weeks before harvesting and weighing. Subsequently, the mice were sacrificed by cervical dislocation according to the approved protocols. The tumor size was estimated according to the formula v= ab2/2, and that of different periods was presented as means ± SD derived from at least three independent experiments. The growth curves of the treatment and control groups were plotted.
Statistical analysis
All the statistical analyses were carried out using the SPSS software program, version 16 (SPSS Inc., Chicago, IL, USA). The quantitative data are presented as means ±SD, derived from three independent tests in a particular assay. P-Values were calculated with Student’s t-test, and p-values <0.05 were considered statistically significant.
Authors’ contributions
JZ and XZ performed the experiments shown in this work, with the help of SL, ZW and ZNH. ZNH, ZWH and XZ designed the experiments. ZNH, ZWH, AG and IE revised the manuscript. XZ conceived the ideal of the study and wrote the manuscript.
ACKNOWLEDGMENTS
We would like to thank Drs Qian Tao, Peter Krammer, Roland Klösel, Bingliang Fang, and Martin Rowe for their gift of the precious reagents, and would like to dedicate the present MS to the late Dr. Eugene Zabarovsky whose persistent efforts have significantly inspired our interests in a 3p tumor suppressor.
CONFLICTS OF INTEREST
The authors do not have anything to declare.
GRANT SUPPORT
Our work was supported by grants from the National Natural Science Foundation of China (NSFC 31170676), Natural Science Foundation of Guangdong Province, China (2015A030313518), Scientific Research Foundation for Returned Overseas Scholars of Guangdong Medical University, China (B2012082); the Sail Plan ‘the Introduction of the Shortage of Top-Notch Talent’ Project (YueRenCaiBan [2014] 1) of Guangdong Province, China to ZNH, and Guangdong Provincial Medical Research Fund to XZ (2014A 267); in part was supported by an International Cancer Research Exchange and Transfer of Technology (ICRETT) fellowship (ICR/2015/391721) from UICC to XZ.
REFERENCES
1. Wei WI, Sham JS. Nasopharyngeal carcinoma. Lancet. 2005; 365:2041–2054.
2. Chang ET and Adami HO. The enigmatic epidemiology of nasopharyngeal carcinoma. Cancer Epidemiol Biomarkers Prevent. 2006; 15:1765–1777.
3. Yu MC, Yuan JM. Epidemiology of nasopharyngeal carcinoma. Semin Cancer Biol. 202; 12:421–429.
4. Xiong W, Zeng ZY, Xia JH, Xia K, Shen SR, Li XL, Hu DX, Tan C, Xiang JJ, Zhou J, Deng H, Fan SQ, Li WF, et al. A susceptibility locus at chromosome 3p21 linked to familial nasopharyngeal carcinoma. Cancer Res. 2004; 64:1972–1974.
5. Bei JX, Li Y, Jia WH, Feng BJ, Zhou G, Chen LZ, Feng QS, Low HQ, Zhang H, He F, Tai ES, Kang T, Liu ET, et al. A genome-wide association study of nasopharyngeal carcinoma identifies three new susceptibility loci. Nat Genet. 2010; 42:599–603.
6. Lin JC, Jan JS, Hsu CY, Liang WM, Jiang RS, Wang WY. Phase III study of concurrent chemoradiotherapy versus radiotherapy alone for advanced nasopharyngeal carcinoma: positive effect on overall and progression-free survival. J Clin Oncol. 2003; 21:631–637.
7. Yu KH, Leung SF, Tung SY, Zee B, Chua DT, Sze WM, Law SC, Kam MK, Leung TW, Sham JS, Lee AW, Au JS, Hui EP, et al. Survival outcome of patients with nasopharyngeal carcinoma with first local failure: a study by the Hong Kong Nasopharyngeal Carcinoma Study Group. Head Neck. 2005; 27:397–405.
8. Lee N, Xia P, Quivey JM, Sultanem K, Poon I, Akazawa C, Akazawa P, Weinberg V, Fu KK. Intensity-modulated radiotherapy in the treatment of nasopharyngeal carcinoma: an update of the UCSF experience. Int J Radiat Oncol Biol Phys. 2002; 53:12–22.
9. Waldron J, Tin MM, Keller A, Lum C, Japp B, Sellmann S, van Prooijen M, Gitterman L, Blend R, Payne D, Liu FF, Warde P, Cummings B, et al. Limitation of conventional two dimensional radiation therapy planning in nasopharyngeal carcinoma. Radiother Oncol. 2003; 68:153–161.
10. Lee AW, Tung SY, Chua DT, Ngan RK, Chappell R, Tung R, Siu L, Ng WT, Sze WK, Au GK, Law SC, O’Sullivan B, Yau TK, et al. Randomized trial of radiotherapy plus concurrent-adjuvant chemotherapy vs radiotherapy alone for regionally advanced nasopharyngeal carcinoma. J. Natl Cancer Inst. 2010; 102:1188–1198.
11. Karin M, Cao Y, Greten FR, Li ZW. NF-κB in cancer: from innocent bystander to major culprit. Nat Rev Cancer. 2002; 2:301–310.
12. Viatour P, Merville MP, Bours V, Chariot A. Phosphorylation of NF-κB and IκB proteins: implications in cancer and inflammation. Trends Biochem Sci. 2005; 30:43–52.
13. Hayden MS, Ghosh S. Shared principles in NF-κB signaling. Cell. 2008; 132:344–62.
14. Rothwarf DM, Zandi E, Natoli G, Karin M. IKK-gamma is an essential regulatory subunit of the IκB kinase complex. Nature. 1998; 395:297–300.
15. Gilmore TD. Mutations in the NF-κB signaling pathway: implications for human disease. Oncogene. 2006; 25:6831–43.
16. Roy N, Deveraux Q, Takahashi R, and Reed J. The c-IAP-1 and c-IAP-2 proteins are direct inhibitors of specific caspases. EMBO J. 1997; 16:6914–6925.
17. Ashkenazi A, Pai RC, Fong S, Leung S, Lawrence DA, Marsters SA, Blackie C, Chang L, McMurtrey AE, Hebert A, DeForge L, Koumenis IL, Lewis D, et al. Safety and antitumor activity of recombinant soluble Apo2 ligand. J Clin Invest. 1995; 104:155–62.
18. Degli-Esposti MA, Dougall WC, Smolak PJ, Waugh JY, Smith CA, Goodwin RG. The novel receptor TRAIL-R4 induces NF-jB and protects against TRAIL-mediated apoptosis, yet retains an incomplete death domain. Immunity. 1997; 7:813–20.
19. Evdokiou A, Bouralexis S, Atkins GJ, Chai F, Hay S, Clayer M, Findlay DM. Chemotherapeutic agents sensitize osteogenic sarcoma cells, but not normal human bone cells, to Apo2L/TRAIL-induced apoptosis. Int J Cancer. 2002; 99:491–504.
20. Farooqi AA, Gadaleta CD, Ranieri G, Fayyaz S, Marech I. New Frontiers in Promoting TRAIL-Mediated Cell Death: Focus on Natural Sensitizers, miRNAs, and Nanotechnological Advancements. Cell Biochem Biophys. 2016; 74:3–10.
21. Herman JG, Baylin SB. Gene silencing in cancer in association with promoter hypermethylation. N Engl J Med. 2003; 349:2042–54.
22. Baylin SB, Chen WY. Aberrant gene silencing in tumor progression: implications for control of cancer. Cold Spring Harb Symp Quant Biol. 2005; 70:427–33.
23. Staber PB, Zhang P, Ye M, Welner RS, Levantini E, Di Ruscio A, Ebralidze AK, Bach C, Zhang H, Zhang J, Vanura K, Delwel R, Yang H, et al. The Runx-PU.1 pathway preserves normal and AML/ETO9a leukemic stem cells. Blood. 2014; 124:2391–9.
24. Hesson LB, Cooper WN, Latif F. Evaluation of the 3p21.3 tumour-suppressor gene cluster. Oncogene. 2007; 26:7283–7301.
25. Ito M, Ito G, Kondo M, Uchiyama M, Fukui T, Mori S, Yoshioka H, Ueda Y, Shimokata K, Sekido Y. Frequent inactivation of RASSF1A, BLU, and SEMA3B on 3p21.3 by promoter hypermethylation and allele loss in non-small cell lung cancer. Cancer Letters. 2005; 225:131–139.
26. Abe M, Ohira M, Kaneda A, Yagi Y, Yamamoto S, Kitano Y, Takato T, Nakagawara A, Ushijima T. CpG island methylator phenotype is a strong determinant of poor prognosis in neuroblastomas. Cancer Res. 2005; 65:828–834.
27. Yi Lo PH, Chung Leung AC, Xiong W, Law S, Duh FM, Lerman MI, Stanbridge EJ, Lung ML. Expression of candidate chromosome 3p21.3 tumor suppressor genes and down-regulation of BLU in some esophageal squamous cell carcinomas. Cancer Lett. 2006; 234:184–192.
28. Agathanggelou A, Dallol A, Zöchbauer-Müller S, Morrissey C, Honorio S, Hesson L, Martinsson T, Fong KM, Kuo MJ, Yuen PW, Maher ER, Minna J, Latif F. Epigenetic inactivation of the candidate 3p21.3 suppressor gene BLU in human cancers. Oncogene. 2003; 22:1580–1588.
29. Marsit CJ, Kim DH, Liu M, Hinds PW, Wiencke JK, Nelson HH, Kelsey KT. Hypermethylation of RASSF1A and BLU tumor suppressor genes in non-small cell lung cancer: implications for tobacco smoking during adolescence. Int J Cancer. 2005; 114:219–223.
30. Lai HC, Lin YW, Chang CC, Wang HC, Chu TW, Yu MH, Chu TY. Hypermethylation of two consecutive tumor suppressor genes, BLU and RASSF1A, located at 3p21.3 in cervical neoplasias. Gynecol Oncol. 2007; 104:629–635.
31. Qiu GH, Tan LK, Loh KS, Lim CY, Srivastava G, Tsai ST, Tsao SW, Tao Q. The candidate tumor suppressor gene BLU, located at the commonly deleted region 3p21.3, is an E2F-regulated, stress-responsive gene and inactivated by both epigenetic and genetic mechanisms in nasopharyngeal carcinoma. Oncogene. 2004; 23:4793–4806.
32. Whang YM, Kim YS, Kim JS, Yoo YD. RASSF1A suppresses the c-Jun-NH2-kinase pathway and inhibits cell cycle progression. Cancer Res. 2005; 65:3682–90.
33. Kim YJ, Yoon SY, Kim JT, Choi SC, Lim JS, Kim JH, Song EY, Lee HG, Choi I, Kim J. NDRG2 suppresses cell proliferation through down-regulation of AP-1 activity in human colon carcinoma cells. Int J Cancer. 2007; 124:7–15.
34. Zhang X, Liu H, Li B, Huang P, Shao J, He Z. Tumor suppressor BLU inhibits proliferation of nasopharyngeal carcinoma cells by regulation of cell cycle, c-Jun N-terminal kinase and the cyclin D1 promoter. BMC Cancer. 2012; 12:267.
35. Park ST, Byun HJ, Kim BR, Dong SM, Park SH, Jang PR, Rho SB. Tumor suppressor BLU promotes paclitaxel antitumor activity by inducing apoptosis through the down-regulation of Bcl-2 expression in tumorigenesis Biochem. Biophys Res Commun. 2013; 435:153–159.
36. Ng P, Graham FL. Construction of first-generation adenoviral vectors. Methods Mol Med. 202; 69:389–414.
37. Kagawa S, He C, Gu J, Koch P, Rha SJ, Roth JA, Curley SA, Stephens LC, Fang B. Antitumor activity and bystander effects of the tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) gene. Cancer Res. 2001; 61:3330–8.
38. Martinez-Lostao L, de Miguel D, Al-Wasaby S, Gallego-Lleyda A, Anel A. Death ligands and granulysin: mechanisms of tumor cell death induction and therapeutic opportunities. Immunother. 2015; 7:883–2.
39. Fernald K, Kurokawa M. Evading apoptosis in cancer. Trends Cell Biol. 2013; 23:620–33.
40. Wennerberg E, Sarhan D, Carlsten M, Kaminskyy VO, D’Arcy P, Zhivotovsky B, Childs R, Lundqvist A. Doxorubicin sensitizes human tumor cells to NK cell- and T-cell-mediated killing by augmented TRAIL receptor signaling. Int J Cancer. 2013; 133:1643–52.
41. Yau WL, Lung HL, Zabarovsky ER, Lerman MI, Sham JS, Chua DT, Tsao SW, Stanbridge EJ, Lung ML. Functional studies of the chromosome 3p21.3 candidate tumor suppressor gene BLU/ZMYND10 in nasopharyngeal carcinoma. Int J Cancer. 2006; 119:2821–6.
42. Ji L, Nishizaki M, Gao B, Burbee D, Kondo M, Kamibayashi C, Xu K, Yen N, Atkinson EN, Fang B, Lerman MI, Roth JA, Minna JD. Expression of several genes in the human chromosome 3p21.3 homozygous deletion region by an adenovirus vector results in tumor suppressor activities in vitro and in vivo. Cancer Res. 2002; 62:2715–20.
43. Scaffidi C, Fulda S, Srinivasan A, Friesen C, Li F, Tomaselli KJ, Debatin KM, Krammer PH, Peter ME. Two CD95 (APO-1/Fas) signaling pathways. EMBO J. 1998; 17:675–1687.
44. Li H, Zhu H, Xu CJ, Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998; 94:491–501.
45. Shelton SN, Shawgo ME, Robertson JD. Cleavage of Bid by executioner caspases mediates feed forward amplification of mitochondrial outer membrane permeabilization during genotoxic stress-induced apoptosis in Jurkat cells. J Biol. Chem. 2009; 284:1247–11255.
46. Kim H, M. Rafiuddin-Shah M, Tu HC, Jeffers JR, Zambetti GP, Hsieh JJ, Cheng EH. Hierarchical regulation of mitochondrion-dependent apoptosis by BCL-2 subfamilies. Nat Cell Biol. 2006; 8:348–1358.
47. Samraj AK, Keil E, Ueffing N, Schulze-Osthoff K, Schmitz I. Loss of caspase-9 provides genetic evidence for the type I/II concept of CD95-mediated apoptosis. J Biol Chem. 2006; 281:29652–29659.
48. Sun XM, Bratton SB, Butterworth M, MacFarlane M, Cohen GM. Bcl-2 and Bcl-xL inhibit CD95-mediated apoptosis by preventing mitochondrial release of Smac/DIABLO and subsequent inactivation of X-linked inhibitor-of-apoptosis protein. J Biol Chem. 2002; 277:11345–51.
49. Li S, Zhao Y, He X, Kim TH, Kuharsky DK, Rabinowich H, Chen J, Du C, Yin XM. Relief of extrinsic pathway inhibition by the Bid-dependent mitochondrial release of Smac in Fas-mediated hepatocyte apoptosis. J Biol Chem 2002; 277:6912–26920.
50. Micheau O, Lens S, Gaide O, Alevizopoulos K, Tschopp J. NF-κB signals induce the expression of c-FLIP. Mol Cell Biol. 2001; 21:299–5305.
51. Chang DW, Xing Z, Pan Y, Algeciras-Schimnich A, Barnhart BC, Yaish-Ohad S, Peter ME, Yang X. cFLIP L is a dual function regulator for caspase-8 activation and CD95-mediated apoptosis. EMBO J. 2002; 21:3704–3714.
52. Perez D, E White. E1A Sensitizes Cells to Tumor Necrosis Factor Alpha by Downregulating c-FLIP s. J Virol. 2003; 77:651–2662.
53. Chen LC, Chung IC, Hsueh C, Tsang NM, Chi LM, Liang Y, Chen CC, Wang LJ, Chang YS. The antiapoptotic protein, FLIP, is regulated by heterogeneous nuclear ribonucleoprotein K and correlates with poor overall survival of nasopharyngeal carcinoma patients. Cell Death Differ. 2010; 17:1463–73.
54. Cahir-McFarland ED, Carter K, Rosenwald A, Giltnane JM, Henrickson SE, Staudt LM, Kieff E. Role of NF-κ B in cell survival and transcription of latent membrane protein 1-expressing or Epstein-Barr virus latency III-infected cells. J Virol. 2004; 78:4108–19.
55. Friboulet L, Pioche-Durieu C, Rodriguez S, Valent A, Souquère S, Ripoche H, Khabir A, Tsao SW, Bosq J, Lo KW, Busson P. Recurrent overexpression of c-IAP2 in EBV-associated nasopharyngeal carcinomas: critical role in resistance to Toll-like receptor 3-mediated apoptosis. Neoplasia. 2008; 10:1183–94.
56. Cheung AK, Ko JM, Lung HL, Chan KW, Stanbridge EJ, Zabarovsky E, Tokino T, Kashima L, Suzuki T, Kwong DT, Chua D, Tsao SW, Lung ML. Cysteine-rich intestinal protein 2 (CRIP2) acts as a repressor of NF-κB-mediated proangiogenic cytokine transcription to suppress tumorigenesis and angiogenesis. Proc Natl Acad Sci U S A. 2011; 108:8390–5.
57. Lo PH, Ko JM, Yu ZY, Law S, Wang LD, Li JL, Srivastava G, Tsao SW, Stanbridge EJ, Lung ML. The LIM domain protein, CRIP2, promotes apoptosis in esophageal squamous cell carcinoma. Cancer Lett. 2012; 316:39–45.
58. Qi H, and Ohh M. The von Hippel-Lindau Tumor Suppressor Protein Sensitizes Renal Cell Carcinoma Cells to Tumor Necrosis Factor-Induced Cytotoxicity By Suppressing the Nuclear Factor-κB-Dependent Antiapoptotic Pathway. Cancer Res. 2003; 63:7076–7080.
59. Meteoglu I, Erdogdu IH, Meydan N, Erkus M, Barutca S. NF-κB expression correlates with apoptosis and angiogenesis in clear cell renal cell carcinoma tissues. J Exp Clin Cancer Res. 2008; 27:53.
60. Hu LF, Eiriksdottir G, Lebedeva T, Kholodniouk I, Alimov A, Chen F, Luo Y, Zabarovsky ER, Ingvarsson S, Klein G, Ernberg I. Loss of heterozygosity on chromosome arm 3p in nasopharyngeal carcinoma. Genes Chromosomes Cancer. 1996; 17:118–26.
61. Chen J, Fu L, Zhang LY, Kwong DL, Yan L, Guan XY. Tumor suppressor genes on frequently deleted chromosome 3p in nasopharyngeal carcinoma. Chin J Cancer. 2012; 31:215–22.
62. Sun W, Guo MM, Han P, Lin JZ, Liang FY, Tan GM, Li HB, Zeng M, Huang XM. Id-1 and the p65 subunit of NF-κB promote migration of nasopharyngeal carcinoma cells and are correlated with poor prognosis. Carcinogenesis. 2012; 33:810–7.
63. Cahir-McFarland ED, Davidson DM, Schauer SL, Duong J, Kieff E. NF-κB inhibition causes spontaneous apoptosis in Epstein-Barr virus-transformed lymphoblastoid cells.Proc Natl Acad Sci U S A. 2000; 97:6055–60.