
INTRODUCTION
Lung adenocarcinoma is a leading cause of cancerous deaths worldwide [1, 2] and great strides have been made to understand its molecular mechanism so that effective cure could be found [3–7]. Traditionally, mutations of the DNA sequence, especially those in cancer driver genes, have been heavily studied to understand the genetic basis of carcinogenesis. However, RNA editing, a post-transcriptional modification event that can flexibly and dynamically change RNA transcripts, could lead to effects similar to genomic mutations and potentially contribute to tumorigenesis as well. In mammals, RNA editing is mainly accomplished by the adenosine deaminase acting on RNA (ADAR) family of enzymes, which modifies adenosine to inosine (A -> I) [8, 9]. RNA editing is enriched in Alu elements [10], which overlaps with many introns and untranslated regions (UTRs) [11], and can modulate RNA structure, RNA splicing and transcript expression [12]. RNA editing has previously been associated with a few cancer types [12–17], such as hepatocellular carcinoma and prostate cancer. Pan-cancer characterization of editing events, including lung adenocarcinoma, has also been carried out in two recent studies [18, 19] which found that RNA editing levels were significantly higher in lung adenocarcinomas than in paired normal tissues. Han et al. also found that ADAR1 expression levels exhibited significantly higher in lung adenocarcinomas than in normal samples, while ADAR2 expression levels were in the opposite direction [19]. Moreover, ADAR1 was reported to be an oncogene undergoing gene amplification-associated activation that affects downstream RNA editing patterns and patient prognosis in non-small-cell lung cancer cells [20]. Although most RNA editing in cancer were likely to be passenger events, a small portion may act as drivers and serve as potential markers for personalized diagnosis and therapy. For example, elevated editing of AZIN1S367G in hepatocellular carcinoma has been reported as oncogenic activity thus might be a potential driver in pathogenesis [13].
To our best knowledge, RNA editome of advanced lung adenocarcinomas, especially metastatic lung adenocarcinomas, has not been systematically characterized and the function of RNA editing in lung adenocarcinomas remains largely unknown. Here, we characterized the RNA editing landscape of the matched normal, primary and metastatic tissues from 24 lung adenocarcinoma patients based on whole genome and transcriptome sequencing and detected a large number of putative RNA editing events and hyper-editing sites, i.e., tumor-specific editing sites or editing sites with significantly higher editing levels in tumors than in adjacent normal samples. We also investigated the possible role of these events in primary and metastatic lung adenocarcinomas and provided new insights to understand the molecular characterization of this disease.
RESULTS
The landscape of RNA editing in 24 lung adenocarcinoma patients
The whole genome and transcriptome sequencing data of the adjacent normal, primary and corresponding lymph node metastatic tissues from 24 Chinese lung adenocarcinoma patients were obtained in a previous study [6]. The average depth of whole genome sequencing (WGS) is 31.9×, 49.6× and 51.2× for normal, primary and metastatic tissues, and the transcriptomes were sequenced to ~77M total reads per sample. We used an in-house RNA editing calling pipeline, which is an improved version of a previously published one [21], to identify 1,071,296 RNA editing sites (RES), with an average of 14,879 per sample (range from 800 to 43,708, Supplementary Table 1). As expected, the dominant editing type is A -> G, average 91.3% in each sample (Figure 1A, Supplementary Figure 1A, Supplementary Table 2), and the proportion of this editing type in primary samples were slightly higher than those in normal or metastatic samples (Supplementary Figure 1C). Most of the editing occurred in Alu regions (average 94.5%, range from 88.1% to 98.7%, Figure 1A, Supplementary Figure 1B and Supplementary Figure 1D, Supplementary Table 3). When looking at the distribution of RNA editing across different genomic regions, it most frequently occurred in introns (average 71.5% in each sample, range from 42.9% to 89.5%), followed by 3’ UTR (average 10.5%, range from 2.1% to 33.0%) and intergenic region (average 9.7%, ranger from 4.2% to 15.4%, Figure 1A, Supplementary Figure 1B, Supplementary Table 3).
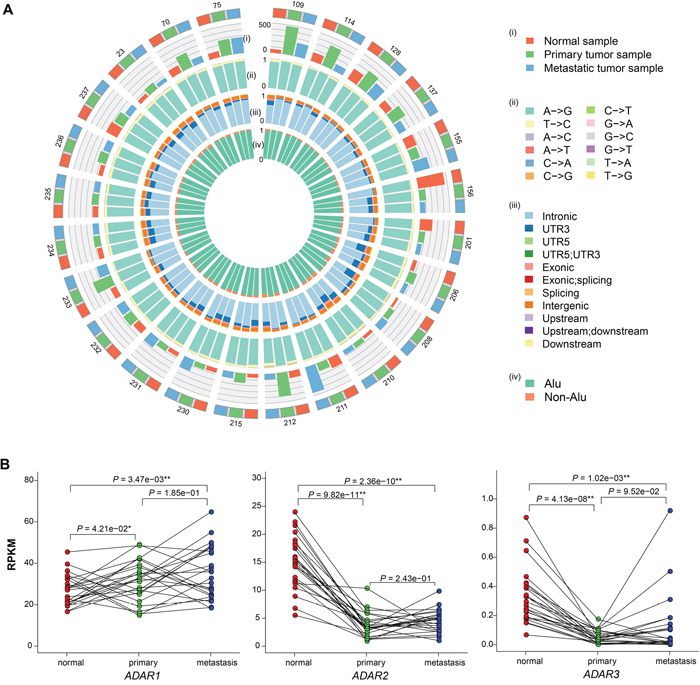
Figure 1: Summary of RNA editing events in adjacent normal, primary and metastatic samples from 24 patients. A. Circos plot depicting the landscape of RNA editing in lung adenocarcinoma. The outermost circle shows the samples for each patient and the numbers stand for the patient ID. Red, green and blue depict normal, primary and metastatic samples, respectively. The (i) and (ii) circle display the editing rate and the proportion of editing type for each sample, respectively. The (iii) and (iv) circle denote the distribution of editing sites in different genomic regions ((iii): intronic, UTR3, UTR5 etc; innermost: Alu and non-Alu). B. Comparison of ADAR family gene expression among adjacent normal, primary and metastatic samples. Red, green and blue points depict normal, primary and metastatic samples, respectively, and lines are drawn to connect samples from the same patient. **, P < 0.01; *, P < 0.05; measured by paired t-test.
To account for sequencing and expression differences among samples, we calculated the editing rate (details in Method). The average editing rate is 121.2 (range: 34.1 – 449.5) for normal samples, 190.4 (79.4 – 473.3) for primary samples and 147.2 (62.0 – 448.5) for metastatic samples (Figure 1A, Supplementary Table 1). Comparing the editing rates among the three groups, the editing rates in primary and metastatic samples are significantly higher than those in normal samples (P = 3.26e-03 and P = 3.46e-02 respectively, Wilcoxon signed rank test). Moreover, the editing rates in primary samples are significantly higher than those in metastatic samples (P = 2.45e-02, Wilcoxon signed rank test). However, no correlation was found between the editing rate and clinical information of patients or the number of SNVs (as analyzed in the previous study, Supplementary Figure 2) or tumor purity (average 0.54 in primary and 0.43 in metastatic samples, Supplementary Figure 3).
ADAR family of enzymes was found to be the primary modulator of the frequency and number of sites edited in some cancers [18, 19] and ADAR1/2 dysregulation demonstrated extremely poor prognoses in non-small-cell lung cancer [20]. Therefore, we calculated the expression of ADAR family of enzymes to investigate the correlation with editing rate (Figure 1B). Compared to normal samples, ADAR1 was expressed significantly higher in primary and metastatic samples (P = 4.21e−02 and P = 3.47e−03, paired t-test), while the opposite was found for ADAR2 and ADAR3 (P = 9.82e−11 and P = 4.13e−08 in primary, P = 2.36e−10 and P = 1.02e−03 in metastatic, paired t-test). The higher expression levels of ADAR1, which was also reported in other solid tumors [19, 22–24], may explain why more RNA editing events occurred in tumor samples than in normal samples. However, there is no significant difference of ADAR1 expression levels between primary and metastatic tumor samples, and the higher RNA editing rate in primary tumor samples could be partly due to the different tissue composition of primary and metastatic samples.
Detection of hyper-editing events
To further investigate cancer relevant editing events, we detected hyper-editing sites (tumor-specific editing sites, which only edited in tumor but not in adjacent normal, or sites with significantly higher editing levels in tumor than adjacent normal from a patient). In total, we identified 67,851 hyper-editing sites in primary tumors (hyper-PT, Supplementary Table 4) and 50,480 hyper-editing sites in metastatic tumors (hyper-MT, Supplementary Table S). These hyper-editing sites exhibited similar distribution to overall RES in terms of editing type and genomic regions (Supplementary Figure 4 versus Figure 1A, Supplementary Figure 5 and Supplementary Figure 6 versus Supplementary Figure 1). To account for possible sequencing and expression differences among samples, we also calculated a hyper-editing rate and found that hyper-editing rates of hyper-PT (average 31.3) were significantly higher than those of hyper-MT (average 22.1, P = 2.29e-02, Wilcoxon signed rank test, Supplementary Figure 4) as well.
Hyper-editing in CDS regions
We first analyzed editing in CDS regions (including core splice sites), which may change the translated amino acid sequences. Altogether, we found 213 hyper-PT and 328 hyper-MT in CDS regions. Comparing the number of hyper-PT and hyper-MT in CDS for the same patient, we found them to be similar for most patients, except three (patient ID 114, 212, 237, Figure 2A). Moreover, there was no obvious difference in the proportion of editing type and genomic distribution between hyper-PT and hyper-MT in CDS (Figure 2B, 2C and 2D). Owing to the lack of sufficient validation on non-A->G editing sites reported in previous studies [21, 22], we only included A -> G hyper-PT and hyper-MT in subsequent analyses.
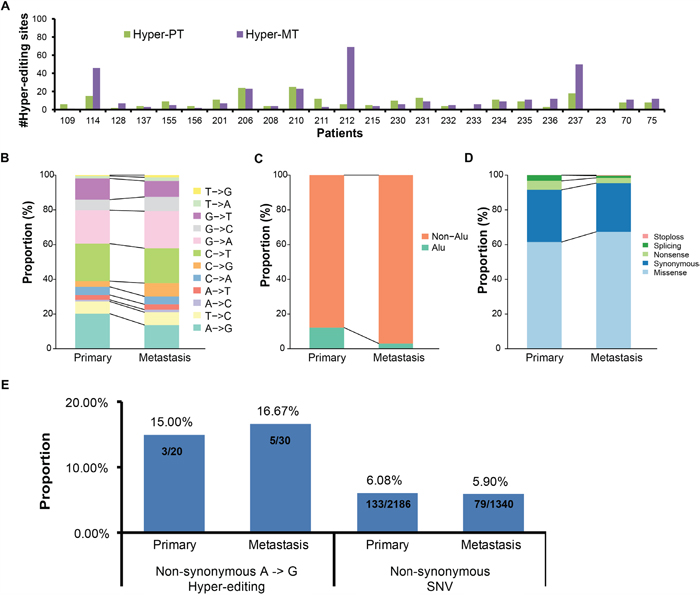
Figure 2: Comparison of hyper-editing sites in CDS regions between primary and metastatic samples. A. The distribution of hyper-editing sites in CDS regions in each sample. B-D. Histograms showing the proportion of editing types (B), the proportion of Alu region (C) and the proportion of mutation types (D) of hyper-editing sites in CDS regions between primary and metastatic samples. E. The proportion of the cancer-related genes with non-synonymous A -> G hyper-editing sites or with non-synonymous SNV.
In total, we found 46 genes with hyper-editing had non-synonymous changes, including 20 genes with A -> G hyper-PT and 30 genes with A -> G hyper-MT (only 4 genes were shared, Supplementary Table 4, 6 and 8, Supplementary Figure 7). Among them, the editing sites chr8:103841636 in AZIN1 (S367G) and chr4:2940026 in NOP14 (I779V), which were previously reported to have significantly elevated editing levels in lung adenocarcinoma from a pan-cancer analysis [19], were detected in two tumors and one tumor, respectively (Table 1). To investigate the role of non-synonymous A -> G hyper-editing, we further compared the proportion of cancer-related genes with non-synonymous A -> G hyper-editing to those with non-synonymous SNVs. We found 15.0% and 16.7% cancer-related genes with non-synonymous hyper-editing in primary and metastatic samples (Table 1), while the proportion of cancer-related genes with non-synonymous SNVs is 6.1% and 5.9% (Figure 2E). This indicates that non-synonymous hyper-editing may play as important a role as non-synonymous SNVs in lung adenocarcinoma, if not more.
Table 1: The selected genes with non-synonymous A -> G hyper-editing
Gene Name |
Chr:Pos |
Editing type |
Allele change |
Amino acid change |
Sample ID |
Class |
Description |
AZIN1 |
chr8:103841636 |
A->G |
c.A1099G |
p.S367G |
137F, 212G |
Reported |
Antizyme Inhibitor 1 |
NOP14 |
chr4:2940026 |
A->G |
c.A2335G |
p.I779V |
212F |
Reported |
NOP14 nucleolar protein |
CHD6 |
chr20:40127981 |
A->G |
c.A869G |
p.D290G |
206F |
CRRG |
Chromodomain helicase DNA binding protein 6 |
TCF3 |
chr19:1615370 |
A->G |
c.A1736G |
p.H579R |
211F |
CGC |
Transcription factor 3 |
ATP1A1 |
chr1:116931630 |
A->G |
c.A743G |
p.N248S |
114F, 114G |
CGC |
ATPase, Na+/K+ transporting, alpha 1 polypeptide |
KDM2A |
chr11:67017933 |
A->G |
c.A1115G |
p.H372R |
235G |
CRRG |
Lysine (K)-specific demethylase 2A |
CSF3R |
chr1:36939477 |
A->G |
c.A373G |
p.I125V |
128G |
CGC |
Colony stimulating factor 3 receptor (granulocyte) |
MSH2 |
chr2:47656925 |
A->G |
c.A1121G |
p.Q374R |
212G |
CGC |
MutS Homolog 2 |
NUMA1 |
chr11:71725974 |
A->G |
c.A2575G |
p.I859V |
210G |
CGC |
Nuclear mitotic apparatus protein 1 |
Note: Cancer Gene Census (CGC); Chromatin remodeling related genes (CRRG). “Reported” stands for gene has been previously reported. Cancer-related genes were found from GCG or TCGA driver genes list or CRRG list.
Interestingly, we found that four cancer-related genes (CSF3R, MSH2, NUMA1 and KDM2A) only hyper-edited in metastatic but not in primary samples (Table 1), suggesting that these alterations may promote metastasis development. Of which, CSF3R, which is a member of the family of cytokine receptors and may also function in some cell surface adhesion or recognition processes, was reported to be related to tumor growth and metastasis [25]. MSH2, a component of the post-replicative DNA mismatch repair system, frequently mutated in melanoma brain metastases but not in primary melanomas, indicating the high genomic instability of metastatic samples [26]. Moreover, MLH1/MSH2 defects were strongly associated with a decreased likelihood of lymph node and distant organ metastases in sporadic colorectal cancer [27]. To further investigate the role of non-synonymous hyper-editing genes, we performed pathway enrichment analysis of these genes and found 3 genes (CSF3R, IL12RB1 and STAT2) in Jak-STAT signaling pathway only hyper-edited in metastatic but not in primary samples (Supplementary Figure 8, Supplementary Table 9). Activated Jak-STAT pathway could promote cellular invasion and migration in cancer, such as in hepatocellular carcinoma [28] and head and neck squamous cell carcinoma [29]. Some drugs have been reported to decrease proliferation and metastatic behavior of tumor cells by modulating Jak-STAT signaling pathway, such as guggulsterone for pancreatic cancer [30].
Hyper-editing in non-coding regions
In our samples, 95.7% of RNA editing took place in non-coding regions, especially introns and UTRs, where they could modulate splicing or RNA structure or gene expression. To investigate the potential correlation between editing and RNA splicing, we first used SPIDEX [31] to predict A -> G hyper-editing sites that may influence splicing. 20 A -> G hyper-PT and 16 A -> G hyper-MT were found, with 77.8% occurring in non-coding regions (Supplementary Table 10, Supplementary Figure 7). Of which, HMOX2 (Heme Oxygenase 2), which belongs to the heme oxygenase family and is related to heme catabolism, was most frequently edited (detected in four primary and three metastatic samples) and harbored three hyper-editing sites (chr16: 4533677, 4533713, 4533730, all located in 5’ UTR of exon3). To investigate the relationship between these three hyper-editing sites and the splicing patterns of HMOX2 (Figure 3A), we calculated the percent spliced in (PSI, detailed in Method) of exon 3 in HMOX2. We found that the PSIs of tumor samples with at least one of three hyper-editing sites were significantly higher than those without them (P = 4.39e−02, Wilcoxon signed rank test, Figure 3B). Moreover, both the expression of exon3 and the transcript that contains exon3 (ENST00000458134) exhibited the same pattern (P = 3.24e−02 and P = 1.55e−02, Wilcoxon signed rank test, Figure 3C and 3D). In addition, we found a weak positive correlation between PSI and the editing index (see Method) of exon3 (Pearson correlation = 0.23, Figure 3E). These results suggested that hyper-editing in 5’ UTR of exon3 could increase the relative usage of exon3 and the expression of the corresponding transcript, and the higher editing index of exon3, the greater the effect.
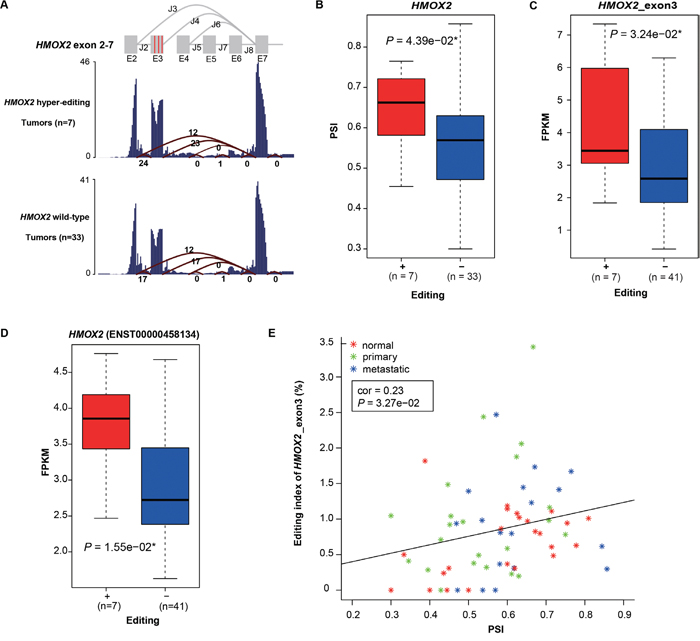
Figure 3: Hyper-editing induced splicing aberration of HMOX2. A. The average RNA-seq read coverage and junction counts are shown. Three red vertical lines in exon3 represent three A -> G hyper-editing sites (from left to right, 4533677, 4533713 and 4533730 in chr16) predicted to affect splicing. Samples with the total number of the splice junction reads supporting J2, J3 and J4 less than 10 are excluded. B and C. Comparison of PSI (B) and expression of exon3 (C) in HMOX2 between two subgroups: tumor samples with at least one of the three hyper-editing sites in HMOX2 (“+”) versus those with none (“-”). Wilcoxon rank sum test was used. D. Box plot showing the expression levels of the corresponding transcript between “+” and “-” groups. P value was calculated by t-test. E. The correlation between the editing index of exon 3 and PSI. Pearson's product-moment correlation was used.
To further discover functional editing sites in non-coding regions, we applied CADD [32] to predicted deleterious editing events. 32 A -> G hyper-editing events were predicted to be deleterious, including 14 hyper-PT and 18 hyper-MT, with 59.4% in non-coding regions (Supplementary Table 11, Supplementary Figure 7). Of which, chr17:8130348, located in the 3’ UTR of CTC1 (CTS Telomere Maintenance Complex Component 1), is the most frequent deleterious hyper-editing site (detected in four primary and one metastatic samples). CTC1 is a component of the CST complex and this complex plays an essential role in protecting telomeres from degradation. To investigate the role of this event, we first compared the editing level of chr17:8130348 among three sample groups, and found that the editing levels in tumor samples were significantly higher than those in normal samples (P = 6.18e−03 and P = 2.10e−02), while the editing levels in primary samples were also higher than metastatic samples (P = 4.90e−02, paired t-test, Figure 4A). Interestingly, the expression of CTC1 in primary samples were significantly lower than normal and metastatic samples (P = 2.44e−03 and P = 6.35e−05, paired t-test, Figure 4B). Moreover, a negative correlation between the editing level of chr17:8130348 and the expression of CTC1 were found (Pearson correlation = -0.38, Figure 4C), which suggests possible regulation of decreasing CTC1 expression through RNA editing. Among cancer patients, those of early stage showed higher CTC1 expression than those of late stage (P = 3.51e−03, t-test, Figure 4D). Clinically, CTC1 down-regulation demonstrated poor recurrence-free survival probability (Log-rank P = 2.89e−02, Figure 4E). These results suggest that when deleterious hyper-editing do not alter amino acid sequences, they may affect gene expression and consequently promote tumor progression or influence clinical outcomes in lung adenocarcinoma.
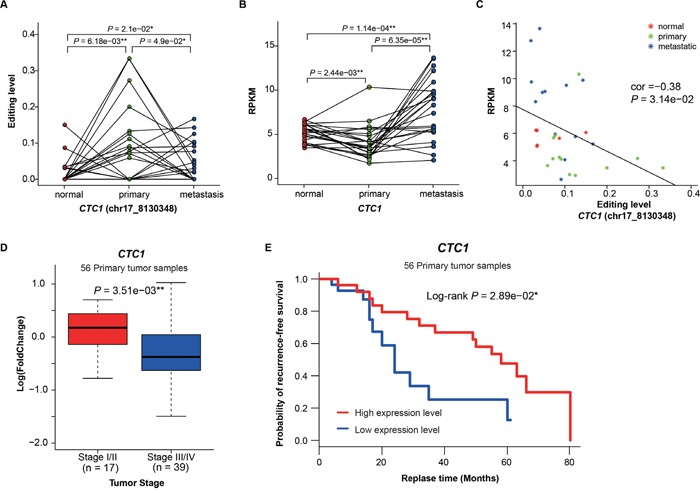
Figure 4: Functional analysis of CTC1 with hyper-editing. A. The editing levels of the deleterious site (chr17:8130348) in CTC1 among normal, primary and metastatic samples. B. The expression of CTC1 among three sample groups. C. The correlation between the editing level of chr17:8130348 and the expression of CTC1. D. CTC1 expression versus tumor stages (P value was calculated by the t-test). E. Kaplan-Meier survival curves showing the relationship between recurrence-free survival probability and CTC1 expression (low: less than median; high: greater than median).
As the above results showed, editing in UTRs, which contain binding sites for regulatory proteins and microRNAs, may induce differential gene expression in lung adenocarcinoma. To further investigate the association between hyper-editing events in UTRs and gene expression, we classified samples into two subgroups: edit+, which contains any A -> G hyper-editing sites in UTRs of a given gene; edit-, contrasting to edit+. Then we calculated the significance of differential expression between the samples of edit+ and edit- for each gene, requiring each group to have at least four samples. In total, we identified 39 and 30 genes having significant expression differences in primary and metastatic samples, respectively (using a threshold of P <= 0.05, t-test, Supplementary Table 12). Among the differentially expressed genes in primary samples, 7 of 39 genes (VHL, GNE, POLH, MAPK13, NOP14, INADL, CDC5L) were reported to be cancer-related in the literature (Supplementary Figure 9, Supplementary Table 13). Of which, VHL (Von Hippel-Lindau syndrome) is a tumor suppressor and expressed significantly lower in edit+ primary samples than in edit- samples (P = 3.78e−02, t-test, Figure 5A). Abnormality of the VHL has been reported to be linked to the development of renal carcinomas [33, 34] and hemangioblastomas [35]. In addition, GNE (Glucosamine (UDP-N-Acetyl)-2-Epimerase/N-Acetylmannosamine Kinase) expressed significantly lower in edit+ primary samples than edit- samples (P = 2.81e−02, t test, Figure 5B). Clinically, we found patients with early stage showed higher GNE expression than those with late stage (P = 4.84e−02, t-test, Figure 5C). We also found patients with low GNE expression levels had poor recurrence-free survival probability (Log-rank P = 1.02e−02, Figure 5D). GNE dysregulation occurred predominantly in pancreatic cancer but also in other malignancies [36]. Among the differentially expressed genes in metastatic samples, we found 7 of 30 genes (MAPK13, XIAP, AHR, SPN, TER1, EMP2, NDUFC2) were cancer-related (Supplementary Figure 10, Supplementary Table 13). Of which, only one, MAPK13 (Mitogen-Activated Protein Kinase 13), was expressed significantly higher in both edit+ primary and metastatic samples than in corresponding edit- samples (Figure 5E, P = 3.3e−02 and P = 1.84e−02, t-test). MAPK13 is a component of the mitogen-activated protein (MAP) kinase family and plays an important role in the development of cancer, such as cholangiocarcinoma [37]. The remaining metastasis-specific genes could be related to metastasis development. Indeed, we found that XIAP (X-linked inhibitor of apoptosis), expressed significantly lower in edit+ than in edit- metastatic samples (P = 3.27e−02, t-test, Supplementary Figure 10), were reported to promote metastasis or tumor recurrence in hepatocellular carcinoma [38] and in papillary thyroid carcinoma [39], and AHR, expressed significantly higher in edit+ than in edit- metastatic samples (P = 5.7e−03, t-test, Supplementary Figure 10), were also reported to be associated with metastasis and may be a potential therapeutic target in the treatment of metastatic breast cancer [40].
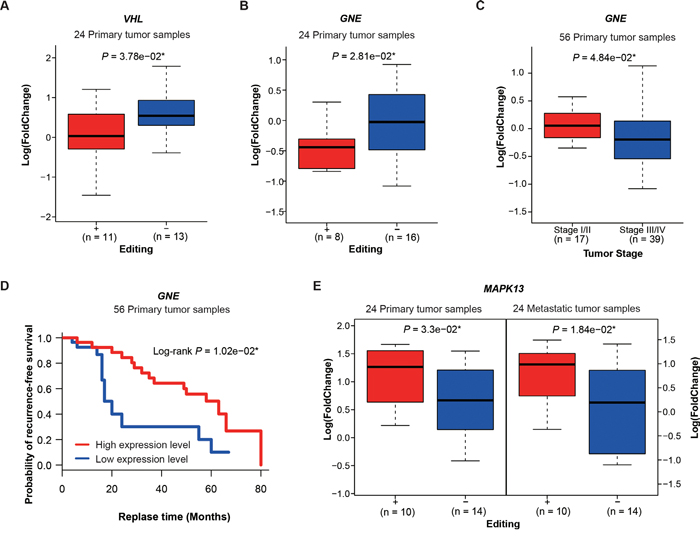
Figure 5: Hyper-editing and differential gene expression. A. Differential expression of VHL between edit+ and edit- primary samples. B. Differential expression of GNE between edit+ and edit- primary samples. C. GNE expression versus tumor stages D. Kaplan-Meier survival curves showing the relationship between recurrence-free survival probability and GNE expression. E. Differential expression of MAPK13 in edit+ and edit- groups for primary and metastatic samples. P values are calculated by t-test.
DISCUSSION
The pathogenicity of advanced lung adenocarcinomas, especially metastatic lung adenocarcinomas, remains largely unknown and RNA editing is an important source of transcriptome diversification in cancer. This study represents a first systematic investigation and characterization of RNA editome in primary and metastatic lung adenocarcinomas based on matched whole genome and transcriptome sequencing data, where we identified 1,071,296 RES, 67,851 hyper-PT and 50,480 hyper-MT sites. We found that editing events played a role in lung adenocarcinomas via changing amino acids, modulating alternative splicing patterns, such as in 5’ UTR of HMOX2, or affecting the expression of genes related to tumor stage and recurrence, such as in 3’ UTRs of CTC1 and GNE. Furthermore, by investigating hyper-edited genes only appeared in metastatic but not primary samples, we discovered genes and pathways that contribute to metastasis development, such as MSH2 and XIAP.
The role of editing events in cancer is still somewhat controversial and further investigations are warranted. One possible role of editing is to regulate alternative splicing. Alternative splicing and RNA editing, which are different post-transcriptional events, have the potential to generate many different mRNAs from the same gene, thus increasing the transcriptome and proteome diversity [41]. Editing could modulate alternative splicing by multiple means: creating the canonical 5’ and 3’ dinucleotide recognition sequences, destroying the 3’ recognition sequence or the internal branch point adenosine, targeting splicing enhancer or suppressor sequences [12]. Currently, little is known about this regulatory mechanism in cancer. A study identified that RNA editing can serve as a mechanism for regulating alternative splicing of ADAR2 in rat [42]. Evidence also exists to indicate a relationship between TPH2 (Tryptophan hydroxylase 2) alternative splicing and RNA editing in psychiatric diseases [43]. In acute myeloid leukemia, editing in the putative branch site of tumor suppressor PTPN6 mRNA, which leads to aberrant intron retention, was suggested to account for PTPN6 functional insufficiency [44]. In this study, we also found that tumors with hyper-editing events in HMOX2 were related to the relative usage of exon and the expression of the corresponding transcript. Although there are no studies showing the relationship between HMOX2 and cancer yet, HMOX1, a paralog of HMOX2, was known as a poor prognostic predictor and its over-expression may increase the metastatic potential of non-small cell lung cancer [45]. Interaction between RNA editing and alternative splicing, which can create a multitude of functionally distinct protein isoforms, might play a crucial role in tumorigenesis and joint analysis of both processes could be a new trend of cancer research.
RNA editing is dynamic and flexible and the editing levels may vary over time and tumor stages, which could be used to investigate cancer progress [19]. Recently, elevated editing of AZIN1 in hepatocellular carcinoma was reported to be related to tumor stages [13]. In this study, we compared RNA editome of primary and metastatic samples and found that RNA editing was significantly more frequent in primary than in paired metastatic samples. We also found a few genes affected by hyper-editing could contribute to metastasis development. Future large-scale studies of lung adenocarcinoma samples with more tumor stages and more complete clinical information should help us to better understand the relationship between RNA editing and lung tumor progression. Ultimately, in-depth functional analysis of RNA editing in cancer will further our understanding of the molecular characteristic of this complex disease and may provide new insights for effective personalized therapies.
MATERIALS AND METHODS
Sequencing data
We obtained the BWA-aligned DNA sequencing data and RNA sequencing data (fastq files, Illumina sequencing) from the adjacent normal tissue, primary tumor and corresponding lymph node metastases tissue of 24 Chinese lung adenocarcinoma patients in a previous study [6]. The method of sampling and sequencing were described in this previous study.
Data preprocessing and reads mapping
The raw reads of RNA sequencing data filtering process as follows: (1) Remove reads with sequence adaptors. (2) Remove reads in which unknown bases are more than 10%. (3) Remove low-quality reads, which have >50% in one read. Tophat2 were chose as the mapper for clean RNA-seq reads due to its high accuracy of alignment. After mapping reads to the reference genome (hg19), Picard (v1.84; http://broadinstitute.github.io/picard/) and GATK (v2.8-1) software were used to remove identical reads (PCR duplicates) and recalibrate base quality, respectively.
In-house RNA editing sites calling pipeline
We used an in-house RNA editing sites calling pipeline, which is an improved version of a previously published one [21], to identify candidate editing sites. The basic principle for identifying an RNA-editing site is that the site must be homozygous for gDNA, while at the same time displaying a mismatch in RNA [46]. Firstly, We summarized the base calls of pre-processed aligned RNA-reads to the human reference in pileup format. Secondly, the initially identified editing sits were then filtered by the following quality-aware steps: (1) The depth, base quality, mapping quality and the frequency of variation were taken into account to do a basic filtering. (2) Statistical test based on binomial distribution B(n, p) were used to distinguish true variants from sequencing errors on every mismatch site [46], where p denotes the background mismatch rate of each transcriptome sequencing, and n denotes sequencing depth on this site. (3) Discard the sites present in dbsnp138. (4) Estimated strand bias and filtered out variants with strand bias based on two-tailed Fisher’s exact test. (5) Estimated and filtered out variants with position bias, such as sites at 3’-end or at 5’-end. (6) Discarded the variation site in simple repeat region or homopolymer region or splicing region. Finally, we summarized the candidate editing sites to DNA bam in pileup format, and only the site was homozygous in gDNA will be kept as a true editing site. Ultimately, all true editing sites were met minimal editing level of 10%, minimal edit bases of 3 and minimal depth of 4×.
Hyper-editing sites and hypo-editing sites calling
For each paired tumor-normal samples, hyper-editing sites were detected in either case as follows. 1) Tumor-specific editing sites, which the sites only edited in tumor but not in adjacent normal from a patient. Namely, a given site only found in tumor samples with a minimal editing level of 10%, minimal edit bases of 3 and minimal depth of 6×, while paired normal samples were covered at least 6× without carrying any edit base. 2) The site showed significantly higher editing level in tumor than adjacent normal from a patient. The difference of editing level must be more than 0.2, and show significantly higher editing level in tumor than in adjacent normal samples (P <= 0.05, fisher test).
Similarly, hypo-editing site was found only in normal samples with a minimal editing level of 10%, minimal edit bases of 3 and minimal depth of 6×, while paired tumor samples were covered at least 6x without carrying any edit base. Or the difference of editing level must be less than -0.2, and show significantly higher editing level in normal than in tumor samples (P <= 0.05, fisher test). The function of hypo-editing sites (Supplementary Table 5 and Supplementary Table 7) were not well known and those sites were ignored in this paper.
Definition
Editing rate is calculated as the total number of editing sites divided by the total length of transcriptome sequencing (minimal covered depth >=4, in Mega base) for a sample. Hyper-editing rate is defined to be the total number of hyper-editing sites divided by the total length of transcriptome sequencing, while hypo-editing rate was calculated as the total number of hypo-editing sites divided by the total length of transcriptome sequencing.
Editing level of a given editing sites is calculated as the variant-supporting reads divided by the total reads. For each paired tumor-normal samples, the difference in editing level of a given site for a patient is defined to be the editing level of this site in tumor sample minus the editing level in normal sample.
Gene expression and different expression
RPKM (reads per kilobase per million mapped reads) was used to calculate the gene expression and the formula is shown as follows:
In which, C is the number of uniquely mapping reads to a given gene, N is the total number of reads that are uniquely aligned to all genes and L stands for the total length of exons from the given gene.
To measure change in the expression level of a gene, different expression of a given gene was calculated as the log (fold change), and the formula is shown as follows:
Of which, RPKMtumor stands for the RPKM of a given gene in tumor sample and RPKMnormal is the RPKM of this gene in normal sample.
Correlation between editing level and expression of gene
For a given editing site, Pearson's product-moment correlation was used to calculated the correlation between editing level and expression of gene. Samples with covered reads lower than 4 and the editing level equal to zero were excluded from this analysis.
Editing-induced splicing aberration
We used SPIDEX [31], which a comprehensive set of mutations and their predicted effects on RNA splicing across the entire human genome, to predict the editing sites with effects on splicing. The dataset was downloaded from the website (http://www.deepgenomics.com/spidex-noncommercial-download). We selected the editing sites with the Z-score >= 2 or Z-score <= -2, which were predicted to more higher effect on splicing regulation.
Analysis of splice variants, the splice junction counts of HMOX2 and the FPKM of exon bins can be performed by SGSeq software package (v1.4.3) available from Bioconductor. We predicted exons and splice junctions from RNA BAM files for given genes using parameters alpha = 1, psi = 0, beta = 0.2, gamma = 0.2.
The percent spliced in (PSI) of HMOX2 was calculated by the formula as follows:
In which, J2 is the splice junction counts of the junction between exon 2 and exon 3 of HMOX2. J3 stands for the splice junction counts of the junction between exon 2 and exon 7. J4 is the splice junction counts of the junction between exon 3 and exon 7 of this gene.
The editing index of exon 3 in HMOX2 measures the averaged editing level across exon 3 region, weighted by their expression. It may be quantified by the ratio of the read number of A-to-G/C/T editing sites with effects on splicing to the total number of reads—nucleotides aligned to a genomic adenosine within exon 3 region. Sites with covered reads lower than 4 were excluded from this analysis.
The deleterious editing sites
Combined Annotation Dependent Depletion (CADD, versions 1.3) is a tool for scoring the deleteriousness of single nucleotide variants in the human genome based on integrating multiple annotations into one metric by contrasting variants that survived natural selection with simulated mutations [32]. The CADD scores data can be downloaded from the website (http://cadd.gs.washington.edu/download). In our work, the editing sites with the PHRED-like scaled C-score >= 20, indicated that these sites are predicted to be the 1% most deleterious substitutions, were detected to be the deleterious editing sites.
Cancer-related genes list
Cancer-related genes can be found from GCG (Cancer Gene Census) genes list or TCGA driver genes list or chromatin remodeling related genes list. CGC genes [47] can be found in the website (http://cancer.sanger.ac.uk/census) and the TCGA pan-cancer driver genes were found in dataset of a previous paper [48]. All chromatin remodeling related genes were found in three databases: HIstome (The Histone Infobase, http://www.actrec.gov.in/histome/), EpiFactors (http://epifactors.autosome.ru/) and CREMOFAC (Chromatin Remodeling Factors, http://www.jncasr.ac.in/cremofac/menuframe.html) database.
Pathway enrichment analysis
Editing genes were use to KEGG pathway enrichment analysis by GSEA database (http://www.broadinstitute.org/gsea/index.jsp). Pathways, which the FDR (False discovery rate) q-value was below 0.05, were selected. To further investigate the role of non-synonymous hyper-editing genes, we performed pathway enrichment analysis of these genes. Two pathways were enriched in metastatic samples, while no pathway enriched in primary samples.
Survival analysis and clinical association analysis
According to the median, we classified 56 primary tumors (including additional 32 primary tumor in Wu et al. paper, the transcriptome data by Ion Proton sequencing) into two groups: high expression level (greater than median) and low expression level (less than median). Kaplan-Meier survival analysis using R package “survival” (http://cran.r-project.org/web/packages/survival/index.html). Log-rank test was used to compare the survival distribution of the above two subgroups.
To analyze the correlation between the expression of a given gene and clinical information, gender, smoking status, age and tumor stages were take into account. T test were used.
ACKNOWLEDGMENTS
We thank Si Qiu (BGI-shenzhen), Xulian Shi (BGI-shenzhen) and Fuqiang Li (BGI-shenzhen) for their valuable suggestions. LJL would like to acknowledge the support of NVIDIA Foundation’s Compute the Cure cancer research grant.
CONFLICTS OF INTEREST
The authors declare no competing financial interests.
Authors’ contributions
LP and DL initiated and designed the study. HX and DL built the RNA editing calling pipeline. LP and HS carried out the data analysis, with valuable suggestions from KW, JH, DX and LJL. JR drew most figures in this paper. LP and LJL wrote the manuscript, which was further revised by DL and KW. All authors read and approved the final manuscript.
REFERENCES
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015; 65: 5-29. doi: 10.3322/caac.21254.
2. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015; 65: 87-108. doi: 10.3322/caac.21262.
3. Ding L, Getz G, Wheeler DA, Mardis ER, McLellan MD, Cibulskis K, Sougnez C, Greulich H, Muzny DM, Morgan MB, Fulton L, Fulton RS, Zhang Q, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008; 455: 1069-75. doi: 10.1038/nature07423.
4. Imielinski M, Berger AH, Hammerman PS, Hernandez B, Pugh TJ, Hodis E, Cho J, Suh J, Capelletti M, Sivachenko A, Sougnez C, Auclair D, Lawrence MS, et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell. 2012; 150: 1107-20. doi: 10.1016/j.cell.2012.08.029.
5. Cancer Genome Atlas Research N. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014; 511: 543-50. doi: 10.1038/nature13385.
6. Wu K, Zhang X, Li F, Xiao D, Hou Y, Zhu S, Liu D, Ye X, Ye M, Yang J, Shao L, Pan H, Lu N, et al. Frequent alterations in cytoskeleton remodelling genes in primary and metastatic lung adenocarcinomas. Nat Commun. 2015; 6: 10131. doi: 10.1038/ncomms10131.
7. Weir BA, Woo MS, Getz G, Perner S, Ding L, Beroukhim R, Lin WM, Province MA, Kraja A, Johnson LA, Shah K, Sato M, Thomas RK, et al. Characterizing the cancer genome in lung adenocarcinoma. Nature. 2007; 450: 893-8. doi: 10.1038/nature06358.
8. Bass BL. RNA editing by adenosine deaminases that act on RNA. Annu Rev Biochem. 2002; 71: 817-46. doi: 10.1146/annurev.biochem.71.110601.135501.
9. Bass BL, Weintraub H. An unwinding activity that covalently modifies its double-stranded RNA substrate. Cell. 1988; 55: 1089-98.
10. Levanon EY, Eisenberg E, Yelin R, Nemzer S, Hallegger M, Shemesh R, Fligelman ZY, Shoshan A, Pollock SR, Sztybel D, Olshansky M, Rechavi G, Jantsch MF. Systematic identification of abundant A-to-I editing sites in the human transcriptome. Nat Biotechnol. 2004; 22: 1001-5. doi: 10.1038/nbt996.
11. Zinshteyn B, Nishikura K. Adenosine-to-inosine RNA editing. Wiley Interdiscip Rev Syst Biol Med. 2009; 1: 202-9. doi: 10.1002/wsbm.10.
12. Dominissini D, Moshitch-Moshkovitz S, Amariglio N, Rechavi G. Adenosine-to-inosine RNA editing meets cancer. Carcinogenesis. 2011; 32: 1569-77. doi: 10.1093/carcin/bgr124.
13. Chen L, Li Y, Lin CH, Chan TH, Chow RK, Song Y, Liu M, Yuan YF, Fu L, Kong KL, Qi L, Li Y, Zhang N, et al. Recoding RNA editing of AZIN1 predisposes to hepatocellular carcinoma. Nat Med. 2013; 19: 209-16. doi: 10.1038/nm.3043.
14. Mo F, Wyatt AW, Sun Y, Brahmbhatt S, McConeghy BJ, Wu C, Wang Y, Gleave ME, Volik SV, Collins CC. Systematic identification and characterization of RNA editing in prostate tumors. PLoS One. 2014; 9: e101431. doi: 10.1371/journal.pone.0101431.
15. Han SW, Kim HP, Shin JY, Jeong EG, Lee WC, Kim KY, Park SY, Lee DW, Won JK, Jeong SY, Park KJ, Park JG, Kang GH, et al. RNA editing in RHOQ promotes invasion potential in colorectal cancer. J Exp Med. 2014; 211: 613-21. doi: 10.1084/jem.20132209.
16. Fumagalli D, Gacquer D, Rothe F, Lefort A, Libert F, Brown D, Kheddoumi N, Shlien A, Konopka T, Salgado R, Larsimont D, Polyak K, Willard-Gallo K, et al. Principles Governing A-to-I RNA Editing in the Breast Cancer Transcriptome. Cell Rep. 2015; 13: 277-89. doi: 10.1016/j.celrep.2015.09.032.
17. Qin YR, Qiao JJ, Chan TH, Zhu YH, Li FF, Liu H, Fei J, Li Y, Guan XY, Chen L. Adenosine-to-inosine RNA editing mediated by ADARs in esophageal squamous cell carcinoma. Cancer Res. 2014; 74: 840-51. doi: 10.1158/0008-5472.CAN-13-2545.
18. Han L, Diao L, Yu S, Xu X, Li J, Zhang R, Yang Y, Werner HM, Eterovic AK, Yuan Y, Li J, Nair N, Minelli R, et al. The Genomic Landscape and Clinical Relevance of A-to-I RNA Editing in Human Cancers. Cancer Cell. 2015; 28: 515-28. doi: 10.1016/j.ccell.2015.08.013.
19. Paz-Yaacov N, Bazak L, Buchumenski I, Porath HT, Danan-Gotthold M, Knisbacher BA, Eisenberg E, Levanon EY. Elevated RNA Editing Activity Is a Major Contributor to Transcriptomic Diversity in Tumors. Cell Rep. 2015; 13: 267-76. doi: 10.1016/j.celrep.2015.08.080.
20. Anadon C, Guil S, Simo-Riudalbas L, Moutinho C, Setien F, Martinez-Cardus A, Moran S, Villanueva A, Calaf M, Vidal A, Lazo PA, Zondervan I, Savola S, et al. Gene amplification-associated overexpression of the RNA editing enzyme ADAR1 enhances human lung tumorigenesis. Oncogene. 2016; 35: 4407-13. doi: 10.1038/onc.2015.469.
21. Peng Z, Cheng Y, Tan BC, Kang L, Tian Z, Zhu Y, Zhang W, Liang Y, Hu X, Tan X, Guo J, Dong Z, Liang Y, et al. Comprehensive analysis of RNA-Seq data reveals extensive RNA editing in a human transcriptome. Nat Biotechnol. 2012; 30: 253-60. doi: 10.1038/nbt.2122.
22. Kang L, Liu X, Gong Z, Zheng H, Wang J, Li Y, Yang H, Hardwick J, Dai H, Poon RT, Lee NP, Mao M, Peng Z, et al. Genome-wide identification of RNA editing in hepatocellular carcinoma. Genomics. 2015; 105: 76-82. doi: 10.1016/j.ygeno.2014.11.005.
23. Pilarsky C, Wenzig M, Specht T, Saeger HD, Grutzmann R. Identification and validation of commonly overexpressed genes in solid tumors by comparison of microarray data. Neoplasia. 2004; 6: 744-50. doi: 10.1593/neo.04277.
24. Jayan GC, Casey JL. Increased RNA editing and inhibition of hepatitis delta virus replication by high-level expression of ADAR1 and ADAR2. J Virol. 2002; 76: 3819-27.
25. Agarwal S, Lakoma A, Chen Z, Hicks J, Metelitsa LS, Kim ES, Shohet JM. G-CSF Promotes Neuroblastoma Tumorigenicity and Metastasis via STAT3-Dependent Cancer Stem Cell Activation. Cancer Res. 2015; 75: 2566-79. doi: 10.1158/0008-5472.CAN-14-2946.
26. Korabiowska M, Konig F, Verheggen R, Schlott T, Cordon-Cardo C, Romeike B, Brinck U. Altered expression and new mutations in DNA mismatch repair genes MLH1 and MSH2 in melanoma brain metastases. Anticancer Res. 2004; 24: 981-6.
27. Park JW, Chang HJ, Jung KH, Kim DY, Sohn DK, Han KS, Hong CW, Lim S-B, Choi HS, Jeong S-Y. Clinicopathologic features of sporadic colorectal cancer with MLH1/MSH2 loss of expression-reduced likelihood of metastases. Journal of the Korean Society of Coloproctology. 2008; 24: 175-83.
28. Saxena NK, Sharma D, Ding X, Lin S, Marra F, Merlin D, Anania FA. Concomitant activation of the JAK/STAT, PI3K/AKT, and ERK signaling is involved in leptin-mediated promotion of invasion and migration of hepatocellular carcinoma cells. Cancer Res. 2007; 67: 2497-507. doi: 10.1158/0008-5472.CAN-06-3075.
29. Lai SY, Childs EE, Xi S, Coppelli FM, Gooding WE, Wells A, Ferris RL, Grandis JR. Erythropoietin-mediated activation of JAK-STAT signaling contributes to cellular invasion in head and neck squamous cell carcinoma. Oncogene. 2005; 24: 4442-9. doi: 10.1038/sj.onc.1208635.
30. Macha MA, Rachagani S, Gupta S, Pai P, Ponnusamy MP, Batra SK, Jain M. Guggulsterone decreases proliferation and metastatic behavior of pancreatic cancer cells by modulating JAK/STAT and Src/FAK signaling. Cancer Lett. 2013; 341: 166-77. doi: 10.1016/j.canlet.2013.07.037.
31. Xiong HY, Alipanahi B, Lee LJ, Bretschneider H, Merico D, Yuen RK, Hua Y, Gueroussov S, Najafabadi HS, Hughes TR, Morris Q, Barash Y, Krainer AR, et al. RNA splicing. The human splicing code reveals new insights into the genetic determinants of disease. Science. 2015; 347: 1254806. doi: 10.1126/science.1254806.
32. Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014; 46: 310-5. doi: 10.1038/ng.2892.
33. Shen C, Kaelin WG, Jr. The VHL/HIF axis in clear cell renal carcinoma. Semin Cancer Biol. 2013; 23: 18-25. doi: 10.1016/j.semcancer.2012.06.001.
34. Vanharanta S, Shu W, Brenet F, Hakimi AA, Heguy A, Viale A, Reuter VE, Hsieh JJ, Scandura JM, Massague J. Epigenetic expansion of VHL-HIF signal output drives multiorgan metastasis in renal cancer. Nat Med. 2013; 19: 50-6. doi: 10.1038/nm.3029.
35. Shankar GM, Taylor-Weiner A, Lelic N, Jones RT, Kim JC, Francis JM, Abedalthagafi M, Borges LF, Coumans JV, Curry WT, Nahed BV, Shin JH, Paek SH, et al. Sporadic hemangioblastomas are characterized by cryptic VHL inactivation. Acta Neuropathol Commun. 2014; 2: 167. doi: 10.1186/s40478-014-0167-x.
36. Kemmner W, Kessel P, Sanchez-Ruderisch H, Moller H, Hinderlich S, Schlag PM, Detjen K. Loss of UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase (GNE) induces apoptotic processes in pancreatic carcinoma cells. FASEB J. 2012; 26: 938-46. doi: 10.1096/fj.11-186700.
37. Tan FL, Ooi A, Huang D, Wong JC, Qian CN, Chao C, Ooi L, Tan YM, Chung A, Cheow PC, Zhang Z, Petillo D, Yang XJ, et al. p38delta/MAPK13 as a diagnostic marker for cholangiocarcinoma and its involvement in cell motility and invasion. Int J Cancer. 2010; 126: 2353-61. doi: 10.1002/ijc.24944.
38. Shi YH, Ding WX, Zhou J, He JY, Xu Y, Gambotto AA, Rabinowich H, Fan J, Yin XM. Expression of X-linked inhibitor-of-apoptosis protein in hepatocellular carcinoma promotes metastasis and tumor recurrence. Hepatology. 2008; 48: 497-507. doi: 10.1002/hep.22393.
39. Gu LQ, Li FY, Zhao L, Liu Y, Chu Q, Zang XX, Liu JM, Ning G, Zhao YJ. Association of XIAP and P2X7 receptor expression with lymph node metastasis in papillary thyroid carcinoma. Endocrine. 2010; 38: 276-82. doi: 10.1007/s12020-010-9384-7.
40. Goode GD, Ballard BR, Manning HC, Freeman ML, Kang Y, Eltom SE. Knockdown of aberrantly upregulated aryl hydrocarbon receptor reduces tumor growth and metastasis of MDA-MB-231 human breast cancer cell line. Int J Cancer. 2013; 133: 2769-80. doi: 10.1002/ijc.28297.
41. Gallo A, Galardi S. A-to-I RNA editing and cancer: from pathology to basic science. RNA Biol. 2008; 5: 135-9.
42. Rueter SM, Dawson TR, Emeson RB. Regulation of alternative splicing by RNA editing. Nature. 1999; 399: 75-80. doi: 10.1038/19992.
43. Grohmann M, Hammer P, Walther M, Paulmann N, Buttner A, Eisenmenger W, Baghai TC, Schule C, Rupprecht R, Bader M, Bondy B, Zill P, Priller J, et al. Alternative splicing and extensive RNA editing of human TPH2 transcripts. PLoS One. 2010; 5: e8956. doi: 10.1371/journal.pone.0008956.
44. Beghini A, Ripamonti CB, Peterlongo P, Roversi G, Cairoli R, Morra E, Larizza L. RNA hyperediting and alternative splicing of hematopoietic cell phosphatase (PTPN6) gene in acute myeloid leukemia. Hum Mol Genet. 2000; 9: 2297-304.
45. Tsai JR, Wang HM, Liu PL, Chen YH, Yang MC, Chou SH, Cheng YJ, Yin WH, Hwang JJ, Chong IW. High expression of heme oxygenase-1 is associated with tumor invasiveness and poor clinical outcome in non-small cell lung cancer patients. Cell Oncol (Dordr). 2012; 35: 461-71. doi: 10.1007/s13402-012-0105-5.
46. Li Q, Wang Z, Lian J, Schiott M, Jin L, Zhang P, Zhang Y, Nygaard S, Peng Z, Zhou Y, Deng Y, Zhang W, Boomsma JJ, et al. Caste-specific RNA editomes in the leaf-cutting ant Acromyrmex echinatior. Nat Commun. 2014; 5: 4943. doi: 10.1038/ncomms5943.
47. Futreal PA, Coin L, Marshall M, Down T, Hubbard T, Wooster R, Rahman N, Stratton MR. A census of human cancer genes. Nat Rev Cancer. 2004; 4: 177-83. doi: 10.1038/nrc1299.
48. Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, Meyerson M, Gabriel SB, Lander ES, Getz G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014; 505: 495-501. doi: 10.1038/nature12912.