
INTRODUCTION
According to the hierarchical model, tumor is formed by a hierarchy of proliferative and progressively differentiated bulk of cells, which originated from a unique subpopulation of cells showing “stem-like” property, cancer stem-like cells (CSCs) [1]. CSCs share many features with normal stem cells, for example, they show self-renewal and differentiation capacity, and have extraordinary proliferation potential. A unique/novel feature of CSCs is that they have the capacity to resist chemotherapy and irradiation due to expression of multidrug resistance related transporters and DNA repair mechanisms [2, 3]. Together with their invasive and metastatic capabilities, CSCs correlate closely with tumor initiation, progression, and recurrence.
Metabolism is the basic process of material circulation and energy exchange in a life entity. At the cellular level, metabolism represents the biochemical reactions supporting the regulation of cellular bioactivities. The metabolic characteristics of cancer cells closely correlated with their malignancy. In general, cancer cells have enhanced anabolism of proteins, lipids, and nucleic acids, and they catabolize glucose in a manner of aerobic glycolysis, a.k.a. the Warburg effect [4], in order to meet the nutrient demands of malignant proliferation [5].
As a special subpopulation, CSCs share general features of metabolic processes with malignant cells; meanwhile, they have certain unique metabolic characteristics that are prominently adapted with their stemness. Metabolic enzymes are main regulators of cellular metabolism. In CSCs, many a metabolic enzyme shows both catalytic activities and transcriptional activities. They not only induce and maintain the metabolic characteristics, but also activate stem-like properties by transcriptional regulation or signal interaction (Figure 1). Such multiple layers of regulation carried out by metabolic enzymes indicate that these molecules can be used as potential targets for separation of CSCs, and detection and treatment of cancer diseases. This review focuses on the metabolic enzymes involved in carbohydrate, protein and lipid metabolism in CSCs, discussing their function in stemness promotion and the involved mechanisms.
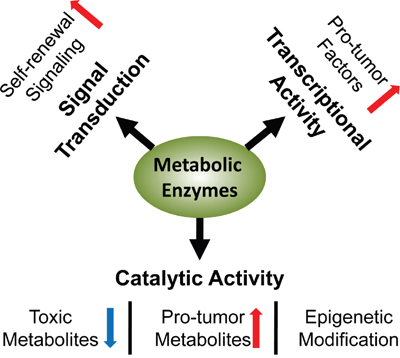
Figure 1: Multifaceted regulation in CSCs involving enzymes. Metabolic enzymes mediate the pro-stemness metabolism by their catalytic activities, and stimulate the expression of pro-tumor factors by their transcriptional activities; they promote the self-renewal signaling by direct interaction or modulation of metabolites.
Altered aerobic glycolysis with branched metabolic pathway in CSCs
Like other malignant cells, CSCs use aerobic glycolysis to satisfy their biosynthesis and energy requirement. However, some glycolytic enzymes show different expression and function in CSCs, inducing an altered aerobic glycolysis matching with stemness. Although CSCs consume more NADH and ADP to produce more glycolysis intermediates, compared with nonstem cancer cells, the two cell populations have the same ATP level [6], which means that the activated glycolysis in CSCs mainly dedicates to biosynthesis, and stimulates downstream branched metabolic pathways (Figure 2).
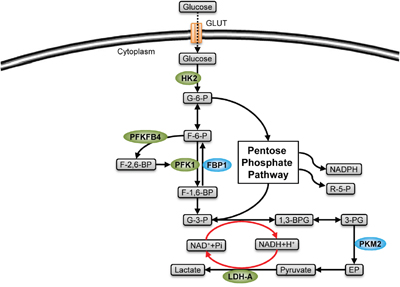
Figure 2: Stemness-related enzymes in glycolysis. Glycolytic enzymes (green) mediate the production of glycolytic metabolites, inducing a pro-stemness metabolism by their catalytic modulation in CSCs. The enzymes shown in green are highly expressed in CSCs, while those shown in blue are expressed in low level (FBP1) or existed in low-activity form (PKM2).
Hexokinase2 (HK2)
Hexokinase is involved in the first rate-limiting reaction of glycolysis, which is the transformation of glucose to glucose-6-phosphate with ATP consumption. Among those isoenzymes of Hexokinase, Hexokinase2 (HK2) has the closest relationship with cancer. HK2 is only expressed in adipose tissue, skeletal muscle, and myocardium of normal human body at low level, but it is highly expressed in embryonic tissues and is the main isoenzyme type in cancer cells.
There are 3 main functions of HK2 in cancer cells. First, HK2 maintains the Warburg effect and its branched pathways, and the inhibition of HK2 activity will obstruct nucleotide production through the pentose phosphate pathway, disturb serine anabolism and decrease the usage of carbon derived from glutamine in the tricarboxylic acid cycle [7]. Second, HK2 combines with the Voltage Dependent Anion Channel (VDAC) on the surface of mitochondria to form a VDAC-HK2 complex, which can inhibit the formation of mitochondrial permeability transition pore, protecting the cancer cell from the release of pro-apoptosis factors induced by mitochondrial damage [8, 9]. Third, HK2 can interact with Akt/mTOR signaling pathway, regulating the growth of cancer cells [10].
HK2 plays a more important role in CSCs. The expression and enzyme activity of HK2 in CD133+ stem and CD133- nonstem cancer cells have been compared. It is shown that although HK2 is expressed less in CD133+ cells, it is almost the only type of isoenzyme, and represents 92.7% total HK enzyme activity in CD133+ CSCs [11]. Research on HK2 inhibitors revealed that they impair not only the metabolic processes, but also the stemness-related properties of CSCs. For example, 3-bromopyruvate (3-BrP), a specific HK2 inhibitor, can suppress the stemness of CSCs in two aspects. First, 3-BrP can reduce the expression of stemness-related molecules like ALDH1, Notch1, Oct-4 and Sox2, weakening CSCs’ self-renewal capacity. Second, 3-BrP can disturb the formation of VDAC-HK2 complex and block NF-κB signaling, increasing the sensitivity of CSCs to chemotherapy [12, 13]. In addition, miR-143 inhibits HK2 expression at the level of transcriptional regulation. It is demonstrated that overexpression of miR-143 can induce the differentiation of glioblastoma stem-like cells, and inhibit their tumorigenic ability under hypoxia [14]. These studies indirectly confirm the significance of HK2 in the maintenance of CSCs’ stem-like properties.
Fructose-1,6-biphosphatase (FBP1) and Phosphofructokinase2 (PFK2)
Phosphofructokinase1 (PFK1) is the second key enzyme of glycolysis, catalyzing the transformation of fructose-6-phosphate to fructose-1,6-biphosphate. Inhibition targeting this process can strongly impair the metabolic adaptation of CSCs.
Fructose-1,6-biphosphatase (FBP1) is the key enzyme of gluconeogenesis, which catalyzes the reversed reaction and competes with branched biosynthesis pathways for substrates. Low expression of FBP1 is beneficial to CSCs. First, it maintains the superiority of glycolysis and induces an increased uptake of glucose, facilitating the production of glycolysis intermediates and the energy supply in CSCs during hypoxia. Second, low expression of FBP1 can inhibit the production of ROS induced by mitochondrial complex1, which protects CSCs from oxidative stress. The mechanism by which CSCs maintain the low expression of FBP1 is not clear. In basal-like breast cancer, it has been demonstrated that the epithelial-mesenchymal transition (EMT) factor Snail can directly bind with the promoter of the gene encoding FBP1, leading to the down regulation of FBP1 expression [15].
Different from FBP1, PFK2 catalyses the production of fructose-2,6-biphosphate, which is the most powerful allosteric activator of PFK1. PFKFB4, an isoenzyme of PFK2 family, is highly expressed in glioma CSCs. Silencing the expression of PFKFB4 by RNA interference can induce the obstruction of glycolysis, creating an increased ratio of AMP/ATP. The high AMP/ATP ratio will activate AMPK and then inhibit the mTOR-related survival signaling pathway, inducing the apoptosis of CSCs [16].
Pyruvate kinase M2 (PKM2)
The third key enzyme of glycolysis is pyruvate kinase (PK), which catalyzes the production of pyruvate and ATP by hydrolysis of PEP. PKM2 is mainly expressed in tumor tissue and existed in a dimer form with low catalytic activity [17]. It has been proven that the PKM2 dimer form possesses special properties that maintain the stemness of CSCs.
The low activity of PKM2 is maintained mainly by phosphotyrosine signaling in CSCs. In CSCs, PKM2 will specifically bind to tyrosine-phosphorylated peptides and release its allosteric activator, fructose-1,6-biphosphate, keeping a low catalytic efficiency. What’s more, CD44, a surface molecule of CSCs, can bind directly with PKM2 at its intracellular domain, synergizing the effect of tyrosine-phosphorylated peptides to maintain the low activity of PKM2, diverting glucose metabolites from energy production to biosynthesis [18, 19].
High activated tetramer form of PKM2 can also form in CSCs. The active upstream glycolysis and obstruction caused by the low activity of PKM2 will lead to the accumulation of glycolytic intermediates, including fructose-1,6-biphosphatase, the allosteric activator of PKM2, inducing the formation of tetramer to promote ATP and pyruvate production [15]. Thus, CSCs can regulate the direction of glycolysis with the variation of PKM2 activation: Low-activity form of PKM2 induces the biosynthesis of ribose and lipid by increasing the upstream glycolytic metabolites; while high-activity form accelerates the pyruvate production, facilitating energy supply and amino acid anabolism.
Apart from its metabolic regulatory function, PKM2 also acts as a transcription factor in stemness-related pathway. Induced by the activation of epidermal growth factor receptor (EGFR), PKM2 can translocate into the nucleus to activate Wnt/β-catenin signaling pathway [20]. In addition, the tetramer form of PKM2 transformed from the dimer form induced by dichloroacetate can silence Oct4 gene, leading to the differentiation of CSCs [21].
Lactic dehydrogenase-A (LDH-A)
Lactic dehydrogenase (LDH) catalyzes the lactate production, which is the transformation of pyruvate to lactate with NAD+ production. LDH-A is mainly expressed in tumor cells. Compared with other isotypes of LDH, LDH-A can catalyze the lactate and NAD+ production more efficiently, which creates a positive feedback in the glycolytic pathway (Figure 2), and inhibits the usage of pyruvate by aerobic oxidation [22]. In non-small cell lung cancer (NSCLC), it has been proven that the lack of LDH-A will enhance the tricarboxylic acid cycle, which produces ROS and causes cell damage, decreasing both the ratio and sphere formation capacity of CSCs [23].
The regulation of pyruvate metabolism is not the only function of LDH-A in CSCs. Studies on NHI, a LDH-A specific inhibitor, show the stemness-related function of LDH in three aspects. First, LDH-A can maintain the formation of clonal spheres in 3D culture and the expression of CSC markers like CD133 and EZH2. Second, LDH-A can facilitate the expression of metalloprotease, which is important for the invasion of CSCs. Third, LDH-A dedicates to the formation of chemoresistance of CSCs, as NHI can stimulate the releasing of deoxycytidine kinase (dCK), which can enhance the anti-tumor efficiency of Gemcitabine [24].
Bioactive lipids and related enzymes are involved in regulation of CSCs
Activated lipid metabolism is a common characteristic of tumor tissues. It can provide cell proliferation with lipid material, and produce bioactive lipids to facilitate pro-tumor signal transduction and gene expression. Many bioactive lipids and lipid metabolic enzymes can induce specific metabolic profiles to maintain CSCs’ stem-like properties (Figure 3).
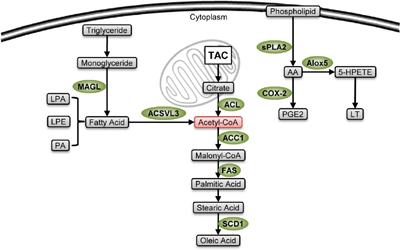
Figure 3: Stemness-related enzymes in lipid metabolism. Lipid metabolic enzymes (green) are highly expressed in CSCs. Their lipid metabolites are used to produce membrane during cellular proliferation, coordinate biosynthesis and energetic supply through acetyl-CoA production (pink), and can maintain stem-like properties. The pro-stemness mechanisms of some of the enzymes, including MAGL, ACSVL3 and SCD1, are not fully understood.
Enzymes involved in lipid synthesis
In CSCs, there is a high expression of enzymes involved in lipid synthesis including ATP citrate lyase (ACL), acetyl-CoA carboxylase1 (ACC1) and fatty acid synthase (FAS). It has been reported that expression of these enzymes is under the control of sterol regulatory element binding protein1 (SREBP1), and either chemical inhibition of these enzymes or transcriptional reduction of their gene expression can enormously impair the stem-like properties [25]. For example, inhibiting ACL by hydroxycitric acid and ACL knockdown lead the same effects including impairment of tumorsphere forming ability, reduction in the expression of stemness marker like c-Myc, Klf4, Oct4, and downregulation of the transcriptional activity of Snail [26]. A specific inhibitor of ACC1 called soraphen A shows a prominent anti-CSCs effect by inhibiting the de novo lipogenesis as it blocks the positive effect of ACC1 on survival and self-renew of CSCs from breast cancer [27]. Resveratrol, which can block the expression of fatty acid synthase, suppresses the growth of CSCs by inhibiting lipid synthesis, and induces cell death by upregulating the expression of pro-apoptosis gene DAPK2 as well as BNIP3 [28].
Enzymes involved in PUFA metabolism
Derivatives of polyunsaturated fatty acids such as prostaglandin, thromboxane and leukotrienes are involved in many important physiological processes. Apart from regulatory roles in inflammatory responses, their influences on stem cell differentiation and proliferation facilitate malignancy development [29].
Arachidonate 5-lipoxygenase (Alox5)
Catalyzed by lipoxygenase, arachidonic acid is converted into hydrogen peroxyeicosatetraenoic acids, which further produce bioactive lipids such as leukotrienes. In murine model of chronic myeloid leukemia induced by BCR-ABL, either transcriptional or chemical inhibition of arachidonate 5-lipoxygenase (Alox5) impairs the survival and asymmetric cell division capacity of leukemia stem cells by downregulation of β-catenin in Wnt signaling, inhibiting leukemia initiation, while the loss of Alox5 exhibits no effect on normal hematopoietic stem cells [30, 31]. Roos et al. further revealed the underlying mechanism by demonstrating that inactive form of Alox5 in association with its catalytic inhibitor CJ-13,610 impeded nuclear translocation of β-catenin by direct interaction [32]. Another inhibitor of Alox5, Nordy, has been found to decrease the expression of stemness related genes including SOX-2, Nanog and Nestin. Such change induces the formation of GFAP expressing mature glioma stem cells, and inhibits the formation of xenograft [33, 34]. These results not only show the function of Alox5 in stemness maintenance, but also indicate that anti-Alox5 therapy may be safe and effective in treatment of cancer.
Cyclooxygenase-2 (COX-2)
Cyclooxygenase (COX) is the key enzyme in the production of prostaglandin from arachidonic acid. COX-2 is the inducible isotype involved in inflammatory responses and tumor diseases. In CSCs, high expression of COX-2 is induced by membrane type-1 matrix metalloproteinase (MT1-MMP) through MAPK/NF-κB signaling in a catalysis-independent manner [35], and is also positively correlated with macrophage migration inhibitory factor (MIF), while miRNA-451 can downregulate COX-2 expression indirectly by targeting MIF gene [36]. In breast CSCs, the expression of COX-2 also positively correlates with Her2 [37, 38]. Prostaglandin E2 catalyzed by COX-2, in conjunction with G-protein coupled receptors such as EP2, EP4 and LGR5, cause inactivation of glycogen synthase kinase 3β to decrease the inhibitory phosphorylation of β-catenin thereby enhancing β-catenin/Wnt signaling to promote the stem-like property of CSCs [39–41]. Meanwhile, COX-2 and its related metabolites can inhibit PI3K/Akt induced apoptosis by inducing the phosphorylation of Akt [42, 43]. Non-steroidal anti-inflammatory drugs (NSAIDs) like celecoxib and indomethacin can serve as COX-2 inhibitors. Apart from their inhibiting effect on COX-2 activity, NSAIDs can also inhibit Akt phosphorylation by activating PPARγ leading to amplification of PTEN signal [43, 44]. Furthermore, NSAIDs can block Wnt signaling [45]. In CSCs, it has been proven that NSAIDs can effectively inhibit tumorsphere formation and self-renewal, and impede chemoresistance [45, 46] while exhibiting no effect on the sensitivity to radiotherapy [47].
Monoacylglycerol lipase (MAGL), Acyl-CoA synthetase VL3 (ACSVL3) and Stearoyl-coAdesaturase1 (SCD1)
There are other stemness related lipid metabolic enzymes, including Monoacylglycerol lipase (MAGL), Acyl-CoA synthetase VL3 (ACSVL3) and Stearoyl-coA desaturase1 (SCD1).
MAGL is expressed more highly in CSCs as compared with nonstem cancer cells and its upregulation is a sign of EMT [48]. MAGL catalyzes the hydrolysis of endocannabinoid 2-arachidonoylglycerol to reverse the inhibition of tumor growth and metastasis induced by endocannabinoid signaling. On the other hand, it catalyzes the production of lipid agonists of G-protein coupled receptors like lysophosphatidic acid (LA), phosphatidic acid (PA) and lysophosphatidylethanolamine (LPE), which act as pro-tumor factors.
ACSVL3 is an isoenzyme of Acyl-CoA synthetase (ACS), which catalyzes acyl-CoA production during β-oxidation of fatty acid. Activation of receptor tyrosine kinase pathways, including HGF/c-Met and EGF/EGFR pathways which promote malignant progression, leads to dramatic upregulation of ACSVL3 expression. This evidence indicates that ACSVL3 is an important metabolite highly required in CSCs [49, 50].
SCD1 is the key enzyme catalyzing synthesis of oleic acid, a monounsaturated fatty acid, from saturated fatty acid. It has been reported that SCD1 inhibitors can induce endoplasmic reticulum stress to disturb protein synthesis, and impair cytoskeleton, indicating that SCD1 is important for maintaining the stabilization of cellular structure during the survival and growth of CSCs [51, 52].
Signal molecules as substrates of protein metabolism in CSCs
Compared with general cancer cells, CSCs have much more active protein anabolism and are more sensitive to amino acid deprivation [53]. Meanwhile, CSCs maintain cyclic utilization of amino acids and regulate their stemness signaling by regulation of proteolysis. Furthermore, amino acid metabolites promote the maintenance of stem-like properties in CSCs.
Enzymes involved in proteolysis
Proteins are degraded by either the ATP-independent cathepsin-lysosome pathway or the ATP-dependent ubiquitin-proteasome pathway (Figure 4). CSCs show activated lysosome pathway to keep cyclic utilization of raw materials for protein synthesis without increasing energy demand. At the same time, they carry on proteasome pathway with low but specific activity, protecting the pro-stemness molecules while selectively degrading the signal molecules which are harmful for stem-like properties by ubiquitin modification.
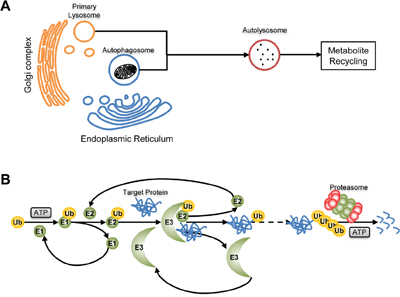
Figure 4: Proteolysis. A. Cathepsin-lysosome pathway. Fusing with Cathepsin-containing primary lysosome, an autophagosome will convert to autolysosome and proteins in it will be degraded to recycle in biosynthesis. B. Ubiquitin-proteasome pathway. Catalyzed by E1, E2 and E3, the target protein will be modified by ubiquitin and finally degraded to peptides by proteosome.
Cathepsin B
With cathepsins, lysosome can transform waste proteins to amino acid materials for protein remanufacture without ATP consumption (Figure 4A). There are natural cathepsin inhibitors in cells regulating the proteolysis activity of lysosome. One of them is called cystatin E/M that is a cathepsin B inhibitor and is expressed by CST6 gene. In CSCs, the promoter of CST6 is highly methylated and there is little cystatin E/M production. Thus, the activity of cathepsin B is maintained at high level to satisfy the active protein metabolism of CSCs [54].
Apart from metabolic regulation, Cathepsin B is also involved in other aspects in CSCs, including induction of radioresistance and maintenance of the stability of cytoskeleton.
Radiation can induce the upregulation of cathepsin B in glioma CSCs. After inhibiting such radiation-enhanced cathepsin B expression by shRNA pUC, c-Met signaling is blocked, leading to the production of DNA damage marker, γH2AX that further induces DNA damage-induced apoptosis [55]. The pUC-induced inhibition of cathepsin B also leads to disassociation of focal adhesion kinase (FAK) and cytoskeletal molecules, which further inhibits PKC/integrin signaling, impairing the adhesion and migration of CSCs [56].
26S Proteasome and ubiquitin chain
Proteasomes specifically recognize and degrade ubiquitinated proteins (Figure 4B). CSCs derived from glioma, breast cancer, lung cancer, hepatoma, pancreatic cancer and osteosarcoma all carry low proteasome activity. When fluorescent proteins are exogenously expressed in cancer cells, the above character induces relative accumulation of fluorescent proteins in CSCs population, which can be used for sorting of CSCs [57–62]. Musashi1, an mRNA binding protein, is the main factor maintaining the low proteasome activity of CSCs. It can downregulate the level of proteasome transcriptional factor NF-YA by directly binding to NF-YA mRNA, leading to the inhibition of proteasome production [63]. Low proteasome activity can induce a static metabolism profile in CSCs to resist stresses, and can also help CSCs to escape from immunological surveillance by reducing the production of antigen peptide [64]. In addition, proteasomes regulate the stem-like property of CSCs via degrading key factors in stemness signaling pathways. Low proteasome activity maintains the stability of transcriptional repressor REST to keep CSCs in a dedifferentiation state [65], decreases the degradation of Notch intracellular domain to sustain Notch signaling [63], and is beneficial to the stability of Nrf2 to induce the expression of anti-oxidation genes [66].
Proteasomes can also maintain stem-like properties of CSCs by specifically degrading ubiquitin modified signal molecules. Protein ubiquitination is a three-step reaction comprising ubiquitin-activating enzyme (E1), ubiquitin-conjugating enzyme (E2) and ubiquitin ligase (E3), and ubiquitin ligase is the key enzyme that catalyzes the direct covalent linkage of ubiquitin with proteins (Figure 4B). In CSCs, ubiquitin ligase supports the stemness by modifying at least three signal molecules. First, it induces the ubiquitinated degradation of receptor-interacting protein1 (RIP1) to inhibit TNFα-induced apoptosis [67]. Second, it induces the ubiquitinated degradation of cyclin E to maintain DNA stability [68]. Third, MYCN is another target of ubiquitin ligase, and its degradation can stimulate asymmetric division [69]. More importantly, many ubiquitin ligase inhibitors show anti-CSCs potentials [70–72]. Apart from ubiquitin ligase, another ubiquitination related enzyme, ubiquitin-specific protease 22, has been discovered as a CSCs marker. It can inhibit gene expression by its ubiquitin hydrolase activity to protect histone H2A and H2B from proteasome degradation [73, 74].
Enzymes involved in amino acid catabolism
Some metabolites produced from amino acid catabolism have special effects to CSCs. And enzymes related with them promote stem-like properties of CSCs through catalyzing the production of these metabolites.
Nitric oxide is produced from arginine with the catalysis of nitric oxide synthase (NOS), and is important for transduction of cell signals (Figure 5A). NOS2, also called inducible NOS (iNOS), is highly expressed in glioma CSCs (GSCs). Catalyzed by NOS2, NO inhibits the expression of the cell-cycle inhibitor cell division autoantigen-1 (CDA1) at transcriptional level to enhance the tumorigenesis capacity of GSCs. An enzymatic inhibitor of NOS2 named BYK191023 can effectively inhibit the survival of CSCs without impairing normal tissues. Because of its high selectivity, ideal pharmacokinetics, perfect bioavailability, and high permeability to pass the blood-brain barrier, BYK191023 may serve as an effective drug in glioma therapy [75].
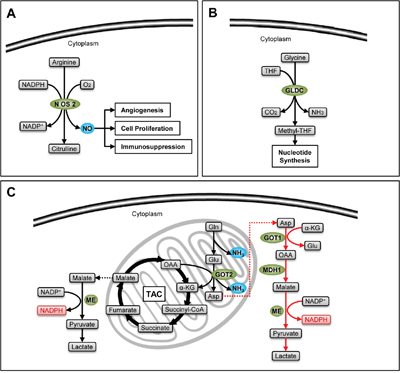
Figure 5: Stemness-related enzymes in amino acid catabolism. A. NO (blue) produced by catalysis of NOS2 sustains the survival of CSCs. B. GLDC increases the production of one carbon units which facilitates nucleotide synthesis in CSCs. C. The classical glutaminolysis (black arrows) increases the TAC influx by the production of α-ketoglutarate, satisfying energetic demand, and produces NH3 (blue), which can neutralize acidic metabolites and induce autophagy, facilitating the survival of CSCs. Meanwhile, the non-canonical glutamine catabolism (red arrows) provides an effective compensation of NADPH (pink), protecting CSCs from oxidative stress.
Some amino acids can produce one carbon units essential for synthesis of purine and pyrimidine. Glycine is one of them. It produces methyl-tetrahydrofolate (methyl-THF) with the catalysis of glycine decarboxylase (GLDC) (Figure 5B). Zhang et al. detected a dramatic upregulation of GLDC in NSCLC CSCs. Interfering GLDC expression with shRNA strongly inhibit the sphere and xenograft formation capacity of CSCs, while overexpression of GLDC can give stemness to 3T3 cells. The group further demonstrated that CSCs activate glycolysis, promoting the usage of pyruvate in biosynthesis to produce amino acids, while at the same time they absorb exogenous amino acids. Such accumulation of amino acids further enhances the production of one carbon units, which satisfies the demand of nucleotides during the growth of CSCs [76].
Glutamine is the precursor of nonessential amino acids and nucleic acids. It can produce GSH and NADPH to maintain redox status, and is involved in many metabolic processes which are important for tumor growth and survival [77]. Catalyzed by glutaminase, glutamine is converted to ammonia and glutamate. In normal cells, glutamate will be further decomposed into α-ketoglutarate (α-KG) by glutamate dehydrogenase1 (GLUD1), participating in TAC, or converted into aspartate by the catalysis of glutamic oxalacetic transaminase2 (GOT2). Recently, Son et al. demonstrate another pathway of glutamine metabolism in pancreatic ductal adenocarcinoma (PDAC). They discovered that glutamate, generated from glutamine, can be converted to oxaloacetic acid by the catalysis of glutamic oxalacetic transaminase1 (GOT1) in cytoplasm, and finally becomes pyruvate with NADPH production by the catalysis of malate dehydrogenase1 (MDH1) and malic enzyme (ME). This process induces an increase of NADPH/NADP+ ratio, protecting cancer cells from oxidative stress [78] (Figure 5C). Such a non-canonical metabolic pathway of glutamine has also been found in CSCs derived from PDAC by Li et al. What’s more, either inhibiting GOT1 expression or depriving glutamine can effectively suppress stem-like properties of PDAC-CSCs, and increase their sensitivity to radiotherapy by the accumulation of ROS [79]. The discovery of the non-canonical metabolic pathway of glutamine may provide new methods for PDAC therapy.
Enzyme-associated metabolic adaptation in CSCs
The expression and activity of metabolic enzymes are highly dynamic. They are correlated with sources of tumors, growth conditions and different regulatory mechanisms [80, 81]. Enzyme-associated metabolism switch in CSCs is a dynamic process which is influenced by tumor microenvironment and stemness related signaling, reflecting the adaptation to tumor microenvironment and different stages of growth and differentiation (Figure 6).
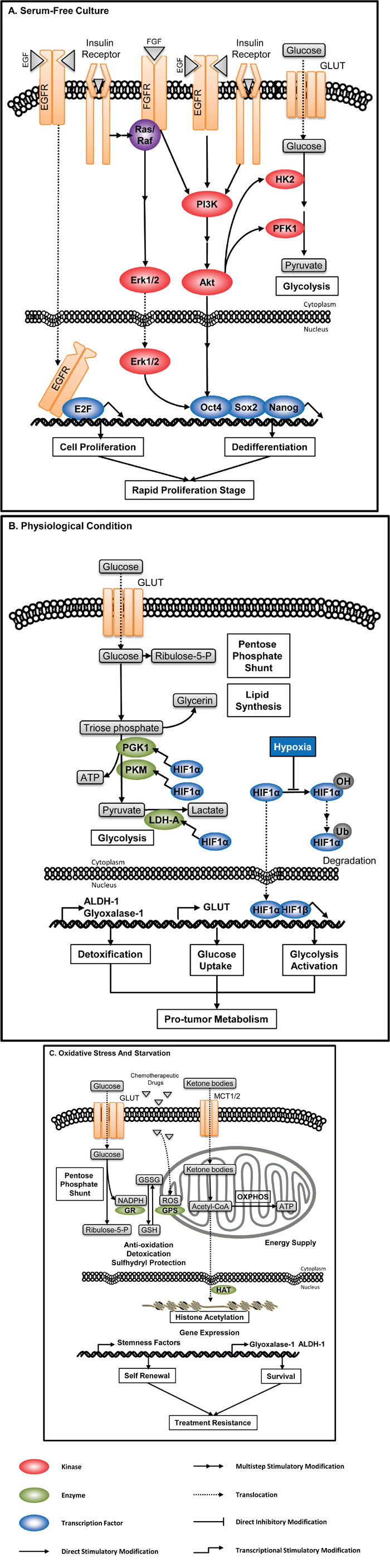
Figure 6: Enzyme-associated metabolic adaptation. Enzyme-mediated metabolic adaptation is a dynamic process, resulting from crosstalk(s) between cellular signaling, metabolic pathways, and microenvironment. A. CSCs in serum-free culture. Growth factors, high glucose level and insulin in artificial culture media activate multiple signaling pathways, inducing expression of stemness-related factors and augmenting glycolytic metabolism. Thus, clonal spheres cultured from serum-free system are not ideal model to represent the metabolic profile of CSCs. B. CSCs under physiological hypoxia. HIF1α is a transcriptional factor activating the expression of many glycolytic enzymes including PGK1, PKM and LDH-A. High level of HIF induced by hypoxia, together with the high expression of GLUT and detoxifying enzymes, mediates the metabolic adaptation under the physiological condition. C. CSCs under stresses. CSCs can modulate dominant metabolic pathways to adapt different environmental stresses. When encountering chemotherapy, CSCs can increase the PPP flux to produce more NADPH, which is involved in eliminating hazardous substances such as ROS, as well as regenerating reductive GSH. Under starvation, CSCs can use ketone bodies to satisfy their energetic demand and induce epigenetic modification to survive.
CSCs in serum-free culture
Activated aerobic glycolysis is the core process in the biosynthesis in CSCs. CSCs, especially of those enriched by serum-free tumorsphere culture, carry much more prominent Warburg effect than general cancer cells [82]. Serum-free culture is one of the main methods to enrich CSCs, and the tumorspheres formed are used to characterize CSCs’ properties including metabolism profiles and involved metabolic enzymes. The number and size of serum-free induced tumorspheres are main evaluation criteria of stem-like properties. The formation of clonal spheres in serum-free culture is mainly induced by exogenous growth factors, insulin and high glucose level in culture medium. These factors activate multiple pro-tumor signaling pathways. For example, growth factors activate PI3K/Akt and MAPK/Erk pathway, which can increase the catalytic activity of glycolytic kinases, stimulate cell proliferation, and maintain dedifferentiation through the activation of three key transcription factors related to pluripotency, Oct4, Sox2 and Nanog. This culture system almost totally inhibits the differentiation of CSCs, and stimulates active cell division. Thus, metabolism profiles of CSCs detected in the serum-free cultures represent the characteristic of CSCs in rapid proliferation stage (Figure 6A).
CSCs under physiological hypoxia
Under physiological conditions, CSCs tend to accumulate in the hypoxia region of a solid tumor, and also show superiority of glycolysis. CSCs sustain rapid glycolysis judged from three aspects. First, CSCs highly express GLUT to guarantee the glucose supply [83–85]. Second, hypoxia protects the proline residue of HIF-1α from hydroxylation, thus decreasing the hydroxylation -induced ubiquitin-mediated degradation of HIF-1α, facilitating the translocation of HIF-1α into nucleus to bind with HIF-1β, which activates a series of gene expression including glycolytic enzymes like PGK1, PKM and LDH-A [24]. Third, CSCs consume metabolites rapidly with active anabolism, preventing the excessive accumulation of glycolytic products, while at the same time highly express Glyoxalase-I [86] and ALDH-1 to eliminate toxic products. The superiority of glycolysis satisfies the nutrient and ATP requirement for CSCs to maintain the growth of tumor and their own population under physiological conditions (Figure 6B).
CSCs under stresses
The superiority of Warburg effect in CSCs is not absolute, as CSCs can regulate the balance between glycolysis, oxidative phosphorylation and pentose phosphate pathway (PPP) to resist environmental stresses (Figure 6C).
Branched pathways for biosynthesis are promoted by activated aerobic glycosis in CSCs. PPP is one of the most important processs of biosynthesis. PPP produces phosphopentose, NADPH and carbon dioxide. It can also produce fructose-6-phosphate and glyceraldehydes-3-phosphate, which participate in TAC, through a series of group-transfer reaction. PPP is significant to cancer cells. On the one hand, PPP produces phosphopentose to satisfy the demand of nucleic acid synthesis during cancer cell proliferation. On the other hand, it produces NADPH, which carries many important functions in biosynthesis: First, NADPH is the hydrogen donor in the production of lipids and amino acids. For example, NADPH provides acetyl-CoA with hydrogen atom to produce fatty acids and cholesterol, and provides hydrogen atom to α-ketoglutarate (α-KG) to produce glutamate. Second, NADPH is also involved in the hydroxylation during the production of cholesterol from squalene. Third, NADPH produced by PPP is the coenzyme of glutathione reductase. It promotes the redox of GSSG to produce GSH, protecting CSCs from ROS-induced cell damage during chemotherapy [19].
TAC in cancer cells can produce carcinogenic metabolites due to the expression of abnormal metabolic enzymes. For example, gene mutations in IDH1 and IDH2 are very common in many cancer diseases. These mutations induce the overexpression of a carcinogenic metabolite, R-2-hydroxyglutarate (R-2-HG). R-2-HG can occupy the binding site of α- ketoglutarate (α-KG) on α-KG dependent dioxygenases like histone demethylation enzymes, inducing a wide range of DNA methylations [87]. It has been proven that such modification is positively correlated with dedifferentiation and self-renewal capability of CSCs [88].
Oxidative phosphorylation, the final process of biological oxidation and the main process of ATP production, may also support stem-like properties by certain means. Bedaquiline, an antibiotic used in the therapy of multi-drug resistant pulmonary tuberculosis, have been proved to have anti-cancer activity. It can specifically bind with mitochondrial ATP-synthase in CSCs, leading to mitochondrial dysfunction and oxidative stress, while is well-tolerated in normal cells [89]. This finding indicates a correlation between chemoresistance and oxidative respiratory chain in CSCs.
It has been found that CSCs can consume extracelluar high-energy metabolites such as pyruvate, lactate and ketone bodies to produce energy through mitochondrial oxidative phosphorylation during nutrient deprivation [90]. In normal human body, ketone bodies are metabolites produced by β oxidation of fatty acids, and serve as a major source of energy in brain and muscles under starvation. Thus, the usage of ketone bodies by CSCs may be an emergency plan to survive from nutrient starvation. Ketone bodies and lactate are more than emergent fuels as they facilitate stem-like properties in many aspects. Martinez-Outschoorn et al. found that ketone bodies and lactate can promote the growth of embryonic stem cells by inducing the production of many stemness-related transcriptional factors, and transform to acetyl-CoA which promotes gene expression by participating in the acetylation of histones [91]. Lamb et al. found that CSCs from in vitro culture of breast cancer cell lines highly express enzymes related to β oxidation. Blocking the uptake of ketone bodies and lactate with specific MCT1/2 inhibitors effectively reduces mammosphere formation [92]. Thus, they indicated that CSCs carry both production and utilization capacity of ketone bodies. However, this phenotype requires further research because it may be barely induced by artificial culture system or just related to some specific tumor origins.
Perspectives
Base on the knowledge about cancer stem-like cells, inhibition of CSCs is the key to treat tumor diseases and prevent tumor recurrence [93]. Compare to conventional chemotherapeutic agents, metabolic enzyme inhibitors have two significant advantages. First, enzyme inhibitors can specifically target the tumorous isoenzymes, while cause little damage to normal tissues; Second, many enzyme inhibitors have been shown to cause direct damage to CSCs or impair their chemoresistance. A case study in a cancer patient with fibrolamellar hepatocellular carcinoma shows the clinical value of 3-bromopyruvate (3-BrP), a HK2 inhibitor targeting the glucose metabolism of cancer cells. TACE treatment with specially formulated 3-BrP efficiently eliminate cancer cells without apparent cyto-toxicity, lengthening the survival time of the patient with an improved life quality [94]. Studies on the CSCs specific metabolic enzymes would help find new targets for CSCs sorting and anti-tumor treatment. Meanwhile, detecting the enzymatic activities and tracing the metabolites can reflect the activity of CSCs in tumor tissues, facilitating the prognostic evaluation of tumor diseases.
Abbreviations
3-BrP, 3-bromopyruvate; α-KG, α-ketoglutarate; ACL, ATP citrate lyase; ACS, Acyl-CoA synthetase; Alox5, arachidonate 5-lipoxygenase; CDA1, cell division autoantigen-1; COX, cyclooxygenase; CSCs, cancer stem-like cells; EMT, epithelial-mesenchymal transition; FAK, focal adhesion kinase; FBP1, fructose-1,6-biphosphatase; GLDC, glycine decarboxylase; GLUD1, glutamate dehydrogenase1; GOT1, glutamic oxalacetic transaminase1; GOT2, glutamic oxalacetic transaminase2; HK2, hexokinase2; IDH, isocitrate dehydrogenase; iNOS, inducible nitric oxide synthase; LA, lysophosphatidic acid; LDH, lactic dehydrogenase; LPE, lysophosphatidyl ethanolamine; MAGL, monoacylglycerol lipase; MCT, monocarboxylate transporter; MDH1, malate dehydrogenase1; ME, malic enzyme; MIF, macrophage migration inhibitory factor; MT1-MMP, membrane type-1 matrix metalloproteinase; NOS, nitric oxide synthase; NSAIDs, non-steroidal anti-inflammatory drugs; NSCLC, non-small cell lung cancer; PA, phosphatidic acid; PDAC, pancreatic ductal adenocarcinoma; PFK1, phosphofructokinase1; PK, pyruvate kinase; PPP, pentose phosphate pathway; R-2-HG, R-2-hydroxyglutarate; SCD1, stearoyl-coA desaturase1; SREBP1, sterol regulatory element binding protein1; TAC, tricarboxylic acid cycle; THF, tetrahydrofolate; VDAC, voltage dependent anion channel.
CONFLICTS OF INTEREST
The authors declare that the article has no conflicts of interest.
GRANT SUPPORT
This work was supported by the Major State Basic Research Development Program of China (973 Program; Grant No.: 2014CB542003), the National Natural Science Foundation of China (NSFC; Grant No.: 81372348) and the Foundation of Zhejiang Scientific Technology Bureau (Grant No.: LY13H200001).
REFERENCES
1. Blagosklonny MV. Cancer stem cell and cancer stemloids: from biology to therapy. Cancer Biol Ther. 2007. 6:1684-90.
2. Visvader JE. Cells of origin in cancer. Nature. 2011. 469:314-22.
3. Levine AJ. Stem cells, aging and cancers. Aging (Albany NY). 2015. 7:457-8. doi: 10.18632/aging.100776.
4. WARBURG O. On the origin of cancer cells. Science. 1956. 123:309-14.
5. Vander HMG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009. 324:1029-33.
6. Wang J, Christison TT, Misuno K, Lopez L, Huhmer AF, Huang Y, Hu S. Metabolomic profiling of anionic metabolites in head and neck cancer cells by capillary ion chromatography with Orbitrap mass spectrometry. Anal Chem. 2014; 86:5116-24.
7. Patra KC, Wang Q, Bhaskar PT, Miller L, Wang Z, Wheaton W, Chandel N, Laakso M, Muller WJ, Allen EL, Jha AK, Smolen GA, Clasquin MF, Robey RB, Hay N. Hexokinase 2 is required for tumor initiation and maintenance and its systemic deletion is therapeutic in mouse models of cancer. Cancer Cell. 2013; 24:213-28. doi: 10.1177/1947601913503341.
8. Wolf A, Agnihotri S, Micallef J, Mukherjee J, Sabha N, Cairns R, Hawkins C, Guha A. Hexokinase 2 is a key mediator of aerobic glycolysis and promotes tumor growth in human glioblastoma multiforme. J Exp Med. 2011; 208:313-26.
9. Krasnov GS, Dmitriev AA, Lakunina VA, Kirpiy AA, Kudryavtseva AV. Targeting VDAC-bound hexokinase II: a promising approach for concomitant anti-cancer therapy. Expert Opin Ther Targets. 2013; 17:1221-33.
10. Roberts DJ, Miyamoto S. Hexokinase II integrates energy metabolism and cellular protection: Akting on mitochondria and TORCing to autophagy. Cell Death Differ. 2014.
11. Gong L, Cui Z, Yu X, Wei Y, Peng J, Leng X. Hexokinase II in CD133+ and CD133- hepatoma BEL-7402 Cells. Pathol Oncol Res. 2012. 18:377-81.
12. Isayev O, Rausch V, Bauer N, Liu L, Fan P, Zhang Y, Gladkich J, Nwaeburu CC, Mattern J, Mollenhauer M, Ruckert F, Zach S, Haberkorn U, Gross W, Schonsiegel F, Bazhin AV, Herr I. Inhibition of glucose turnover by 3-bromopyruvate counteracts pancreatic cancer stem cell features and sensitizes cells to gemcitabine. Oncotarget. 2014; 5:5177-89. doi: 10.18632/oncotarget.2120.
13. Wintzell M, Lofstedt L, Johansson J, Pedersen AB, Fuxe J, Shoshan M. Repeated cisplatin treatment can lead to a multiresistant tumor cell population with stem cell features and sensitivity to 3-bromopyruvate. Cancer Biol Ther. 2012; 13:1454-62.
14. Zhao S, Liu H, Liu Y, Wu J, Wang C, Hou X, Chen X, Yang G, Zhao L, Che H, Bi Y, Wang H, Peng F, Ai J. miR-143 inhibits glycolysis and depletes stemness of glioblastoma stem-like cells. Cancer Lett. 2013; 333:253-60.
15. Dong C, Yuan T, Wu Y, Wang Y, Fan TW, Miriyala S, Lin Y, Yao J, Shi J, Kang T, Lorkiewicz P, St Clair D, Hung MC, Evers BM, Zhou BP. Loss of FBP1 by Snail-mediated repression provides metabolic advantages in basal-like breast cancer. Cancer Cell. 2013; 23:316-31.
16. Goidts V, Bageritz J, Puccio L, Nakata S, Zapatka M, Barbus S, Toedt G, Campos B, Korshunov A, Momma S, Van Schaftingen E, Reifenberger G, Herold-Mende C, Lichter P, Radlwimmer B. RNAi screening in glioma stem-like cells identifies PFKFB4 as a key molecule important for cancer cell survival. Oncogene. 2012; 31:3235-43.
17. Christofk HR, Vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, Wei R, Fleming MD, Schreiber SL, Cantley LC. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature. 2008; 452:230-3.
18. Christofk HR, Vander HMG, Wu N, Asara JM, Cantley LC. Pyruvate kinase M2 is a phosphotyrosine-binding protein. Nature. 2008; 452:181-6.
19. Tamada M, Nagano O, Tateyama S, Ohmura M, Yae T, Ishimoto T, Sugihara E, Onishi N, Yamamoto T, Yanagawa H, Suematsu M, Saya H. Modulation of glucose metabolism by CD44 contributes to antioxidant status and drug resistance in cancer cells. Cancer Res. 2012; 72:1438-48.
20. Yang W, Xia Y, Ji H, Zheng Y, Liang J, Huang W, Gao X, Aldape K, Lu Z. Nuclear PKM2 regulates beta-catenin transactivation upon EGFR activation. Nature. 2011; 480:118-22.
21. Morfouace M, Lalier L, Oliver L, Cheray M, Pecqueur C, Cartron PF, Vallette FM. Control of glioma cell death and differentiation by PKM2-Oct4 interaction. Cell Death Dis. 2014; 5:e1036.
22. Miao P, Sheng S, Sun X, Liu J, Huang G. Lactate dehydrogenase A in cancer: a promising target for diagnosis and therapy. IUBMB Life. 2013. 65:904-10.
23. Xie H, Hanai J, Ren JG, Kats L, Burgess K, Bhargava P, Signoretti S, Billiard J, Duffy KJ, Grant A, Wang X, Lorkiewicz PK, Schatzman S, Bousamra M 2nd, Lane AN, Higashi RM, Fan TW, Pandolfi PP, Sukhatme VP, Seth P. Targeting lactate dehydrogenase—a inhibits tumorigenesis and tumor progression in mouse models of lung cancer and impacts tumor-initiating cells. Cell Metab. 2014; 19:795-809.
24. Maftouh M, Avan A, Sciarrillo R, Granchi C, Leon LG, Rani R, Funel N, Smid K, Honeywell R, Boggi U, Minutolo F, Peters GJ, Giovannetti E. Synergistic interaction of novel lactate dehydrogenase inhibitors with gemcitabine against pancreatic cancer cells in hypoxia. Br J Cancer. 2014; 110:172-82.
25. Pandey PR, Xing F, Sharma S, Watabe M, Pai SK, Iiizumi-Gairani M, Fukuda K, Hirota S, Mo YY, Watabe K. Elevated lipogenesis in epithelial stem-like cell confers survival advantage in ductal carcinoma in situ of breast cancer. Oncogene. 2013; 32:5111-22.
26. Hanai JI, Doro N, Seth P, Sukhatme VP. ATP citrate lyase knockdown impacts cancer stem cells in vitro. Cell Death Dis. 2013; 4:e696.
27. Corominas-Faja B, Cuyas E, Gumuzio J, Bosch-Barrera J, Leis O, Martin AG, Menendez JA. Chemical inhibition of acetyl-CoA carboxylase suppresses self-renewal growth of cancer stem cells. Oncotarget. 2014; 5:8306-16. doi: 10.18632/oncotarget.2059.
28. Pandey PR, Okuda H, Watabe M, Pai SK, Liu W, Kobayashi A, Xing F, Fukuda K, Hirota S, Sugai T, Wakabayashi G, Koeda K, Kashiwaba M, Suzuki K, Chiba T, Endo M, Fujioka T, Tanji S, Mo YY, Cao D, Wilber AC, Watabe K. Resveratrol suppresses growth of cancer stem-like cells by inhibiting fatty acid synthase. Breast Cancer Res Treat. 2011; 130:387-98.
29. Das UN. Essential fatty acids and their metabolites as modulators of stem cell biology with reference to inflammation, cancer, and metastasis. Cancer Metastasis Rev. 2011; 30:311-24.
30. Chen Y, Hu Y, Zhang H, Peng C, Li S. Loss of the Alox5 gene impairs leukemia stem cells and prevents chronic myeloid leukemia. Nat Genet. 2009. 41:783-92.
31. Chen Y, Li D, Li S. The Alox5 gene is a novel therapeutic target in cancer stem cells of chronic myeloid leukemia. Cell Cycle. 2009. 8:3488-92.
32. Roos J, Oancea C, Heinssmann M, Khan D, Held H, Kahnt AS, Capelo R, la Buscato E, Proschak E, Puccetti E, Steinhilber D, Fleming I, Maier TJ, Ruthardt M. 5-Lipoxygenase is a candidate target for therapeutic management of stem cell-like cells in acute myeloid leukemia. Cancer Res. 2014; 74:5244-55.
33. Wang B, Yu SC, Jiang JY, Porter GW, Zhao LT, Wang Z, Tan H, Cui YH, Qian C, Ping YF, Bian XW. An inhibitor of arachidonate 5-lipoxygenase, Nordy, induces differentiation and inhibits self-renewal of glioma stem-like cells. Stem Cell Rev. 2011; 7:458-70.
34. Yang X, Cui W, Yu S, Xu C, Chen G, Gu A, Li T, Cui Y, Zhang X, Bian X. A synthetic dl-nordihydroguaiaretic acid (Nordy), inhibits angiogenesis, invasion and proliferation of glioma stem cells within a zebrafish xenotransplantation model. PLoS One. 2014; 9:e85759.
35. Annabi B, Laflamme C, Sina A, Lachambre MP, Beliveau R. A MT1-MMP/NF-kappaB signaling axis as a checkpoint controller of COX-2 expression in CD133+ U87 glioblastoma cells. J Neuroinflammation. 2009. 6:8.
36. Bitarte N, Bandres E, Boni V, Zarate R, Rodriguez J, Gonzalez-Huarriz M, Lopez I, Javier Sola J, Alonso MM, Fortes P, Garcia-Foncillas J. MicroRNA-451 is involved in the self-renewal, tumorigenicity, and chemoresistance of colorectal cancer stem cells. Stem Cells. 2011; 29:1661-71.
37. Wang KH, Kao AP, Chang CC, Lee JN, Hou MF, Long CY, Chen HS, Tsai EM. Increasing CD44+/CD24(-) tumor stem cells, and upregulation of COX-2 and HDAC6, as major functions of HER2 in breast tumorigenesis. Mol Cancer. 2010; 9:288.
38. Kanojia D, Zhou W, Zhang J, Jie C, Lo PK, Wang Q, Chen H. Proteomic profiling of cancer stem cells derived from primary tumors of HER2/Neu transgenic mice. Proteomics. 2012; 12:3407-15.
39. Castellone MD, Teramoto H, Williams BO, Druey KM, Gutkind JS. Prostaglandin E2 promotes colon cancer cell growth through a Gs-axin-beta-catenin signaling axis. Science. 2005. 310:1504-10.
40. Al-Kharusi MR, Smartt HJ, Greenhough A, Collard TJ, Emery ED, Williams AC, Paraskeva C. LGR5 promotes survival in human colorectal adenoma cells and is upregulated by PGE2: implications for targeting adenoma stem cells with NSAIDs. Carcinogenesis. 2013; 34:1150-7.
41. Majumder M, Xin X, Liu L, Girish GV, Lala PK. Prostaglandin E2 receptor EP4 as the common target on cancer cells and macrophages to abolish angiogenesis, lymphangiogenesis, metastasis, and stem-like cell functions. Cancer Sci. 2014. 105:1142-51.
42. Leng J, Han C, Demetris AJ, Michalopoulos GK, Wu T. Cyclooxygenase-2 promotes hepatocellular carcinoma cell growth through Akt activation: evidence for Akt inhibition in celecoxib-induced apoptosis. Hepatology. 2003. 38:756-68.
43. Chu TH, Chan HH, Kuo HM, Liu LF, Hu TH, Sun CK, Kung ML, Lin SW, Wang EM, Ma YL, Cheng KH, Lai KH, Wen ZH, Hsu PI, Tai MH. Celecoxib suppresses hepatoma stemness and progression by up-regulating PTEN. Oncotarget. 2014; 5:1475-90. doi: 10.18632/oncotarget.1745.
44. Moon CM, Kwon JH, Kim JS, Oh SH, Jin Lee K, Park JJ, Pil Hong S, Cheon JH, Kim TI, Kim WH. Nonsteroidal anti-inflammatory drugs suppress cancer stem cells via inhibiting PTGS2 (cyclooxygenase 2) and NOTCH/HES1 and activating PPARG in colorectal cancer. Int J Cancer. 2014; 134:519-29.
45. Deng Y, Su Q, Mo J, Fu X, Zhang Y, Lin EH. Celecoxib downregulates CD133 expression through inhibition of the Wnt signaling pathway in colon cancer cells. Cancer Invest. 2013. 31:97-102.
46. Cho DY, Lin SZ, Yang WK, Lee HC, Hsu DM, Lin HL, Chen CC, Liu CL, Lee WY, Ho LH. Targeting cancer stem cells for treatment of glioblastoma multiforme. Cell Transplant. 2013; 22:731-9.
47. Pang LY, Gatenby EL, Kamida A, Whitelaw BA, Hupp TR, Argyle DJ. Global gene expression analysis of canine osteosarcoma stem cells reveals a novel role for COX-2 in tumour initiation. PLoS One. 2014. 9:e83144.
48. Nomura DK, Lombardi DP, Chang JW, Niessen S, Ward AM, Long JZ, Hoover HH, Cravatt BF. Monoacylglycerol lipase exerts dual control over endocannabinoid and fatty acid pathways to support prostate cancer. Chem Biol. 2011; 18:846-56.
49. Li Y, Li A, Glas M, Lal B, Ying M, Sang Y, Xia S, Trageser D, Guerrero-Cazares H, Eberhart CG, Quinones-Hinojosa A, Scheffler B, Laterra J. c-Met signaling induces a reprogramming network and supports the glioblastoma stem-like phenotype. Proc Natl Acad Sci U S A. 2011; 108:9951-6.
50. Sun P, Xia S, Lal B, Shi X, Yang KS, Watkins PA, Laterra J. Lipid metabolism enzyme ACSVL3 supports glioblastoma stem cell maintenance and tumorigenicity. BMC Cancer. 2014; 14:401.
51. Ben-David U, Gan QF, Golan-Lev T, Arora P, Yanuka O, Oren YS, Leikin-Frenkel A, Graf M, Garippa R, Boehringer M, Gromo G, Benvenisty N. Selective elimination of human pluripotent stem cells by an oleate synthesis inhibitor discovered in a high-throughput screen. Cell Stem Cell. 2013; 12:167-79.
52. Noto A, Raffa S, De Vitis C, Roscilli G, Malpicci D, Coluccia P, Di Napoli A, Ricci A, Giovagnoli MR, Aurisicchio L, Torrisi MR, Ciliberto G, Mancini R. Stearoyl-CoA desaturase-1 is a key factor for lung cancer-initiating cells. Cell Death Dis. 2013; 4:e947.
53. Lamb R, Harrison H, Smith DL, Townsend PA, Jackson T, Ozsvari B, Martinez-Outschoorn UE, Pestell RG, Howell A, Lisanti MP, Sotgia F. Targeting tumor-initiating cells: Eliminating anabolic cancer stem cells with inhibitors of protein synthesis or by mimicking caloric restriction. Oncotarget. 2015; 6:4585-601. doi: 10.18632/oncotarget.3278.
54. Qiu J, Ai L, Ramachandran C, Yao B, Gopalakrishnan S, Fields CR, Delmas AL, Dyer LM, Melnick SJ, Yachnis AT, Schwartz PH, Fine HA, Brown KD, Robertson KD. Invasion suppressor cystatin E/M (CST6): high-level cell type-specific expression in normal brain and epigenetic silencing in gliomas. Lab Invest. 2008; 88:910-25.
55. Malla RR, Gopinath S, Alapati K, Gorantla B, Gondi CS, Rao JS. uPAR and cathepsin B inhibition enhanced radiation-induced apoptosis in gliomainitiating cells. Neuro Oncol. 2012. 14:745-60.
56. Alapati K, Gopinath S, Malla RR, Dasari VR, Rao JS. uPAR and cathepsin B knockdown inhibits radiation-induced PKC integrated integrin signaling to the cytoskeleton of glioma-initiating cells. Int J Oncol. 2012. 41:599-610.
57. Vlashi E, Lagadec C, Chan M, Frohnen P, McDonald AJ, Pajonk F. Targeted elimination of breast cancer cells with low proteasome activity is sufficient for tumor regression. Breast Cancer Res Treat. 2013. 141:197-203.
58. Pan J, Zhang Q, Wang Y, You M. 26S proteasome activity is down-regulated in lung cancer stem-like cells propagated in vitro. PLoS One. 2010. 5:e13298.
59. Adikrisna R, Tanaka S, Muramatsu S, Aihara A, Ban D, Ochiai T, Irie T, Kudo A, Nakamura N, Yamaoka S, Arii S. Identification of pancreatic cancer stem cells and selective toxicity of chemotherapeutic agents. Gastroenterology. 2012; 143:234-45.e7.
60. Tamari K, Hayashi K, Ishii H, Kano Y, Konno M, Kawamoto K, Nishida N, Koseki J, Fukusumi T, Hasegawa S, Ogawa H, Hamabe A, Miyo M, Noguchi K, Seo Y, Doki Y, Mori M, Ogawa K. Identification of chemoradiation-resistant osteosarcoma stem cells using an imaging system for proteasome activity. Int J Oncol. 2014; 45:2349-54.
61. Lagadec C, Vlashi E, Bhuta S, Lai C, Mischel P, Werner M, Henke M, Pajonk F. Tumor cells with low proteasome subunit expression predict overall survival in head and neck cancer patients. BMC Cancer. 2014; 14:152.
62. Muramatsu S, Tanaka S, Mogushi K, Adikrisna R, Aihara A, Ban D, Ochiai T, Irie T, Kudo A, Nakamura N, Nakayama K, Tanaka H, Yamaoka S, Arii S. Visualization of stem cell features in human hepatocellular carcinoma reveals in vivo significance of tumor-host interaction and clinical course. Hepatology. 2013; 58:218-28.
63. Lagadec C, Vlashi E, Frohnen P, Alhiyari Y, Chan M, Pajonk F. The RNA-binding protein Musashi-1 regulates proteasome subunit expression in breast cancer- and glioma-initiating cells. Stem Cells. 2014. 32:135-44.
64. Vlashi E, Kim K, Lagadec C, Donna LD, McDonald JT, Eghbali M, Sayre JW, Stefani E, McBride W, Pajonk F. In vivo imaging, tracking, and targeting of cancer stem cells. J Natl Cancer Inst. 2009; 101:350-9.
65. Zhang P, Lathia JD, Flavahan WA, Rich JN, Mattson MP. Squelching glioblastoma stem cells by targeting REST for proteasomal degradation. Trends Neurosci. 2009. 32:559-65.
66. Achuthan S, Santhoshkumar TR, Prabhakar J, Nair SA, Pillai MR. Drug-induced senescence generates chemoresistant stemlike cells with low reactive oxygen species. J Biol Chem. 2011. 286:37813-29.
67. Bellail AC, Olson JJ, Yang X, Chen ZJ, Hao C. A20 ubiquitin ligase-mediated polyubiquitination of RIP1 inhibits caspase-8 cleavage and TRAIL-induced apoptosis in glioblastoma. Cancer Discov. 2012. 2:140-55.
68. Kossatz U, Breuhahn K, Wolf B, Hardtke-Wolenski M, Wilkens L, Steinemann D, Singer S, Brass F, Kubicka S, Schlegelberger B, Schirmacher P, Manns MP, Singer JD, Malek NP. The cyclin E regulator cullin 3 prevents mouse hepatic progenitor cells from becoming tumor-initiating cells. J Clin Invest. 2010; 120:3820-33.
69. Izumi H, Kaneko Y. Trim32 Facilitates Degradation of MYCN on Spindle Poles and Induces Asymmetric Cell Division in Human Neuroblastoma Cells. Cancer Res. 2014. 74:5620-30.
70. Chung S, Suzuki H, Miyamoto T, Takamatsu N, Tatsuguchi A, Ueda K, Kijima K, Nakamura Y, Matsuo Y. Development of an orally-administrative MELK-targeting inhibitor that suppresses the growth of various types of human cancer. Oncotarget. 2012; 3:1629-40. doi: 10.18632/oncotarget.790.
71. Chan CH, Morrow JK, Li CF, Gao Y, Jin G, Moten A, Stagg LJ, Ladbury JE, Cai Z, Xu D, Logothetis CJ, Hung MC, Zhang S, Lin HK. Pharmacological inactivation of Skp2 SCF ubiquitin ligase restricts cancer stem cell traits and cancer progression. Cell. 2013; 154:556-68.
72. Di K, Linskey ME, Bota DA. TRIM11 is overexpressed in high-grade gliomas and promotes proliferation, invasion, migration and glial tumor growth. Oncogene. 2013. 32:5038-47.
73. Zhang XY, Pfeiffer HK, Thorne AW, McMahon SB. USP22, an hSAGA subunit and potential cancer stem cell marker, reverses the polycomb-catalyzed ubiquitylation of histone H2A. Cell Cycle. 2008. 7:1522-4.
74. Chipumuro E, Henriksen MA. The ubiquitin hydrolase USP22 contributes to 3'-end processing of JAK-STAT-inducible genes. FASEB J. 2012. 26:842-54.
75. Eyler CE, Wu Q, Yan K, MacSwords JM, Chandler-Militello D, Misuraca KL, Lathia JD, Forrester MT, Lee J, Stamler JS, Goldman SA, Bredel M, McLendon RE, Sloan AE, Hjelmeland AB, Rich JN. Glioma stem cell proliferation and tumor growth are promoted by nitric oxide synthase-2. Cell. 2011; 146:53-66.
76. Zhang WC, Shyh-Chang N, Yang H, Rai A, Umashankar S, Ma S, Soh BS, Sun LL, Tai BC, Nga ME, Bhakoo KK, Jayapal SR, Nichane M, Yu Q, Ahmed DA, Tan C, Sing WP, Tam J, Thirugananam A, Noghabi MS, Pang YH, Ang HS, Mitchell W, Robson P, Kaldis P, Soo RA, Swarup S, Lim EH, Lim B. Glycine decarboxylase activity drives non-small cell lung cancer tumor-initiating cells and tumorigenesis. Cell. 2012; 148:259-72.
77. Mohamed A, Deng X, Khuri FR, Owonikoko TK. Altered glutamine metabolism and therapeutic opportunities for lung cancer. Clin Lung Cancer. 2014. 15:7-15.
78. Son J, Lyssiotis CA, Ying H, Wang X, Hua S, Ligorio M, Perera RM, Ferrone CR, Mullarky E, Shyh-Chang N, Kang Y, Fleming JB, Bardeesy N, Asara JM, Haigis MC, DePinho RA, Cantley LC, Kimmelman AC. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature. 2013; 496:101-5.
79. Li D, Fu Z, Chen R, Zhao X, Zhou Y, Zeng B, Yu M, Zhou Q, Lin Q, Gao W, Ye H, Zhou J, Li Z, Liu Y, Chen R. Inhibition of glutamine metabolism counteracts pancreatic cancer stem cell features and sensitizes cells to radiotherapy. Oncotarget. 2015; 6:31151-63. doi: 10.18632/oncotarget.5150.
80. Guidoni L, Ricci-Vitiani L, Rosi A, Palma A, Grande S, Luciani AM, Pelacchi F, di Martino S, Colosimo C, Biffoni M, De Maria R, Pallini R, Viti V. 1H NMR detects different metabolic profiles in glioblastoma stem-like cells. NMR Biomed. 2014; 27:129-45.
81. Saga I, Shibao S, Okubo J, Osuka S, Kobayashi Y, Yamada S, Fujita S, Urakami K, Kusuhara M, Yoshida K, Saya H, Sampetrean O. Integrated analysis identifies different metabolic signatures for tumor-initiating cells in a murine glioblastoma model. Neuro Oncol. 2014; 16:1048-56.
82. Vincent Z, Urakami K, Maruyama K, Yamaguchi K, Kusuhara M. CD133-positive cancer stem cells from Colo205 human colon adenocarcinoma cell line show resistance to chemotherapy and display a specific metabolomic profile. Genes Cancer. 2014. 5:250-60. doi: 10.18632/genesandcancer.23.
83. Shibuya K, Okada M, Suzuki S, Seino M, Seino S, Takeda H, Kitanaka C. Targeting the facilitative glucose transporter GLUT1 inhibits the self-renewal and tumor-initiating capacity of cancer stem cells. Oncotarget. 2015; 6:651-61. doi: 10.18632/oncotarget.2892.
84. Flavahan WA, Wu Q, Hitomi M, Rahim N, Kim Y, Sloan AE, Weil RJ, Nakano I, Sarkaria JN, Stringer BW, Day BW, Li M, Lathia JD, Rich JN, Hjelmeland AB. Brain tumor initiating cells adapt to restricted nutrition through preferential glucose uptake. Nat Neurosci. 2013; 16:1373-82.
85. Micucci C, Orciari S, Catalano A. Hyperglycemia promotes K-Ras-induced lung tumorigenesis through BASCs amplification. PLoS One. 9:e105550.
86. Takeuchi M, Kimura S, Kuroda J, Ashihara E, Kawatani M, Osada H, Umezawa K, Yasui E, Imoto M, Tsuruo T, Yokota A, Tanaka R, Nagao R, Nakahata T, Fujiyama Y, Maekawa T. Glyoxalase-I is a novel target against Bcr-Abl+ leukemic cells acquiring stem-like characteristics in a hypoxic environment. Cell Death Differ. 2010; 17:1211-20.
87. Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, Ito S, Yang C, Wang P, Xiao MT, Liu LX, Jiang WQ, Liu J, Zhang JY, Wang B, Frye S, Zhang Y, Xu YH, Lei QY, Guan KL, Zhao SM, Xiong Y. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011; 19:17-30.
88. Turcan S, Fabius AW, Borodovsky A, Pedraza A, Brennan C, Huse J, Viale A, Riggins GJ, Chan TA. Efficient induction of differentiation and growth inhibition in IDH1 mutant glioma cells by the DNMT Inhibitor Decitabine. Oncotarget. 2013; 4:1729-36. doi: 10.18632/oncotarget.1412.
89. Fiorillo M, Lamb R, Tanowitz HB, Cappello AR, Martinez-Outschoorn UE, Sotgia F, Lisanti MP. Bedaquiline, an FDA-approved antibiotic, inhibits mitochondrial function and potently blocks the proliferative expansion of stem-like cancer cells (CSCs). Aging (Albany NY). 2016. 8:1593-607. doi: 10.18632/aging.100983.
90. Cuyas E, Corominas-Faja B, Menendez JA. The nutritional phenome of EMT-induced cancer stem-like cells. Oncotarget. 2014; 5:3970-82.
91. Martinez-Outschoorn UE, Prisco M, Ertel A, Tsirigos A, Lin Z, Pavlides S, Wang C, Flomenberg N, Knudsen ES, Howell A, Pestell RG, Sotgia F, Lisanti MP. Ketones and lactate increase cancer cell “stemness,” driving recurrence, metastasis and poor clinical outcome in breast cancer: achieving personalized medicine via Metabolo-Genomics. Cell Cycle. 2011; 10:1271-86.
92. Lamb R, Harrison H, Hulit J, Smith DL, Lisanti MP, Sotgia F. Mitochondria as new therapeutic targets for eradicating cancer stem cells: Quantitative proteomics and functional validation via MCT1/2 inhibition. Oncotarget. 2014. 5:11029-37. doi: 10.18632/oncotarget.2789.
93. Blagosklonny MV. Target for cancer therapy: proliferating cells or stem cells. Leukemia. 2006. 20:385-91.
94. Ko YH, Verhoeven HA, Lee MJ, Corbin DJ, Vogl TJ, Pedersen PL. A translational study “case report” on the small molecule “energy blocker” 3-bromopyruvate (3BP) as a potent anticancer agent: from bench side to bedside. J Bioenerg Biomembr. 2012. 44:163-70.