
INTRODUCTION
Type 2 diabetes mellitus (T2DM) accounts for 90% of diabetes [1]. T2DM is not due to insufficient use of insulin but due to insufficient insulin secretion and insufficient insulin action. Hospitalization rate for urinary tract infection (UTI) caused by diabetes is over twice as much as those caused by other factors [2]. Damage to the genitourinary system caused by diabetic neuropathy results in bladder dysfunction, and increases the probability of UTI [3]. High levels of urine glucose (UGLU) can favor a proper microenvironment for UTI due to increased bacterial overgrowth [4]. Female patients are known to have higher prevalence of UTI than males [5], which may be associated with the anatomical and structural differences in the urethra between genders.
General clinical practice dictates diagnosing UTI with a standard urine culture (SUC), then prescribing proper antibiotics based on the culture results. UTI is diagnosed when a known uropathogen surpasses 105 colony-forming units (CFUs) per milliliter [6]. The premise of this diagnosis is based on the hypothesis that the bladder is sterile. SUC limitations include: 90% of bacteria cannot be cultured via standard culture media [7], SUC technique favors fast-growing bacteria but cannot consider the fastidious pathogens [8], and SUC techniques are unable to detect the presence of bacteria embedded within biofilms [9]. Fastidious pathogens may not lead to a UTI diagnosis because they are below the 105 CFU/mL threshold, but can still cause urinary disorders [4].
Recent 16S rRNA sequencing results indicate that the urinary tract possesses bacteria, regardless of a patient’s present urinary tract symptoms [10-23]. The main bacteria of females with asymptomatic bacteriuria are Lactobacillus [11, 12, 18, 22], Gardnerella [11, 13, 18], and Prevotella [11, 12]. Increased diversity of urinary microbiota in females undergoing stress urinary incontinence surgery was correlated with patients’ hormonal status and body mass index (BMI) [22]. Urinary microbiota in women with urgency urinary incontinence (UUI) was affected by UUI episodes, treatment response, and risk of post-treatment UTI [20]. Because microorganisms are present in healthy urinary systems, administering antibiotics to treat a UTI may be inappropriate. Antibiotics may kill the beneficial microbiota in the bladder, promote harmful bacteria to multiply, and result in dysbiosis.
With the occurrence of T2DM, the function of the urinary system can be impaired, as indicated by the urine composition, showing bladder dysfunction, increased UGLU, and renal urine net acid excretion [24-26]. We studied whether the difference in urinary microbiota between healthy controls (HCs) and T2DM patients can be detected, and if these differences are influenced by patients’ conditions, such as fasting blood glucose (FBG), UGLU, BMI, etc.
RESULTS
Cohort description
The women in both cohorts were similar in age, marital status, menstrual status, BMI, co-occurrence of coronary heart disease and hypertension, water intake, and asymptomatic bacteriuria (p > 0.05). T2DM cohort had higher FBG, UGLU, higher occurrence of hyperlipidemia, and more UTIs in the last year (p < 0.05) (Table 1).
Table 1: Descriptive data of participants
Parameter |
Value for cohort (na)b or statistic |
|||
HCs |
T2DM |
p-valuec |
||
Age group (yrs) [no.(%)] d |
1.00 |
|||
26-35 |
2 (2.86) |
2 (2.86) |
||
36-45 |
6 (8.57) |
6 (8.57) |
||
46-55 |
11 (15.71) |
11 (15.71) |
||
56-65 |
17 (24.3) |
17 (24.3) |
||
66-75 |
23 (32.9) |
23 (32.9) |
||
76 and above |
11 (15.7) |
11 (15.7) |
||
Marital status [no. (%)]d |
1.00 |
|||
Living with a spouse |
64 (91.4) |
64 (91.4) |
||
Not living with a spouse |
6 (8.6) |
6 (8.6) |
||
Menstrual status [no. (%)]d |
1.00 |
|||
Premenopausal |
11 (15.7) |
11 (15.7) |
||
Postmenopausal |
54 (77.1) |
54 (77.1) |
||
Hysterectomy |
5 (7.1) |
5 (7.1) |
||
Body mass index (kg/m2) |
23.10 ± 4.49 |
23.87 ± 3.65 |
0.27 |
|
Co-occurrence of disease [no. (%)] |
||||
Coronary Heart Disease |
8 (11.42) |
13 (18.57) |
0.34 |
|
Hypertension |
23 (32.86) |
35 (50.00) |
0.06 |
|
Hyperlipidemia |
7 (10.00) |
18 (25.71) |
0.03 |
|
Diabetes duration (yrs) |
N/A |
9.77 (± 7.49) |
||
FBG (mmol/L) |
5.22 (± 0.61) |
7.83 (± 2.35) |
0.00 |
|
FBG > 10 mmol/L [no. (%)] |
N/A |
12 (17.1) |
||
UGLU POS [no. (%)] e |
0(0.00) |
14 (20.00) |
0.00 |
|
Taking metformin |
N/A |
70 (100.00) |
||
UTIs [no. (%)] f |
7 (10.00) |
27 (38.57) |
0.00 |
|
Water intake g |
2484.24 ± 99.20 |
2526.04 ± 108.02 |
0.78 |
|
Asymptomatic bacteriuria with E. coli POS [no. (%)]e |
5 (7.14) |
6 (8.57) |
1.00 |
|
a no. of subjects.
b Mean ± SD or no.(%).
c Pearson’s chi-square and Fisher’s exact tests were used with categorical variables. Independent t-test was used with continuous variables.
d Variables used for individual matching.
e POS indicates positive.
f UTIs indicates episodes of UTI in the last year.
g The amount of water intake included drinking water intake and dietary fluid intake, which was examined by the Chinese Food Frequency Questionnaire.
Sequencing data
From 140 samples, a total of 8,602,818 high quality reads were produced, with a median read length of 441bp (range from 423 to 491). Those reads accounted for 78.91% of the valid reads (11,181,603 total sequences), with an average of 61,448 reads (range from 13,419 to 308,123) per barcoded sample for downstream analysis. The total number of unique sequences from the two cohorts was 2,501,986, and represented all phylotypes.
Good’s coverage indicated sufficient depth for the investigation of T2DM-associated urinary microbiota (Figure 1A). Both Shannon and Simpson indices illustrated the urinary microbiota diversity was lower in T2DM patients than in HCs (Table 2 and Table S1; Figure 1B and 1C). In the richness indices, ACE and Chao1 were lower in T2DM patients than those in HCs (Table 2 and Table S1). Interestingly, when correlation analyses between microbiota diversity and richness indices and participants’ age were conducted, no significant correlations were found (Table S2). A Venn diagram demonstrated 31,024 of the total 82,904 OTUs were shared between the two cohorts (Figure 1D). To measure the extent of the similarity of microbial communities, beta diversity was calculated using unweighted UniFrac and principal coordinate analysis (Figure 1E). The heatmap showed that the two cohorts were partially clustered (Figure S1).
Table 2: Comparison of richness and diversity estimation in HC and T2DM patients’ urine microbiota
Parametera |
HCs |
T2DM |
p-value |
No. of reads |
4,621,299 |
3,981,519 |
0.16 |
No. of OTUs b |
2692 |
1708 |
0.00 |
ACE c |
5446 |
3869 |
0.00 |
Chao1 |
2336 |
1500 |
0.00 |
Shannon |
5.08 |
4.05 |
0.03 |
Simpson |
0.72 |
0.67 |
0.23 |
Observed species |
986 |
589 |
0.00 |
PD whole tree |
97 |
64 |
0.00 |
a parameters calculated by QIIME software;
b the operational taxonomic units (OTUs) were defined at the 97% similarity level;
c ACE indicates Abundance-based Coverage Estimator.
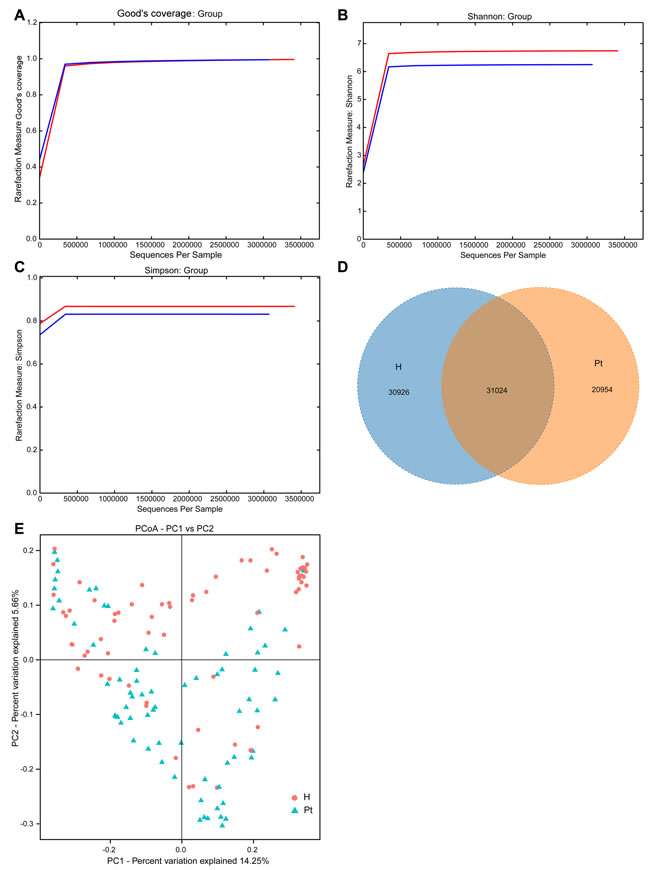
Figure 1: Structural comparison of urinary microbiota between two cohorts. A. The Good’s coverage was used to assess sequencing depth. B. and C. The Shannon and Simpson Rarefaction curves were applied to estimate diversity. D. Venn diagram demonstrating overlap of OTUs in the urinary microbiota between the two cohorts. E. Principal coordinate analysis plot of the urinary microbiota based on the unweighted UniFrac metric. Red and blue lines and dots represent healthy controls and T2DM patients, respectively. H and Pt stand for healthy controls and T2DM patients, respectively.
Associations of urinary microbiota and T2DM
At the phylum level, the predominant sequences in the HCs were from Proteobacteria (58.01%), Firmicutes (22.41%), Bacteroidetes (9.33%), Actinobacteria (4.76%), and Acidobacteria (1.31%). The abundant sequences for the T2DM group belonged to Proteobacteria (51.63%), Firmicutes (24.31%), Bacterioidetes (13.07%), Actinobacteria (7.49%), and Thermi (0.80%). When the relative abundance of bacterial phylum was compared, Chloroflexi, Nitrospirae, and Gemmatimonadetes were more abundant in the HCs than those from the T2DM cohort (data not shown).
At the genus level, HCs were mainly assigned to Prevotella (12.67%), Blautia (7.24%), and Klebsiella (7.07%). The most abundant genus was also Prevotella (18.76%) in T2DM patients, followed by Lactobacillus (12.15%) and Shuttleworthia (6.42%). Genera with different relative abundances between the two cohorts are listed in Figure 2, and their proportions are listed in Table S3 (p < 0.05). The relative abundance of Gardnerella was only 0.0018% and 0.0017% in the HCs and T2DM patients, respectively. Moreover, it only presented in 3 HCs and 5 T2DM patients. Only 18.57% (13/70) of the HCs and 17.14% (12/70) of the T2DM subjects presented Escherichia (p > 0.05).
The significantly different bacterial species are listed in Figure S2. The abundance of Akkermansia muciniphila was significantly higher in the HCs than T2DM subjects. Interestingly, Escherichia coli was not significantly more abundant in patients than HCs (0.008 ± 0.010 vs. 0.006 ± 0.009, p > 0.05).
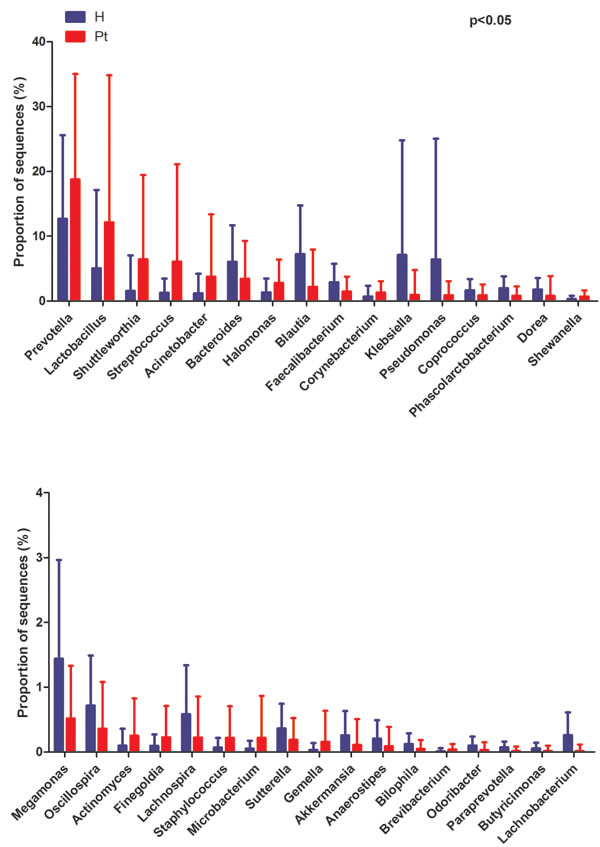
Figure 2: Genus-level OTUs different between the two cohorts (Mean ± SD). Welch’s t-test was used to compare the abundance at the bacterial genus level between HCs and T2DM patients. The different genera were assigned only to those presenting a minimum variation at a significant level [p (corrected) < 0.05)]. H and Pt represent healthy controls and T2DM patients, respectively.
T2DM associated biomarkers
To identify the specific bacteria taxa associated with T2DM, the urinary microbiota in the two cohorts were compared using LEfSe. A cladogram representative of the structure of the urinary microbiota and their predominant bacteria is shown in Figure 3A and 3B; the greatest differences in taxa between two cohorts are displayed. The data suggested that dysbiosis was extensive in T2DM patients. Actinobacteria, Flavobacteriales, and Flavobacteria could be used as potential distinguishing biomarkers.
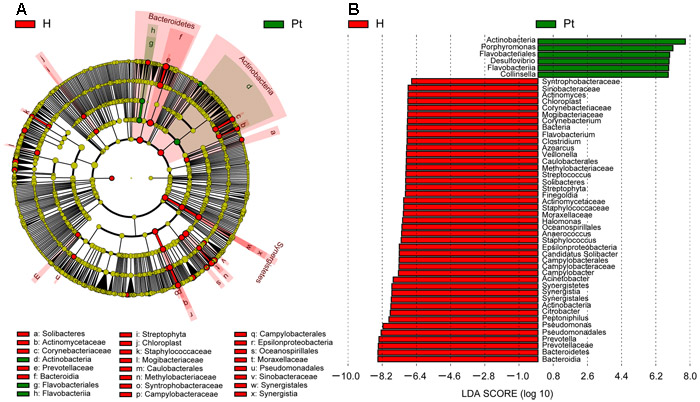
Figure 3: Cladogram showing differentially abundant taxa of microbiota. A. LEfSe cladogram showed the most differentially abundant taxa between the two cohorts. Taxonomic cladogram obtained from LEfSe analysis of 16S sequences. Taxa enriched for HCs in red; T2DM enriched taxa in Green. The brightness of each dot is proportional to its effect size. B. Only taxa meeting an LDA threshold > 1.8 are shown. H and Pt represent healthy controls and T2DM patients, respectively.
Urinary microbiota affected by participants’ conditions
The relative abundance of Actinobacteria and Lactobacillus increased with FBG and UGLU in the T2DM cohort (Figure S3A, S3B, S4A, S4B). In contrast, A. muciniphila was lower in the T2DM patients with FBG > 10 mmol/L and UGLU positive results (Figure S5A and S5B).
The elderly (> = 65 years old) HCs had significantly lower abundance of Lactobacillus than the non-elderly (< 65 years old) HCs [38], but the elderly T2DM patients did not show significantly decreased Lactobacillus abundance compared to the non-elderly T2DM patients. The healthy elderly had significantly lower abundance of Lactobacillus than the elderly T2DM patients. In contrast, the healthy non-elderly subjects did not show a significantly lower abundance than the non-elderly T2DM patients (Figure 4A). The HC subgroup with the lowest Lactobacillus abundance was the post-menopause subgroup, and the lowest one from the T2DM cohort was the hysterectomy subgroup (Figure 4B). Moreover, the abundance of Lactobacillus in the HCs was negatively correlated to the years after menopause (r = -0.33, p < 0.01). No correlation was found in the T2DM patients (r = -0.06, p > 0.05).
A. muciniphila was most abundant in the obese HC group, and least abundant in the obese T2DM group (Figure S6A). Actinobacteria was not only enriched in T2DM patients, but also increased with their BMI (Figure S6B).
No difference was noted between the asymptomatic bacteriuria samples with E. coli, and the non-asymptomatic bacteriuria samples (0.02 ± 0.01, 0.01 ± 0.01, p > 0.05). In addition, the abundance of the E. coli was correlated with water intake (r = 0.26, p < 0.01). No correlation with UTIs was found (r = 0.11, p > 0.05).
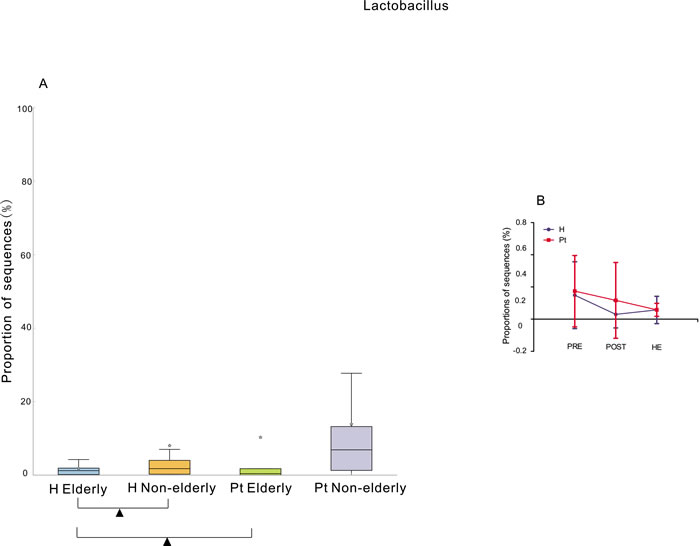
Figure 4: Relative abundance of Lactobacillus associated with age and menstrual status. A. Box plot showing the distribution in the proportion of Lactobacillus assigned to samples from H elderly, H non-elderly, Pt elderly, and Pt non-elderly. Triangles represent a significant difference was found between H elderly and H non-elderly, and between H elderly and Pt elderly. The median value is shown as a line within the box, and the mean value as a star. B. The difference among pre-menopausal status (PRE), hysterectomy (HE), and post-menopausal (POST) status groups. ANOVA test was applied, and Benjamini-Hochberg FDR was used as a correction approach to control the false discovery rate, p (corrected) < 0.05 was considered significant. H and Pt mean healthy controls and T2DM patients, respectively.
Urinary microbiota associated with metabolism
We observed that HCs and T2DM patients had differing patterns of enrichment in terms of clusters of orthologous group categories. Carbohydrate and amino acid metabolism was damaged in T2DM patients (Figure 5), and they were positively correlated with Chao1 and ACE (r = 0.53, p < 0.01: r = 0.56, p < 0.01; r = 0.54, p < 0.01: r = 0.58, p < 0.01).
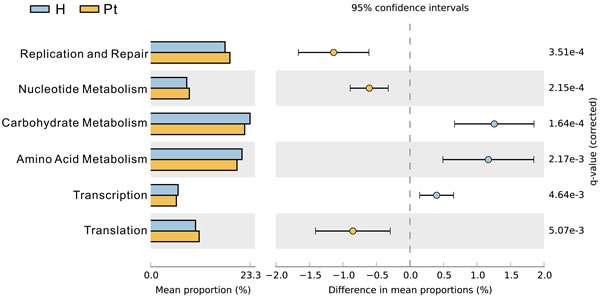
Figure 5: Clusters of orthologous group categories. Clusters of orthologous group categories reveal metabolic functions that were enriched in urinary microbiota from the two cohorts. P values were based on White’s nonparameteric t-test with the Benjamini-Hochberg FDR false discovery rate correction approach.
DISCUSSION
In the present study, a reduction of urinary microbial diversity and overall richness in T2DM patients was detected in contrast to HCs (Figure 1B, 1C, and Table 2). The alteration was similar to previous studies that compared the urinary microbiota of HCs to UUI and interstial cystitis patients [13, 18], which also indicated that non-healthy patients had reduced diversity and richness. Additionally, it was similar to another study in which the urinary microbiota richness was lower in UTI patients than non-UTI subjects [21]. However, it was dissimilar to a recent study, which revealed that UUI patients had slightly higher diversity than controls [28]. The diversity was not correlated with age (Table S2), and was inconsistent with a previous study in which women over 70 had 75% fewer bacteria than women aged 20-49 [15]. The Venn diagram demonstrated less bacterial phylotypes in T2DM patients compared to HCs (Figure 1D). It also suggested the occurrence of T2DM has altered urinary microbiota composition. The principal coordinate analysis and heatmap showed that most of the samples from the HCs and T2DM patients can be clustered together (Figure 1E and Figure S1), although not completely clustered. The inter-individual variation suggests that other factors may affect urinary microbiota besides T2DM. This phenomenon of inter-individual variation is also demonstrated by recent studies on urinary microbiota [10, 12, 16, 17, 28].
The most abundant phylum in both cohorts was Proteobacteria, as opposed to Firmicutes, which has commonly been found to be the most abundant bacteria [11, 13, 18, 22, 28]. The relative abundance of Proteobacteria was higher in the HCs than the T2DM cohort. This result is in contrast to similar studies which found the HCs to have lower relative Proteobacteria abundance when compared to non-healthy controls [21].
In the present study, the most abundant bacteria for both cohorts were Prevotella, while Lactobacillus was the second most abundant bacteria. In previous studies, Lactobacillus was the most abundant genus, while Prevotella was the second and/or third most abundant, or even less [11, 18, 20]. This shift may be associated with ethnicity, since there is a difference between Caucasian and Chinese gut microbiota [29]. Prevotella was enriched in T2DM patients (Figure 2), and similar studies have shown Prevotella is higher in non-healthy patients when compared to controls [13, 18]. Prevotella has been considered pathogenic in the vaginal microbiome [30], and future studies should explore whether Prevotella also acts pathogenic in the infections in diabetes [31].
Lactobacillus was increased in T2DM patients (Figure 2), and abundance increased with patients’ FBG and UGLU levels (Figure S4A, S4B). Lactobacillus is probiotic, and can produce organic acid and hydrogen peroxide that create an inhospitable environment for pathogenic bacteria. Treatment with Lactobacillus can reduce the incidence of UTIs [32], and can reduce FBG, homocysteine, and interleukin-6 which play roles in preventing infection for diabetic patients [33-35]. In addition, the higher proportion of Lactobacillus can increase insulin secretion [36]. Therefore, increased Lactobacillus might be considered a protective effect on T2DM patients from infections and reducing blood glucose.
The elderly and postmenopausal participants from both the HC and T2DM cohorts had a lower abundance of Lactobacillus (Figure 4A, 4B). This is similar to Karstens et al. study, in which Lactobacillus dominated samples from premenopausal female [28], and higher frequency of Lactobacillus was detected in samples from premenopausal female and postmenopausal female on exogenous hormone therapy [22]. The level of free glycogen in post-menopausal women is lower than that in pre-menopausal women, caused by the reduction of estrogen [37], and levels of free glycogen are associated with the levels of Lactobacillus in the vagina [37, 38]. The abundance of Lactobacillus in urine can be affected by the levels of vaginal Lactobacillus, because the urinary meatus is near the vagina. Thus, the reduction of Lactobacillus might be caused by the decline of estrogen with the arrival of menopause [39]. However, Lactobacillus did not decrease with the number of years after menopause in the T2DM cohort. These trends might suggest that T2DM inhibited the reduction of Lactobacillus, or reducing estrogen levels in elderly diabetes patients only plays a limited role in regulating the reproduction of Lactobacillus. Future study focusing on whether Lactobacillus in T2DM patient responds differently from HCs or estrogen regulating the reproduction of Lactobacillus in T2DM is different from HCs is needed.
The T2DM hysterectomy subgroup had the lowest Lactobacillus among pre-menopause, hysterectomy, and post-menopause patients (Figure 4B), while this was not the case in controls. This might be due to vaginal microbiota dysbiosis caused by hysterectomy, including declined Lactobacillus [40]. Adjacent to the urinary tract is the reproductive system, and it is possible that Lactobacillus of urine migrates from the vagina.
A decrease of Akkermansia and A. muciniphila in T2DM patients was found (Figure 2 and Figure S2), and the abundance of A. muciniphila decreased with patients’ FBG and UGLU (Figure S5A, S5B). Intestinal studies have also shown that A. muciniphila was involved in glucose homeostasis [41], but its abundance was not correlated with BMI (Figure S6A). This suggests that A. muciniphila cannot be considered as “lean” bacteria in urine.
Actinobacteria, a representative member of phyla in healthy female urine [15], was higher in T2DM patients than HCs. Moreover, it has been identified as a biomarker distinguishing T2DM and HCs (Figure 3A, 3B), and has a higher gut prevalence in obese patients when compared to lean subjects [42]. Our study also demonstrated that Actinobacteria increased in patients with a higher BMI (Figure S6B). Actinobacteria increased with patients’ FBG and UGLU (Figure S3A, S3B), so it could be used as a predictor of diabetic progress.
Since E. coli can adhere better in diabetic females than in healthy subjects [43], the abundance of E. coli in T2DM patients’ urine should be higher than controls. However, no difference was found in the abundance of E. coli between the two cohorts. Additionally, no difference was detected between E. coli positive and negative samples in T2DM patients. It is possible that Lactobacillus, or other probiotic bacteria, in urine could inhibit the growth of E. coli [44]. The high abundance of Lactobacillus in patients might impair the ability of E. coli to adhere to uroepithelial cells.
Gardnerella, a genus representing one of the major urotypes in previous studies [11,18,20], had decreased abundance and prevalence in both cohorts, despite being a major component of vaginal microbiota [45]. The low abundance and prevalence might be related to the MMSU technique designed in our study. Because a genuine midstream urine was obtained, the risk of Gardnerella contaminating the specimen was low. The abundance of Escherichia was less than a previous study that collected urine with transurethral catheter or suprapubic aspiration [14]. This result might suggest that the MMSU can avoid bacterial contamination.
Carbohydrate and amino acid metabolism was damaged in T2DM patients (Figure 5), and correlated with patients’ bacterial diversity. This suggests that if carbohydrate and amino acid metabolism is improved in patients, urine microbiota composition will be enhanced. Moreover, therapy based on urinary microbiota can be used to evaluate and modulate a patient’s metabolism.
Considering urine samples are at risk of contamination by bacteria of the female vagina and gut [14], we modified the aseptic technique used in the transurethral catheter. In addition, we designed a four-tube collection method which guaranteed that true midstream urine could be obtained. A previous study described that the rate of urine bacterial DNA detected for sequencing was about 33%-86% [16, 20, 46]. We applied magnetic-based beads DNA extraction method with adding lysis buffer [23] and only 1.43% (2/140) failed to provide sufficient DNA for sequence.
Our study did contain some limitations, such as including only female T2DM patients, so the influences of sex hormones on urinary microbiota could not be ruled out. Participants could not recall whether they took antibiotics over the last 6 months, and we could not collect the information in their medical records. We cannot judge long term effects of antibiotics on urinary microbiota [47]. Lastly, all the T2DM participants were treated with metformin, so we were unable to compare the effects of insulin and metformin treatment on the urinary microbiota (since the intestinal microbiota is affected by metformin) [48]. A useful future study would involve exploring the urinary microbiota of participants taking these two medications.
CONCLUSIONS
We demonstrated that microbiota dysbiosis may be associated with T2DM. Secondly, the relative abundance of some key bacteria in T2DM patients was different than in the HCs, and the relative abundancies were affected by the patients’ characteristics. Lastly, there was an interdependency between urine microbiota and the patients’ metabolism. Future studies should focus on how the urinary microbiota affects patient’s characteristics such as FBG and UGLU.
MATERIALS AND METHODS
Study design
The matched case-control study enrolled 70 patients with T2DM patients and 70 HCs from June 2015 to January 2016 from the Department of Endocrinology, the First Affiliated Hospital, School of Medicine, Zhejiang University (Table 1). The diagnostic criteria for T2DM are based on recommendations from the World Health Organization: fasting blood glucose (FBG) ≥ 7.0 mmol/L, or 2-h plasma glucose ≥ 11.0 mmol/L [49]. HCs were from local communities, had never been diagnosed with diabetes, and had a FBG ≤ 6.1 mmol/L [49]. The HCs were matched with the T2DM patients for age, marital status, and menstrual status (Table 1). A senior nurse assessed weather the participants met the inclusion criteria for the T2DM and HC cohorts. Written informed consent was obtained from participants prior to enrollment, with the approval of the Ethics Committee of the First Affiliated Hospital, School of Medicine, Zhejiang University (Reference Number: 295). The following criteria from participants’ medical records and/or complaints were used to exclude subjects: UTI in the previous month; use of antibiotics, probiotics, prebiotics, or synbiotics in the previous 3 months; unable to complete the questionnaire; menstruation; urinary incontinence; known anatomic urinary tract abnormalities (e.g. cystoceles, hydronephrosis, renal atrophy, or neurogenic bladder); urinary catheter.
Sample collection and processing
Before urine sampling, the participants were instructed to use the modified mid-stream urine (MMSU) collection technique which was composed of disinfection techniques and four-tube collection methods (Supplemental Protocol 1). The first urine of the day was collected, immediately placed on ice, transferred to the laboratory within 15 minutes, and stored at -80 °C [50]. Urine from Tube 2 and Tube 3 were used for urinalysis and SUC. Based on the guidelines established by the China Ministry of Health, urine culture was the reference method to determine specimen contamination [51].
Asymptomatic bacteriuria is defined as the presence of two consecutive MMSU specimens with isolations of the same bacterial strain at > 105 CFU/mL [52]. If asymptomatic bacteriuria was confirmed, the second urine sample was used for bacterial sequencing. Additionally, FBG was measured on the same day as urine sample collection. After the participants were recruited into the HC or T2DM cohort, T2DM patients were divided into two subgroups: FBG ≤ 10 mmol/L group (well controlled group) and FBG > 10 mmol/L group (poor controlled group) [53]. A self-report questionnaire was used to collect demographic characteristics, and the Chinese Food Frequency Questionnaire was used to examine water intake [54].
DNA extraction, PCR, and MiSeq sequencing
Total DNA was extracted from the pellet of urine from Tubes 2 and 3, and 40 mL of urine was aspirated from each tube , separated into three sections, and injected into three 15 mL sterile centrifuge tubes. Each tube was pelleted by centrifugation at 4,000 × g for 15 min at 4 °C. 10 mL of the supernatant was decanted, and the pellet was obtained by centrifugation for 15 min at 4,000 × g at 4 °C. The pellet was transferred into a 2 mL sterile centrifugation tube which contained 500 µL of lysis buffer [23]. The tube was kept at -80 °C until DNA extraction. Magnetic bead isolation of genomic DNA from bacteria was performed per the manufacturer’s protocol with minor modifications (Supplemental Protocol 2) [23]. The concentration of extracted DNA was determined by using a Nanodrop ND-1000 spectrophotometer (Thermo Electron Corporation, USA); its integrity and size were checked by 1.0 % agarose gel electrophoresis containing 0.5 mg/mL ethidium bromide. The DNA complex was placed at -20 °C until PCR amplification. Two of the 140 samples failed to provide sufficient DNA for sequencing, so we recruited two subjects who had the same attributes as the former ones, and obtained sufficient DNA for sequencing. The 16S rRNA gene V3-V4 regions were PCR-amplified from microbial genome DNA (forward primer, 5ʹ-ACTCCTACGGGAGGCAGCAG-3ʹ; reverse primer, 5ʹ-GGACTACHVGGGTWTCTAAT-3ʹ) [55]. Negative DNA extraction controls (lysis buffer and kit reagents only) were amplified and sequenced as contamination controls. The amplicons were normalized, pooled, and sequenced on the Illumina MiSeq platform using a V3 reagent kit with 2x300 cycles.
Bioinformatic and statistical analysis
Sequencing reads were processed using QIIME (version 1.9.0), and included additional quality trimming, demultiplexing, and taxonomic assignments. Profiling of predictive urine microbiota was analyzed by using PiCRUSt based on 13 August 2013 Greengenes database [56]. KW rank sum test and pairwise Wilcoxon test were used for the identification of the different markers, and LDA was used to score each feature in the LEfSe analysis. Index of alpha diversity was calculated with QIIME based on sequence similarity at 97%. Beta diversity was measured by unweighted UniFrac distance, which was also calculated by QIIME. Hierarchical clustering was performed, and a heatmap was generated using a Spearman’s rank correlation coefficient as a distance measure and a customized script developed in the R statistical package. The output file was further analyzed using Statistical Analysis of Metagenomic Profiles software package (version 2.1.3) [57].
To obtain insight into the possible functional pathways that differ between T2DM and HCs, we used PiCRUSt to calculate contributions of various OTUs to known biological pathways using Kyoto Encyclopedia of Genes and Genomes (KEGG) databases [56]. The pathways that were nonprokaryotic, had fewer than 2 sequences in each cohort, or had a difference in mean proportions less than 0.1% was excluded from analysis [23].
Statistical analysis was performed using the SPSS data analysis program (version 21.0) and Statistical Analysis of Metagenomic Profiles software. For continuous variables, independent t-test, Welch’s t- test, White’s nonparametric t-test, and Mann-Whitney U-test were applied. For categorical variables between groups, using either the Pearson chi-square or Fisher’s exact test, depending on assumption validity. For taxon among subgroups, ANOVA test was applied (Tukey-Kramer was used in Post-hoc test, Effect size was Eta-squared) with Benjamini-Hochberg FDP false discovery rate correction [58, 59]. All tests of significance were two-sided, and p < 0.05, or corrected p < 0.05, was considered statistically significant.
Accession number
The sequence data from this study are deposited in the GenBank Sequence Read Archive with accession number SRP 087709.
ACKNOWLEDGMENTS
We gratefully acknowledge the volunteers who participated in our study. We thank English Edit OT for editing this manuscript and Chunmao Han for his valuable comments on this study.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
GRANT SUPPORT
This study was supported by the Opening Foundation of the State Key Laboratory for Diagnosis and Treatment of Infectious Diseases (grant number: 2015KF05).
Authors contribution
LJL, WW, YHX, and FPL conceived and designed the study. ZXL generated the sequencing data. FPL, LZ, and JPZ collected the samples. QY and FPL conducted urine cultures and the urinalysis. FPL, LXL, HFL, and HZ extracted the bacterial DNA. FPL, ZXL, YHX, and LXL analyzed the data, carried out the computational analysis, interpreted the data, and drafted the manuscript.
Editorial note
This paper has been accepted based in part on peer-review conducted by another journal and the authors’ response and revisions as well as expedited peer-review in Oncotarget.
References
1. Rubino F. Is type 2 diabetes an operable intestinal disease? A provocative yet reasonable hypothesis. Diabetes Care. 2008; 31 Suppl 2:S290-S296.
2. Korbel L and Spencer JD. Diabetes mellitus and infection: an evaluation of hospital utilization and management costs in the United States. J Diabetes Complications. 2015; 29(2):192-195.
3. Fu AZ, Iglay K, Qiu Y, Engel S, Shankar R and Brodovicz K. Risk characterization for urinary tract infections in subjects with newly diagnosed type 2 diabetes. J Diabetes Complications. 2014; 28(6):805-810.
4. Wilke T, Boettger B, Berg B, Groth A, Mueller S, Botteman M, Yu S, Fuchs A and Maywald U. Epidemiology of urinary tract infections in type 2 diabetes mellitus patients: An analysis based on a large sample of 456,586 German T2DM patients. J Diabetes Complications. 2015; 29(8):1015-1023.
5. Sewify M, Nair S, Warsame S, Murad M, Alhubail A, Behbehani K, Al-Refaei F and Tiss A. Prevalence of Urinary Tract Infection and Antimicrobial Susceptibility among Diabetic Patients with Controlled and Uncontrolled Glycemia in Kuwait. J Diabetes Res. 2016; 2016:6573215.
6. Vasudevan R. Urinary Tract Infection: An Overview of the Infection and the Associated Risk Factors. J Microbiol Exp. 2014; 1(2):1-15.
7. Hilt E, McKinley K, Pearce MM, Rosenfeld AB, Zilliox MJ, Mueller ER, Brubaker L, Gai X, Wolfe AJ and Schreckenberger P. Urine is not sterile: use of enhanced urine culture techniques to detect resident bacterial flora in the adult female bladder. J Clin Microbiol. 2014; 52(3):871-876.
8. Siddiqui H, Lagesen K, Nederbragt AJ, Eri LM, Jeansson SL and Jakobsen KS. Pathogens in Urine from a Female Patient with Overactive Bladder Syndrome Detected by Culture-independent High Throughput Sequencing: A Case Report. Open Microbiol J. 2014; 8:148-153.
9. Schneeweiss J, Koch M and Umek W. The human urinary microbiome and how it relates to urogynecology. Int Urogynecol J. 2016; 27(9):1307-1312.
10. Nelson DE, Van D, Dong Q, Revanna KV, Fan B, Easwaran S, Sodergren E, Weinstock GM, Diao L and Fortenberry JD. Characteristic male urine microbiomes associate with asymptomatic sexually transmitted infection. PLoS One. 2010; 5(11):e14116.
11. Siddiqui H, Nederbragt AJ, Lagesen K, Jeansson SL and Jakobsen KS. Assessing diversity of the female urine microbiota by high throughput sequencing of 16S rDNA amplicons. BMC Microbiol. 2011;(11):244.
12. Fouts DE, Pieper R, Szpakowski S, Pohl H, Knoblach S, Suh MJ, Huang ST, Ljungberg I, Sprague BM, Lucas SK, Torralba M, Nelson KE and Groah SL. Integrated next-generation sequencing of 16S rDNA and metaproteomics differentiate the healthy urine microbiome from asymptomatic bacteriuria in neuropathic bladder associated with spinal cord injury. J Transl Med. 2012; 10:174.
13. Siddiqui H, Lagesen K, Nederbragt AJ, Jeansson SL and Jakobsen KS. Alterations of microbiota in urine from women with interstitial cystitis. BMC Microbiol. 2012;(12):205.
14. Wolfe AJ, Toh E, Shibata N, Rong R, Kenton K, Fitzgerald M, Mueller ER, Schreckenberger P, Dong Q, Nelson DE and Brubaker L. Evidence of uncultivated bacteria in the adult female bladder. J Clin Microbiol. 2012; 50(4):1376-1383.
15. Lewis DA, Brown R, Williams J, White P, Jacobson SK, Marchesi JR and Drake MJ. The human urinary microbiome; bacterial DNA in voided urine of asymptomatic adults. Front Cell Infect Microbiol. 2013; 3:41.
16. Fricke WF, Maddox C, Song Y and Bromberg JS. Human microbiota characterization in the course of renal transplantation. Am J Transplant. 2014; 14(2):416-427.
17. Nienhouse V, Gao X, Dong Q, Nelson DE, Toh E, McKinley K, Schreckenberger P, Shibata N, Fok CS, Mueller ER, Brubaker L, Wolfe AJ and Radek KA. Interplay between bladder microbiota and urinary antimicrobial peptides: mechanisms for human urinary tract infection risk and symptom severity. PLoS One. 2014; 9(12):e114185.
18. Pearce MM, Hilt EE, Rosenfeld AB, Zilliox MJ, Thomas-White K, Fok C, Kliethermes S, Schreckenberger PC, Brubaker L, Gai X and Wolfe AJ. The female urinary microbiome: a comparison of women with and without urgency urinary incontinence. Mbio. 2014; 5(4):e1214-e1283.
19. Willner D, Low S, Steen JA, George N, Nimmo GR, Schembri MA and Hugenholtz P. Single clinical isolates from acute uncomplicated urinary tract infections are representative of dominant in situ populations. Mbio. 2014; 5(2):e1013-e1064.
20. Pearce MM, Zilliox MJ, Rosenfeld AB, Thomas-White KJ, Richter HE, Nager CW, Visco AG, Nygaard IE, Barber MD, Schaffer J, Moalli P, Sung VW, Smith AL, Rogers R, Nolen TL and Wallace D, et al. The female urinary microbiome in urgency urinary incontinence. Am J Obstet Gynecol. 2015; 213(3):341-347.
21. Santiago-Rodriguez TM, Ly M, Bonilla N and Pride DT. The human urine virome in association with urinary tract infections. Front Microbiol. 2015; 6:14.
22. Thomas-White KJ, Kliethermes S, Rickey L, Lukacz ES, Richter HE, Moalli P, Zimmern P, Norton P, Kusek JW, Wolfe AJ and Brubaker L. Evaluation of the urinary microbiota of women with uncomplicated stress urinary incontinence. Am J Obstet Gynecol. 2016; pii:S0002-9378(16):30521-30522.
23. Shoskes DA, Altemus J, Polackwich AS, Tucky B, Wang H and Eng C. The Urinary Microbiome Differs Significantly Between Patients With Chronic Prostatitis/Chronic Pelvic Pain Syndrome and Controls as Well as Between Patients With Different Clinical Phenotypes. Urology. 2016; 92:26-32.
24. Cameron MA, Maalouf NM, Adams-Huet B, Moe OW and Sakhaee K. Urine composition in type 2 diabetes: predisposition to uric acid nephrolithiasis. J Am Soc Nephrol. 2006; 17(5):1422-1428.
25. Cho AJ, Kim SJ, Lee YK, Song YR, Oh J, Kim SG, Seo JW, Yoon JW, Koo JR, Kim HJ and Noh JW. Effect of post-voiding urine volume on progression of renal function decline in patients with type 2 diabetes. Diabetes Res Clin Pract. 2015; 109(1):164-169.
26. Yousri NA, Mook-Kanamori DO, Selim MM, Takiddin AH, Al-Homsi H, Al-Mahmoud KA, Karoly ED, Krumsiek J, Do KT, Neumaier U, Mook-Kanamori MJ, Rowe J, Chidiac OM, McKeon C, Al MW and Kader SA, et al. A systems view of type 2 diabetes-associated metabolic perturbations in saliva, blood and urine at different timescales of glycaemic control. Diabetologia. 2015; 58(8):1855-1867.
27. Raccah D, Miossec P, Esposito V, Niemoeller E, Cho M and Gerich J. Efficacy and safety of lixisenatide in elderly ( ≥65 years old) and very elderly ( ≥75 years old) patients with type 2 diabetes: an analysis from the GetGoal phase III programme. Diabetes/Metabolism Research and Reviews. 2015; 31(2):204-211.
28. Karstens L, Asquith M, Davin S, Stauffer P, Fair D, Gregory WT, Rosenbaum JT, McWeeney SK and Nardos R. Does the Urinary Microbiome Play a Role in Urgency Urinary Incontinence and Its Severity? Front Cell Infect Microbiol. 2016; 6:78.
29. Prideaux L, Kang S, Wagner J, Buckley M, Mahar JE, De Cruz P, Wen Z, Chen L, Xia B, van Langenberg DR, Lockett T, Ng SC, Sung JJ, Desmond P, McSweeney C and Morrison M, et al. Impact of ethnicity, geography, and disease on the microbiota in health and inflammatory bowel disease. Inflamm Bowel Dis. 2013; 19(13):2906-2918.
30. Xia Q, Cheng L, Zhang H, Sun S, Liu F, Li H, Yuan J, Liu Z and Diao Y. Identification of vaginal bacteria diversity and it’s association with clinically diagnosed bacterial vaginosis by denaturing gradient gel electrophoresis and correspondence analysis. Infect Genet Evol. 2016; 44:479-486.
31. Casqueiro J, Casqueiro J and Alves C. Infections in patients with diabetes mellitus: A review of pathogenesis. Indian J Endocrinol Metab. 2012; 16 Suppl 1(7):S27-S36.
32. Stapleton AE, Au-Yeung M, Hooton TM, Fredricks DN, Roberts PL, Czaja CA, Yarova-Yarovaya Y, Fiedler T, Cox M and Stamm WE. Randomized, placebo-controlled phase 2 trial of a Lactobacillus crispatus probiotic given intravaginally for prevention of recurrent urinary tract infection. Clin Infect Dis. 2011; 52(10):1212-1217.
33. Kano H, Kita J, Makino S, Ikegami S and Itoh H. Oral administration of Lactobacillus delbrueckii subspecies bulgaricus OLL1073R-1 suppresses inflammation by decreasing interleukin-6 responses in a murine model of atopic dermatitis. J Dairy Sci. 2013; 96(6):3525-3534.
34. Lin CH, Lin CC, Shibu MA, Liu CS, Kuo CH, Tsai FJ, Tsai CH, Hsieh CH, Chen YH and Huang CY. Oral Lactobacillus reuteri GMN-32 treatment reduces blood glucose concentrations and promotes cardiac function in rats with streptozotocin-induced diabetes mellitus. Br J Nutr. 2014; 111(4):598-605.
35. Hariri M, Salehi R, Feizi A, Mirlohi M, Ghiasvand R and Habibi N. A randomized, double-blind, placebo-controlled, clinical trial on probiotic soy milk and soy milk: effects on epigenetics and oxidative stress in patients with type II diabetes. Genes Nutr. 2015; 10(6):52.
36. Simon MC, Strassburger K, Nowotny B, Kolb H, Nowotny P, Burkart V, Zivehe F, Hwang JH, Stehle P, Pacini G, Hartmann B, Holst JJ, MacKenzie C, Bindels LB, Martinez I and Walter J, et al. Intake of Lactobacillus reuteri improves incretin and insulin secretion in glucose-tolerant humans: a proof of concept. Diabetes Care. 2015; 38(10):1827-1834.
37. Mirmonsef P, Modur S, Burgad D, Gilbert D, Golub ET, French AL, McCotter K, Landay AL and Spear GT. Exploratory comparison of vaginal glycogen and Lactobacillus levels in premenopausal and postmenopausal women. Menopause. 2015; 22(7):702-709.
38. Muhleisen AL and Herbst-Kralovetz MM. Menopause and the vaginal microbiome. Maturitas. 2016; 91:42-50.
39. Mirmonsef P, Hotton AL, Gilbert D, Burgad D, Landay A, Weber KM, Cohen M, Ravel J and Spear GT. Free glycogen in vaginal fluids is associated with Lactobacillus colonization and low vaginal pH. PLoS One. 2014; 9(7):e102467.
40. Song SJ, Niu ZQ, University SM and Hospital SD. Vaginal micro ecological condition analysis after total hysterectomy. Proceeding of Clinical Medicine. 2015; 24(10):731-734.
41. Everard A, Belzer C, Geurts L, Ouwerkerk JP, Druart C, Bindels LB, Guiot Y, Derrien M, Muccioli GG, Delzenne NM, de Vos WM and Cani PD. Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc Natl Acad Sci USA. 2013; 110(22):9066-9071.
42. Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, Sogin ML, Jones WJ, Roe BA, Affourtit JP, Egholm M, Henrissat B, Heath AC, Knight R and Gordon JI. A core gut microbiome in obese and lean twins. Nature. 2009; 457(7228):480-484.
43. Gupta A, Dwivedi M, Mahdi AA, Nagana GG, Khetrapal CL and Bhandari M. Inhibition of adherence of multi-drug resistant E. coli by proanthocyanidin. Urol Res. 2012; 40(2):143-150.
44. Davoodabadi A, Soltan DM, Lashani E and Tajabadi EM. Antimicrobial Activity of Lactobacillus spp. Isolated From Fecal Flora of Healthy Breast-Fed Infants Against Diarrheagenic Escherichia coli. Jundishapur J Microbiol. 2015; 8(12):e27852.
45. Ling Z, Kong J, Liu F, Zhu H, Chen X, Wang Y, Li L, Nelson KE, Xia Y and Xiang C. Molecular analysis of the diversity of vaginal microbiota associated with bacterial vaginosis. BMC Genomics. 2010; 11:488.
46. Thomas-White K, Brady B, Wolfe AJ and Mueller ER. The Bladder Is Not Sterile: History and Current Discoveries on the Urinary Microbiome. Curr Bladder Dysfunct Rep. 2016; 11(1):18-24.
47. Dethlefsen L and Relman DA. Incomplete recovery and individualized responses of the human distal gut microbiota to repeated antibiotic perturbation. Proc Natl Acad Sci USA. 2011; 108 Suppl 1:4554-4561.
48. Mardinoglu A, Boren J and Smith U. Confounding Effects of Metformin on the Human Gut Microbiome in Type 2 Diabetes. Cell Metab. 2016; 23(1):10-12.
49. Alberti KG and Zimmet PZ. Definition, diagnosis and classification of diabetes mellitus and its complications. Part 1: diagnosis and classification of diabetes mellitus provisional report of a WHO consultation. Diabet Med. 1998; 15(7):539-553.
50. Nickel JC, Stephens A, Landis JR, Mullins C, van Bokhoven A, Lucia MS and Ehrlich GD. Assessment of the Lower Urinary Tract Microbiota during Symptom Flare in Women with Urologic Chronic Pelvic Pain Syndrome: A MAPP Network Study. J Urol. 2016; 195(2):356-362.
51. Ministry of Health, China. Nosocomial infection diagnosis standard. Natl Med J China. 2001; 81(5):314-320.
52. Varli M, Guruz H, Aras S, Yalcin A, Atli T and Turgay M. Asymptomatic bacteriuria among the elderly living in the community: Prevalence, risk factors and characteristics. Eur Geriatr Med. 2012; 3(2):87-91.
53. Groeneveld Y, Petri H, Hermans J and Springer M. An assessment of structured care assistance in the management of patients with type 2 diabetes in general practice. Scand J Prim Health Care. 2001; 19(1):25-30.
54. Zhao W, Huang Z, Zhang X, He L, Willett W, Wang J, Hasegawa K and Chen J. Reproducibility and Validity of a Chinese Food Frequency Questionnaire. Biomed Environ Sci. 2010; 23S:1-38.
55. Fadrosh DW, Ma B, Gajer P, Sengamalay N, Ott S, Brotman RM and Ravel J. An improved dual-indexing approach for multiplexed 16S rRNA gene sequencing on the Illumina MiSeq platform. Microbiome. 2014; 2(1):6.
56. Langille MG, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, Clemente JC, Burkepile DE, Vega TR, Knight R, Beiko RG and Huttenhower C. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol. 2013; 31(9):814-821.
57. Parks DH, Tyson GW, Hugenholtz P and Beiko RG. STAMP: statistical analysis of taxonomic and functional profiles. Bioinformatics. 2014; 30(21):3123-3124.
58. Parks DH and Beiko RG. Identifying biologically relevant differences between metagenomic communities. Bioinformatics. 2010; 26(6):715-721.
59. White JR, Nagarajan N and Pop M. Statistical methods for detecting differentially abundant features in clinical metagenomic samples. PLoS Comput Biol. 2009; 5(4):e1000352.