
INTRODUCTION
Ovarian clear cell carcinoma (OCCC) is an aggressive histological subtype of epithelial ovarian cancer (EOC) with a higher rate of de novo resistance to platinum-based chemotherapy than high-grade serous EOCs [1, 2]. OCCCs have been shown to constitute a distinct subtype of EOCs histologically and genetically. Unlike high-grade serous carcinomas, OCCCs usually lack TP53 mutations [3] and germline or somatic BRCA1 or BRCA2 mutations [4]. In contrast, OCCCs are characterised by the presence of ARID1A mutations [5], activating PIK3CA mutations [6], loss of PTEN expression [6, 7], and amplification of PPM1D [8]. It should be noted, however, that there is evidence to suggest that OCCCs constitute a heterogeneous group of cancers at the genetic level, exemplified by the existence of subgroups harbouring distinct constellations of gene copy number alterations [9] and a varied repertoire of somatic mutations [5].
Despite the progress in the understanding of the molecular basis of OCCCs, patients with this disease are still managed with chemotherapy based on platinum salts and taxanes. Given the reported aggressive clinical behaviour and generally poor response rates to these conventional chemotherapy regimens, patients with OCCCs have been shown to have a worse outcome than those with other types of EOCs [9, 10]. It should be noted, however, that the heterogeneity of OCCCs is also apparent in terms of its response to specific therapeutic agents, as there are models of OCCCs that have been shown to be sensitive to platinum salts [2].
Poly(ADP) ribose polymerase (PARP) inhibitors constitute a new class of targeted therapeutic agents that have shown great promise in preclinical studies and phase I/ II clinical trials based on the principle of synthetic lethality [11, 12], PARP inhibitors selectively target cells that lack competent homologous recombination (HR) DNA repair in the presence of DNA double-strand breaks (DSBs) [13]. PARP inhibition leads to an impairment of the ability of cells to repair DNA single-strand breaks by base excision DNA repair. In cells treated with PARP inhibitors, single-strand breaks are not repaired and during S-phase, these lead to replication fork stalling and collapse, ultimately resulting in DNA DSBs [13]. In normal cells, these DSBs are repaired by HR DNA repair. In cancers with dysfunctional HR, such as those lacking BRCA1 [14], BRCA2 [14], PTEN [15] and RAD51D [16] function, PARP inhibitor-induced DSBs cannot be corrected by HR, and are repaired by error prone mechanisms (e.g. non-homologous end-joining), which result in high levels of genetic instability and eventually cell death. Defects in HR can be associated with inactivation of multiple components of the HR pathway [17, 18]. In particular, BRCA1 and BRCA2 loss of function have been shown to lead to dysfunctional HR in cancer cells [15, 18] whereas the association between PTEN loss of function and dysfunctional HR is more controversial [19]. MRE11 loss of expression has recently been shown to be associated with increased PARP inhibitor sensitivity in prostate, colorectal and endometrial cancer cells [20, 21].
Consistent with the results from preclinical models for the PARP-inhibitor BMN-673 [22, 23], clinical trials testing the efficacy of PARP inhibitors, have yielded promising results in patients with advanced BRCA1 or BRCA2 hereditary breast and ovarian cancers [24–31], but also in sporadic high-grade serous EOC [32, 33], which often harbour somatic inactivation of genes involved in HR DNA repair [34]. Pre-clinical studies [15] and clinical evidence from the analysis of a patient with advanced endometrial cancer [35] suggests that tumours with loss of PTEN expression, a recurrent aberration in OCCC [6, 7], may be sensitive to PARP inhibitors.
Given that OCCCs are heterogeneous at the molecular level and that a subgroup of these cancers are sensitive to platinum-based chemotherapy, we hypothesised that a subset of OCCCs may have dysfunctional HR DNA repair of DNA DSBs and that this defect would result in sensitivity to platinum salts and PARP inhibitors. To address these hypotheses, we analysed a panel of 12 OCCC cell lines and a series of 50 primary OCCCs i) to define the prevalence of dysfunctional HR DNA repair, ii) to determine if subgroups of OCCCs are sensitive to platinum salts and/or the PARP inhibitor BMN-673, iii) to determine if HR DNA repair defects would be associated with loss of PTEN function, and iv) to determine the prevalence of PTEN loss of expression in a cohort of primary OCCCs.
RESULTS
A subset of OCCC cells is unable to elicit RAD51 foci in the presence of DNA DSBs
We sought to determine if OCCC cells would harbour defects in HR. The inability of cancer cells to elicit RAD51 foci in the presence of DNA DSBs was used as a surrogate for dysfunctional HR as previously described [14, 15, 41]. We first assessed the ability of OCCC cells to elicit RAD51 foci formation in the presence of ionising radiation-induced DNA DSBs (Figure 1A–1C). As expected, SUM149 and CAPAN1 cells were unable to elicit RAD51 foci following exposure to DNA damaging agents, given that they harbour deleterious BRCA1 and BRCA2 mutations, respectively (Figure 1A and 1B) [14, 15, 41]. Although none of the OCCC cells completely lacked the ability to elicit RAD51 foci, KOC-7c, TOV-21G, KK, RMG-1, and SMOV-2 cells had significantly lower proportions of RAD51-positive nuclei in response to ionising radiation than HR competent cancer cells (i.e. SKBR3 and SUM44, Mann-Whitney U Test, p < 0.01, Figure 1A and 1B). These findings suggest that a subset of OCCC cells may lack competent HR DNA repair of DSBs.
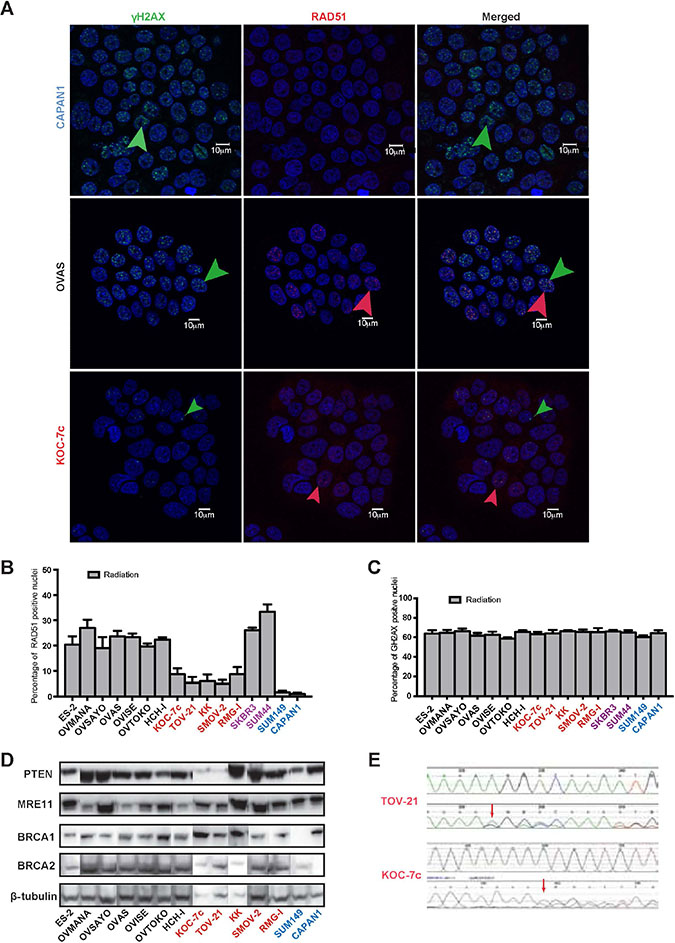
Figure 1: A subset of OCCC cells is unable to elicit RAD51 foci in the presence of DNA DSBs. A panel of 12 OCCC cell lines were subjected to RAD51 foci formation assays to determine their homologous recombination (HR) function in the presence of DNA double strand breaks (DSBs). Cell lines were treated with ionising radiation before immunofluorescent detection of γH2AX foci (markers of DNA DSBs) and RAD51 foci (markers of competent HR) was undertaken (A). Akin to CAPAN1 (BRCA2-mutant) and SUM149 (BRCA1-mutant) control cells, five OCCC cell lines showed reduced RAD51 foci formation (i.e. TOV-21G, KOC-7c, SMOV-2, RMG-1, and KK) (B). The remaining seven OCCC cell lines displayed RAD51 foci formation at similar levels to HR-competent SKBR3 and SUM44 cells (B). All cell lines showed similar levels of γH2AX foci formation (C). Western blot analysis of PTEN, MRE11, BRCA1 and BRCA2 in 12 OCCC cell lines. Beta-tubulin was employed as loading control. Two cell lines (TOV-21G and KOC-7c) showed loss of PTEN expression (D). PTEN Sanger sequencing traces of TOV-21G (top) and KOC-7c (bottom), which both harboured a PTEN frameshift mutation (c.795het_delA in TOV-21G and c.968het_delA in KOC-7c), red arrows, (E). The top sequencing trace in each panel represents the wild type sequence, while the cell line sequence showing the mutation is shown below it. In all panels, cell lines with reduced RAD51 foci formation (HR defective) are marked in red, those with normal RAD51 foci formation (HR competent) are marked in black, those with BRCA1 or BRCA2 loss of function are marked in blue, and those previously documented to be HR competent in purple.
A subset of OCCC cells unable to elicit RAD51 foci in the presence of DSBs harbours PTEN gene mutations
As BRCA1, BRCA2, PTEN and MRE11 loss has been associated with dysfunctional HR and/or PARP inhibitor sensitivity, we assessed the expression of BRCA1, BRCA2, PTEN and MRE11 in 12 OCCC cell lines included in this study by western blotting. All OCCC cells expressed similar levels of BRCA1, BRCA2 and MRE11 protein (Figure 1D). In addition, all OCCC cells with competent HR as defined by the RAD51 foci formation assay expressed PTEN protein. Out of the five cell lines with reduced RAD51 foci in the presence of DSBs, however, two, TOV-21G and KOC-7c, showed loss of PTEN expression (Figure 1D) and harboured PTEN frameshift mutations (TOV-21G, c.795_795delA; KOC-7c, c.968_968delA; Figure 1E). None of the cell lines tested displayed PTEN homozygous deletions and both PTEN-null cell lines (TOV-21G and KOC-7c) harboured two copies of the PTEN locus as defined by FISH (data not shown). No BRCA1 was detected in these cell lines (Supplementary Table S3). Taken together, these data suggest that a subset of OCCC cell lines harbour defects in HR, some of which are associated with loss of PTEN expression. Consistent with these data, when a cohort of 50 primary OCCC tumour samples were assessed for PTEN loss of expression by immunohistochemistry (Figure 2), 5/49 analysable cases were found to harbour PTEN loss (10%, Supplementary Table S1). This is less frequent than previously reported for OCCCs (27.5–37.5%) [7, 43].
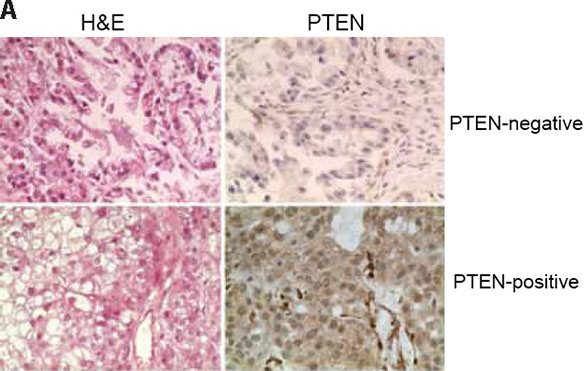
Figure 2: PTEN expression in primary OCCCs. Using a validated antibody to PTEN, and previously reported scoring systems, PTEN expression was assessed in a panel of 50 primary OCCCs in a previously constructed tissue microarray. Cases were reviewed by at least two pathologists and discordant scores reassessed on whole tissue sections. Complete loss of PTEN expression was identified in 5/49 analysable cases (10%). Representative haematoxylin & eosin micrographs (left) and PTEN immunohistochemistry (right) of a PTEN-negative (top) and a PTEN-positive OCCC.
Sensitivity of OCCC cell lines to cisplatin and BMN-673 is associated with HR functional status
Given that a subset of OCCC cells was unable to elicit RAD51 foci in the presence of DNA DSBs, we sought to define whether these cell lines would show a higher sensitivity to cisplatin and PARP inhibitors than the remaining OCCCs. Interestingly, cell lines with low levels of RAD51 foci formation upon treatment with ionising radiation were sensitive to cisplatin and BMN-673 (Figure 3A and 3B), with survival fractions 50 (SF50s) similar to those of the BRCA2-mutant CAPAN1 cells, with the exception of KOC-7c. On the other hand, OCCC cells able to elicit RAD51 foci upon treatment with ionising radiation were relatively resistant to cisplatin and BMN-673 (Figure 3A and 3B).
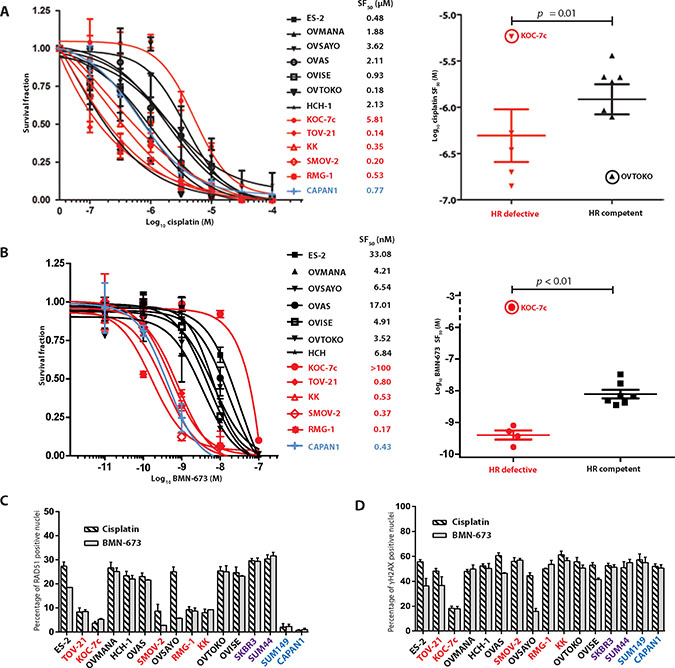
Figure 3: Sensitivity of OCCC cell lines to cisplatin and BMN-673 can be predicted by HR functional status and PTEN status. Given the recognised sensitivity of cells with defects in HR to agents that cause DNA double strand breaks, the panel of 12 OCCC cell lines were subjected to treatment with cisplatin or the PARP inhibitor BMN-673. With the exception of KOC-7c, all HR defective cell lines showed dose-response curves to cisplatin (A) and BMN-673 (B) with sensitive profiles, while all HR competent cell lines (with the exception of OVTOKO) showed dose-response curves with resistant profiles. CAPAN1 cells were used as controls for this experiment. When outliers were excluded (KOC-7c, circled in scatter plots, A and B as well as OVSAYO circled in scatter plot B), HR defective cell lines had cisplatin and BMN-673 SF50 concentrations that were significantly lower than those of HR competent cell lines (Mann-Whitney U test, p < 0.01) Assessment of RAD51 (C) and γH2AX (D) foci following treatment with cisplatin and BMN-673 demonstrated that HR defective cell lines had a lower rate of RAD51 foci formation compared to HR competent cells (C). KOC-7c, did not form γH2AX to the same degree as other treated cells (D), suggesting cisplatin and BMN-673 may be subject to drug efflux. In all panels, HR defective cell lines are marked in red, HR competent cell lines are marked in black, those with BRCA1/2 mutations are marked in blue, and those previously documented to be HR competent in purple. Scatter plots in A and B are annotated with the mean and standard error of mean (SEM).
Given that KOC-7c cells elicited relatively low numbers of RAD51 foci upon treatment with ionising radiation, but displayed a relative resistance to cisplatin and BMN-673 (Figure 3A and 3B), we sought to define the ability of these cells to elicit RAD51 and γH2AX foci upon treatment with these agents. Upon treatment with cisplatin and BMN-673, KOC-7c, TOV-21G, KK, RMG-1, and SMOV-2 cells displayed significantly lower proportions of RAD51-positive nuclei than the remaining OCCC cells (7.1 (SD +/− 1.37) vs 18.8 (SD +/− 3.31), p < 0.05) (Figure 3C). Importantly, however, the assessment of γH2AX foci formation upon treatment with cisplatin or BMN-673 in these cells revealed a lower proportion of γH2AX-positive nuclei in KOC-7c cells than in the remaining OCCC cell lines (17.2 vs 43.9 (SD +/− 7.68), p < 0.05) (Figure 3D). These observations provide strong circumstantial evidence that cisplatin and BMN-673 are not causing DNA damage in KOC-7c cells.
DISCUSSION
OCCCs have been shown to constitute a heterogeneous group of tumours in terms of their genetic features [9] and response to therapy [44]. Here we demonstrate that OCCCs are also heterogeneous in regards to their ability to elicit competent HR DNA repair. A subset of OCCC cells exhibited impaired HR DNA repair of DSBs, as determined by RAD51 foci formation assays, and were shown to be sensitive to cisplatin and BMN-673. In addition, our results also provided evidence to suggest that PTEN loss of function may be associated with impaired HR DNA repair in a subset of OCCC cells and was found to be lost in 10% of primary OCCCs.
The role of PTEN loss of function in HR DNA repair of DSBs is controversial. Studies have failed to demonstrated a significant association between PTEN loss of function [45, 46] and either sensitivity to PARP inhibitors or HR defects have not yet provided a definite mechanism for this association [15, 47]. Possible mechanisms include the role of PTEN as a modulator of RAD51 transcription [48]. Importantly, the impact of PTEN loss of function in HR DNA repair of DSBs may be context dependent. While in endometrial cancer [15, 35] and gliomas [47] PTEN loss of function results in impaired HR DNA repair and sensitivity to PARP inhibitors, similar effects have not been observed in non-small cell lung cancer and prostate cancer [20, 49]. Endocrine factors, in particular estrogen levels may also play a role, in sensitizing PTEN-null endometrial cancer cells to PARP inhibitors [50]. It is also plausible that the chronology of the loss of PTEN function may determine its impact on HR DNA repair. In endometrial cancers, loss of PTEN is an early event, whereas in prostate cancer, there is evidence to suggest that PTEN loss is a late event [19]. More recent data in prostate cancer suggest that the effects of PTEN loss on cell growth and response to PARP inhibition are context dependent [51]; in a series of in vitro and in vivo experiments using pten heterozygous, pten null, with or without tp53 loss, mouse embryonic fibroblasts (MEFs) or mice, it was demonstrated that in a p53 proficient setting, treatment of PTEN–deficient cells with PARP inhibitors leads to a senescence response. By comparison, upon loss of p53, the response is more apoptotic. Furthermore, the group demonstrated that treatment with PARP inhibitor leads to AKT activation through the PI3K cascade, which can be mitigated with combination therapy with dual PI3K inhibitors and PARP inhibitors.
Our results are consistent with those of previous reports suggesting that PTEN loss of function is associated with defective HR-mediated repair of DNA DSBs, as defined by RAD51 foci formation [15, 18]. Both OCCC cell lines studied here that harbour PTEN loss of function (TOV-21G and KOC-7c) exhibit impaired HR DNA repair as determined by RAD51 foci formation. In the light of the recent data in prostate cancer discussed above, it is possible that these two events (PTEN loss of function and impaired HR DNA repair) may not be causally linked in isolation; PTEN loss of function may be a bystander event in these HR defective OCCC cell lines, or a hitherto as yet undiscovered epistatic interaction may be required for PTEN loss of function to be mechanistically linked to impaired HR DNA repair. Further functional studies assessing PTEN loss of function in OCCC are required to clarify this issue. Given that aberrations in several other genes in the HR pathway can lead to defects in HR [18], it is plausible that other HR DNA repair-related genes may be lost in a subgroup of OCCCs [24, 25, 52]. Through a reanalysis of the results by Tan et al. [9], we have ruled out EMSY amplification [41] as a potential cause of HR deficiency in these cells, as none of them harboured this genetic aberration (data not shown). Further studies to define the basis of the deficient HR DNA repair in these cells are warranted.
Contrary to the observations derived from the experiments performed with BMN-673, where all BMN-673-sensitive cell lines were HR DNA deficient, OVTOKO cells exhibited an exquisite sensitivity to cisplatin, despite being HR competent. The fact that OVTOKO was resistant to BMN-673 suggests this cisplatin sensitivity is not mediated by a defect in HR, and that impaired HR-mediated repair of DNA DSBs is not the only determinant of cisplatin sensitivity [53, 54].
This study has several limitations. Although the OCCC models tested in this study have been shown to recapitulate the genetic and transcriptomic features of primary OCCCs [8, 9], seven of the 12 cell lines assessed in this study had cisplatin SF50s comparable to those CAPAN1 (sub-micromolar concentrations). Hence, there might be an enrichment for chemotherapy–sensitive OCCCs in this panel of cell lines.
In conclusion, we have identified a subset of OCCC cell lines that harbour defects in HR-mediated repair of DNA DSBs. This preclinical study provides a rationale for the assessment of HR deficiency in vivo to potentially identify a role of PARP inhibitors in this histological subset.
MATERIALS AND METHODS
Further details of all the methods described here are found in the Supplementary Methods.
RAD51 and γH2AX foci assessment
To determine if cancer cells would have dysfunctional HR repair of DNA DSBs, Nuclear γ-H2AX and RAD51 foci formation was employed as previously described [15]. Details of the methods used are described in the Supplementary Methods. To determine HR status, RAD51 foci formation in response to 10 Gy of ionizing radiation was assessed. The differential response of HR competent and HR defective cells to either 10 μM cisplatin for 6 hrs or 10 μM BMN-673 for 12 hrs was then determined.
Immunoblotting
Western blotting was performed as previously described [36], using anti-β-tubulin (ab6046, Abcam, Cambridge, UK), anti-PTEN (138G6, Cell Signaling Technology, Danvers, MA, USA), anti-BRCA2 (Ab-1 [OP95], Calbiochem/Merck, Nottingham, UK) and anti-BRCA1 (C-20, Santa Cruz Biotechnology) antibodies.
Sanger sequencing
Sequencing of the full-length cDNA of PTEN was performed for all cell lines as previously described [8]. Mutations were confirmed by a repeat PCR from a new cDNA from the cell line harbouring the mutation and by sequencing of forward and reverse strands.
Assessment of PTEN copy number in OCCC cells
To determine PTEN copy number in OCCC cells and if any OCCC cells harboured homozygous deletions of PTEN, fluorescence in situ hybridisation (FISH) was performed as previously described [9, 15]. The Vysis PTEN/CEP10 dual colour FISH probe (Abbott Molecular, Des Plaines, IL, USA) was hybridised to slides as previously described [37].
Cisplatin sensitivity assays
Cisplatin sensitivity assays were carried out in 96 well plates, in triplicate, as previously described [15]. OCCC cells were grown for at least seven days, using CAPAN1 as a HR DNA repair-deficient and cisplatin- sensitive control [38–40]. Cells were treated with DMSO (control) or cisplatin (serial dilutions; 10–7M to 10–4M) (Sigma Aldrich). CellTiter Glo Luminescent Cell Viability Assay (Promega, UK) was used according to manufacturer’s instructions to determine the survival fraction of cisplatin vs DMSO-treated cells.
BMN-673 sensitivity assays
BMN-673 sensitivity was determined using colony formation assays (CFAs) in six well plates in triplicate, as previously described [15]. Cells were treated with 10–7M to 10–11M BMN-673 every 2 days. After 14 days cells were fixed and cell number quantified with a sulphorhodamine B assay, as previously described [15, 41].
Patient samples and tumour characteristics
Formalin-fixed paraffin-embedded (FFPE) samples from 50 consecutive primary OCCCs were retrieved from the pathology files of The Royal Marsden Hospital, London, The Edinburgh Royal Infirmary, The Royal Hospital Group, Belfast and The Hammersmith Hospital, London, as previously described [8]. Patient demographics and tumour characteristics are summarised in Supplementary Table S1, in line with REMARK guidelines (Supplementary Table S2). Samples were anonymised prior to analysis and the study approved by local ethical committees of the authors’ institutions. All cases were reviewed and diagnosis of clear cell carcinoma confirmed as previously described [8]. Tissue microarrays (TMAs) were constructed from paraffin blocks with triplicate 0.6 mm tumour cores, containing 50 samples and normal tissue controls, as previously described [9]. Surgical and adjuvant therapy are described in Supplementary Table S1.
Immunohistochemistry (IHC)
Representative 3 μm-thick sections of the TMA described above were subjected to immunohistochemistry (IHC) using an antibody against PTEN (6H2.1 Dilution 1:100, Antigen retrieval, Dako target retrieval solution, pH9, 20 min 97C° DAKO, Glostrup, Denmark). The protocol for PTEN immunohistochemical analysis was previously validated [15]. Immunohistochemical analysis was interpreted by at least two pathologists (AC-F, AG and/ or JRF) using a semi-quantitative scoring system. Nuclear and cytoplasmic PTEN expression was scored using the Quick Score method [42], with cases defined as PTEN negative if nuclear expression was absent (i.e. Quick Score = 0). Cases with discordant scores between replicate cores, or those with missing cores from the TMA were assessed on full sections by the three pathologists on a multi-headed microscope and a consensus score for each was rendered.
Statistical analysis
All statistical analyses were carried out in Prism 6.05 (Graphpad Software Inc., California, USA). Details of statistical analyses are described in the Supplementary Methods. A two-tailed p value < 0.05 was considered significant.
CONFLICTS OF INTEREST
None.
FINANCIAL SUPPORT
This study was funded in part by Breakthrough Breast Cancer Research. KJD is the recipient of a Swiss National Science Foundation fellowship. PMW is funded by a Wellcome Trust Clinical Research Fellowship grant. We acknowledge NHS funding to the NIHR Biomedical Research Centre. Research reported in this publication was supported in part by a Cancer Center Support Grant of the National Institutes of Health/National Cancer Institute (Grant No. P30CA008748). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The study sponsors had no involvement in the design of this perspective, the literature review, data interpretation, writing of the manuscript or the decision to submit it for publication.
REFERENCES
1. du Bois A, Luck HJ, Meier W, Adams HP, Mobus V, Costa S, Bauknecht T, Richter B, Warm M, Schroder W, Olbricht S, Nitz U, Jackisch C, et al. A randomized clinical trial of cisplatin/paclitaxel versus carboplatin/paclitaxel as first-line treatment of ovarian cancer. J Natl Cancer Inst. 2003; 95:1320–1329.
2. Tan DS, Kaye S. Ovarian clear cell adenocarcinoma: a continuing enigma. J Clin Pathol. 2007; 60:355–360.
3. Bowtell DD. The genesis and evolution of high-grade serous ovarian cancer. Nat Rev Cancer. 2010; 10:803–808.
4. Zhang S, Royer R, Li S, McLaughlin JR, Rosen B, Risch HA, Fan I, Bradley L, Shaw PA, Narod SA. Frequencies of BRCA1 and BRCA2 mutations among 1,342 unselected patients with invasive ovarian cancer. Gynecol Oncol. 2011; 121:353–357.
5. Jones S, Wang TL, Shih Ie M, Mao TL, Nakayama K, Roden R, Glas R, Slamon D, Diaz LA, Jr., Vogelstein B, Kinzler KW, Velculescu VE, et al. Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science. 2010; 330:228–231.
6. Kuo KT, Mao TL, Jones S, Veras E, Ayhan A, Wang TL, Glas R, Slamon D, Velculescu VE, Kuman RJ, Shih Ie M. Frequent activating mutations of PIK3CA in ovarian clear cell carcinoma. Am J Pathol. 2009; 174:1597–1601.
7. Ho CM, Lin MC, Huang SH, Huang CJ, Lai HC, Chien TY, Chang SF. PTEN promoter methylation and LOH of 10q22-23 locus in PTEN expression of ovarian clear cell adenocarcinomas. Gynecol Oncol. 2009; 112:307–313.
8. Tan DS, Lambros MB, Rayter S, Natrajan R, Vatcheva R, Gao Q, Marchio C, Geyer FC, Savage K, Parry S, Fenwick K, Tamber N, Mackay A, et al. PPM1D is a potential therapeutic target in ovarian clear cell carcinomas. Clin Cancer Res. 2009; 15:2269–2280.
9. Tan DS, Iravani M, McCluggage WG, Lambros MB, Milanezi F, Mackay A, Gourley C, Geyer FC, Vatcheva R, Millar J, Thomas K, Natrajan R, Savage K, et al. Genomic analysis reveals the molecular heterogeneity of ovarian clear cell carcinomas. Clin Cancer Res. 2011; 17:1521–1534.
10. Lee YY, Kim TJ, Kim MJ, Kim HJ, Song T, Kim MK, Choi CH, Lee JW, Bae DS, Kim BG. Prognosis of ovarian clear cell carcinoma compared to other histological subtypes: A meta-analysis. Gynecol Oncol. 2011.
11. Mullard A. European regulators approve first PARP inhibitor. Nat Rev Drug Discov. 2014; 13:877–877.
12. FDA News Release , FDA approves Lynparza to treat advanced ovarian cancer. http://wwwfdagov/NewsEvents/Newsroom/PressAnnouncements/ucm427554htm. 2014.
13. Ashworth A. A synthetic lethal therapeutic approach: poly (ADP) ribose polymerase inhibitors for the treatment of cancers deficient in DNA double-strand break repair. J Clin Oncol. 2008; 26:3785–3790.
14. Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, Martin NM, Jackson SP, Smith GC, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005; 434:917–921.
15. Dedes KJ, Wetterskog D, Mendes-Pereira AM, Natrajan R, Lambros MB, Geyer FC, Vatcheva R, Savage K, Mackay A, Lord CJ, Ashworth A, Reis-Filho JS. PTEN deficiency in endometrioid endometrial adenocarcinomas predicts sensitivity to PARP inhibitors. Science translational medicine. 2010; 2:53ra75.
16. Loveday C, Turnbull C, Ramsay E, Hughes D, Ruark E, Frankum JR, Bowden G, Kalmyrzaev B, Warren-Perry M, Snape K, Adlard JW, Barwell J, Berg J, et al. Germline mutations in RAD51D confer susceptibility to ovarian cancer. Nat Genet. 2011; 43:879–882.
17. McCabe N, Turner NC, Lord CJ, Kluzek K, Bialkowska A, Swift S, Giavara S, O’Connor MJ, Tutt AN, Zdzienicka MZ, Smith GC, Ashworth A. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly (ADP-ribose) polymerase inhibition. Cancer Res. 2006; 66:8109–8115.
18. Dedes KJ, Wilkerson PM, Wetterskog D, Weigelt B, Ashworth A, Reis-Filho JS. Synthetic lethality of PARP inhibition in cancers lacking BRCA1 and BRCA2 mutations. Cell Cycle. 2011; 10:1192–1199.
19. Hunt CR, Gupta A, Horikoshi N, Pandita TK. Does PTEN loss impair DNA double-strand break repair by homologous recombination? Clinical cancer research. 2012; 18:920–922.
20. Fraser M, Zhao H, Luoto KR, Lundin C, Coackley C, Chan N, Joshua AM, Bismar TA, Evans A, Helleday T, Bristow RG. PTEN deletion in prostate cancer cells does not associate with loss of RAD51 function: implications for radiotherapy and chemotherapy. Clin Cancer Res. 2012; 18:1015–1027.
21. Vilar E, Bartnik CM, Stenzel SL, Raskin L, Ahn J, Moreno V, Mukherjee B, Iniesta MD, Morgan MA, Rennert G, Gruber SB. MRE11 deficiency increases sensitivity to poly (ADP-ribose) polymerase inhibition in microsatellite unstable colorectal cancers. Cancer Res. 2011; 71:2632–2642.
22. Shen Y, Rehman FL, Feng Y, Boshuizen J, Bajrami I, Elliott R, Wang B, Lord CJ, Post LE, Ashworth A. BMN 673, a novel and highly potent PARP1/2 inhibitor for the treatment of human cancers with DNA repair deficiency. Clin Cancer Res. 2013; 19:5003–5015.
23. Koppensteiner R, Samartzis EP, Noske A, von Teichman A, Dedes I, Gwerder M, Imesch P, Ikenberg K, Moch H, Fink D, Stucki M, Dedes KJ. Effect of MRE11 loss on PARP-inhibitor sensitivity in endometrial cancer in vitro. PLoS One. 2014; 9:e100041.
24. Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, Mortimer P, Swaisland H, Lau A, O’Connor MJ, Ashworth A, Carmichael J, Kaye SB, et al. Inhibition of poly (ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009; 361:123–134.
25. Fong PC, Yap TA, Boss DS, Carden CP, Mergui-Roelvink M, Gourley C, De Greve J, Lubinski J, Shanley S, Messiou C, A’Hern R, Tutt A, Ashworth A, et al. Poly(ADP)-ribose polymerase inhibition: frequent durable responses in BRCA carrier ovarian cancer correlating with platinum-free interval. J Clin Oncol. 2010; 28:2512–2519.
26. Audeh MW, Carmichael J, Penson RT, Friedlander M, Powell B, Bell-McGuinn KM, Scott C, Weitzel JN, Oaknin A, Loman N, Lu K, Schmutzler RK, Matulonis U, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: a proof-of-concept trial. Lancet. 2010; 376:245–251.
27. Tutt A, Robson M, Garber JE, Domchek SM, Audeh MW, Weitzel JN, Friedlander M, Arun B, Loman N, Schmutzler RK, Wardley A, Mitchell G, Earl H, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trial. Lancet. 2010; 376:235–244.
28. McLornan DP, List A, Mufti GJ. Applying synthetic lethality for the selective targeting of cancer. N Engl J Med. 2014; 371:1725–1735.
29. Oza AM, Cibula D, Benzaquen AO, Poole C, Mathijssen RH, Sonke GS, Colombo N, Spacek J, Vuylsteke P, Hirte H, Mahner S, Plante M, Schmalfeldt B, et al. Olaparib combined with chemotherapy for recurrent platinum-sensitive ovarian cancer: a randomised phase 2 trial. The Lancet Oncology. 2015; 16:87–97.
30. Kaufman B, Shapira-Frommer R, Schmutzler RK, Audeh MW, Friedlander M, Balmana J, Mitchell G, Fried G, Stemmer SM, Hubert A, Rosengarten O, Steiner M, Loman N, et al. Olaparib Monotherapy in Patients With Advanced Cancer and a Germline BRCA1/2 Mutation. Journal of clinical oncology. 2015; 33:244–250.
31. Deeks ED. Olaparib: first global approval. Drugs. 2015; 75:231–240.
32. Ledermann JA, Harter P, Gourley C, Friedlander M, Vergote IB, Rustin GJ, Scott C, Meier W, Shapira-Frommer R, Safra T, Matei D, Macpherson E, Watkins C, et al. Phase II randomized placebo-controlled study of olaparib (AZD2281) in patients with platinum-sensitive relapsed serous ovarian cancer (PSR SOC). J Clin Oncol. 2011; 29.
33. Gelmon KA, Tischkowitz M, Mackay H, Swenerton K, Robidoux A, Tonkin K, Hirte H, Huntsman D, Clemons M, Gilks B, Yerushalmi R, Macpherson E, Carmichael J, et al. Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: a phase 2, multicentre, open-label, non-randomised study. Lancet Oncol. 2011; 12:852–861.
34. TCGA. Integrated genomic analyses of ovarian carcinoma. Nature. 2011; 474:609–615.
35. Forster MD, Dedes KJ, Sandhu S, Frentzas S, Kristeleit R, Ashworth A, Poole CJ, Weigelt B, Kaye SB, Molife LR. Treatment with olaparib in a patient with PTEN-deficient endometrioid endometrial cancer. Nat Rev Clin Oncol. 2011; 8:302–306.
36. Natrajan R, Lambros MB, Rodriguez-Pinilla SM, Moreno-Bueno G, Tan DS, Marchio C, Vatcheva R, Rayter S, Mahler-Araujo B, Fulford LG, Hungermann D, Mackay A, Grigoriadis A, et al. Tiling path genomic profiling of grade 3 invasive ductal breast cancers. Clin Cancer Res. 2009; 15:2711–2722.
37. Lambros MB, Simpson PT, Jones C, Natrajan R, Westbury C, Steele D, Savage K, Mackay A, Schmitt FC, Ashworth A, Reis-Filho JS. Unlocking pathology archives for molecular genetic studies: a reliable method to generate probes for chromogenic and fluorescent in situ hybridization. Lab Invest. 2006; 86:398–408.
38. Edwards SL, Brough R, Lord CJ, Natrajan R, Vatcheva R, Levine DA, Boyd J, Reis-Filho JS, Ashworth A. Resistance to therapy caused by intragenic deletion in BRCA2. Nature. 2008; 451:1111–1115.
39. Bhattacharyya A, Ear US, Koller BH, Weichselbaum RR, Bishop DK. The breast cancer susceptibility gene BRCA1 is required for subnuclear assembly of Rad51 and survival following treatment with the DNA cross-linking agent cisplatin. J Biol Chem. 2000; 275:23899–23903.
40. Hastak K, Alli E, Ford JM. Synergistic chemosensitivity of triple-negative breast cancer cell lines to PARP inhibition, gemcitabine and cisplatin. Cancer Res. 2010; 70:7970–7980.
41. Wilkerson PM, Dedes KJ, Wetterskog D, Mackay A, Lambros MB, Mansour M, Frankum J, Lord CJ, Natrajan R, Ashworth A, Reis-Filho JS. Functional characterisation of EMSY gene amplification in human cancer. J Pathol. 2011; 225:29–42.
42. Harvey JM, Clark GM, Osborne CK, Allred DC. Estrogen receptor status by immunohistochemistry is superior to the ligand-binding assay for predicting response to adjuvant endocrine therapy in breast cancer. J Clin Oncol. 1999; 17:1474–1481.
43. Hashiguchi Y, Tsuda H, Inoue T, Berkowitz RS, Mok SC. PTEN expression in clear cell adenocarcinoma of the ovary. Gynecol Oncol. 2006; 101:71–75.
44. Itamochi H, Kigawa J, Sultana H, Iba T, Akeshima R, Kamazawa S, Kanamori Y, Terakawa N. Sensitivity to anticancer agents and resistance mechanisms in clear cell carcinoma of the ovary. Jpn J Cancer Res. 2002; 93:723–728.
45. Ihnen M, zu Eulenburg C, Kolarova T, Qi JW, Manivong K, Chalukya M, Dering J, Anderson L, Ginther C, Meuter A, Winterhoff B, Jones S, Velculescu VE, et al. Therapeutic potential of the poly (ADP-ribose) polymerase inhibitor rucaparib for the treatment of sporadic human ovarian cancer. Mol Cancer Ther. 2013; 12:1002–1015.
46. Miyasaka A, Oda K, Ikeda Y, Wada-Hiraike O, Kashiyama T, Enomoto A, Hosoya N, Koso T, Fukuda T, Inaba K, Sone K, Uehara Y, Kurikawa R, et al. Anti-tumor activity of olaparib, a poly (ADP-ribose) polymerase (PARP) inhibitor, in cultured endometrial carcinoma cells. BMC Cancer. 2014; 14:179.
47. McEllin B, Camacho CV, Mukherjee B, Hahm B, Tomimatsu N, Bachoo RM, Burma S. PTEN loss compromises homologous recombination repair in astrocytes: implications for glioblastoma therapy with temozolomide or poly (ADP-ribose) polymerase inhibitors. Cancer Res. 2010; 70:5457–5464.
48. Shen WH, Balajee AS, Wang J, Wu H, Eng C, Pandolfi PP, Yin Y. Essential role for nuclear PTEN in maintaining chromosomal integrity. Cell. 2007; 128:157–170.
49. Pappas G, Zumstein LA, Munshi A, Hobbs M, Meyn RE. Adenoviral-mediated PTEN expression radiosensitizes non-small cell lung cancer cells by suppressing DNA repair capacity. Cancer Gene Ther. 2007; 14:543–549.
50. Janzen DM, Paik DY, Rosales MA, Yep B, Cheng D, Witte ON, Kayadibi H, Ryan CM, Jung ME, Faull K, Memarzadeh S. Low levels of circulating estrogen sensitize PTEN-null endometrial tumors to PARP inhibition in vivo. Mol Cancer Ther. 2013; 12:2917–2928.
51. Gonzalez-Billalabeitia E, Seitzer N, Song SJ, Song MS, Patnaik A, Liu XS, Epping MT, Papa A, Hobbs RM, Chen M, Lunardi A, Ng C, Webster KA, et al. Vulnerabilities of PTEN-TP53-deficient prostate cancers to compound PARP-PI3K inhibition. Cancer discovery. 2014; 4:896–904.
52. Tan DS, Rothermundt C, Thomas K, Bancroft E, Eeles R, Shanley S, Ardern-Jones A, Norman A, Kaye SB, Gore ME. “BRCAness” syndrome in ovarian cancer: a case-control study describing the clinical features and outcome of patients with epithelial ovarian cancer associated with BRCA1 and BRCA2 mutations. J Clin Oncol. 2008; 26:5530–5536.
53. Lin C, Zhao XY, Li L, Liu HY, Cao K, Wan Y, Liu XY, Nie CL, Liu L, Tong AP, Deng HX, Li J, Yuan Z, et al. NOXA-Induced Alterations in the Bax/Smac Axis Enhance Sensitivity of Ovarian Cancer Cells to Cisplatin. PLoS One. 2012; 7:e36722.
54. Paul I, Savage KI, Blayney JK, Lamers E, Gately K, Kerr K, Sheaff M, Arthur K, Richard DJ, Hamilton PW, James JA, O’Byrne KJ, Harkin DP, et al. PARP inhibition induces BAX/BAK-independent synthetic lethality of BRCA1-deficient non-small cell lung cancer. J Pathol. 2011; 224:564–574.