
Introduction
The class I phosphoinositide 3-kinase (PI3K) pathway is frequently altered in cancer via genetic alterations in several gene pathway members, contributing to uncontrolled cell proliferation and survival [1, 2]. Class IA consists of heterodimers of catalytic subunits (p110α, p110β and p110δ encoded by PIK3CA, PIK3CB and PIK3CD respectively) and adaptor proteins (p85α and p85β encoded by PIK3R1 and PIK3R2 respectively) [3] which phosphorylate phosphatidylinositol (4,5)-bisphosphate (PtdIns(4,5)P2) to phosphatidylinositol (3,4,5)-trisphosphate (PtdIns(3,4,5)P3). This reaction is opposed by PTEN (phosphatase and tensin homolog), which de-phosphorylates PtdIns(3,4,5)P3 to PtdIns(4,5)P2, hence limiting the effects of class I PI3K activity [4]. In cancer, activating PIK3CA mutations are frequent [5, 6] while PIK3CB is mutated less often. So far, two activating mutations in PIK3CB have been characterized [7, 8], one of which has been discovered in a few cancer types by whole exome sequencing [8]. p110β can however promote oncogenic transformation when over-expressed [9] and has also been shown to be the key isoform mediating tumorigenesis in PTEN-deficient breast and prostate cancers [10-14]. A study by Juric et al further highlighted the importance of p110β in tumorigenesis [15] by showing that PIK3CA mutant tumors, which were initially sensitive to p110α inhibition, eventually developed resistance due to acquired loss of PTEN. Resistance to p110α inhibition could however be overcome when treatment with a p110β inhibitor was introduced. Other studies have however shown that certain tissues with PTEN loss become dependent on p110α rather than p110β [16]. These contrasting studies indicate the importance of studying isoform-dependence associated with PTEN-loss in each tissue as this may have significant therapeutic implications.
Endometrial cancer is the most common gynecological malignancy in developed countries. Endometrial tumors have been traditionally divided into two groups, type I and type II, according to clinical, pathologic and molecular features. About 80% of diagnosed cases are comprised of the histologic subtype endometrioid endometrial cancer (EEC) and are classified as type I. These tumors are more often estrogen-dependent, linked to obesity, low grade and stage and with good prognosis if treated early. On the other hand, type II, or non-endometrioid endometrial cancer (NEEC), are usually estrogen-independent with serous, clear cell or undifferentiated morphology, high grade and stage and with poor prognosis. Recent whole exome sequencing and integrative genomic profiling led to a molecular-based sub-classification of EEC and NEEC tumors [17-19]. The PI3K pathway is the most frequently altered pathway in EEC with more than 80% of tumors harboring somatic alterations in at least one gene member of the pathway, including high frequency mutations in PTEN, PIK3CA and PIK3R1 and low frequency in AKT and PIK3R2 [20-22]. Loss-of-function mutation of the tumor suppressor gene PTEN is the most common genetic event in EEC and occurs as an early event in 18-50% of lesions with atypical hyperplasia [23-25]. PIK3CA is frequently mutated in 10-39% of EEC but in contrast to PTEN has a higher frequency in high grade, aggressive, invasive and less differentiated tumors [24, 26, 27]. PIK3CA gene amplification can also account for other mechanisms for PI3K pathway activation and was found to correlate with a PI3K activation profile which segregated more frequently to a group of aggressive and invasive tumors, notably in NEECs. In contrast to PIK3CA, mutation events are rare in PIK3CB with 2.3% in endometrial cancer according to data from COSMIC (release v72 http://cancer.sanger.ac.uk/cosmic [28], including a recently characterized oncogenic mutation in its catalytic domain [8]). PIK3CB mRNA levels were found to be elevated in endometrial tumors compared to normal tissue in a few patient samples [29]. Overexpression of the p110β isoform is thus a possible explanation for the oncogenic properties of the wild type form of this isoform, but this is largely unknown particularly in endometrial cancer. Considering that PTEN loss and PI3K pathway activation are known key drivers of carcinogenesis in endometrial cancer, we hypothesized that p110β could play a significant role particularly in PTEN-deficient tumors. We therefore explored the cellular function and signaling properties of p110β compared to those of p110α in a panel of PTEN-positive and PTEN-deficient endometrial carcinoma cell lines. Finding that the protein levels of p110β, but not p110α, were upregulated in most endometrial carcinoma cell lines, we then demonstrated the distinct contribution of p110α and p110β to cell survival, proliferation and signaling depending on the presence of PTEN and PIK3CA mutations, using selective pharmacological inhibitors. Furthermore, the potential clinical relevance for these findings were substantiated by exploring an extensively clinically annotated patient cohort with 234 samples ranging from precursors through different stages of dedifferentiation during tumorigenesis demonstrating an increase in PIK3CB mRNA levels in early endometrioid lesions that associated with a high cell cycle progression score and decreased survival.
Results
p110β levels are elevated in endometrial cancer cell lines and increase from precursors to invasive lesions in clinical samples correlating with reduced survival
We have examined the protein levels of the class I PI3K catalytic (p110α and p110β) and regulatory subunits (p85α and p85β), PTEN, Akt and phosphorylated Akt on serine 473 (p-S473-Akt) and threonine 308 (p-T308-Akt) in whole cell extracts of 7 endometrial cancer cell lines versus a non-tumor immortalized endometrial cell line (EM). In actively growing cells, the levels of p110β were elevated in endometrial cancer cell lines compared to EM cells, independently of their mutational status, whereas the levels of p110α demonstrated less change (Figure 1A-1B). The levels of p85α in the cancer cells were comparable to EM cells but lower levels of p85β were observed in several of the cancer cell lines. To determine the PI3K signaling status, we examined the levels of p-S473-Akt and p-T308-Akt versus total Akt in actively growing cells (Figure 1C). Consistent with other studies [30], all cell lines with PTEN mutations (MFE-296) and exhibiting protein loss (MFE-319, RL-95-2 and Ishikawa) displayed high p-Akt levels (Figure 1C-1D – c.f. quantifications in Supplementary Figure S1). Interestingly, MFE-280 cells, which express PTEN but harbor mutations in PIK3CA, demonstrated low levels of p-S473/T308-Akt (Figure 1C-1D), consistent with the notion that PTEN loss and PIK3CA mutation can have different effects on Akt activation. Low p-Akt levels have previously been observed in breast cancer cell lines with PIK3CA mutations, demonstrating possible Akt-independent effects of some mutations in PIK3CA [31-33]. We also analyzed the mRNA levels of PIK3CB compared to PIK3CA in a cohort of endometrial cancer patients, including 18 with complex atypical hyperplasia (CAH), 174 primary tumors and 42 metastatic lesions. PIK3CB mRNA levels were significantly increased from precursor lesions with CAH to grade 1 EEC lesions and remained constant in higher grades, NEEC and metastatic tumors (Figure 2A). These results suggest that increased levels of PIK3CA/p110β contribute to the early phase of endometrial tumorigenesis. In contrast, a distinct pattern was seen for PIK3CA mRNA levels being significantly elevated in NEEC (Figure 2B). Furthermore, elevated levels of both PIK3CA and PIK3CB were also reflected in lower disease specific survival (Figure 2C-2D).
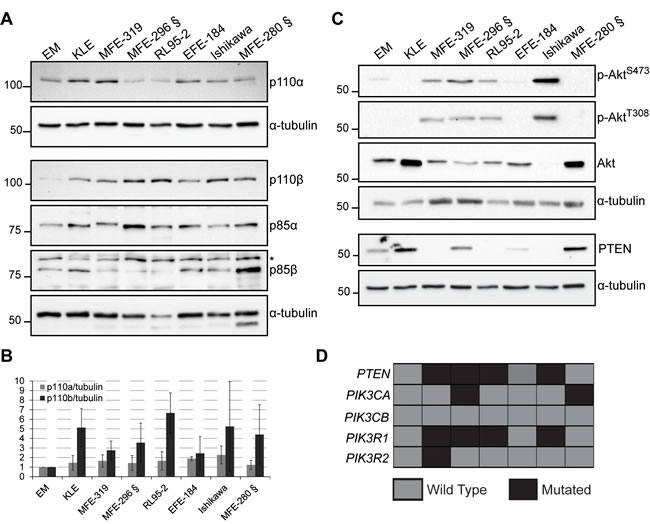
Figure 1: p110β levels are elevated in endometrial cancer cell lines. A. and C. Whole cell extracts obtained from actively growing cells analyzed by Western immunoblotting with the indicated antibodies. α-tubulin was used as a loading control. § indicate PIK3CA mutant cell lines. * indicate a non-specific band. B. p110β/α-tubulin and p110α/α-tubulin ratios shown as fold increase compared to EM cells from 3-4 independent experiments with standard deviations. D. Chart showing the mutational status of each gene and for each cell line (gray, WT; black, mutated gene).
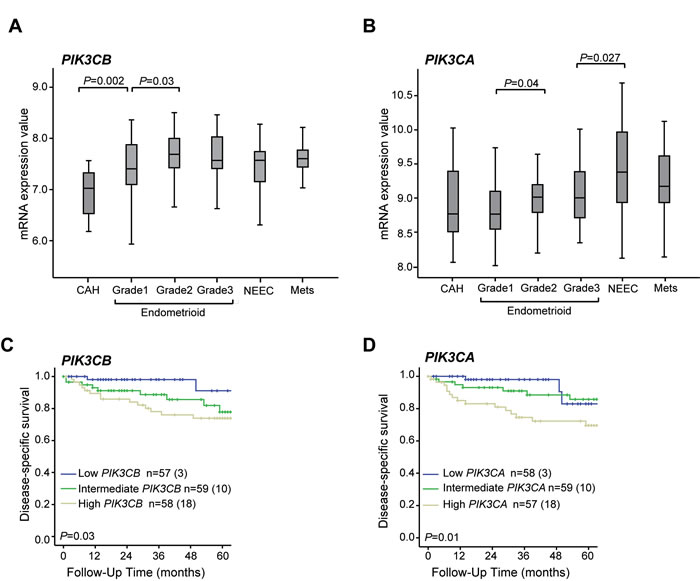
Figure 2: PIK3CB mRNA levels increase from precursors to low grade endometrial carcinoma lesions which associates with lower survival. Box-plots showing PIK3CB A. and PIK3CA B. mRNA levels in relation to histological type and grade. Significant Kruskall Wallis test p-values are given above the relevant box plots. CAH: Complex atypical hyperplasia (n = 18). Endometrioid grade 1 (n = 48), grade 2 (n = 53), grade 3 (n = 39). NEEC: Non endometrioid endometrial carcinoma (n = 34). Mets: Metastatic lesions (n = 42). Kaplan-Meier survival curves shown for patients with high, medium and low mRNA levels according to tertile limits for PIK3CB C. and PIK3CA D. Numbers in brackets represent number of disease specific deaths in each group.
PIK3CA mutant endometrial cancer cells respond to p110α inhibition by a decrease in cell survival
PTEN-deficient cancer cells have been suggested to rely upon p110β signaling for their sustained cell proliferation and tumorigenesis [10, 13]. To investigate whether endometrial cancer cell functions are also influenced by PTEN expression status and if they are dependent upon p110α and/or p110β, we evaluated the cell viability of PTEN-positive (EM, KLE and MFE-280) and PTEN-deficient cell lines (MFE-319 and RL95-2) following treatment with the selective p110α (A66) and p110β (TGX-221) inhibitors (Figure 3A) [34]. Inhibition of p110α had no effect in PTEN-deficient cells and had only a marginal effect in KLE and EM cells with WT PTEN. In contrast, a dose-dependent decrease in cell viability was induced in MFE-280 cells harboring an activating mutation in PIK3CA (H1047Y), reaching an SF50 (50% surviving cell fraction) at < 1 µM. All cell lines had little response to inhibition of p110β with TGX-221 and required ≥ 10 µM to reach an SF50. To investigate if the observed decrease in cell viability in MFE-280 cells was due to an induction of apoptosis, cells were analyzed by Western immunoblotting for poly(ADP-ribose) polymerase 1 (PARP) cleavage following treatment with A66 and TGX-221 (Figure 3B-3C). Incubation with A66 and TGX-221 had no effect in KLE, MFE-319 or RL95-2 cells on PARP cleavage (Figure 3B and Supplementary Figure S2A-B). In contrast, PARP cleavage was induced by A66 in a dose-dependent manner in MFE-280 cells as cleavage was apparent at 1 µM and more strongly at 10 µM (Figure 3B-3C). Inhibition of p110β with TGX-221 had a much weaker effect on PARP cleavage in these cells (Figure 3B-3C). Interestingly, A66 and TGX-221 induced similar effects on the production of high molecular weight DNA fragments but without the generation of oligosomal DNA fragments (Figure 3D), perhaps indicative of the induction of an alternative type of cell death as previously observed in other studies [35-37]. These results suggest that p110α inhibition preferentially triggers a decrease in cell viability and induces cell death in PIK3CA mutant MFE-280 cells, and not in PIK3CA WT KLE cells. PTEN-deficient cells were in contrast resistant to both p110α and p110β inhibition.
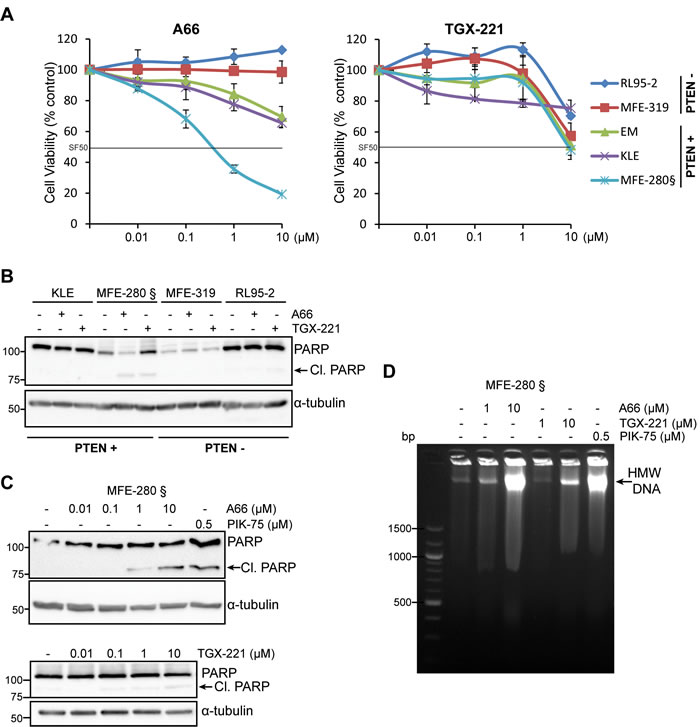
Figure 3: PIK3CA mutation and PTEN expression status influence differently the sensitivity of p110α and p110β inhibitors to cell survival in endometrial carcinoma cells. A. Cell viability determined by MTS assay in PTEN-positive (PTEN+) (EM, KLE and MFE280) and PTEN-deficient (PTEN-) cell lines (RL95-2 and MFE-319) of which only MFE-280 harbor mutated PIK3CA (§), treated with 0.01-10 µM of A66 or TGX-221 for 72 h. Data is presented for each cell line as a % relative to DMSO control (mean of 3 independent experiments in triplicates + SDs). Surviving cell fraction of 50% (SF50) threshold is indicated as a black line. B. Whole cell extracts were obtained from cells treated with 10 µM of the p110α inhibitor A66 or p110β inhibitor TGX-221 for 24 h and analyzed by Western immunoblotting for PARP cleavage and α-tubulin levels. C. Whole cell extracts were obtained from MFE-280 cell treated with 0.01-10 µM of A66 or TGX-221 and 0.5 µM PIK-75 for 24 h and analyzed by Western immunoblotting for PARP cleavage and α-tubulin levels. D. Agarose electrophoresis of high molecular weight DNA fragmentation of MFE-280 floating cells collected following treatment with 1-10 µM of A66 or TGX-221 and 0.5 µM PIK-75 for 24 h.
p110β inhibition impairs cell proliferation in PTEN-deficient cells but with variable responses
p110β has previously been shown to regulate cell proliferation [10, 38] particularly in PTEN-deficient tumors [13]. Since p110β inhibition did not induce PARP cleavage in PTEN-deficient cells, we tested if an effect could be apparent on cell proliferation, using the highest concentration of TGX-221 (10 µM) which showed a 50% decrease in cell viability in some cell lines (Figure 3A). While no significant decrease in cell number was observed for the PTEN-positive cell lines, p110β inhibition induced a significant decrease in the number of cells in the PTEN-deficient cell lines RL95-2 and MFE-319 following 4 and 3 days of treatment respectively (Figure 4A). However, after 4 days of treatment, the cell division rate of MFE-319 cells returned to that of control cells. This was reflected by a significant increase in total doubling time in RL95-2 but not in MFE-319 cells following TGX-221 treatment (Figure 4B). p110β has also been shown to contribute to G1 to S phase progression [39, 40] and we therefore evaluated if p110β inhibition affected the cell cycle distribution of the PTEN-deficient cells. As shown in Figure 4C, RL95-2, but not MFE-319 cells, demonstrated an increase in the percentage of cells in G1 phase and a concomitant decrease in the percentage of cells in S and G2/M phases compared to control cells. These results demonstrate that p110β can contribute to cell proliferation by regulating G1 to S phase progression to a certain extent but only in a subset of PTEN-deficient cells.
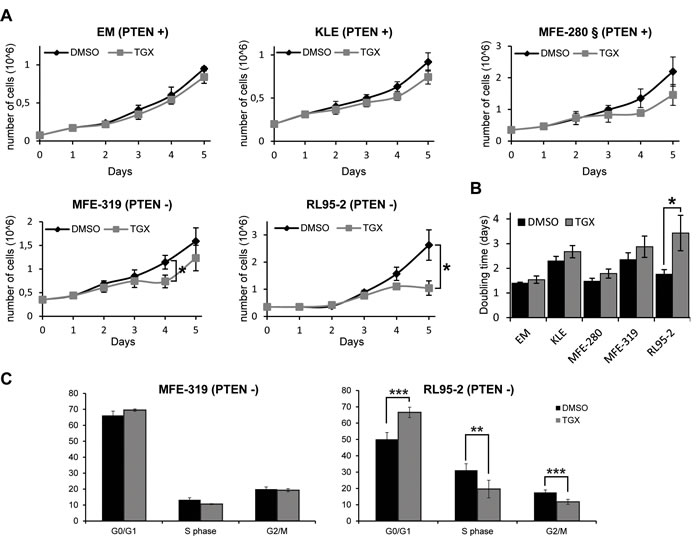
Figure 4: Inhibition of p110β delays cell proliferation in PTEN-deficient endometrial cancer cells. A. Cell lines were plated at day 0 and treated with DMSO (black line) or with 10 µM TGX-221 (grey line) at day 1 and day 3 and the number of cells were counted at all the indicated days. Cell numbers are presented as means of 3 independent experiments in triplicates for each day + SDs, t-test, * P < 0.05. The PIK3CA mutant cell line MFE-280 is marked with §. B. Doubling time calculated from data acquired in (A) for each cell line. C. MFE-319 and RL95-2 cells were treated with DMSO or 10 µM TGX-221 for 24 h and the percentage of cells in each phase of the cell cycle was determined by flow cytometry. Data are means +/- SDs of 3-4 independent experiments and Student’s t tests were performed comparing TGX-221-treated cells to DMSO-treated cells for each cell cycle phase with ** P < 0.01 and *** P < 0.001.
High PIK3CB mRNA levels correlate with a high cell cycle progression gene signature score in clinical samples
To further explore the clinical relevance of the observed contribution of p110β activity in cell proliferation in cell line studies, we analyzed potential phenotypic links between mRNA levels of both PIK3CB and PIK3CA in clinically annotated endometrial carcinoma samples with a cell cycle progression (CCP) score established from 31 CC genes [41]. High PIK3CB mRNA levels were significantly associated with high level of CCP signature score and high protein levels of the proliferation cell nuclear antigen (PCNA) for all primary tumors and notably within the endometrioid subgroup (Figure 5A-5B). In contrast, PIK3CA mRNA levels did not associate with measures for cell proliferation when explored for all primary tumors or the endometrioid subgroup (Figure 5C-5D). These results suggest that PIK3CB/p110β contributes to endometrial tumorigenesis by influencing the cell cycle regulation of endometrial cancer cells. In contrast, poor disease specific survival observed with high PIK3CA levels (Figure 2D) is more likely due to other PI3K-mediated cellular processes than proliferation.
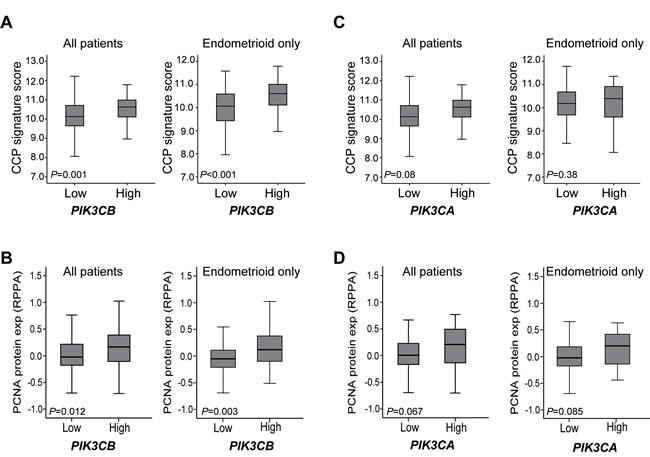
Figure 5: High PIK3CB mRNA levels associates with high cell proliferation markers in primary tumors. Box-plots are shown for the cell cycle progression (CCP) signature score in relation to low versus high PIK3CB A. or PIK3CA mRNA levels C. and for PCNA protein levels in relation to low versus high PIK3CB B. or PIK3CA mRNA levels D., either in all primary tumors or endometrioid subgroup tumors.
PTEN positive and deficient endometrial cancer cells respond differently to p110α and p110β inhibition on Akt and S6 signaling
To correlate cellular function with PI3K pathway signaling, we determined the effect of both p110α and p110β inhibitors on AKT (p-S473-Akt) and mTORC1 (p-S240/S244-S6) signaling (Figure 6). All cell lines, whether PTEN-positive with low p-S473-Akt or PTEN-deficient with high p-S473-Akt, responded to p110α inhibition with a decrease in Akt basal activity. Interestingly, the best response to p110α inhibition, measured both by decreased p-Akt and cell viability, was observed in the PIK3CA mutant MFE-280 cells. Inhibition of p110β also led to a decrease in p-S473-Akt in all cells except the PTEN-deficient cell line MFE-319. Response to p110α inhibition on S6 signaling was varied from a low response in PTEN-positive cells to no response at all in PTEN-negative cells. Response to p110β inhibition on S6 signaling was even more varied and independent of PTEN expression status. The PTEN-deficient RL95-2 cells were responsive while MFE-319 cells were resistant. The PTEN-positive cells KLE were very responsive while MFE-280 cells were resistant.
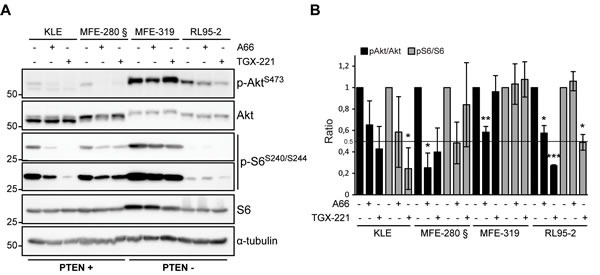
Figure 6: Heterogeneous response on Akt and S6 signaling following p110α and p110β inhibition. A. Whole cell extracts were obtained from cells treated with 10 µM of A66 or TGX-221 for 24 h and analyzed by Western immunoblotting for pS473 Akt, Akt, pS240/244 S6 ribosomal protein (short and long exposures are shown), S6 and α-tubulin levels. B. Quantification of pS473-Akt/Akt and pS240/244-S6/S6 ratios relative to DMSO-treated controls for each cell line from at least 3 independent experiments. Standard deviations and significance level for difference (T-test compared to DMSO-treated cells: *P < 0.05, **P < 0.01, ***P < 0.001) are shown for each condition.
Discussion
All class I PI3K isoforms can produce PtdIns(3,4,5)P3, the levels of which are controlled by at least the action of PTEN. In lesions with loss of PTEN, tumorigenicity is thought to rely on the activity of not only p110β but also of p110α depending on the tissue type [10, 13, 16, 42]. In EEC, PTEN is the most commonly mutated gene [43] but little is known however about the exact contribution of p110β in endometrial tumor development particularly in relation to PTEN loss. In this study, we demonstrate that p110β is highly expressed in endometrial cancer cell lines and that its mRNA levels increase in clinical samples during the initial steps of endometrial carcinogenesis for the endometrioid subtype. In contrast, high PIK3CA mRNA levels were observed in non-endometrioid tumors, which would appear to be consistent with previous findings linking PIK3CA gene amplification to aggressive endometrial tumors of non-endometrioid histology [44]. In addition, different roles were found for the two isoforms in this study, as supported by the association of high mRNA levels of PIK3CB, but not PIK3CA, with markers for high proliferation (CCP score and PCNA levels), notably in endometrioid tumors. Interestingly, the CCP score was reported to increase early in grades 1 and 2 compared to CAHs [45], at the same stage of tumor development when PIK3CB mRNA levels are found to be increased in this study. Hence, high PIK3CB mRNA levels may provide a proliferative advantage in early stages of tumorigenesis. Using pharmacological inhibition of p110β, our cell line studies also pointed to a role for p110β in cell proliferation since its inhibition delayed cell proliferation by arresting cells in G1 in a PTEN-deficient cancer cell line, RL95-2. Consistently, p110β has previously been shown to be localized in the nucleus [40, 46, 47] and to mediate S phase entry, control DNA replication and loading of PCNA on chromatin [40]. In functional studies of nuclear p110β [40, 48], TGX-221 was previously used at high concentration (30 µM), which was shown to be specific for p110β and not p110α [40]. In this study, inhibition of proliferation was only apparent with 10 µM of the inhibitor, which may be due to high levels of p110β in endometrial cancer cell lines as well as its subcellular localization spanning not only the cytoplasm but also the nucleus in other cell lines [46, 47] and the endometrial cancer cells used in this study (data not shown). Although we cannot exclude off-target effects at 10 µM, an extensive study of TGX-221 at that concentration on a large panel of lipid and protein kinases by Jamieson et al showed high specificity for p110β [34].
The reported association between p110β level and activity with cell proliferation suggests further clinical testing of endometrial tumors with PTEN mutations for potential benefit from p110β selective inhibitors to delay tumor growth. This is also particularly relevant in light of a recent study showing response to inhibition of p110β in PTEN-deficient breast cancer cells, resistant to p110α inhibition [15]. However, we observed that PTEN-deficient cancer cells can respond with different efficacy following p110β inhibition (MFE-319 versus RL95-2 cells) and resistance to treatment was observed in MFE-319 cells (Figure 4). This would be consistent with a recent study by Schwartz et al showing that p110α was reactivated following p110β selective inhibition in PTEN-deficient cells [49]. The efficacy of p110β inhibition may hence have limited durability and combined or sequential treatment targeting p110β and p110α may be necessary in tumors with PTEN loss. Other genetic factors may also influence the efficacy of p110β inhibition, such as the presence of RAS mutations, since the RAS-MEK and PI3K pathways are known to converge [50]. Indeed RL95-2 cells harbor an activating mutation in H-Ras (Q61H), which may explain the necessity of high concentration of TGX-221. PTEN-deficient cells with RAS mutation may benefit from a combination of p110β and RAS/MEK pathway inhibition. The cause for resistance to p110β inhibition in MFE-319 cells is not yet known but it is worth noticing that these cells harbor a mutation in the fibroblast growth factor receptor 2 (S252W) shown previously to be oncogenic and to occur with high frequency in endometrial cancer [51, 52]. However, although FGFR signaling is known to activate the PI3K pathway, MFE-319 cells were shown to be insensitive to FGFR inhibition [53]. In any case, identifying the molecular mechanisms of resistance to p110β inhibition in a subset of PTEN-deficient tumors is crucial to design clinical therapeutic strategies with long-term efficacy. In addition, this would anticipate understanding of the results of a phase I/IIa clinical trial with a selective p110β inhibitor in carcinomas with PTEN loss, including endometrial cancer (NCT01458067) and which has recently been completed [54].
The PTEN-positive MFE-280 cells harboring a PIK3CA mutation in its catalytic domain (H1047Y) responded to selective p110α inhibition with a decrease in cell viability and an increase in cell death while PIK3CA WT and PTEN-deficient cell lines were resistant. The observation that these cells were more sensitive to p110α inhibition than WT PIK3CA is in agreement with the studies of Torbett et al [12] showing that breast cancer cells with PIK3CA mutations in their catalytic domain responded to the p110α inhibitor PIK-75, but not PTEN-deficient cells. Consistently, T47D breast cancer cells with the H1047R mutation in PIK3CA were sensitive to the p110α inhibitor BYL719 measured by cell viability assay, but exhibited a lower response when PTEN was knocked down [15]. We also showed that inhibition of p110β had little effect on cell viability and required >10 µM TGX-221 to reach 50% cell survival in most endometrial cancer cell lines studied, including PTEN-deficient cells. This is in agreement with the work of Weigelt et al [30] in endometrial cancer cells but in contrast to studies in breast and prostate cancer [10, 11, 13], arguing that p110β does not contribute to cell survival in endometrial cancer cells. In this study, we showed that the PIK3CA mutant cell line MFE-280 reached an SF50 of 10 µM in cell viability assay and was the only cancer cell line where TGX-221 induced cell death, albeit weakly, and only at the highest dose at 10 µM, hence with low efficacy. This may suggest an active cooperation between p110α and p110β. A recent study has indeed showed that following serum stimulation, activated p110α associates with a fraction of p110β and leads to p110β activation [55]. Accordingly, the activating mutation in PIK3CA found in MFE-280 cells may allow the recruitment of p110β to p110α and its subsequent activation. The resulting activation of both isoforms would hence allow these cells to respond to inhibitors selective for either isoform. The association of p110β with p110α would implicate that both isoforms can engage in similar cellular function, in this case cell survival, and respond to similar external cues. On the other hand, the remaining fraction of p110β not associated with p110α could still engage in other cellular functions including proliferation.
Different effects on PI3K isoform-specific signaling were demonstrated to be PTEN context dependent for p110α and p110β selective inhibition. Decrease in p-Akt was observed with variable efficacy with both p110α and p110β inhibitors, independently of PTEN status and despite their differential effects on cell viability. In contrast, a different response to p110α inhibition was apparent for p-S6 which was lower in PTEN-positive but not in PTEN-deficient cells. This would be consistent with a study in breast cancer cells where persistence of mTORC1 signaling via p-S240/S244-S6 was shown to be a marker of resistance to p110α inhibition [56] although this study did not show any correlation with PTEN status. p110β inhibition decreased p-S6 in RL95-2 cells but not in MFE-319 displaying a more transient effect of p110β inhibition on cell proliferation. The difference in mTORC1/S6 signaling sensitivity to p110β inhibition in PTEN-deficient cells seemed to be correlated with the difference in p-S6 levels, i.e. with a higher response in RL95-2 cells that have the lowest p-S6 levels and no response in MFE-319 with the highest levels. In addition, decrease in p-S6 by p110β inhibition correlated with a decrease in cell proliferation in RL95-2 cells. MFE-319 cells may thus have acquired resistance to both p110α and p110β inhibitors via mTORC1 hyper-activation.
In conclusion, our results demonstrate for the first time that high levels of PIK3CB/p110β associate with the early stages of endometrial tumorigenesis and increased cell proliferation, suggesting a proliferative advantage. In cell lines, p110β plays a role in a PTEN-dependent context in cell proliferation but not in cell survival. However, the benefits of p110β inhibition may not be lasting and lead to resistance in some PTEN-deficient endometrial tumors. This may be relevant for the design of more effective and combined targeted therapy in future clinical trial in endometrial cancer featuring loss-of-function mutations in PTEN.
Materials and methods
Reagents
Antibodies used in Western immunoblotting are the following: anti-Akt (2920), -S473-Akt (9271), -pT308-Akt (C31E5E), -p110α (4249), -p110β (3011), -PARP (2730), -S6 (2217) and -pS240/S244-S6 (5364) from Cell Signaling; anti-p85α (05-212) from Millipore; anti-p85β (S3089) from Epitomics; anti-PTEN (7974) from Santa Cruz and anti-α-tubulin (T5168) from Sigma-Aldrich. Horse radish peroxidase-conjugated secondary antibodies were from Life Technologies. The PI3K selective inhibitors targeting p110α were from Selleck Chemicals (A66) and Millipore (PIK-75) and targeting p110β (TGX-221) from Cayman chemicals [34, 57].
Cell lines and cell culture conditions
Seven endometrial cancer cell lines were chosen to represent different status for PI3K/Akt pathway activation and PTEN protein expression, as shown in this study and by others [21, 30, 43, 58]. Cancer cell lines were obtained from ATCC (KLE, RL95-2), DSMZ Germany (MFE-296, MFE-319, EFE-184 and MFE-280) and Sigma-Aldrich (Ishikawa). EM-E6/E7-hTERT (EM), a non-transformed endometrial cell line isolated from glandular endometrial tissue and immortalized with E6/E7 and human TERT [59, 60], was a gift from Prof PM Pollock (University of Queensland, Australia). All cancer cells were authenticated by short tandem repeat (STR) DNA profiling (IdentiCell Service, Dept. Molecular Medicine, Aarhus University Hospital, Denmark for all cancer cell lines). EM cells were confirmed to have a unique STR profile as shown in Table 1 (MD Anderson Cancer Center, USA). All cancer cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and antibiotics (100 IU/ml penicillin and 100 μg/ml streptomycin). EM cells were cultured in DMEM/Ham’s F12 supplemented with Insulin-Transferrin-Selenium, 10% FBS and antibiotics and changed to DMEM containing 10% FBS and antibiotics 24 h before harvest. Cells were harvested when they reached a maximum of 80% confluence.
Table 1: STR profile of EM-E6/E7-hTERT cells
Marker |
Allele(s) |
| AMEL | X |
| CSF1PO | 11,12 |
| D13S317 | 8,9 |
| D16S539 | 10,11 |
D18S51 |
15 |
| D21S11 | 31.2,32 |
| D3S1358 | 15,17 |
| D5S818 | 12,13 |
| D7S820 | 10,12 |
| D8S1179 | 10,13 |
| FGA | 21,23 |
| TH01 | 6,7 |
| TPOX | 8,11 |
| vWA | 14 |
Cell viability assay, cell counts and flow cytometry
For cell viability, cells were seeded in 96-well plates with 1000 or 3000 cells/well depending on the cell line, grown for 24 h and treated with inhibitors for 72 h. Cells were then assayed with the CellTiter 96 AQueous One Solution Cell Proliferation Assay (Promega) by incubation with 20 µL of 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium for 1 h and reading at 490 nm. For cell counts, cells were plated in 6-well plates, grown for 24 h, treated and counted following trypsinization at the same time point every day with a BioRad TC10 automated cell counter. For flow cytometry, 2 x105 cells were seeded in 6-well plates, treated for 24 h and processed as described previously [61]. DNA was stained with 100 μg/mL propidium iodide and the cell cycle distribution was analyzed using a BD Biosciences Accuri C6 flow cytometer. For DNA fragmentation assays, cells were plated in 6-well plates, treated for 24 h and the floating cells were collected, lysed in 40 µL DMSO and 40 µL Tris-EDTA pH 7.4 supplemented with 2% SDS, according to Suman et al [62] and half was loaded on a 2% agarose gel.
Whole cell extracts, subcellular fractionation and Western immunoblotting
Whole cell extracts were prepared in radioimmunoprecipitation assay (RIPA) lysis buffer (50 mM Tris pH 8.0, 0.5% deoxycholic acid, 150 mM NaCl, 1% NP-40, 0.1% SDS) supplemented with 5 mM NaF, 2 mM Na3VO4 and 1x Sigma Protease Inhibitor Cocktail. Subcellular fractionation was carried out according to O’Caroll et al. [63] and nuclear pellets were lysed in RIPA. RL95-2 cells required an additional syringing step of the nuclear pellet resuspended in wash buffer to avoid cytoplasmic contamination. Equal amount of proteins were resolved by SDS-PAGE, immunoblotted as described previously [64] and detected by enhanced chemiluminescence using the SuperSignal West Pico Chemiluminescent Substrate (Pierce) and visualized with a BioRad ChemiDocTM Xrs+.
Patient series
Fresh frozen tissue was collected from patients diagnosed with endometrial cancer at Haukeland University hospital during the period from 2001-2013 and include a total of 234 clinical samples with 18 endometrial cancer precursor lesions (CAH), 174 primary tumors and 42 metastases. Clinical data were collected as described earlier [25, 45]. The patient cohort used for p110β immunohistochemistry is described in detail in Tangen et al [45]. This study was conducted in line with Norwegian legislation and international demands for ethical review, approved by the Norwegian Data Inspectorate, Norwegian Social Sciences Data Services and the Western Regional Committee for Medical and Health Research Ethics (NSD15501; REK 052.01). Patients signed an informed consent form.
Microarray analyses and reverse phase protein array
Fresh frozen tissue collected in parallel from 18 CAH, 174 primary and 42 metastatic lesions was used for RNA extraction as previously reported [25, 45]. The majority of samples had tumor purity above 80% with a threshold inclusion set at >50%. Samples were gross dissected when necessary to achieve this level of tumor purity. mRNA was extracted from fresh frozen tissue using the RNeasy Mini Kit (Qiagen). Samples were hybridized to Agilent Whole Human Genome Microarrays (G4112F), scanned and normalized as previously reported [65]. mRNA values for PIK3CA (probe A_23_P92057) and PIK3CB (probe A_23_P346969) were extracted from the microarray dataset. For survival analyses using mRNA values, values were grouped in tertiles, according to similarity in survival and considering the size of the subgroups and the number of events in each category. The CCP signature score was calculated as previously described [45] using the gene expression signature of 31 CCP genes reported by Cuzick et al. [41]. For CCP analyses, mRNA values were further grouped in high and low, where the two lowest tertiles were defined as low, and the highest tertile was defined as high. Microarray data are publicly available at ArrayExpress (accession number E-MTAB-2532). Reverse phase protein array for PCNA protein levels was performed as previously described [45].
Statistical analyses
For clinical samples, statistical analyses were performed using the software package SPSS 22 (SPSS Inc, Chicago, IL) and significance was defined as p < 0.05. Correlations between groups were evaluated using the Mann-Whitney U test for continuous variables. Survival analyses were done using the Kaplan-Meier (product-limit) method. Date of primary surgery was set as entry date and time of death due to endometrial cancer as endpoint (disease specific survival). Survival between groups was compared with log-rank test for trend. For cell lines data, Student’s unpaired t test with unequal variance was used.
Abbreviations
CAH: complex atypical hyperplasia; CCP: cell cycle progression; EEC: endometrioid endometrial cancer; NEEC: non-endometrioid endometrial cancer; PI3K: phosphoinositide 3-kinase; PTEN: phosphatase and tensin homolog; PARP: poly(ADP-ribose) polymerase 1; PtdIns(3,4,5)P3: phosphatidylinositol (3,4,5)-trisphosphate; SF: survival fraction.
Acknowledgments
We thank Ellen Valen, Britt Edvardsen and Kadri Madissoo for technical assistance.
Conflict of interest
The authors disclose no potential conflicts of interest.
Funding
This work was supported by grants from the Norwegian Cancer Society to AEL and from Helse Vest, the University of Bergen, the Norwegian Cancer Society (Harald Andersen Legat) and the Research Council of Norway to HBS as well as from Bergen Medisinske Forskningsstiftelse to CK.
Author contributions
• Conception and design: HB Salvesen and AE Lewis
• Acquisition of data. T Karlsson, AE Lewis, C Krakstad and I Løberg Tangen
• Analysis of data: T Karlsson, C Krakstad, I Løberg Tangen, EA Hoivik, HB Salvesen, P Pollock and AE Lewis
• Writing, review and/or revision of the manuscript: T Karlsson, C Krakstad, HB Salvesen, P Pollock and AE Lewis.
Editorial note
This paper has been accepted based in part on peer-review conducted by another journal and the authors’ response and revisions as well as expedited peer-review in Oncotarget.
Dedicated
This article is dedicated to the memory of Professor Helga B Salvesen and her valuable contribution to research in gynecology.
References
1. Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer. 2009; 9(8):550-562.
2. Fruman DA and Rommel C. PI3K and cancer: lessons, challenges and opportunities. Nat Rev Drug Discov. 2014; 13(2):140-156.
3. Vanhaesebroeck B, Stephens L and Hawkins P. PI3K signalling: the path to discovery and understanding. Nat Rev Mol Cell Biol. 2012; 13(3):195-203.
4. Chalhoub N and Baker SJ. PTEN and the PI3-kinase pathway in cancer. Annu Rev Pathol. 2009; 4:127-150.
5. Kok K, Geering B and Vanhaesebroeck B. Regulation of phosphoinositide 3-kinase expression in health and disease. Trends Biochem Sci. 2009; 34(3):115-127.
6. Martini M, De Santis MC, Braccini L, Gulluni F and Hirsch E. PI3K/AKT signaling pathway and cancer: an updated review. Annals of medicine. 2014; 46(6):372-383.
7. Dbouk HA, Khalil BD, Wu H, Shymanets A, Nurnberg B and Backer JM. Characterization of a tumor-associated activating mutation of the p110beta PI 3-kinase. PLoS One. 2013; 8(5):e63833.
8. Pazarentzos E, Giannikopoulos P, Hrustanovic G, St John J, Olivas VR, Gubens MA, Balassanian R, Weissman J, Polkinghorn W and Bivona TG. Oncogenic activation of the PI3-kinase p110beta isoform via the tumor-derived PIK3Cbeta(D1067V) kinase domain mutation. Oncogene. 2016; 35(9):1198-1205.
9. Kang S, Denley A, Vanhaesebroeck B and Vogt PK. Oncogenic transformation induced by the p110beta, -gamma, and -delta isoforms of class I phosphoinositide 3-kinase. Proc Natl Acad Sci U S A. 2006; 103(5):1289-1294.
10. Jia S, Liu Z, Zhang S, Liu P, Zhang L, Lee SH, Zhang J, Signoretti S, Loda M, Roberts TM and Zhao JJ. Essential roles of PI(3)K-p110beta in cell growth, metabolism and tumorigenesis. Nature. 2008; 454(7205):776-779.
11. Ni J, Liu Q, Xie S, Carlson C, Von T, Vogel K, Riddle S, Benes C, Eck M, Roberts T, Gray N and Zhao J. Functional characterization of an isoform-selective inhibitor of PI3K-p110beta as a potential anticancer agent. Cancer Discov. 2012; 2(5):425-433.
12. Torbett NE, Luna-Moran A, Knight ZA, Houk A, Moasser M, Weiss W, Shokat KM and Stokoe D. A chemical screen in diverse breast cancer cell lines reveals genetic enhancers and suppressors of sensitivity to PI3K isoform-selective inhibition. Biochem J. 2008; 415(1):97-110.
13. Wee S, Wiederschain D, Maira SM, Loo A, Miller C, deBeaumont R, Stegmeier F, Yao YM and Lengauer C. PTEN-deficient cancers depend on PIK3CB. Proc Natl Acad Sci U S A. 2008; 105(35):13057-13062.
14. Edgar KA, Wallin JJ, Berry M, Lee LB, Prior WW, Sampath D, Friedman LS and Belvin M. Isoform-specific phosphoinositide 3-kinase inhibitors exert distinct effects in solid tumors. Cancer research. 2010; 70(3):1164-1172.
15. Juric D, Castel P, Griffith M, Griffith OL, Won HH, Ellis H, Ebbesen SH, Ainscough BJ, Ramu A, Iyer G, Shah RH, Huynh T, Mino-Kenudson M, et al. Convergent loss of PTEN leads to clinical resistance to a PI(3)Kalpha inhibitor. Nature. 2014.
16. Berenjeno IM, Guillermet-Guibert J, Pearce W, Gray A, Fleming S and Vanhaesebroeck B. Both p110alpha and p110beta isoforms of PI3K can modulate the impact of loss-of-function of the PTEN tumour suppressor. Biochem J. 2012; 442(1):151-159.
17. Liang H, Cheung LW, Li J, Ju Z, Yu S, Stemke-Hale K, Dogruluk T, Lu Y, Liu X, Gu C, Guo W, Scherer SE, Carter H, et al. Whole-exome sequencing combined with functional genomics reveals novel candidate driver cancer genes in endometrial cancer. Genome Res. 2012; 22(11):2120-2129.
18. Cancer Genome Atlas Research N, Kandoth C, Schultz N, Cherniack AD, Akbani R, Liu Y, Shen H, Robertson AG, Pashtan I, Shen R, Benz CC, Yau C, Laird PW, et al. Integrated genomic characterization of endometrial carcinoma. Nature. 2013; 497(7447):67-73.
19. Hong B, Le Gallo M and Bell DW. The mutational landscape of endometrial cancer. Curr Opin Genet Dev. 2015; 30:25-31.
20. O’Hara AJ and Bell DW. The genomics and genetics of endometrial cancer. Adv Genomics Genet. 2012; 2012(2):33-47.
21. Cheung LW, Hennessy BT, Li J, Yu S, Myers AP, Djordjevic B, Lu Y, Stemke-Hale K, Dyer MD, Zhang F, Ju Z, Cantley LC, Scherer SE, et al. High frequency of PIK3R1 and PIK3R2 mutations in endometrial cancer elucidates a novel mechanism for regulation of PTEN protein stability. Cancer Discov. 2011; 1(2):170-185.
22. Rudd ML, Price JC, Fogoros S, Godwin AK, Sgroi DC, Merino MJ and Bell DW. A unique spectrum of somatic PIK3CA (p110alpha) mutations within primary endometrial carcinomas. Clinical cancer research. 2011; 17(6):1331-1340.
23. Mutter GL, Lin MC, Fitzgerald JT, Kum JB, Baak JP, Lees JA, Weng LP and Eng C. Altered PTEN expression as a diagnostic marker for the earliest endometrial precancers. Journal of the National Cancer Institute. 2000; 92(11):924-930.
24. Hayes MP, Wang H, Espinal-Witter R, Douglas W, Solomon GJ, Baker SJ and Ellenson LH. PIK3CA and PTEN mutations in uterine endometrioid carcinoma and complex atypical hyperplasia. Clinical cancer research. 2006; 12(20 Pt 1):5932-5935.
25. Berg A, Hoivik EA, Mjos S, Holst F, Werner HM, Tangen IL, Taylor-Weiner A, Gibson WJ, Kusonmano K, Wik E, Trovik J, Halle MK, Oyan AM, et al. Molecular profiling of endometrial carcinoma precursor, primary and metastatic lesions suggests different targets for treatment in obese compared to non-obese patients. Oncotarget. 2015; 6:1327-39. doi: 10.18632/oncotarget.2675.
26. Catasus L, Gallardo A, Cuatrecasas M and Prat J. PIK3CA mutations in the kinase domain (exon 20) of uterine endometrial adenocarcinomas are associated with adverse prognostic parameters. Mod Pathol. 2008; 21(2):131-139.
27. Konopka B, Janiec-Jankowska A, Kwiatkowska E, Najmola U, Bidzinski M, Olszewski W and Goluda C. PIK3CA mutations and amplification in endometrioid endometrial carcinomas: relation to other genetic defects and clinicopathologic status of the tumors. Hum Pathol. 2011; 42(11):1710-1719.
28. Forbes SA, Bindal N, Bamford S, Cole C, Kok CY, Beare D, Jia M, Shepherd R, Leung K, Menzies A, Teague JW, Campbell PJ, Stratton MR, et al. COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2011; 39(Database issue):D945-950.
29. An HJ, Cho NH, Yang HS, Kwak KB, Kim NK, Oh DY, Lee SW, Kim HO and Koh JJ. Targeted RNA interference of phosphatidylinositol 3-kinase p110-beta induces apoptosis and proliferation arrest in endometrial carcinoma cells. J Pathol. 2007; 212(2):161-169.
30. Weigelt B, Warne PH, Lambros MB, Reis-Filho JS and Downward J. PI3K pathway dependencies in endometrioid endometrial cancer cell lines. Clin Cancer Res. 2013; 19(13):3533-3544.
31. Stemke-Hale K, Gonzalez-Angulo AM, Lluch A, Neve RM, Kuo WL, Davies M, Carey M, Hu Z, Guan Y, Sahin A, Symmans WF, Pusztai L, Nolden LK, et al. An integrative genomic and proteomic analysis of PIK3CA, PTEN, and AKT mutations in breast cancer. Cancer research. 2008; 68(15):6084-6091.
32. Vasudevan KM, Barbie DA, Davies MA, Rabinovsky R, McNear CJ, Kim JJ, Hennessy BT, Tseng H, Pochanard P, Kim SY, Dunn IF, Schinzel AC, Sandy P, et al. AKT-independent signaling downstream of oncogenic PIK3CA mutations in human cancer. Cancer Cell. 2009; 16(1):21-32.
33. Gasser JA, Inuzuka H, Lau AW, Wei W, Beroukhim R and Toker A. SGK3 mediates INPP4B-dependent PI3K signaling in breast cancer. Molecular cell. 2014; 56(4):595-607.
34. Jamieson S, Flanagan JU, Kolekar S, Buchanan C, Kendall JD, Lee WJ, Rewcastle GW, Denny WA, Singh R, Dickson J, Baguley BC and Shepherd PR. A drug targeting only p110alpha can block phosphoinositide 3-kinase signalling and tumour growth in certain cell types. Biochem J. 2011; 438(1):53-62.
35. Brown DG, Sun XM and Cohen GM. Dexamethasone-induced apoptosis involves cleavage of DNA to large fragments prior to internucleosomal fragmentation. J Biol Chem. 1993; 268(5):3037-3039.
36. Oberhammer F, Wilson JW, Dive C, Morris ID, Hickman JA, Wakeling AE, Walker PR and Sikorska M. Apoptotic death in epithelial cells: cleavage of DNA to 300 and/or 50 kb fragments prior to or in the absence of internucleosomal fragmentation. EMBO J. 1993; 12(9):3679-3684.
37. Solovyan VT, Bezvenyuk ZA, Salminen A, Austin CA and Courtney MJ. The role of topoisomerase II in the excision of DNA loop domains during apoptosis. J Biol Chem. 2002; 277(24):21458-21467.
38. Benistant C, Chapuis H and Roche S. A specific function for phosphatidylinositol 3-kinase alpha (p85alpha-p110alpha) in cell survival and for phosphatidylinositol 3-kinase beta (p85alpha-p110beta) in de novo DNA synthesis of human colon carcinoma cells. Oncogene. 2000; 19(44):5083-5090.
39. Marques M, Kumar A, Cortes I, Gonzalez-Garcia A, Hernandez C, Moreno-Ortiz MC and Carrera AC. Phosphoinositide 3-kinases p110alpha and p110beta regulate cell cycle entry, exhibiting distinct activation kinetics in G1 phase. Mol Cell Biol. 2008; 28(8):2803-2814.
40. Marques M, Kumar A, Poveda AM, Zuluaga S, Hernandez C, Jackson S, Pasero P and Carrera AC. Specific function of phosphoinositide 3-kinase beta in the control of DNA replication. Proc Natl Acad Sci U S A. 2009; 106(18):7525-7530.
41. Cuzick J, Swanson GP, Fisher G, Brothman AR, Berney DM, Reid JE, Mesher D, Speights VO, Stankiewicz E, Foster CS, Moller H, Scardino P, Warren JD, et al. Prognostic value of an RNA expression signature derived from cell cycle proliferation genes in patients with prostate cancer: a retrospective study. Lancet Oncol. 2011; 12(3):245-255.
42. Schmit F, Utermark T, Zhang S, Wang Q, Von T, Roberts TM and Zhao JJ. PI3K isoform dependence of PTEN-deficient tumors can be altered by the genetic context. Proc Natl Acad Sci U S A. 2014; 111(17):6395-6400.
43. Oda K, Stokoe D, Taketani Y and McCormick F. High frequency of coexistent mutations of PIK3CA and PTEN genes in endometrial carcinoma. Cancer research. 2005; 65(23):10669-10673.
44. Salvesen HB, Carter SL, Mannelqvist M, Dutt A, Getz G, Stefansson IM, Raeder MB, Sos ML, Engelsen IB, Trovik J, Wik E, Greulich H, Bo TH, et al. Integrated genomic profiling of endometrial carcinoma associates aggressive tumors with indicators of PI3 kinase activation. Proc Natl Acad Sci U S A. 2009; 106(12):4834-4839.
45. Tangen IL, Werner HM, Berg A, Halle MK, Kusonmano K, Trovik J, Hoivik EA, Mills GB, Krakstad C and Salvesen HB. Loss of progesterone receptor links to high proliferation and increases from primary to metastatic endometrial cancer lesions. European journal of cancer. 2014; 50(17):3003-3010.
46. Kumar A, Redondo-Munoz J, Perez-Garcia V, Cortes I, Chagoyen M and Carrera AC. Nuclear but not cytosolic phosphoinositide 3-kinase beta has an essential function in cell survival. Mol Cell Biol. 2011; 31(10):2122-2133.
47. Karlsson T, Altankhuyag A, Dobrovolska O, Turcu DC and Lewis AE. A polybasic motif in ErbB3-binding protein 1 (EBP1) has key functions in nucleolar localization and polyphosphoinositide interaction. Biochem J. 2016; 473(14):2033-2047.
48. Kumar A, Fernandez-Capetillo O and Carrera AC. Nuclear phosphoinositide 3-kinase beta controls double-strand break DNA repair. Proc Natl Acad Sci U S A. 2010; 107(16):7491-7496.
49. Schwartz S, Wongvipat J, Trigwell CB, Hancox U, Carver BS, Rodrik-Outmezguine V, Will M, Yellen P, de Stanchina E, Baselga J, Scher HI, Barry ST, Sawyers CL, et al. Feedback Suppression of PI3Kalpha Signaling in PTEN-Mutated Tumors Is Relieved by Selective Inhibition of PI3Kbeta. Cancer Cell. 2015; 27(1):109-122.
50. McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Franklin RA, Montalto G, Cervello M, Libra M, Candido S, Malaponte G, Mazzarino MC, Fagone P, Nicoletti F, et al. Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR cascade inhibitors: how mutations can result in therapy resistance and how to overcome resistance. Oncotarget. 2012; 3:1068-1111. doi: 10.18632/oncotarget.659.
51. Dutt A, Salvesen HB, Chen TH, Ramos AH, Onofrio RC, Hatton C, Nicoletti R, Winckler W, Grewal R, Hanna M, Wyhs N, Ziaugra L, Richter DJ, et al. Drug-sensitive FGFR2 mutations in endometrial carcinoma. Proc Natl Acad Sci U S A. 2008; 105(25):8713-8717.
52. Byron SA, Gartside MG, Wellens CL, Mallon MA, Keenan JB, Powell MA, Goodfellow PJ and Pollock PM. Inhibition of activated fibroblast growth factor receptor 2 in endometrial cancer cells induces cell death despite PTEN abrogation. Cancer research. 2008; 68(17):6902-6907.
53. Konecny GE, Kolarova T, O’Brien NA, Winterhoff B, Yang G, Qi J, Qi Z, Venkatesan N, Ayala R, Luo T, Finn RS, Kristof J, Galderisi C, et al. Activity of the fibroblast growth factor receptor inhibitors dovitinib (TKI258) and NVP-BGJ398 in human endometrial cancer cells. Mol Cancer Ther. 2013; 12(5):632-642.
54. Dillon LM and Miller TW. Therapeutic targeting of cancers with loss of PTEN function. Curr Drug Targets. 2014; 15(1):65-79.
55. Perez-Garcia V, Redondo-Munoz J, Kumar A and Carrera AC. Cell activation-induced phosphoinositide 3-kinase alpha/beta dimerization regulates PTEN activity. Molecular and cellular biology. 2014; 34(18):3359-3373.
56. Elkabets M, Vora S, Juric D, Morse N, Mino-Kenudson M, Muranen T, Tao J, Campos AB, Rodon J, Ibrahim YH, Serra V, Rodrik-Outmezguine V, Hazra S, et al. mTORC1 Inhibition Is Required for Sensitivity to PI3K p110α Inhibitors in PIK3CA-Mutant Breast Cancer. Science translational medicine. 2013; 5(196):196ra199-196ra199.
57. Jackson SP, Schoenwaelder SM, Goncalves I, Nesbitt WS, Yap CL, Wright CE, Kenche V, Anderson KE, Dopheide SM, Yuan Y, Sturgeon SA, Prabaharan H, Thompson PE, et al. PI 3-kinase p110beta: a new target for antithrombotic therapy. Nat Med. 2005; 11(5):507-514.
58. Dedes KJ, Wetterskog D, Mendes-Pereira AM, Natrajan R, Lambros MB, Geyer FC, Vatcheva R, Savage K, Mackay A, Lord CJ, Ashworth A and Reis-Filho JS. PTEN deficiency in endometrioid endometrial adenocarcinomas predicts sensitivity to PARP inhibitors. Science translational medicine. 2010; 2(53):53ra75.
59. Kyo S, Nakamura M, Kiyono T, Maida Y, Kanaya T, Tanaka M, Yatabe N and Inoue M. Successful immortalization of endometrial glandular cells with normal structural and functional characteristics. The American journal of pathology. 2003; 163(6):2259-2269.
60. Mizumoto Y, Kyo S, Ohno S, Hashimoto M, Nakamura M, Maida Y, Sakaguchi J, Takakura M, Inoue M and Kiyono T. Creation of tumorigenic human endometrial epithelial cells with intact chromosomes by introducing defined genetic elements. Oncogene. 2006; 25(41):5673-5682.
61. Lewis AE, Wong BC, Langman MJ and Eggo MC. Protein kinase C inhibition induces DNA fragmentation in COLO 205 cells which is blocked by cysteine protease inhibition but not mediated through caspase-3. Exp Cell Res. 2003; 289(1):1-10.
62. Suman S, Pandey A and Chandna S. An improved non-enzymatic “DNA ladder assay” for more sensitive and early detection of apoptosis. Cytotechnology. 2012; 64(1):9-14.
63. O’Carroll SJ, Mitchell MD, Faenza I, Cocco L and Gilmour RS. Nuclear PLCbeta1 is required for 3T3-L1 adipocyte differentiation and regulates expression of the cyclin D3-cdk4 complex. Cell Signal. 2009; 21(6):926-935.
64. Aumo L, Rusten M, Mellgren G, Bakke M and Lewis AE. Functional roles of protein kinase A (PKA) and exchange protein directly activated by 3’,5’-cyclic adenosine 5’-monophosphate (cAMP) 2 (EPAC2) in cAMP-mediated actions in adrenocortical cells. Endocrinology. 2010; 151(5):2151-2161.
65. Wik E, Raeder MB, Krakstad C, Trovik J, Birkeland E, Hoivik EA, Mjos S, Werner HM, Mannelqvist M, Stefansson IM, Oyan AM, Kalland KH, Akslen LA, et al. Lack of estrogen receptor-alpha is associated with epithelial-mesenchymal transition and PI3K alterations in endometrial carcinoma. Clin Cancer Res. 2013; 19(5):1094-1105.