
INTRODUCTION
Cancer immunogene therapy is a promising alternative to conventional cancer therapy [1–3]. Cancer immunogene therapy focuses on efficient transfer of immune stimulatory genes such as chemokines, costimulators, and cytokines into tumor cells to induce antitumor immune responses [4–6]. A critical hurdle to successful cancer immunogene therapy is tumor-induced immunosuppression, which attenuates the potency of immunotherapeutics by anergy or tolerance.
Interleukin (IL)-12 is one of the most effective and promising cytokine for cancer immunotherapy. IL-12, which is secreted by activated macrophages and dendritic cells (DCs), facilitates T helper type 1 (Th1) differentiation and enhances the cytotoxicity of natural killer (NK) cells and cytotoxic T lymphocytes (CTLs) [7–9]. Preclinical evaluation of IL-12 shows induction of potent antitumor immunity in murine tumor models [10, 11]. However, clinical trials of recombinant IL-12 cytokine therapy do not yield satisfactory results due to tumor-induced immunosuppression and the transient nature of systemically administered IL-12. The tumors of patients in the clinical trials were composed of heterogeneous tumor cell populations that actively recruited or produced immunosuppressive factors, generating a highly immunosuppressive tumor microenvironment [9, 12–15]. Therefore, a novel strategy is required to improve the therapeutic efficacy of IL-12 in clinical environments. We previously demonstrated that cancer-specific expression and amplification of the IL-12 gene mediated by oncolytic adenovirus (Ad) maintains IL-12 expression at therapeutic doses for a prolonged period in murine melanoma models, resulting in potent antitumor efficacy [16–20].
Transforming growth factor-β (TGF-β) and regulatory T (Treg) cells are major contributors to the formation of immunosuppressive networks in tumor tissues that attenuate the potency of immunotherapeutics. TGF-β suppresses activation, maturation, and differentiation of immune cells such as CTLs, NK cells, and DCs [21]. TGF-β is integral to the maintenance and generation of immunosuppressive Treg cells that inhibit the antitumor immune functions of tumor-specific CD8+ and CD4+ T cells by cell-cell contact and production of immunosuppressive cytokines such as IL-10 or TGF-β [22, 23]. These immunosuppressive attributes are frequently observed in clinical tumors and inhibit the induction of effective antitumor immune responses [24, 25]. Weakly immunogenic tumor models have these immunosuppressive attributes of clinical tumors, making them useful for evaluating anticancer immunotherapeutics.
Decorin (DCN), a prototype member of a small leucine-rich proteoglycan family, is a ubiquitous component of the extracellular matrix (ECM) that regulates diverse functions through interaction with ECM components. DCN suppresses the biological activity of TGF-β by preventing TGF-β binding to its receptor [26–30]. DCN inhibits primary tumor growth and metastasis by decreasing TGF-β-induced immunosuppression [30]. Thus, inhibition of TGF-β expression in combination with immune stimulatory cytokines could induce antitumor immunity by overcoming tumor-mediated immunosuppression.
In this study, we generated an oncolytic Ad co-expressing IL-12 and DCN (RdB/IL12/DCN). The purpose was to suppress TGF-β-mediated immunosuppression in tumor microenvironments for enhanced induction of IL-12-mediated antitumor immune responses. In a weakly immunogenic murine breast cancer model, RdB/IL12/DCN elicited a potent antitumor effect by restoring antitumor immune function in a tumor milieu. Oncolytic Ad-mediated suppression of TGF-β expression in tumor tissues correlated with enhanced induction of antitumor immune responses as evidenced by enhanced infiltration of T cells into tumor tissue treated with RdB/IL12/DCN compared with a cognate control oncolytic Ad expressing a single therapeutic gene (RdB/DCN or RdB/IL12). In addition, we demonstrated that oncolytic Ad-mediated DCN expression enhanced the distribution of oncolytic Ad within tumor tissues, contributing to efficient transgene expression and the antitumor efficacy of RdB/IL12/DCN.
RESULTS
Expression of IL-12 and DCN by RdB/IL12/DCN
To construct an oncolytic Ad co-expressing IL-12 and DCN, IL-12 and DCN genes were placed in the E1 and E3 region of oncolytic Ad, RdB, respectively (Figure 1A). To assess IL-12 expression mediated by the Ad, U343 cells were treated with RdB, RdB/IL12, RdB/DCN, or RdB/IL12/DCN at multiplicity of infection (MOI) 1 or 3. IL-12 secretion was dose-dependently elevated in cancer cells infected with either RdB/IL12 or RdB/IL12/DCN (Figure 1B; P < 0.001). DCN was analyzed by western blot. Cancer cells treated with RdB/DCN or RdB/IL12/DCN efficiently expressed DCN, whereas no expression was observed in the RdB-treated or RdB/IL12-treated groups (Figure 1C). These results demonstrated that IL-12 and DCN were efficiently expressed by RdB/IL12/DCN.
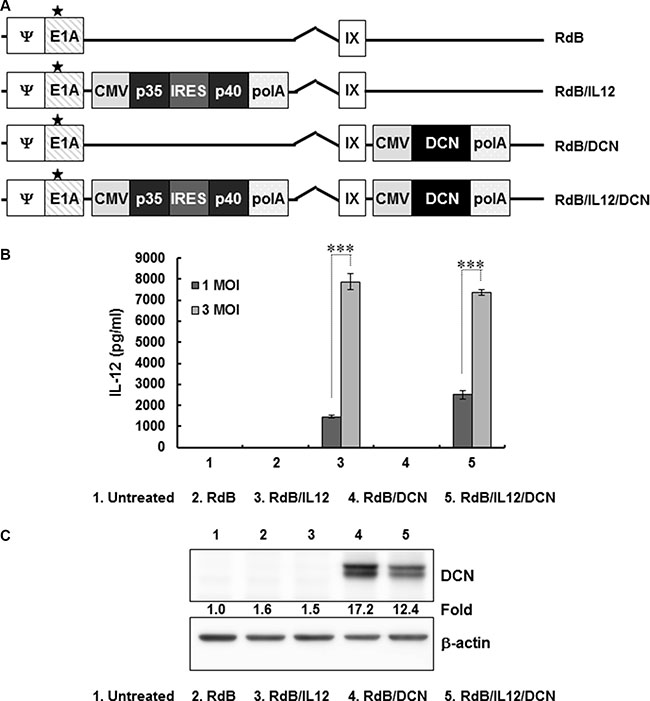
Figure 1: Characterization of oncolytic adenovirus (Ad) vectors expressing interleukin (IL)-12 and/or decorin (DCN) (A) Schematic representation of the genomic structures of oncolytic Ads. RdB has genes for mutated E1A and lacks E1B19 kD, E1B55 kD, and E3 genes. RdB/IL12 and RdB/DCN has genes for IL-12 in the E1 and DCN in the E3 region of RdB. RdB/IL12/DCN has genes for IL-12 in the E1 and DCN in the E3 region of RdB. Asterisk, mutation in the Rb binding site of E1A. (B, C) IL-12 and DCN expression in Ad-permissive U343 cells after infection with RdB, RdB/IL12, RdB/DCN, or RdB/IL12/DCN. Cell supernatants were harvested at 48 hr after infection and IL-12 expression was quantified by ELISA. Representative western blot of DCN using lysates harvested at 48 hr after infection. ELISA data are mean ± SD of triplicate experiments. ***P < 0.001.
Antitumor efficacy of RdB/IL12/DCN
Most types of cancer are weakly immunogenic due to antigen masking or overall immunosuppression [24, 25]. The therapeutic efficacy of IL-12-mediated cancer gene therapy is markedly attenuated in a weakly immunogenic 4T1 tumor model [31, 32]. TGF-β is the most potent immunosuppressive cytokine at inhibiting IL-12-induced antitumor immune responses by interfering with the IL-12 signaling pathway essential to interferon (IFN)-γ production. This effect results in the formation of an immunosuppressive network in tumors [33]. TGF-β inhibits the proliferation, differentiation, and function of macrophages, T cells, B cells, and NK cells [34–37]. Previously, we demonstrated that a DCN-expressing oncolytic Ad suppresses TGF-β expression in keloid fibroblasts [38]. We hypothesized that oncolytic Ad-mediated DCN expression would ameliorate TGF-β-induced immunosuppression in a tumor microenvironment, leading to enhanced induction of the antitumor immune response mediated by IL-12.
To assess the therapeutic potential of RdB/IL12/DCN in a weakly immunogenic 4T1 orthotopic breast cancer model, tumor-bearing mice were injected intratumorally with phosphate-buffered saline (PBS), RdB, RdB/IL12, RdB/DCN, or RdB/IL12/DCN. Tumors treated with PBS grew rapidly and average tumor volume reached 1065.6 ± 71.3 mm3 by day 17 after initial treatment (Figure 2A). Inhibition of tumor growth was 29.3% for treatment with RdB (754.3 ± 59.7 mm3), 50.2% with RdB/IL12 (531.7 ± 85.0 mm3), 37.3% with RdB/DCN (668.8 ± 80.1 mm3), and 78.2% with RdB/IL12/DCN (233.1 ± 36.4 mm3) compared with PBS-treated control tumors (P < 0.05 for RdB, P < 0.01 for RdB/DCN, P < 0.001 for RdB/IL12 and RdB/IL12/DCN versus PBS). Both of the IL-12-expressing oncolytic Ads (RdB/IL12 and RdB/IL12/DCN) showed similar tumor growth inhibition up to day 9 after initial treatment. In RdB/IL12-treated tumors, significant tumor regrowth was observed, however, at later time periods. In contrast, RdB/IL12/DCN-treated tumors continued to show tumor growth inhibition during later time periods, suggesting that oncolytic Ad-mediated expression of DCN functioned as an adjuvant to IL-12 and enhanced the antitumor efficacy of oncolytic Ad (P < 0.05, RdB/IL12/DCN versus RdB/IL12 by 17 days).
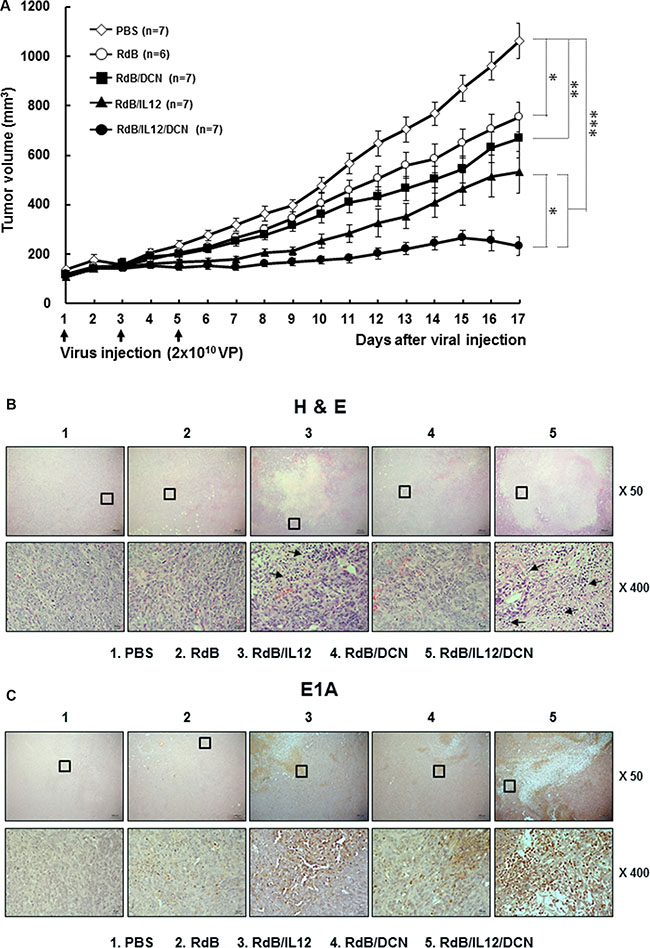
Figure 2: Antitumor effect of oncolytic Ads in an orthotopic breast cancer model. (A) Established orthotopic 4T1 tumors were injected with RdB (open circles), RdB/DCN (filled squares), RdB/IL12 (filled triangles), or RdB/IL12/DCN (filled circles) at 2 × 1010 VP on days 1, 3, and 5, with PBS control (open diamonds). Tumor growth was monitored daily. Arrow, treatment administration. Data are mean ± SD of tumor volume by group (n = 6–7 mice). *P < 0.05, **P < 0.01, or ***P < 0.001. (B and C) Histology and immunohistochemistry of tumor tissues treated with PBS, RdB, RdB/IL12, RdB/DCN, or RdB/IL12/DCN. Tumor tissues were collected from mice at 6 days after final treatment. Paraffin sections were stained with hematoxylin and eosin (H&E) or anti-Ad E1A. Arrows, immune cell infiltration into tumor tissues. Images are representatives of results from three independent experiments. Original magnification: × 50 with × 400 magnification of the boxed area.
To further assess antitumor efficacy mediated by RdB/IL12/DCN, tumor tissues were examined by histology and immunohistochemistry. A reduction was observed in viable tumor cells along with extensive necrotic regions in tumors treated with RdB/IL12 or RdB/IL12/DCN compared with tumors treated with PBS, RdB, or RdB/DCN (Figure 2B). RdB/IL12/DCN-treated tumors showed larger necrotic areas and more immune cell infiltration than RdB/IL12-treated tumors. This result implied that intratumoral expression of DCN facilitated induction of the cytokine-mediated antitumor immune response. In addition, higher expression of E1A, a viral replication marker, was observed in RdB/IL12/DCN-treated sections compared with RdB/IL12-treated sections (Figure 2C). This result suggested that intratumoral expression of DCN enhanced viral distribution and replication in tumor tissues. These results demonstrated that RdB/IL12/DCN induced potent and prolonged tumor growth inhibition by enhancing immune cell infiltration and viral dispersion within the tumor bed.
Upregulation of IL-12, DCN, IFN-γ, TNF-α, and MCP-1 by RdB/IL12/DCN
To evaluate the effect of oncolytic Ad-mediated immune-modulation of gene expression in tumor microenvironments, intratumoral cytokines were measured in tumor tissues. IL-12 levels were not detectable in tumors treated with PBS, RdB, or RdB/DCN (Figure 3A). However, tumors treated with RdB/IL12 or RdB/IL12/DCN showed high IL-12 levels (239.0 ± 3.0 pg/mg and 238.0 ± 2.0 pg/mg, respectively; P < 0.01). In addition, RdB/DCN or RdB/IL12/DCN-treated tumors exhibited significantly higher DCN expression than those treated with either RdB or RdB/IL12 (Figure 3B). These results implied that RdB/IL12/DCN expressed both therapeutic genes at high levels in tumor tissues.
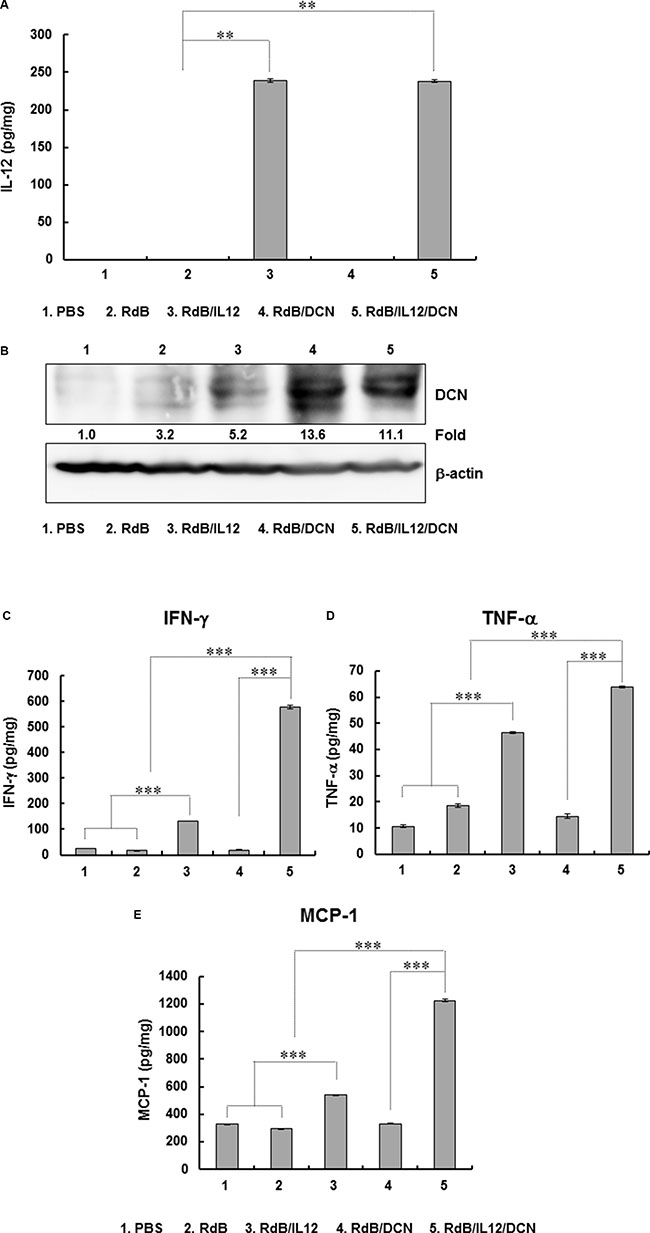
Figure 3: Expression of IL-12, DCN, IFN-γ, TNF-α, and MCP-1 in tumor tissues. Tissues were obtained at 6 days after final viral treatment for quantitation of (A) IL-12, (B) DCN, (C) IFN-γ, (D) TNF-α, and (E) MCP-1. Experiments were in triplicate and repeated three times (A, C, D, E) DCN expression is representative of three independent experiments (B) Data points are mean expression ± SD for IL-12, IFN-γ, TNF-α, and MCP-1 for each tumor. **P < 0.01 or ***P < 0.001.
IL-12 induces a Th1 immune response by promoting secretion of IFN-γ and tumor necrosis factor (TNF)-α from T and NK cells [59–61]. Therefore, we examined the expression of IFN-γ and TNF-α in tumor tissues treated with oncolytic Ads. RdB/IL12/DCN-inoculated tumors showed significantly higher levels of IFN-γ (579.1 ± 6.0 pg/mg) than tumors treated with PBS (23.9 ± 0.1 pg/mg), RdB (16.0 ± 0.2 pg/mg), RdB/DCN (19.4 ± 0.4 pg/mg), or RdB/IL12 (131.1 ± 0.7 pg/mg) (P < 0.001) (Figure 3C and 3D). Likewise, RdB/IL12/DCN-treated tumors exhibited significantly higher TNF-α expression (63.7 ± 0.3 pg/mg) than those treated with PBS (10.6 ± 0.4 pg/mg), RdB (18.6 ± 0.8 pg/mg), RdB/DCN (14.4 ± 0.8 pg/mg), or RdB/IL12 (46.4 ± 0.4 pg/mg) (P < 0.001). Of interest, the expression of both IFN-γ and TNF-α in tumor tissues treated with RdB/IL12/DCN was significantly greater than tumors treated with RdB/IL-12 (P < 0.001), although no significant difference was observed in IL-12 expression between tumors treated with the two oncolytic Ads (Figure 3A). These results suggested that IL-12-induced Th1 immune responses were enhanced by concurrent intratumoral expression of DCN.
Intratumoral expression of IFN-γ or TNF-α induces monocyte chemoattractant protein-1 (MCP-1) production, which enhances T cell infiltration into tumor tissues [39–41]. RdB/IL12/DCN-treated tumors exhibited significantly higher expression of MCP-1 (1224.4 ± 11.1 pg/mg) than tumors treated with PBS (328.0 ± 3.0 pg/mg), RdB (295.5 ± 3.4 pg/mg), RdB/DCN (329.6 ± 2.9 pg/mg), or RdB/IL12 (537.9 ± 3.9 pg/mg) (Figure 3E). A strong positive correlation was seen for expression of MCP-1, IFN-γ, and TNF-α (P < 0.001). These results suggested that intratumoral expression of Th1 cytokines (IFN-γ and TNF-α) and the T cell-recruiting chemokine MCP-1 were significantly enhanced by co-expression of IL-12 and DCN.
Efficient induction of tumor-specific immunity by RdB/IL12/DCN
To assess the tumor-specific immune response mediated by oncolytic Ads, splenocytes were harvested from tumor-bearing mice at 6 days following last treatment and co-cultured with irradiated 4T1 cells. Splenocytes were evaluated for IFN-γ-secreting immune cells by IFN-γ ELISPOT. The number of IFN-γ-secreting immune cells was markedly elevated in RdB/IL12/DCN-treated mice compared with mice treated with PBS, RdB, RdB/IL12 or RdB/DCN (Figure 4A). Furthermore, IFN-γ-producing immune cells were recovered from RdB/IL12/DCN-treated mice significantly more frequently (188.7 ± 28.2) than from mice treated with PBS (11.7 ± 3.5), RdB (94.0 ± 2.6), RdB/IL12 (124.7 ± 17.2), or RdB/DCN (26.3 ± 8.9) (Figure 4B; P < 0.001 versus PBS, P < 0.05 versus RdB and RdB/IL12, P < 0.01 versus RdB/DCN). To further examine tumor-specific immunity driven by oncolytic Ads, IFN-γ and TNF-α were assessed in culture supernatants of splenocytes co-cultured with irradiated 4T1 cells. RdB/IL12/DCN-treated groups exhibited significantly higher IFN-γ and TNF-α than groups treated with PBS, RdB, RdB/IL12, or RdB/DCN (P < 0.001) (Figure 4C and 4D), suggesting that RdB/IL12/DCN induced a potent tumor-specific adaptive immune response.
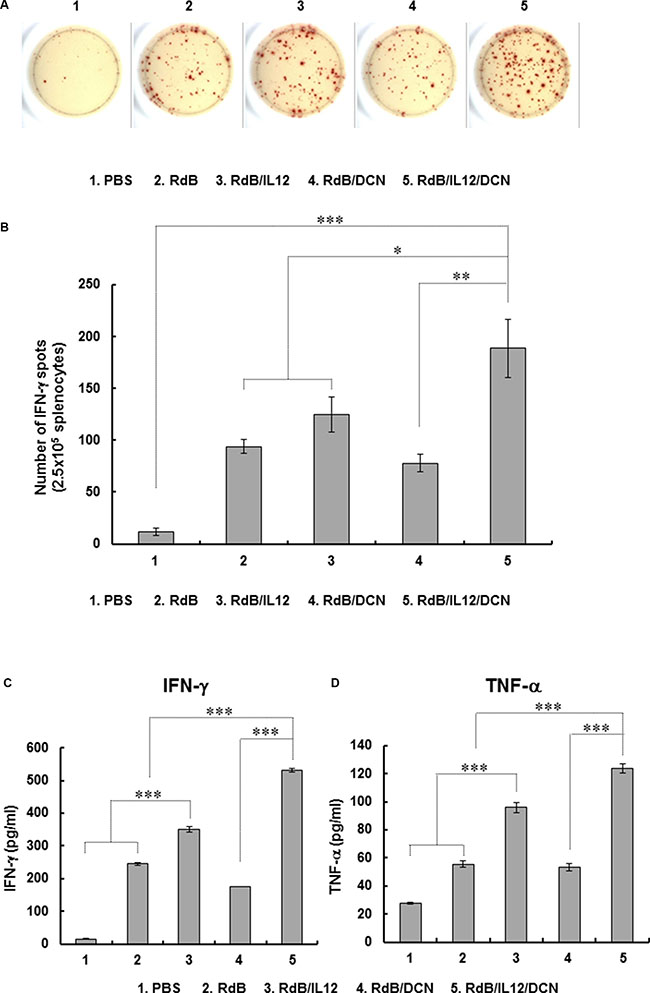
Figure 4: Assessment of tumor-specific immunity. Splenocytes were collected from mice treated with PBS, RdB, RdB/IL12, RdB/DCN, or RdB/IL12/DCN at 6 days after final treatment and co-incubated with pre-irradiated 4T1 cells for 1 day. Assays were IFN-γ ELISPOT. (A) Spot-forming cell response. Images are representatives of results from three independent experiments. (B) Number of spots for 2.5 × 105 splenocytes. Values are mean spot number ± SD for triplicates representative of three independent experiments. *P < 0.05, **P < 0.01, or ***P < 0.001. (C, D) Quantification of IFN-γ and TNF-α from tumor-specific immune cells. Splenocytes were co-cultured with pre-irradiated 4T1 cells for 2 days. IFN-γ and TNF-α were evaluated in co-cultured supernatants by cytometric bead array (CBA) mouse inflammation kits. Data points are mean ± SD of triplicates representative of three independent experiments. ***P < 0.001.
Downregulation of intratumoral TGF-β expression level by RdB/IL12/DCN
To assess the effect of oncolytic Ads on expression of immunosuppressive TGF-β, we first evaluated the effect of green fluorescent protein (GFP) and DCN-expressing replication-incompetent Ad (dE1/GFP/DCN) on TGF-β expression in cancer cells. Treating 4T1 cells with dE1/GFP/DCN resulted in significant, dose-dependent inhibition of TGF-β expression compared to treatment with cognate control replication-incompetent Ad (dE1/GFP) (Figure 5A; P < 0.01). At 200 MOI, dE1/GFP/DCN completely suppressed TGF-β expression, implying that Ad-mediated DCN expression can efficiently inhibit TGF-β production in cancer cells.
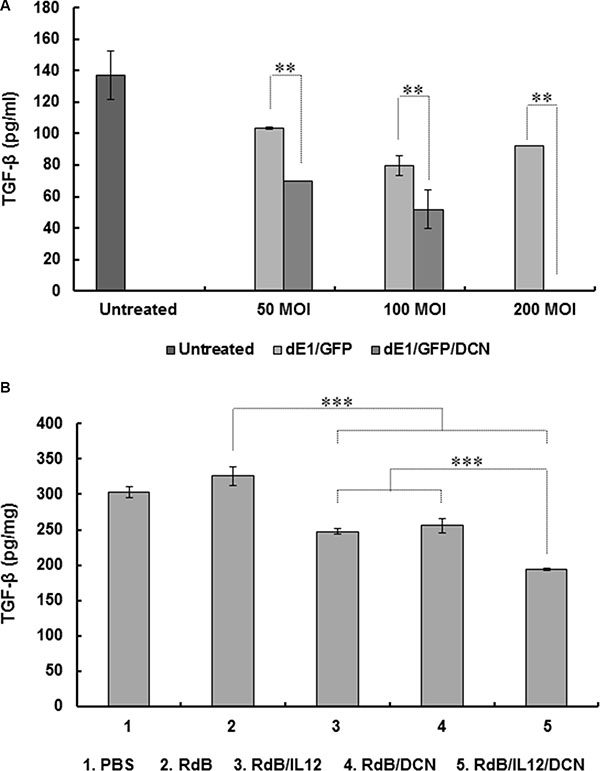
Figure 5: Inhibition of TGF-β expression by DCN-expressing Ad in vitro and in vivo. (A) 4T1 cells were transduced with replication-incompetent dE1/GFP or dE1/GFP/DCN at indicated MOIs and TGF-β expression measured from culture supernatants at 48 hr after transduction using ELISA. Values are mean ± SD of triplicates representative of three independent experiments. **P < 0.01 compared with dE1/GFP. (B) Established 4T1 tumors were treated with PBS, RdB, RdB/IL12, RdB/DCN, or RdB/IL12/DCN, and harvested from mice at 5 days after final treatment. ELISA was performed to estimate TGF-β in tumor tissues. Data points are mean ± SD of triplicates representative of three independent experiments. ***P < 0.001 compared with RdB, RdB/IL12 or RdB/DCN.
To further assess the effect of oncolytic Ad-mediated DCN expression on suppression of TGF-β secretion in tumor microenvironments, expression of TGF-β in tumor tissues was assessed following administration of PBS control or oncolytic Ads. TGF-β expression was significantly attenuated in tumors treated with RdB/IL12, RdB/DCN, or RdB/IL12/DCN compared with tumors treated with RdB (Figure 5B), indicating that either transgene alone efficiently inhibited secretion of immunosuppressive TGF-β (P < 0.001). Importantly, RdB/IL12/DCN-treated tumors showed significantly attenuated TGF-β expression compared to tumors treated with RdB/IL12 or RdB/DCN (P < 0.001), suggesting that both IL-12 and DCN functioned as adjuvants for efficient suppression of TGF-β. These results suggested that co-expression of IL-12 and DCN efficiently downregulated immunosuppressive TGF-β expression in tumor tissue, leading to antitumor efficacy and tumor-specific immunity.
Decreased Treg cells and enhanced accumulation of cytotoxic T cells in RdB/IL12/DCN-treated tumors
CD4+CD25+Foxp3+ Treg cells are key contributors to TGF-β-mediated immunosuppression in tumor microenvironments [42, 43]. To assess the effects of oncolytic Ads on the CD4+CD25+Foxp3+ Treg cell population, we examined draining lymph nodes (DLNs) from tumor-bearing mice by fluorescence-activated cell sorting (FACS). By gating for CD4+ cells and analyzing expression of CD25 and Foxp3, we found that the Treg population was significantly reduced in DLNs from mice treated with RdB/IL12/DCN compared to mice treated with PBS, RdB, RdB/IL12, or RdB/DCN (Figure 6A; P < 0.001 versus PBS and RdB, P < 0.01 versus RdB/IL12 and RdB/DCN).
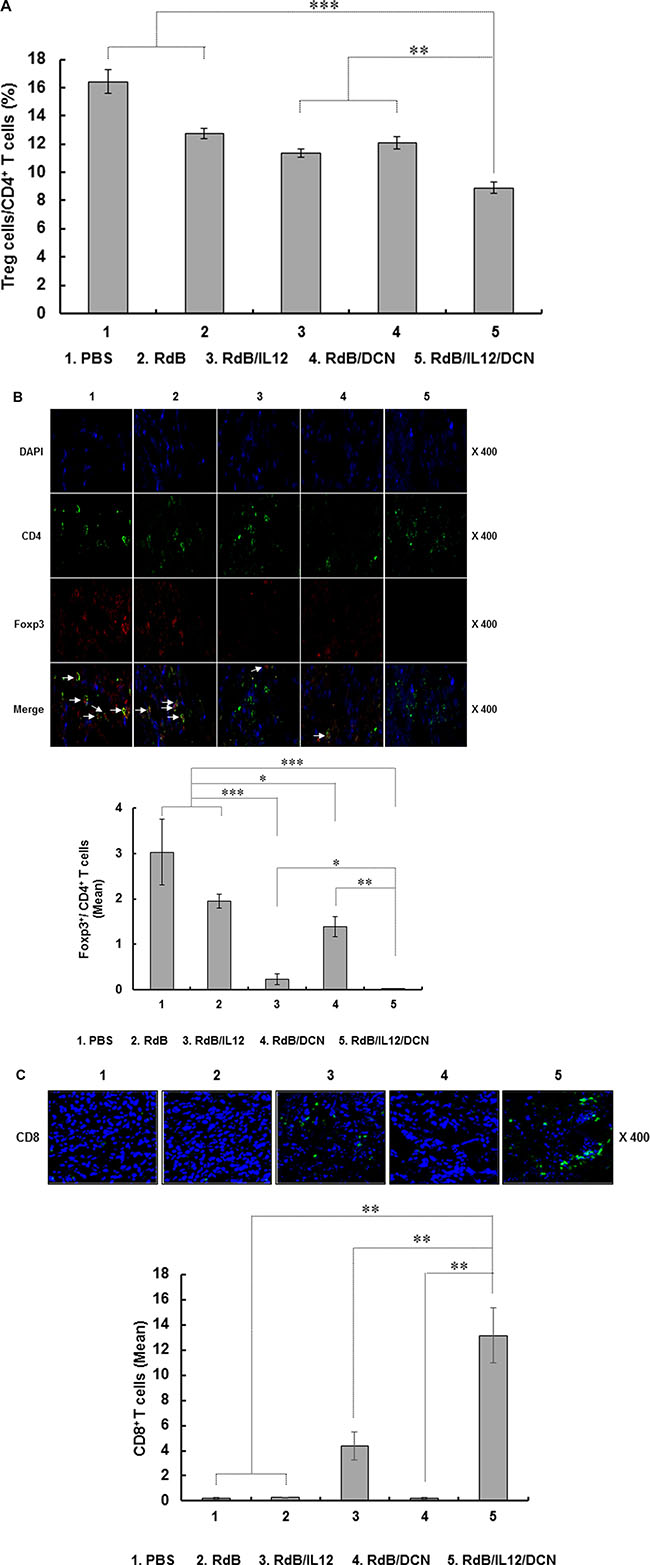
Figure 6: Proportion of Treg and quantification of CD8+ T cells. DLNs and tumor tissues were collected from mice treated with PBS, RdB, RdB/IL12, RdB/DCN, or RdB/IL12/DCN at 6 days after final viral treatment. (A) Population of Treg cells in DLNs from mice were analyzed by flow cytometry. Gating was for CD4+ T cells and analysis for CD25+ and Foxp3+ cells. Data points are mean ± SD of triplicate experiments with at least three mice per group. Similar results were obtained from at least two separate experiments. **P < 0.01, ***P < 0.001. (B) Cryosections of tumor tissues stained with anti-CD4 (green) and anti-Foxp3 (red) monoclonal antibodies. Arrows, T cells co-expressing CD4 and Foxp3 in tumors. Ratio of Foxp3+/CD4+ T cells was assessed by ImageJ software. The relative mean intensity of CD4- or Foxp3-positive cells were quantified from three independent fields within microscope images for each experimental group. Data are representative of three independent experiments. *P < 0.05, **P < 0.01, or ***P < 0.001. (C) Cryosections of tumor tissues were incubated with antibody against CD8 (green). Mean ± SD of CD8+ T cells was quantified by ImageJ software. The mean intensity of three different images was quantified for each sample (CD8+ T cells/field). Images are representative of three independent experiments. Original magnification: × 400. **P < 0.01.
To assess intratumoral infiltration of Treg cells, tumor tissues were examined by immunofluorescence double staining with anti-CD4 and anti-Foxp3 antibodies (Abs). Foxp3+ Treg cells were attenuated following treatment with RdB/IL12 (P < 0.001), RdB/DCN (P < 0.05), or RdB/IL12/DCN (P < 0.001) compared to treatment with PBS or RdB (Figure 6B). More importantly, the ratio of Treg cells in the CD4+ T cell population was lower in tumor tissues treated with RdB/IL12/DCN than in tissues treated with RdB/IL12 (P < 0.05) or RdB/DCN (P < 0.01). This result suggested that accumulation of immunosuppressive Treg cells in tumors was attenuated more by co-expression of IL-12 and DCN than by expressing either gene alone.
Accumulation of Treg cells correlates with reduced infiltration of CD8+ T cells in tumors and poor prognosis for cancer patients [44–46]. To evaluate the density of tumor-infiltrating CD8+ T cells following oncolytic Ad treatments, tumor tissues were assessed by immunohistochemistry using CD8-specific Abs. Markedly higher quantities of CD8+ T cells were found in RdB/IL12/DCN-treated tumors compared with tumors treated with PBS, RdB, RdB/IL12 or RdB/DCN (P < 0.01) (Figure 6C), suggesting that the decreased population of Treg cells correlated with enhanced infiltration of CD8+ T cells into tumor tissues. Collectively, these results demonstrated that an oncolytic Ad co-expressing IL-12 and DCN alleviated TGF-β-mediated immunosuppression and reduced infiltration of immunosuppressive Treg cells in tumor microenvironments, facilitating IL-12-mediated T cell antitumor immunity.
DISCUSSION
Immunogene therapy focuses on regulating the antitumor immune response using immune stimulatory factors such as cytokines and costimulatory factors to activate and recruit immune cells to tumor tissues [47, 48]. In clinical tumor microenvironments, interaction between cancer, immune, and stromal cells contributes to the formation and maintenance of an immunosuppressive network that promotes tumor growth and attenuates the efficacy of immunogene therapy [3, 49, 50]. Therefore, combining immune modulators with factors capable of amending the immunosuppressive tumor microenvironment is critical for inducing an optimal antitumor immune response.
IL-12 induces a potent antitumor immune response by promoting Th1 differentiation of CD4+ T cells and activating CTL- and NK cell-mediated cytotoxicity (51). However, systemic administration of IL-12 induces severe side effects in both preclinical animal models and clinical trials [52–55]. To attenuate IL-12-mediated systemic toxicities, the IL-12 gene has been incorporated into viral [56–58] and nonviral [8, 59, 60] vectors [59, 61–63], resulting in improved safety and therapeutic efficacy. In particular, cytokine-expressing oncolytic Ads demonstrate potent therapeutic outcomes as viral replication amplifies expression of cytokines, tumor cell lysis, and release of tumor-associated antigens in situ [16-20].
Tumor-induced immunosuppression is another critical hurdle of IL-12-mediated cancer immunotherapy in clinical trials. Clinical tumors are often weakly immunogenic or nonimmunogenic due to minimal expression of tumor antigens, downregulated expression of MHC class I molecules and antigen presentation capability, T cell anergy, and immunosuppression; this leads to low immunotherapy efficacy [24, 25]. In tumor-induced immunosuppression, TGF-β overexpression and accumulation of Treg cells in tumor tissues are integral components of tumor immune evasion mechanisms [21, 42, 64]. To overcome immunosuppression in tumor microenvironments mediated by Treg cells and TGF-β, we generated an oncolytic Ad co-expressing IL-12 and DCN. Our aim was to enhance IL-12-mediated induction of the antitumor immune response through DCN-mediated downregulation of TGF-β activity in a weakly immunogenic murine 4T1 orthotopic breast cancer model that mimics clinical tumors.
Earlier reports demonstrated that the core structure of DCN is composed of 10 central leucine-rich repeats that suppress TGF-β activity by preventing receptor binding. DCN also negatively regulates the stability of critical downstream effectors in the insulin-like growth factor receptor I signaling pathway, indirectly inhibiting TGF-β receptor signaling [30, 65–67]. Consistent with these reports, DCN-expressing Ads attenuated TGF-β expression in vitro and in vivo, suggesting that Ad-mediated DCN expression can functionally block TGF-β expression (Figure 5A and 5B). Further, in weakly immunogenic tumors, treatment with oncolytic Ad coexpressing IL-12 and DCN elicited more antitumor effects than treatment with an oncolytic Ad expressing IL-12 or DCN alone (Figure 2A). Of note, RdB/IL12/DCN-treated mice had larger necrotic regions and viral dispersion in tumors than mice treated with RdB/DCN or RdB/IL12 (Figure 2B and 2C). These results are in good agreement with our previous report that DCN-expressing oncolytic Ad induces apoptosis and ECM degradation, leading to enhanced viral dispersion in tumor tissues [26, 68]. RdB/IL12/DCN-treated tumor tissues showed more infiltration of immune cells than tissues treated with RdB/IL12. This result suggested that DCN-mediated degradation of ECM and downregulation of TGF-β activity facilitated infiltration of immune cells into tumors for enhanced induction of an antitumor immune response. Together, these results demonstrated that co-expression of IL-12 and DCN by a single oncolytic Ad vector induced a potent antitumor effects by inducing extensive necrosis, efficient infiltration of immune cells, and proficient viral distribution in tumor tissues.
RdB/IL12/DCN-treated tumors exhibited similar expression of IL-12 as tumors treated with RdB/IL12 (Figure 3A). However, both IFN-γ and TNF-α expression was significantly higher in RdB/IL12/DCN-treated tumor tissues (Figure 3C and 3D). Of note, the expression of TGF-β in RdB/IL12/DCN-treated tumor tissues was significantly lower than in tissues treated with RdB, RdB/IL12 or RdB/DCN, indicating that IL-12 and DCN may function as adjuvant to restore the immune stimulatory function of IL-12 and attenuate immunosuppressive TGF-β expression (Figure 5B). These findings are in good agreement with a previous report demonstrating that IL-12-activated human T cells are inhibited by TGF-β, which interferes with an IL-12-mediated signaling pathway [69].
Increased expression of Th1 cytokines (IFN-γ and TNF-α) and attenuation of TGF-β expression in tumor microenvironments positively correlated with enhanced tumor-specific adaptive immune responses. The evidence was the significantly elevated secretion of IFN-γ or TNF-α by tumor-specific immune cells of RdB/IL12/DCN-treated mice compared with RdB/IL-12-treated mice (Figure 4C and 4D). These observations were supported by the presence of markedly higher IFN-γ-secreting cells in the spleen of mice treated with RdB/IL12/DCN compared with that of RdB/IL12 (Figure 4A and 4B). These findings suggested that RdB/IL12/DCN may create a tumor microenvironment more favorable for activation and generation oftumor-specific immune cells through DCN-mediated amelioration of a immunosuppressive tumor network and IL-12-mediated upregulation of Th1 cytokines.
High expression of TGF-β in a tumor microenvironment suppresses DC differentiation and function, leading to generation of immature myeloid DCs that promote Treg cell proliferation [22, 70]. In addition, TGF-β promotes conversion of CD4+CD25- T cells into Treg cells [71–73]. Therefore, inhibition of TGF-β expression could restore antitumor immune function in a tumor milieu by reduction of the Treg cell population. Consistent with these reports, RdB/IL12/DCN-treated mice exhibited significantly fewer Treg cells in both DLN and tumor tissues compared with mice treated with RdB, RdB/IL12 or RdB/DCN. This finding positively correlated with TGF-β expression (Figure 5B), implying that oncolytic Ad-mediated suppression of TGF-β inhibited the accumulation of immunosuppressive Treg cells in DLN and tumor tissues (Figure 6A and 6B). These results suggested that oncolytic Ad-mediated expression of DCN ameliorated the immunosuppressive tumor microenvironment via downregulation of TGF-β expression and the Treg cell population, leading to potent induction of an antitumor immune response.
High MCP-1 levels, which can be caused by elevated Th1 cytokine expression, promote recruitment and activation of T cells, resulting in tumor regression [40, 41, 74, 75]. In good agreement with these reports, RdB/IL12/DCN-treated tumors, which exhibited the highest MCP-1 levels, showed the highest infiltration of CD8+ T cells (Figures 3E and 6C). An increase in intratumoral MCP-1, IFN-γ, and TNF-α expression was well correlated with more infiltration of CD8+ T cells into tumor tissues treated with oncolytic Ads. These results demonstrated that oncolytic Ad-mediated co-expression of IL-12 and DCN induced an antitumor immune response in a weakly immunogenic tumor by downregulation of immunosuppression, active expression of cytokines, and enhanced influx of immune effector T cells. This effect resulted in an effective IL-12-mediated Th1 antitumor immune response.
Collectively, these results demonstrated that combining IL-12 and DCN is a promising candidate strategy to induce a potent antitumor immune response for treating highly immunosuppressive clinical tumors by overcoming Treg-mediated immunosuppression.
MATERIALS AND METHODS
Cell lines and cultures
U343 (human glioma cell line) and 4T1 (murine breast carcinoma cell line) were from the American Type Culture Collection (ATCC, Manassas, VA, USA). U343 cells were cultured in high-glucose Dulbecco’s Modified Eagle’s Medium (DMEM, Gibco BRL, Grand Island, NY, USA) and 4T1 cells in RPMI-1640 (Gibco BRL) containing 10% fetal bovine serum (FBS, Gibco BRL). All cell lines were cultured at 37°C in a humidified atmosphere 5% CO2 and 95% air.
Mice
Female BALB/c mice, 6- to 8-week-old, were from Charles River Japan, Inc. (Yokohama, Japan) and maintained in a laminar air-flow cabinet with specific pathogen-free conditions. All facilities were approved by the Association for Assessment and Accreditation of Laboratory Animal Care. All animal studies were performed according to the institutionally approved protocols of Hanyang University.
Construction and generation of Ad
Replication-incompetent Ad expressing GFP) and/or DCN (dE1/GFP and dE1/GFP/DCN) was generated as described previously [26] (Supplementary Figure S1). To construct an oncolytic Ad co-expressing IL-12 at the E1 region and DCN at the E3 region, respectively, the pSP72-E3/DCN Ad E3 shuttle vector [26] was linearized and co-transformed with SpeI-digested RdB/IL12 Ad total vector [76] into Escherichia coli BJ5183, generating an RdB/IL12/DCN Ad total plasmid. Ads used in this study were propagated in 293 and purified by CsCl gradient centrifugation as described previously [77]. Ads were stored at –80°C until use. Numbers of viral particles (VPs) were calculated from optical density measurements at 260 nm (OD260), where absorbance 1 (OD260 = 1) was equivalent to 1.1 × 1012 VP/ml.
Quantification of IL-12 and DCN expression
U343 cells were plated onto 6-well plates at 1 × 105 cells/well, and infected with RdB, RdB/IL12, RdB/DCN, or RdB/IL12/DCN at MOI 1 or 3. At 48 hr after infection, supernatants were obtained. IL-12 was determined by ELISA according to manufacturer’s instructions (IL-12 ELISA kit: Endogen, Woburn, MA, USA). DCN was determined by western blot analysis as previously described [76]. In brief, at 48 hr after infection of U343 cells with RdB, RdB/IL12, RdB/DCN, or RdB/IL12/DCN at MOI 3, proteins from cell extracts were separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to PVDF membranes (RPN 303F, Amersham, Arlington Heights, IL). Membranes were incubated with primary anti-DCN Ab (R&D Systems, Minneapolis, MN) or anti-β-actin Ab (Cell Signaling Technology, Beverly, MA), then horseradish peroxidase-conjugated secondary Ab (Cell Signaling Technology).
For evaluating IL-12 and DCN expression in tumor tissues, tissues were collected from mice treated with Ad at 6 days after final viral treatment. Tumor tissues were homogenized (ART-MICCRA D-8; ART modern Labortechnik, Munchen, Germany) in ice-cold RIPA buffer (Elipis Biotech, Taejeon, South Korea) with a proteinase inhibitor cocktail (Sigma-Aldrich). Homogenates were centrifuged in a high-speed microcentrifuge for 10 min and total protein was determined using BCA protein assay reagent kits (Pierce, Rockford, IL). Levels of IL-12 in the tumor tissue extract and serum were measured by ELISA (Supplementary Figure S3). DCN expression in the tumor tissue extract was assessed by western blot analysis. DCN expression was semiquantitatively analyzed using ImageJ software (version 1.50b; U.S. National Institutes of Health, Bethesda, MD).
Quantification of TGF-β expression
For transduction, 4T1 cells were plated onto 6-well plates at 1 × 105 cells, then transduced with dE1/GFP or dE1/GFP/DCN at MOI 50, 100, or 200. At 48 hr after transduction, supernatants were collected. To remove endogenously produced TGF-β, medium was replaced with serum-free RPMI 1640 media 24 hr before harvest time points. TGF-β expression was determined by ELISA according to the manufacturer’s protocol for TGF-β ELISA kits (R&D Systems). For assessing TGF-β expression in a tumor milieu, tumor tissues were collected from mice treated with PBS, RdB, RdB/IL12, RdB/DCN, or RdB/IL12/DCN at 5 days after final treatment. Total protein was prepared as described above and TGF-β was measured by ELISA. ELISA results were normalized to total protein concentration in each group and calculated as picograms per milligrams of total.
Antitumor effects of oncolytic Ad co-expressing IL-12 and DCN
To compare antitumor effects of RdB, RdB/IL12, RdB/DCN, or RdB/IL12/DCN, an orthotopic breast cancer model was established by subcutaneously injecting 4T1 cells (1 × 106) into fat pads of 6- to 7-week-old female BABL/c mice. When average tumor volume reached 110–120 mm3, mice were randomly sorted into groups (PBS, RdB, RdB/IL12, RdB/DCN, and RdB/IL12/DCN) with similar mean tumor volumes and treatment was initiated. The first treatment day was designated as day 1. Ad or PBS was administrated intratumorally (2 × 1010 VP per tumor in 20 μL PBS with 1% DMSO) on days 1, 3, and 5. Tumor growth was monitored daily by electronic caliper measure as volume = 0.523 LW2 (L = length of tumor, W = width).
Quantification of MCP-1, IFN-γ, and TNF-α expression
Tumor tissues were harvested from mice treated with oncolytic Ad at 6 days after last viral treatment. MCP-1, IFN-γ, and TNF-α in tumor tissue extracts or serum (Supplementary Figure S3) were measured by cytometric bead array (CBA) mouse inflammation kits (BD Biosciences). Results were normalized to total protein concentration per tumor and calculated as picograms per milligram of total protein.
To measure IFN-γ and TNF-α released by tumor-specific immune cells, spleens were obtained aseptically from tumor-bearing mice at 6 days following last viral treatment. Unicellular splenocytes were prepared as previously described [19]. Prepared splenocytes (1.5 × 106) were co-cultured with irradiated 4T1 (1.5 × 105 cells; 5 Gy) for 2 days in the presence of recombinant mouse IL-2 (100 units/mL, R&D Systems). Culture supernatants were harvested and analyzed using CBA mouse inflammation kits (BD Biosciences).
IFN-γ ELISPOT assay in splenocytes
To assess tumor-specific IFN-γ-producing immune cell populations, IFN-γ ELISPOT assays were performed. Six days following last Ad injection, spleens were harvested aseptically from mice, and unicellular splenocytes prepared as previously described [19]. Prepared spleen cells (2.5 × 105) were co-cultured with pre-irradiated 4T1 (5.0 × 103 cells; 5 Gy) for 24 hr in the presence of recombinant mouse IL-2 (100 units/mL). IFN-γ ELISPOT assays were performed according to the manufacturer’s specifications for IFN-γ ELISPOT kits (BD Biosciences). Colored spots were quantitatively analyzed by a computer-based immunospot system (AID Elispot Reader System, Version 3.4, Autoimmun Diagnostika GmbH, Strassberg, Germany).
Fluorescence-activated cell sorting analysis
For the assessment Treg cell populations by FACS, DLNs were harvested at 6 days following final viral treatment of 4T1 tumor-bearing mice. Cells were pretreated with saturating anti-CD16/CD32 Ab (Biolegend, San Diego, CA) in staining buffer (2% FBS, 0.02% sodium azide in PBS) to block cellular Fc receptors. Cells were stained extracellularly with peridinin chlorophyll protein-CY5.5-conjugated anti-CD4 Ab (BD Biosciences) and phycoerythrin-conjugated anti-CD25 Ab (eBioscience, San Diego, CA). Cells were then permeabilized with Foxp3 fixation/permeabilization buffer (eBioscience) according to the supplier’s protocol and stained with allophycocyanin-conjugated anti-Foxp3 Ab (eBioscience). Samples were analyzed using a BD Biosciences BD FACScanto II flow cytometry analyzer and FACSDiva software (BD Biosciences).
Histological and immunohistochemical analysis
Tumor tissues were harvested from mice at 6 days after final treatment and fixed in 10% formalin, processed for paraffin embedding, and cut into 5-mm sections. Sections were stained with hematoxylin and eosin (H&E) and examined by light microscopy. Tumor sections were also immunostained with rabbit anti-Ad E1A polyclonal Ab (Santa Cruz Biotechnology) to assess viral replication and spreading. Slides were counterstained with Meyer’s hematoxylin. To detect lymphocytes, tumor tissues were dehydrated with 30% sucrose at 4°C, frozen in OCT compound (Sakura Finetec, Torrance, CA), and cut into 10-mm sections. Cryosections were incubated with rat anti-mouse CD4 monoclonal Ab (BD Biosciences), rat anti-mouse CD8 monoclonal Ab, or rabbit anti-Foxp3 monoclonal Ab (Abcam, Cambridge, UK). After incubating with primary Ab at 4°C overnight, sections were incubated with Alexa Fluor 488-conjugated anti-rat IgG (Invitrogen, Carlsbad, CA, USA) or Alexa Fluor 568-conjugated anti-rabbit IgG as secondary Abs for 1 hr. For counterstaining, samples were incubated with 4,6-diamidino-2-phenylindole (DAPI, Sigma-Aldrich) and semiquantitatively analyzed by ImageJ software.
Statistical analysis
Data were expressed as mean ± standard deviation (SD). Statistical significance was determined by two-tailed Student t-test (SPSS 13.0 software; SPSS, Chicago, IL). *P < 0.05, **P < 0.01 and ***P < 0.001 were considered statistically significant.
CONFLICTS OF INTEREST
No potential conflicts of interest were disclosed.
GRANT SUPPORT
This work was supported by grants from the National Research Foundation of Korea (2015R1A2A1A13027811, 2013M3A9D3045879, and 2016M3A9B5942352).
REFERENCES
1. Kruger C, Greten TF, Korangy F. Immune based therapies in cancer. Histol Histopathol. 2007; 22:687–696.
2. Nagai H, Hara I, Horikawa T, Oka M, Kamidono S, Ichihashi M. Elimination of CD4(+) T cells enhances anti-tumor effect of locally secreted interleukin-12 on B16 mouse melanoma and induces vitiligo-like coat color alteration. J Invest Dermatol. 2000; 115:1059–1064.
3. Croci DO, Zacarias Fluck MF, Rico MJ, Matar P, Rabinovich GA, Scharovsky OG. Dynamic cross-talk between tumor and immune cells in orchestrating the immunosuppressive network at the tumor microenvironment. Cancer Immunol Immunother. 2007; 56:1687–1700.
4. Bubenik J. Genetically modified tumour vaccines carrying inserted genes for immunoregulatory molecules. Folia Biol (Praha). 1996; 42:295–304.
5. Colombo MP, Rodolfo M. Tumor cells engineered to produce cytokines or cofactors as cellular vaccines: do animal studies really support clinical trials? Cancer Immunol Immunother. 1995; 41:265–270.
6. Pardoll DM. New strategies for enhancing the immunogenicity of tumors. Curr Opin Immunol. 1993; 5:719–725.
7. Del Vecchio M, Bajetta E, Canova S, Lotze MT, Wesa A, Parmiani G, Anichini A. Interleukin-12: biological properties and clinical application. Clin Cancer Res. 2007; 13:4677–4685.
8. Fewell JG, Matar MM, Rice JS, Brunhoeber E, Slobodkin G, Pence C, Worker M, Lewis DH, Anwer K. Treatment of disseminated ovarian cancer using nonviral interleukin-12 gene therapy delivered intraperitoneally. J Gene Med. 2009; 11:718–728.
9. Wang Y, Fan KT, Li JM, Waller EK. The regulation and activity of interleukin-12. Front Biosci (Schol Ed). 2012; 4:888–899.
10. Colombo MP, Trinchieri G. Interleukin-12 in anti-tumor immunity and immunotherapy. Cytokine Growth Factor Rev. 2002; 13:155–168.
11. Wigginton JM, Wiltrout RH. IL-12/IL-2 combination cytokine therapy for solid tumours: translation from bench to bedside. Expert Opin Biol Ther. 2002; 2:513–524.
12. Weiss JM, Back TC, Scarzello AJ, Subleski JJ, Hall VL, Stauffer JK, Chen X, Micic D, Alderson K, Murphy WJ, Wiltrout RH. Successful immunotherapy with IL-2/anti-CD40 induces the chemokine-mediated mitigation of an immunosuppressive tumor microenvironment. Proc Natl Acad Sci USA. 2009; 106:19455–19460.
13. Rabinovich GA, Gabrilovich D, Sotomayor EM. Immunosuppressive strategies that are mediated by tumor cells. Annu Rev Immunol. 2007; 25:267–296.
14. Zwirner NW, Croci DO, Domaica CI, Rabinovich GA. Overcoming the hurdles of tumor immunity by targeting regulatory pathways in innate and adaptive immune cells. Curr Pharm Des. 2010; 16:255–267.
15. Malvicini M, Ingolotti M, Piccioni F, Garcia M, Bayo J, Atorrasagasti C, Alaniz L, Aquino JB, Espinoza JA, Gidekel M, Scharovsky OG, Matar P, Mazzolini G. Reversal of gastrointestinal carcinoma-induced immunosuppression and induction of antitumoural immunity by a combination of cyclophosphamide and gene transfer of IL-12. Mol Oncol. 2011; 5:242–255.
16. Zhang SN, Choi IK, Huang JH, Yoo JY, Choi KJ, Yun CO. Optimizing DC vaccination by combination with oncolytic adenovirus coexpressing IL-12 and GM-CSF. Mol Ther. 2011; 19:1558–1568.
17. Huang JH, Zhang SN, Choi KJ, Choi IK, Kim JH, Lee MG, Kim H, Yun CO. Therapeutic and tumor-specific immunity induced by combination of dendritic cells and oncolytic adenovirus expressing IL-12 and 4-1BBL. Mol Ther. 2010; 18:264–274.
18. Choi KJ, Zhang SN, Choi IK, Kim JS, Yun CO. Strengthening of antitumor immune memory and prevention of thymic atrophy mediated by adenovirus expressing IL-12 and GM-CSF. Gene Ther. 2012; 19:711–723.
19. Choi IK, Lee JS, Zhang SN, Park J, Sonn CH, Lee KM, Yun CO. Oncolytic adenovirus co-expressing IL-12 and IL-18 improves tumor-specific immunity via differentiation of T cells expressing IL-12Rbeta2 or IL-18Ralpha. Gene Ther. 2011; 18:898–909.
20. Choi IK, Li Y, Oh E, Kim J, Yun CO. Oncolytic adenovirus expressing IL-23 and p35 elicits IFN-gamma- and TNF-alpha-co-producing T cell-mediated antitumor immunity. PLoS One. 2013; 8:e67512.
21. Pinkas J, Teicher BA. TGF-beta in cancer and as a therapeutic target. Biochem Pharmacol. 2006; 72:523–529.
22. Zou W. Regulatory T cells, tumour immunity and immunotherapy. Nat Rev Immunol. 2006; 6:295–307.
23. Curiel TJ. Tregs and rethinking cancer immunotherapy. J Clin Invest. 2007; 117:1167–1174.
24. Foss FM. Immunologic mechanisms of antitumor activity. Semin Oncol. 2002; 29:5–11.
25. Whelan MC, Casey G, MacConmara M, Lederer JA, Soden D, Collins JK, Tangney M, O'Sullivan GC. Effective immunotherapy of weakly immunogenic solid tumours using a combined immunogene therapy and regulatory T-cell inactivation. Cancer Gene Ther. 2010; 17:501–511.
26. Choi IK, Lee YS, Yoo JY, Yoon AR, Kim H, Kim DS, Seidler DG, Kim JH, Yun CO. Effect of decorin on overcoming the extracellular matrix barrier for oncolytic virotherapy. Gene Ther. 2010; 17:190–201.
27. Faust SM, Lu G, Wood SC, Bishop DK. TGFbeta neutralization within cardiac allografts by decorin gene transfer attenuates chronic rejection. J Immunol. 2009; 183:7307–7313.
28. Shintani K, Matsumine A, Kusuzaki K, Morikawa J, Matsubara T, Wakabayashi T, Araki K, Satonaka H, Wakabayashi H, Iino T, Uchida A. Decorin suppresses lung metastases of murine osteosarcoma. Oncol Rep. 2008; 19:1533–1539.
29. Nili N, Cheema AN, Giordano FJ, Barolet AW, Babaei S, Hickey R, Eskandarian MR, Smeets M, Butany J, Pasterkamp G, Strauss BH. Decorin inhibition of PDGF-stimulated vascular smooth muscle cell function: potential mechanism for inhibition of intimal hyperplasia after balloon angioplasty. Am J Pathol. 2003; 163:869–878.
30. Goldoni S, Iozzo RV. Tumor microenvironment: Modulation by decorin and related molecules harboring leucine-rich tandem motifs. Int J Cancer. 2008; 123:2473–2479.
31. Rakhmilevich AL, Janssen K, Hao Z, Sondel PM, Yang NS. Interleukin-12 gene therapy of a weakly immunogenic mouse mammary carcinoma results in reduction of spontaneous lung metastases via a T-cell-independent mechanism. Cancer Gene Ther. 2000; 7:826–838.
32. Rakhmilevich AL, Hooper AT, Hicklin DJ, Sondel PM. Treatment of experimental breast cancer using interleukin-12 gene therapy combined with anti-vascular endothelial growth factor receptor-2 antibody. Mol Cancer Ther. 2004; 3:969–976.
33. Pardoux C, Ma X, Gobert S, Pellegrini S, Mayeux P, Gay F, Trinchieri G, Chouaib S. Downregulation of interleukin-12 (IL-12) responsiveness in human T cells by transforming growth factor-beta: relationship with IL-12 signaling. Blood. 1999; 93:1448–1455.
34. Ranges GE, Figari IS, Espevik T, Palladino MA, Jr. Inhibition of cytotoxic T cell development by transforming growth factor beta and reversal by recombinant tumor necrosis factor alpha. J Exp Med. 1987; 166:991–998.
35. Kehrl JH, Wakefield LM, Roberts AB, Jakowlew S, Alvarez-Mon M, Derynck R, Sporn MB, Fauci AS. Pillars Article: production of transforming growth factor beta by human T lymphocytes and its potential role in the regulation of T cell growth. J Exp Med. 1986. 163:1037–1050. J Immunol. 2014; 192:2939–2952.
36. Ruegemer JJ, Ho SN, Augustine JA, Schlager JW, Bell MP, McKean DJ, Abraham RT. Regulatory effects of transforming growth factor-beta on IL-2- and IL-4-dependent T cell-cycle progression. J Immunol. 1990; 144:1767–1776.
37. Su HC, Leite-Morris KA, Braun L, Biron CA. A role for transforming growth factor-beta 1 in regulating natural killer cell and T lymphocyte proliferative responses during acute infection with lymphocytic choriomeningitis virus. J Immunol. 1991; 147:2717–2727.
38. Lee WJ, Ahn HM, Roh H, Na Y, Choi IK, Lee JH, Kim YO, Lew DH, Yun CO. Decorin-expressing adenovirus decreases collagen synthesis and upregulates MMP expression in keloid fibroblasts and keloid spheroids. Exp Dermatol. 2015; 24:591–597.
39. Wang H, Nemoto-Sasaki Y, Kondo T, Akiyama M, Mukaida N. Potential involvement of monocyte chemoattractant protein (MCP)-1/CCL2 in IL-4-mediated tumor immunity through inducing dendritic cell migration into the draining lymph nodes. Int Immunopharmacol. 2003; 3:627–642.
40. Zhang T, Somasundaram R, Berencsi K, Caputo L, Gimotty P, Rani P, Guerry D, Swoboda R, Herlyn D. Migration of cytotoxic T lymphocytes toward melanoma cells in three-dimensional organotypic culture is dependent on CCL2 and CCR4. Eur J Immunol. 2006; 36:457–467.
41. Franciszkiewicz K, Boissonnas A, Boutet M, Combadiere C, Mami-Chouaib F. Role of chemokines and chemokine receptors in shaping the effector phase of the antitumor immune response. Cancer Res. 2012; 72:6325–6332.
42. Wang Y, Ma Y, Fang Y, Wu S, Liu L, Fu D, Shen X. Regulatory T cell: a protection for tumour cells. J Cell Mol Med. 2012; 16:425–436.
43. Whiteside TL. What are regulatory T cells (Treg) regulating in cancer and why? Semin Cancer Biol. 2012; 22:327–334.
44. Gao Q, Qiu SJ, Fan J, Zhou J, Wang XY, Xiao YS, Xu Y, Li YW, Tang ZY. Intratumoral balance of regulatory and cytotoxic T cells is associated with prognosis of hepatocellular carcinoma after resection. J Clin Oncol. 2007; 25:2586–2593.
45. Fu J, Xu D, Liu Z, Shi M, Zhao P, Fu B, Zhang Z, Yang H, Zhang H, Zhou C, Yao J, Jin L, Wang H, et al. Increased regulatory T cells correlate with CD8 T-cell impairment and poor survival in hepatocellular carcinoma patients. Gastroenterology. 2007; 132:2328–2339.
46. Suzuki H, Chikazawa N, Tasaka T, Wada J, Yamasaki A, Kitaura Y, Sozaki M, Tanaka M, Onishi H, Morisaki T, Katano M. Intratumoral CD8(+) T/FOXP3 (+) cell ratio is a predictive marker for survival in patients with colorectal cancer. Cancer Immunol Immunother. 2010; 59:653–661.
47. Dranoff G. Cytokines in cancer pathogenesis and cancer therapy. Nat Rev Cancer. 2004; 4:11–22.
48. Podhajcer OL, Lopez MV, Mazzolini G. Cytokine gene transfer for cancer therapy. Cytokine Growth Factor Rev. 2007; 18:183–194.
49. Zou W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat Rev Cancer. 2005; 5:263–274.
50. Yaguchi T, Sumimoto H, Kudo-Saito C, Tsukamoto N, Ueda R, Iwata-Kajihara T, Nishio H, Kawamura N, Kawakami Y. The mechanisms of cancer immunoescape and development of overcoming strategies. Int J Hematol. 2011; 93:294–300.
51. Weiss JM, Subleski JJ, Wigginton JM, Wiltrout RH. Immunotherapy of cancer by IL-12-based cytokine combinations. Expert Opin Biol Ther. 2007; 7:1705–1721.
52. Orange JS, Salazar-Mather TP, Opal SM, Spencer RL, Miller AH, McEwen BS, Biron CA. Mechanism of interleukin 12-mediated toxicities during experimental viral infections: role of tumor necrosis factor and glucocorticoids. J Exp Med. 1995; 181:901–914.
53. Lotze MT, Zitvogel L, Campbell R, Robbins PD, Elder E, Haluszczak C, Martin D, Whiteside TL, Storkus WJ, Tahara H. Cytokine gene therapy of cancer using interleukin-12: murine and clinical trials. Ann N Y Acad Sci. 1996; 795:440–454.
54. Cohen J. IL-12 deaths: explanation and a puzzle. Science. 1995; 270:908.
55. Tugues S, Burkhard SH, Ohs I, Vrohlings M, Nussbaum K, Vom Berg J, Kulig P, Becher B. New insights into IL-12-mediated tumor suppression. Cell Death Differ. 2015; 22:237–246.
56. Quetglas JI, Ruiz-Guillen M, Aranda A, Casales E, Bezunartea J, Smerdou C. Alphavirus vectors for cancer therapy. Virus Res. 2010; 153:179–196.
57. Tahara H, Zitvogel L, Storkus WJ, Zeh HJ, 3rd, McKinney TG, Schreiber RD, Gubler U, Robbins PD, Lotze MT. Effective eradication of established murine tumors with IL-12 gene therapy using a polycistronic retroviral vector. J Immunol. 1995; 154:6466–6474.
58. Quetglas JI, Rodriguez-Madoz JR, Bezunartea J, Ruiz-Guillen M, Casales E, Medina-Echeverz J, Prieto J, Berraondo P, Hervas-Stubbs S, Smerdou C. Eradication of liver-implanted tumors by Semliki Forest virus expressing IL-12 requires efficient long-term immune responses. J Immunol. 2013; 190:2994–3004.
59. Lucas ML, Heller L, Coppola D, Heller R. IL-12 plasmid delivery by in vivo electroporation for the successful treatment of established subcutaneous B16.F10 melanoma. Mol Ther. 2002; 5:668–675.
60. Mendiratta SK, Quezada A, Matar M, Wang J, Hebel HL, Long S, Nordstrom JL, Pericle F. Intratumoral delivery of IL-12 gene by polyvinyl polymeric vector system to murine renal and colon carcinoma results in potent antitumor immunity. Gene Ther. 1999; 6:833–839.
61. Heinzerling LM, Feige K, Rieder S, Akens MK, Dummer R, Stranzinger G, Moelling K. Tumor regression induced by intratumoral injection of DNA coding for human interleukin 12 into melanoma metastases in gray horses. J Mol Med (Berl). 2001; 78:692–702.
62. Caruso M, Pham-Nguyen K, Kwong YL, Xu B, Kosai KI, Finegold M, Woo SL, Chen SH. Adenovirus-mediated interleukin-12 gene therapy for metastatic colon carcinoma. Proc Natl Acad Sci USA. 1996; 93:11302–11306.
63. Nasu Y, Bangma CH, Hull GW, Lee HM, Hu J, Wang J, McCurdy MA, Shimura S, Yang G, Timme TL, Thompson TC. Adenovirus-mediated interleukin-12 gene therapy for prostate cancer: suppression of orthotopic tumor growth and pre-established lung metastases in an orthotopic model. Gene Ther. 1999; 6:338–349.
64. Flavell RA, Sanjabi S, Wrzesinski SH, Licona-Limon P. The polarization of immune cells in the tumour environment by TGFbeta. Nat Rev Immunol. 2010; 10:554–567.
65. Sofeu Feugaing DD, Gotte M, Viola M. More than matrix: the multifaceted role of decorin in cancer. Eur J Cell Biol. 2013; 92:1–11.
66. Hildebrand A, Romaris M, Rasmussen LM, Heinegard D, Twardzik DR, Border WA, Ruoslahti E. Interaction of the small interstitial proteoglycans biglycan, decorin and fibromodulin with transforming growth factor beta. Biochem J. 1994; 302:527–534.
67. Iozzo RV, Buraschi S, Genua M, Xu SQ, Solomides CC, Peiper SC, Gomella LG, Owens RC, Morrione A. Decorin antagonizes IGF receptor I (IGF-IR) function by interfering with IGF-IR activity and attenuating downstream signaling. J Biol Chem. 2011; 286:34712–34721.
68. Na Y, Choi JW, Kasala D, Hong J, Oh E, Li Y, Jung SJ, Kim SW, Yun CO. Potent antitumor effect of neurotensin receptor-targeted oncolytic adenovirus co-expressing decorin and Wnt antagonist in an orthotopic pancreatic tumor model. J Control Release. 2015; 220:766–782.
69. Ahn HJ, Maruo S, Tomura M, Mu J, Hamaoka T, Nakanishi K, Clark S, Kurimoto M, Okamura H, Fujiwara H. A mechanism underlying synergy between IL-12 and IFN-gamma-inducing factor in enhanced production of IFN-gamma. J Immunol. 1997; 159:2125–2131.
70. Ghiringhelli F, Puig PE, Roux S, Parcellier A, Schmitt E, Solary E, Kroemer G, Martin F, Chauffert B, Zitvogel L. Tumor cells convert immature myeloid dendritic cells into TGF-beta-secreting cells inducing CD4+CD25+ regulatory T cell proliferation. J Exp Med. 2005; 202:919–929.
71. Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, McGrady G, Wahl SM. Conversion of peripheral CD4+CD25– naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med. 2003; 198:1875–1886.
72. Fantini MC, Becker C, Monteleone G, Pallone F, Galle PR, Neurath MF. Cutting edge: TGF-beta induces a regulatory phenotype in CD4+CD25- T cells through Foxp3 induction and down-regulation of Smad7. J Immunol. 2004; 172:5149–5153.
73. Wan YY, Flavell RA. Identifying Foxp3-expressing suppressor T cells with a bicistronic reporter. Proc Natl Acad Sci USA. 2005; 102:5126–5131.
74. Tsuchiyama T, Nakamoto Y, Sakai Y, Mukaida N, Kaneko S. Optimal amount of monocyte chemoattractant protein-1 enhances antitumor effects of suicide gene therapy against hepatocellular carcinoma by M1 macrophage activation. Cancer Sci. 2008; 99:2075–2082.
75. Grosso JF, Herbert LM, Owen JL, Lopez DM. MUC1/sec-expressing tumors are rejected in vivo by a T cell-dependent mechanism and secrete high levels of CCL2. J Immunol. 2004; 173:1721–1730.
76. Lee YS, Kim JH, Choi KJ, Choi IK, Kim H, Cho S, Cho BC, Yun CO. Enhanced antitumor effect of oncolytic adenovirus expressing interleukin-12 and B7-1 in an immunocompetent murine model. Clin Cancer Res. 2006; 12:5859–5868.
77. Kim J, Kim JH, Choi KJ, Kim PH, Yun CO. E1A- and E1B-Double mutant replicating adenovirus elicits enhanced oncolytic and antitumor effects. Hum Gene Ther. 2007; 18:773–786.
78. Kim J, Kim PH, Nam HY, Lee JS, Yun CO, Kim SW. Linearized oncolytic adenoviral plasmid DNA delivered by bioreducible polymers. J Control Release. 2012; 158:451–460.