

INTRODUCTION
Prostate cancer is the most frequently diagnosed solid malignancy in American men with an estimated 220,800 new cases and 27,540 deaths in 2015 [1]. More than 90% of prostate cancer-related mortality results from widespread metastatic cancer, which initially respond to androgen-deprivation therapy, but subsequently become androgen-independent [2, 3].
In contrast to other cancer types, in vitro cultures of human prostatic cells have been limited in availability and scope. Three frequently used spontaneously established cell lines, PC-3, DU145 and LNCaP, all derived from metastases, do not span the range of prostate cancer phenotypes and are not representative of primary adenocarcinomas of the prostate [4]. Patient-derived xenograft (PDX) models are often easier to establish from aggressive, high-grade and metastatic tumors as compared to primary tumors that are slow growing and likely non-metastatic [5–7]. Development of a PDX model can take anywhere from 2 to 12 months with engraftment rates typically from 2% to 50% depending on the tumor type. This limits the ability to use such cancer cell lines and PDXs for predicting responses to drug-, radiation-, or immuno-therapies. Progress in the field has been hindered by the absence of appropriate models of human-derived prostate cancer cells, precluding investigation of transforming alterations and development of treatment approaches. For this reason, primary cultures of malignant prostatic cells and normal, preferably donor-matched, epithelial counterparts grown under identical conditions are needed.
Over the past 20 years, many of the technical hurdles involved in growing primary cultures of human prostatic epithelial cells have been overcome, and a variety of methods have been reported for epithelial cell cultures from radical prostatectomy specimens [4]. However, a lingering question relates to the types of cells grown from prostatectomy specimens and whether they can appropriately represent the epithelial components of normal and tumor prostate tissues. In vitro, tumor drug sensitivity screening requires cellular models that are clinically relevant and potentially useful for providing information for the optimization of drug treatment. The normal prostate gland epithelium contains three primary differentiated cell types: luminal, basal and neuroendocrine cells [8]. Luminal cells are columnar epithelial cells that express secretory proteins including prostate-specific antigen (PSA) and markers, such as cytokeratin 8 (KRT8), KRT18, NKX3.1, and high levels of androgen receptor (AR). Basal cells are localized beneath the luminal layer and express markers including KRT5, KRT14 and TP63, but express low levels of AR. A rare third cell type, of neuroendocrine origin, expresses endocrine markers such as synaptophysin and chromogranin A, and does not express AR. There is evidence in support of a basal stem cell population in the prostate [9]. In particular, subpopulations of basal cells, isolated using cell-surface markers, display bi-potentiality and self-renewal in explant culture and tissue grafts [10, 11]. Luminal cells were considered as precursors of human prostate adenocarcinoma based on cancer histological characterization showing expansion of luminal cells and disappearance of basal cells. However, Goldstein et al. showed that histological characterization does not correlate with the cellular origin of the disease and that basal cells can form a prostate tumor in immunodeficient mice with histological and molecular characteristics of prostate cancer [12]. In the mouse model, both luminal and basal cells seem to be the targets for transformation and they induce different phenotypes [13]. In primary human cancer, the luminal phenotype is typical, with glands, AR positivity, and absence of basal cells. However, there is a difference between the outcome phenotype and the actual cell that was transformed [14]. Therefore, basal cells can give rise to tumors of luminal phenotype and basal cells seem to be the efficient target for transformation [11–15].
Recently, exciting technological developments have changed the landscape of possibilities for generating in vitro human cancer models. These include 2D conditional reprogramming (CR) cultures [16, 17], as well as 3D organoid cultures [18–25]. Organoid culture models work well for normal prostate cells and advanced prostate cancers [26–28], and the CR technology additionally allows cultures to be established from primary tumors. CR cells cultured from normal epithelium are morphologically undifferentiated and express adult stem cell markers, but can fully differentiate when placed into in vivo or in vitro conditions that mimic their natural environment [17]. Using CR technology, we were able to identify a patient-specific drug therapy for a rare disease, aggressive recurrent respiratory papillomatosis [29], and others have used the technique for studies of targeted therapy-resistant lung cancer [30], for prostate [31–33] and other types of epithelial cells [34–37].
Previously we generated donor-matched normal/tumor cell lines from a variety of tissue types including breast, lung, colon, and prostate specimens using the CR technology [16, 17]. These included 7 matched normal and tumor prostate CR cell cultures, of which tumor-derived cultures referred to as GUMC-30 in this study (GUMC-29 are matched normal cells) retained tumorigenic potential in SCID mice. These novel cell strains were established from normal and tumor tissues from the same patient without introduction of viral and/or cellular genes. In this study we demonstrate that both, normal and tumor prostate epithelial cells, GUMC-29 and GUMC-30, proliferate indefinitely in CR conditions and predominantly express markers of basal cells in 2D (2-dimensional) culture. However, the tumor cells exhibit an increase in a number of luminal markers when established as xenografts in mice, therefore, further suggesting that the basal-like cell population serves as the origin for prostate tumor, in agreement with previous reports [38, 39]. Exome DNA sequencing of the matched normal and tumor pairs reveals significant differences in several signaling pathways, some of which correspond to those discovered in comprehensive analyses of genetic changes in primary prostate cancer specimens. The ability to rapidly establish cell lines from both normal and cancer prostate biospecimens provides a unique platform for identifying the genetic and molecular events of early prostate cancer and will hopefully enable the development of new approaches for early therapeutic intervention.
RESULTS AND DISCUSSION
Growth properties of the matched normal and tumor CR cells (GUMC-29 and GUMC-30 cells)
Cells of non-malignant and malignant regions of a prostatectomy specimen were grown in keratinocyte serum-free medium (K-SFM) and then transferred to CR conditions to generate immortalized cultures as previously described [8]. The normal CR cells are now designated as GUMC-29 and the tumor CR cells GUMC-30. When grown under CR conditions, both GUMC-29 and GUMC-30 exhibited the typical morphologic characteristics of epithelial cells and were microscopically indistinguishable (Figure 1A). In addition to having the same morphologic appearance, both normal and tumor cells displayed identical growth rates and did not show any evidence of slowed growth or senescence (Figure 1B). However, the GUMC-30 cells formed large colonies in agarose, indicating their anchorage-independent growth properties (Figure 1C). Furthermore, when injected subcutaneously into immunodeficient mice, the GUMC-30 cells formed large tumors within 2 weeks. The tumor size induced by the GUMC-30 cells ranged from 220-2000mm3 (Figure 1E) with one animal not forming a tumor. Histopathologic examination revealed poorly differentiated adenocarcinoma (Figure 1D). The GUMC-29 cells did not grow is soft agar and did not form xenograft tumors (Figures 1C-E).
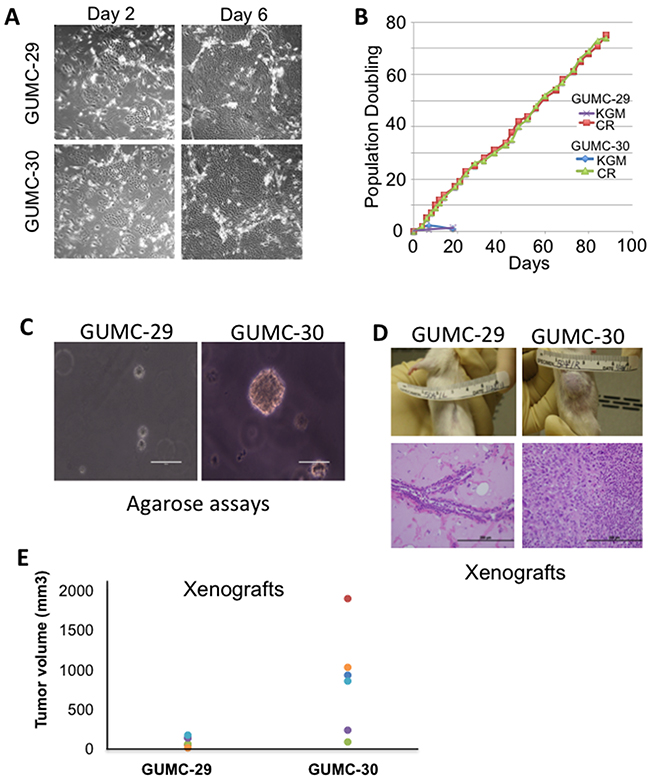
Figure 1: Establishment and biological characterizations of cell cultures from matched normal and tumor prostate specimens. A. Prostate tissues from a patient with Gleason7 prostate cancer were harvested and digested with trypsin-collagenase, as described previously [16, 17, 61]. The initial cultures were established in keratinocyte growth medium (Invitrogen) and then were plated on a feeder layer of irradiated (3000 rad) Swiss 3T3 cells (J2 subclone) and grown in F medium containing 10 μmol/L ROCK inhibitor (Y-27632). Small colonies were observed after 2 days (left panel). At day 6 (right panel), the typical large islands of epithelial cells were observed that compressed the surrounding feeder cells. B. The prostate cells were passaged under CR conditions (F medium containing feeders and Y-27632) or in KGM. Total cell number was recorded at each passage, and the population doubling rate was determined. Only cells grown under CR conditions continued to proliferate past day 20. C. Growth in soft agar. GUMC-29 and GUMC-30 CRs were plated in 0.3% agarose in conditioned media and overlaid with 0.6% agar. Only tumor CRs were able to form viable colonies. D. GUMC-29 and GUMC-30 were mixed with matrigel and injected subcutaneously into adult male SCID mice. Histopathology of the tumor sample exhibited well differentiated human prostate cancer. E. Tumor volumes are shown as measured using verneir calipers. GUMC-29 and -30 cells were grown as spheres for 5-days and then injected on the opposite flanks of nude athymic mice. Paired T-test p-value was 0.018.
To confirm the human origin of the xenograft and its relationship to the injected GUMC-30 cells, we performed STR analysis (Table 1). Both the GUMC-29 and GUMC-30 cultures showed identical STR patterns as did the GUMC-30-induced xenograft tumors. There are no cell lines in the ATCC database that match the STR results shown.
Table 1: STR analysis of prostate GUMC-29 and GUMC-30 cells
Cell ID |
GUMC-29 |
GUMC-30 |
GUMC-30 xenograft |
||||
|---|---|---|---|---|---|---|---|
MARKER |
Allele1 |
Allele2 |
Allele1 |
Allele2 |
Allele1 |
Allele2 |
ATCC MATCH |
AMEL |
X |
Y |
X |
Y |
X |
Y |
no match |
CSF1PO |
10 |
12 |
10 |
12 |
10 |
12 |
|
D13S317 |
12 |
14 |
12 |
14 |
12 |
14 |
|
D16S539 |
11 |
12 |
11 |
12 |
11 |
12 |
|
D18S51 |
12 |
15 |
12 |
15 |
12 |
15 |
|
D19S433 |
14 |
17.2 |
14 |
17.2 |
14 |
17.2 |
|
D21S11 |
29 |
30 |
29 |
30 |
29 |
30 |
|
D2S1338 |
18 |
25 |
18 |
25 |
18 |
25 |
|
D3S1358 |
17 |
18 |
17 |
18 |
17 |
18 |
|
D5S818 |
9 |
12 |
9 |
12 |
9 |
12 |
|
D7S820 |
9 |
11 |
9 |
11 |
9 |
11 |
|
D8S1179 |
12 |
13 |
12 |
13 |
12 |
13 |
|
FGA |
18 |
26 |
18 |
26 |
18 |
26 |
|
TH01 |
6 |
9.3 |
6 |
9.3 |
6 |
9.3 |
|
TPOX |
9 |
11 |
9 |
11 |
9 |
11 |
|
vWA |
14 |
17 |
14 |
17 |
14 |
17 |
|
We interpret these results as that CR conditions allow immortalization of both normal and tumor primary prostate cells while preserving their tumorigenic potential.
Different properties of the matched normal and tumor CR cells compared to cancer cell lines
To evaluate prostate marker expression in GUMC-29 and GUMC-30, we utilized qRT-PCR and flow cytometry techniques. Compared to the established cancer cell line LNCaP, both CR lines expressed high levels of basal cell markers KRT14, KRT5 and TP63, low to negligible levels of luminal cell markers such as AR and NKX3.1, and similar levels of other luminal markers such as AMACR, ACPP and KRT8 (Figure 2A). Importantly, other spontaneously immortalized prostate lines, DU145 and PC3, do not express basal cell markers TP63, KRT5, and KRT14 and are similar to LNCaP cells in this respect (Figure 2B). However, these cells express very low levels of luminal cell markers AR and NKX3.1 comparable to GUMC-29/30. Interestingly, AMACR, ACPP, and KRT8 levels are lower in PC3 and DU145 compared to GUMC-29/30. Analysis of prostate marker expression in the primary prostate cells PrEC showed that these cells express high levels of basal markers TP63 and KRT14, but very low levels of all luminal markers. These data suggest that both phenotypes, basal and luminal are present in CR cultures. Notably, there are no significant differences in the levels of expression of any prostate differentiation markers between GUMC-29 and GUMC-30, suggesting that the normal and tumor cells were inherently similar or that the CR conditions converted them to a similar and less-differentiated state.
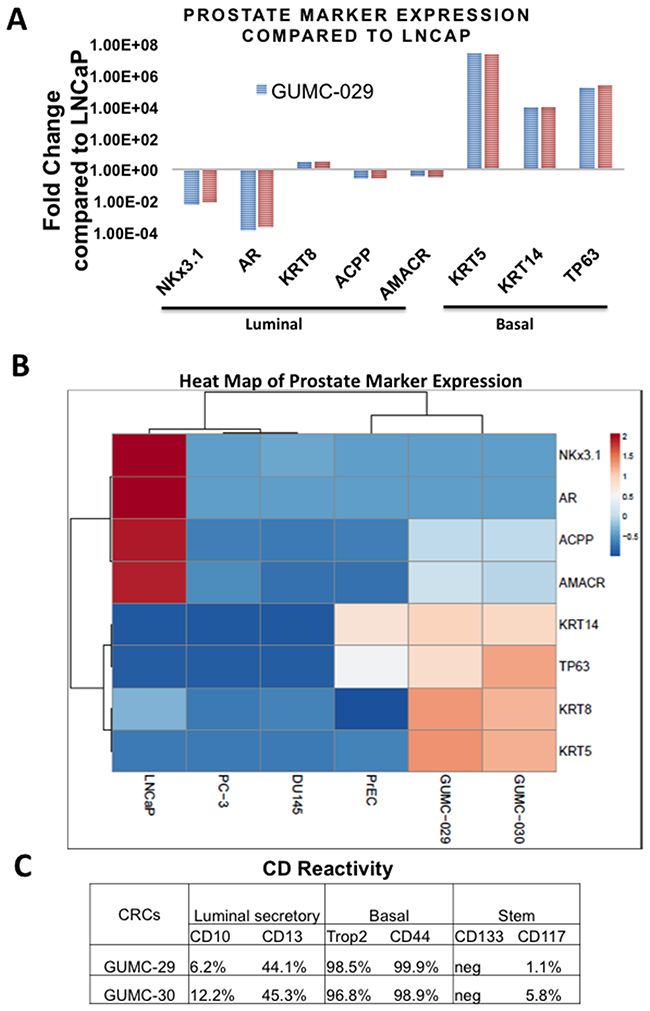
Figure 2: Characterization of prostate epithelial cells markers using qRT-PCR and Flow Cytometry. A. Results of quantitative Reverse Transcription PCR (qRT-PCR) performed on total RNA extracted from 3 biological replicas of CR prostate cultures compared to LNCaP for basal cell markers: KRT5, KRT14, TP63 and luminal cell markers: AR, AMACR, ACPP, NKX3.1, and KLK3. B. Heatmap of prostate marker expression determined by qRT-PCR in GUMC-29/30, LNCaP, PC3, DU145, and PrEC. C. The number of cells expressing markers of luminal secretory, basal, and stem cells was measured using flow cytometry.
CR cells are “transit-amplifying” cells
Flow cytometry analysis of surface CD markers in GUMC-29/30 demonstrated that nearly all normal and tumor cells express basal cell markers, CD44 and Trop2 (CD49f) (Figure 2C and Supplementary Figure 1). Interestingly, about 45% of the normal and tumor cells also expressed the luminal CD13 marker, while only a small portion (6-12%) expressed the CD10 luminal marker. We found a population of CD117-expressing cells in the prostate cultures, considered to be prostate stem cells [10].
Approximately 5% cells of the GUMC-30 cancer cell population were CD117-positive, while only 1% of the GUMC-29 normal cells were CD117-positive. No CD133-positive cells were detected in CR cultures.
Taken together, the results of Flow Cytometry and RT-PCR analyses suggest that cells growing under CR conditions resemble “transit-amplifying” cells, which express markers of both basal and luminal cells and can differentiate into either phenotype upon receiving an appropriate stimulus [40–42]. Using skin and tracheal cells, we have previously shown that transit-amplifying-like cells from CR cultures can ultimately differentiate into mature epithelium containing both basal and luminal cells [43].
Differences in gene expression between normal and tumor CR cells
To study differences in gene expression, we performed Affymetrix GeneChip microarray. We identified 87 genes differentially regulated in GUMC-30 compared to GUMC-29 (Figure 3A and Supplementary Data Set 1). Genes involved in cellular development, growth and proliferation, and metabolism were identified. RT-PCR confirmed differential expression of selected genes (Figure 3B). Some of the genes may play a role in prostate tumorigenesis, for example CXCL11 (Chemokine (C-X-C Motif) Ligand 11), NNMT (Nicotinamide N-methyltransferase) and CDK2 have been reported to be over-expressed in prostate cancer and CDKN1C is repressed in breast and prostate cancer [44–47]. Further studies will investigate the role of these proteins in prostate tumorigenesis.
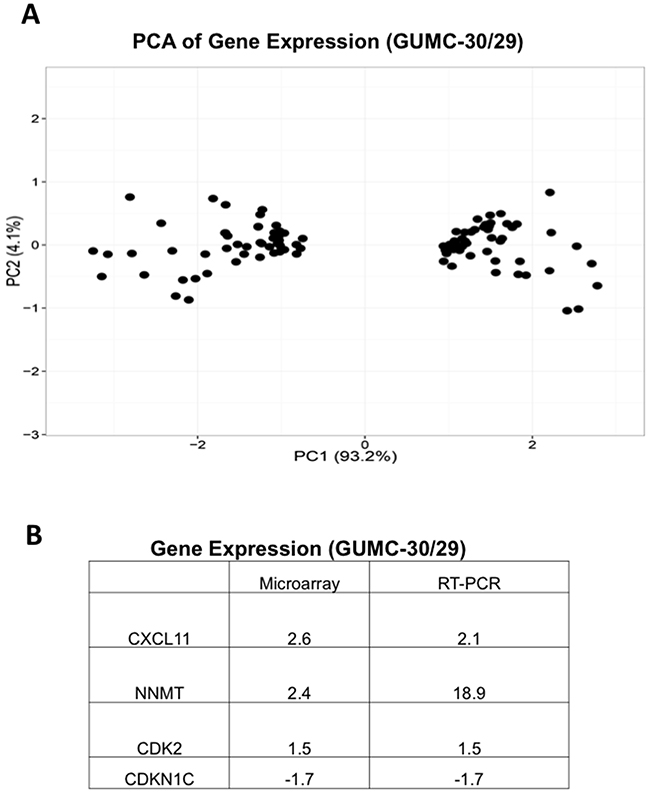
Figure 3: Differential gene expression in GUMC-29 and GUMC-30. A. PCA analysis of differential gene expression in normal and tumor CR cells detected by microarray. B. Differential expression of selected genes was confirmed using qRT-PCR analyses.
Differential sensitivity of matched CR cells to docetaxel
To test the patient-derived CR cells for potential use for drug screening, we exposed matched CR cells to a commonly used chemotherapeutic agent, docetaxel. As shown in Supplementary Figure 2, the IC50 values of docetaxel to GUMC-29 and GUMC-30 were 1.79 µM and 0.29 µM, respectively. These data suggested that the matched normal and tumor CR cells can be used to aid drug selection for the individual patients or as a system that enables high throughput drug discovery. Indeed, we were able to individualize drug therapy for a patient with an aggressive lung tumor using CR technology [29]. Others have used our technique for studying and developing therapies for targeted therapy-resistant lung cancer [30]. Most recently, our technology has been used to screen for new drugs or to discover new targets [33, 48–50].
Re-expression of androgen receptor in normal and tumor CR cells under differentiation conditions
Since GUMC-30 cells formed adenocarcinoma when injected into immunodeficient mice (Figure 1D,E), we hypothesized that this process may be accompanied by changes in prostate marker expression. qRT-PCR assays demonstrated that luminal cell markers, including AR and NKX3.1, were up-regulated 10- to 100-fold in the xenograft-derived tissues when compared to the GUMC-30 adherent cell cultures (Figure 4). In contrast, basal cell markers, including TP63, KRT14, and KRT5, dramatically decreased in the xenografts (Figure 4A). These data indicate that the tumorigenic GUMC-30 cells are somewhat plastic in their expression profiles and that the in vivo xenograft conditions favor a more differentiated, luminal-like cell phenotype. These data further support previous observations that basal/trans-amplifying cells may form prostate adenocarcinomas in immunodeficient mice [51–55], and, therefore, represent important target cells in prostate cancer.
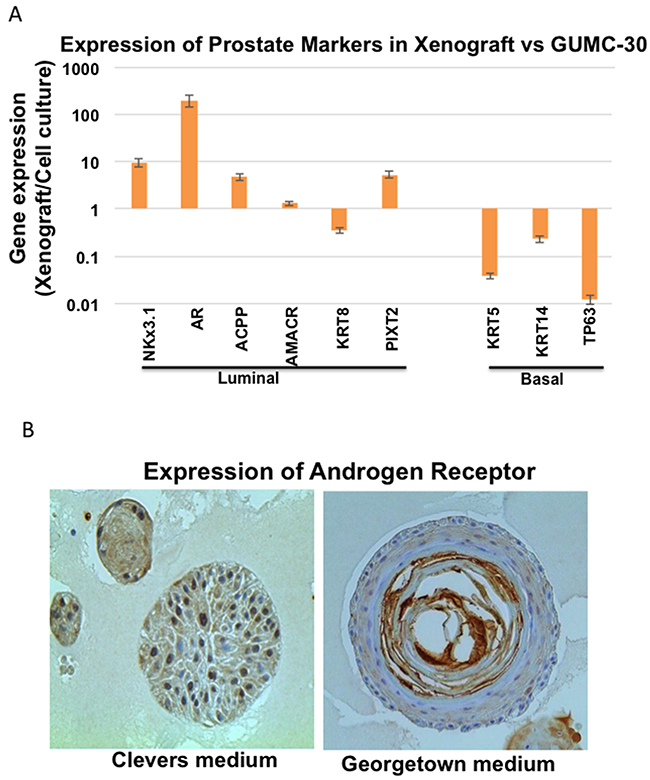
Figure 4: Differential gene expression in tumor CRs and xenografted tumor. A. Expression of luminal and basal epithelial markers were measured using qRT-PCR in GUMC-30 cultured under CR conditions and GUMC-30 xenograft tumors. Expression of most luminal markers increased in xenograft tumor compared to CRs, while basal markers significantly decreased in the tumor xenograft compared to CR. B. Organoid cultures were established as described in Materials and Methods. Organoids were fixed overnight with 4% paraformaldehyde at room temperature and the following day with 70% ethanol. Fixed organoids were embedded in paraffin for sectioning at 5 µm before probed with anti-AR (Santa Cruz, sc-816) for immunohistochemistry. Images of growing organoids were acquired using EVOS FL Cell Imaging System (Invitrogen) for microscopic imaging.
As shown in Figure 4B, normal CR cells from PrEC (Lonza) also re-expressed androgen receptor when they were cultured in organoid conditions with both Clevers’ medium and Georgetown medium (CR medium or F medium with Y-27632).
These data suggested that both, normal and tumor CR cells, maintain some differential potential and their original benign and malignant phenotypes in in vitro or in vivo differentiation environments.
Karyotype and exome sequence analysis of the matched normal and tumor CR cells
To further characterize these matched prostate CR cells at the cytogenetic and molecular levels, we performed karyotype analyses followed by exome sequencing, including SNPs and INDELs. The karyotype analyses revealed that approximately 10% of the GUMC-30 cells carry an additional copy of chromosome 13 (Figure 5A). We generated a Circos plot of the variants detected by sequencing analysis (Figure 5B). Our exome sequencing analysis has identified a total of 815 variants present only in the GUMC-30 sample. These variants were found to associate with 756 genes with unique HGNC gene symbols. Consensus genotype calls were generated using the UnifiedGenotyper tool from GATK (v3.5) and annotated using the Annovar package. Different basic types of annotations were separated into several tables based on the effects of a variant on gene, transcript, and protein sequence, such as an amino acid change, frameshift, or promoter region alteration, etc. (Figure 6A) based on GRCh37 (Supplementary Data set 2). We compared our variant gene list with the publicly available 2755 genes prostate cancer list [56]. Using Venn diagram analysis we identified a set of 133 genes common to the both lists (Figure 7) among which 11 genes belong to the KEGG cancer pathway gene set (Figure 6B, Supplementary Data Set 3), including FN1, CASP8, HIF1a, SMAD2, RAC1, and RET [51–55].
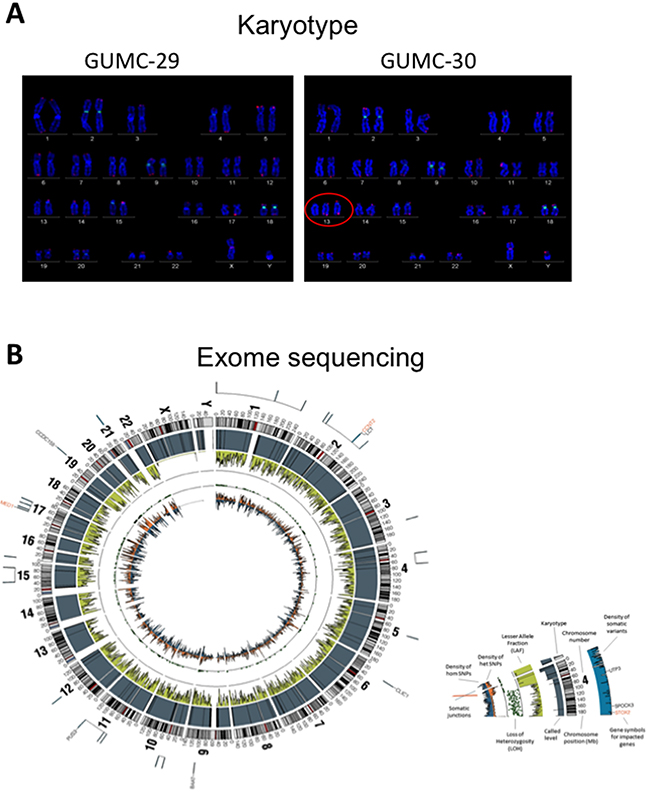
Figure 5: Karyotype and exome sequence analysis of the prostate CR cells. Both GUMC-29 and GUMC-30 CRs were subjected to karyotyping and exome-sequencing analyses. A. Roughly 10% of population of GUMC-30 cells exhibited trisomy at chromosome 13, while the remainder of GUMC-30 cells and 100% GUMC-29 harbor normal karyotypes. B. Exome-sequencing was performed on both GUMC-29 and GUMC-30 using the CG platform as showing a Circos visualization of variations detected in the Genome, along with other associated data. Density of hom SNPs represents the density of high confidence homozygous SNPs in 1Mb windows, arbitrarily scaled in a histogram with the Y-axis pointing inward; Density of het SNPs represents the density of heterozygous SNPs in 1Mb windows, arbitrarily scaled in a histogram with Y-axis pointing outward; Lesser Allele Fraction (LAF) represents the single-sample LAF estimate for 100 kb windows, with Y-axis scale of 0 to 0.5, pointing inward. Estimates are based on read counts at called heterozygous loci; Called Ploidy represents the called ploidy from a segmentation file, which partition the reference genome into regions of distinct ploidy levels, giving the estimated ploidy, the average and relative adjusted coverage for each segment, and measures of confidence in the called segments. They are arbitrarily scaled with the Y-axis pointing inward; Karyotype represents the standard Circos ideogram depicting chromosome position and chromosome number.

Figure 6: SNPs and INDELs and pathway analysis in tumor CR cells. A. Numbers of SNP and INDELs, plus pathways, that differ in GUMC-30 versus GUMC-29. B. KEGG pathway analysis of cancer pathways genes.
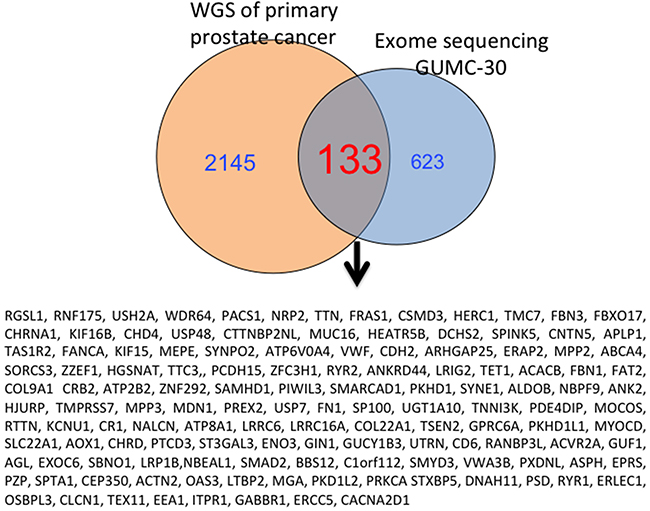
Figure 7: Overlapping genes from GUMC-30 and Garraway’s study with NGS are shown. 133 Mutated genes in tumor CR cells were presented in result from a previous study with NGS from the patients with primary prostate cancer.
Genomics is a useful tool for cancer research that has largely focused on basic mutational catalogs in primary tumors. Given the complexity of primary prostate cancer, it is necessary to deepen the structural characterization of cancer genomics and comprehensive functional characterization of cancer cells. This will help further understanding of basic and translational biology of human prostate cancer. The matched normal and tumor CR cells from the same patients provide a novel functional platform for these studies.
Selection of tumor CR cells from mixed normal and tumor population
Considering that CR technique supports growth of both normal and tumor cells and that all clinical tissue specimens are mixtures of normal, tumor and stromal cells, we wanted to select out pure tumor cells from mixed cultures.
Various culture conditions were tested to identify factors that allowed proliferation of tumor cells, but did not support expansion of normal cells. We found that culturing CRs in DMEM without serum for 3 days led to differentiation of normal GUMC-29 cells, while supporting a “fibroblast”-like or mesenchymal morphologic phenotype of GUMC-30 (Figure 8A). Interestingly, CR conditions reversed “fibroblast”- or mesenchymal-like cells back to epithelial morphology. To further validate this approach, GFP-labeled GUMC-30 cells were mixed with normal HFKs in equal amounts and were first grown in DMEM without serum for 7 days. Only “green” tumor CR cells grew after 7 days of selection (Figure 8B, 8C).
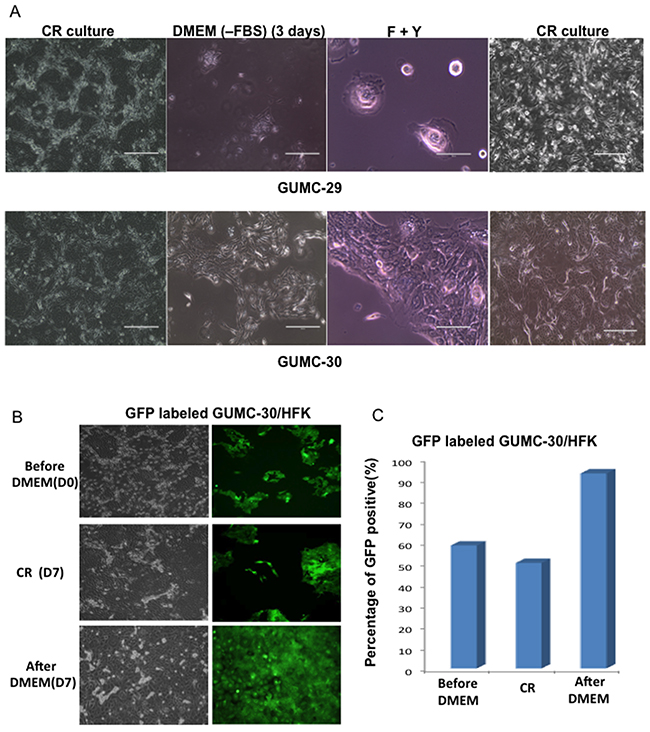
Figure 8: Selection of Tumor CR cells. GUMC-29 and -30 under CRC condition were changed to DMEM without FBS for 3 days and media were replaced with F medium plus Y-27632 for 5 days, lastly leftover were re-plated in CRC condition A. normal CRCs (GUMC-29) entered complete senescence stage without any recovery once the cells were transferred back to CR condition while tumor CRCs (GUMC-30) were able to recover from selection process and formed typical epithelial colonies as usual The mixed culture with GFP-labeled GUMC-30 and non-labeled normal foreskin keratinocytes (HFK) (50:50) underwent the same selection protocol, only GFP positive cells presented in culture after selection B and C.
This selection procedure may be particularly important for primary cancer studies. Currently, CR technology is the only method by which expansion of primary prostate cancer cells is obtained. The organoid protocol can be used for establishing cultures from normal mouse and human prostate tissue with efficiency > 95%. However, advanced prostate cancers yield only about 15 – 20% efficiency [26–28]. Growing organoid cultures derived from primary prostate cancers has not been successful, possibly due to overgrowth by normal prostate epithelium present within each sample [26–28].
If this is truly the case, pre-selection procedures will be important. We are currently working to develop selection methods that allow “purification” of tumor for selective expansion as CR cells with different approaches summarized in a recently accepted paper by Nature Protocols [17].
Micro-heterogeneity
Finally, given that primary prostate cancer is highly heterogeneous disease, it is important to establish a functional method to study tumor heterogeneity. We used a tissue sample collected from a patient with primary prostate cancer at Georgetown Lombardi Cancer Center for preparing a single cell suspension that was diluted to 1 cell/200 µl FY medium, and plated 100 µl volumes per well of ten 96-well plates. We established 9 clones from single cell cultures using CR technology. Our data suggests that clones have different invasive abilities (Supplementary Figure 4). Comprehensive genomics and functional studies (especially response to drugs) on these clones will help further understanding heterogeneity of primary prostate cancer.
Summary
We have generated matched normal and tumor cell cultures from a patient with primary prostate adenocarcinoma. These cultures, while exhibiting similar morphologies and division rates in CR conditions, were different in their ability for anchorage-independent growth and capacity to induce tumor formation in immunodeficient mice. The normal and tumor cells also displayed differences in karyotype, gene expression, and exome sequences. Some of the genetic mutations have been noted in previous comprehensive analyses, including genes involved in pathways that regulate cell attachment, cell apoptosis, and HIF signaling. We provide a novel functional platform for cancer biology, discovery of biomarkers and anti-cancer drugs, and cancer precision medicine.
MATERIALS AND METHODS
Generation of primary cell cultures
The non-malignant tissue (RC-123N) and malignant tissue (RC-123T) used for generating the primary cells were obtained from a radical prostectomy of a 57-year old European American patient. Samples were obtained according to an approved Internal Review Board (IRB) protocol from the Walter Reed Army Medical Center and the Uniformed Services University of the Health. This patient was clinically staged with T3b adenocarcinoma with Gleason grade 3+4=7. Non-malignant cells were derived from histopathologically confirmed non-cancerous regions of the prostate. The method for generating primary prostate cell cultures has been previously described [57]. Briefly, the tumor and non-malignant tissues, obtained by an experienced pathologist, were minced into 1-2 mm small fragments with a sterile scalpel. The tissue was then placed into type 1 collagen-treated dishes (Becton-Dickinson, Boston, MA) containing growth medium and were allowed to attach to the bottom surface of the culture dishes. The cells were incubated for approximately one week at 37˚C in 5% CO2 until reaching semi-confluency. Aliquots of the primary cultures were then frozen and stored in liquid nitrogen. For serial passages, routine trypsinization was performed weekly in collagen-treated culture dishes, with the split ratio of cells being 1:2. The cells were grown in Keratinocyte serum-free medium (K-SFM) supplemented with bovine pituitary extract and recombinant epidermal growth factor (Life Technologies, Inc., Gaithersburg, MD).
Generation of conditionally reprogrammed cells, GUMC-29 and GUMC-30
Cells were seeded on a feeder layer of lethally irradiated (30 Gy) J2 fibroblasts in F medium. The F medium consisted of 25% Ham’s F-12 nutrient mix (Life Technologies) and 75% complete DMEM, supplemented with 25 ng/mL hydrocortisone, 5 mg/mL insulin, 0.1 nmol/L cholera toxin (Sigma-Aldrich, St. Louis, MO), 250 ng/mL Fungizone (Thermo Fisher Scientific, Waltham, MA), 0.125 ng/mL epidermal growth factor, and 10 mg/mL gentamicin (Life Technologies). In most experiments, cells were cultured in the presence of the ROCK inhibitor Y-27632 at a final concentration of 5 µmol/L (Enzo Life Sciences, Farmingdale, NY). In the absence of feeder cells, cells were grown either in F medium containing Y-27632 or in keratinocyte growth medium (KGM) (Life Technologies).
Passaging CR cells
To remove J2 feeder cells, prostate co-cultures were rinsed with Dulbecco’s PBS and treated with 0.05% trypsin-EDTA (Life Technologies) for 30 seconds at room temperature. The culture vessel was then gently rocked until the feeder cells detached, and the feeder cells were removed by aspiration. The prostate epithelial cells were washed with Dulbecco’s PBS and treated with trypsin-EDTA for 3 to 5 minutes at 37 C. The prostate epithelial cells were detached by gentle tapping, and trypsin was neutralized by adding Dulbecco’s PBS containing 10% fetal bovine serum. After centrifugation, the prostate epithelial cells were suspended in F medium and plated on freshly irradiated feeder cells.
Selection of tumor CR cells
GuMC-29, GUMC-30 or mixed CR cells with GFP-labeled GUMC-30 and non-labeled normal foreskin keratinocytes (HFK) from above CR condition were initially selected in DMEM without FBS for 3 days, then in F medium with Y-27632 for 5 days. All the leftover were placed back in CR condition.
Affymetrix microarray analysis
Total RNA was extracted using an RNeasy Kit (Qiagen, Valencia, CA, USA). RNA labeling and hybridization were performed according to Affymetrix standard protocol for one-cycle target labeling. Fragmented cRNA was hybridized in triplicate to Affymetrix GeneChip HG-U133A 2.0 arrays (Affymetrix, Santa Clara, CA). Affymetrix data analysis included pre-processing of the probe-level Affymetrix data (CEL files). We applied RMA for background adjustment, quantile method for normalization, and the ‘median polish’ for summarization. The triplicate arrays representing the same subject were averaged. The random variance model implemented in BRB-ArrayTools (NCI, Bethesda, MD) was used for this analysis [48] Pathway analysis was performed with Database for Annotation, Visualization and Integrated Discovery (DAVID) [58].
Quantitative RT-PCR
Total RNA was extracted from 3 biological replicates of CR prostate cultures, including GUMC-29, GUMC-30, and established cell lines LNCaP, DU145, PC3 as well as primary PrEC cells using RNeasy kit (Qiagene) as previously described [59]. One microgram of total RNA was reverse-transcribed using the High Capacity cDNA Reverse Transcription Kit (Life Technologies) in 20 µl of reaction mixture. Two microliters of reverse transcription reaction corresponding to 100 ng of total RNA were taken into quantitative real-time PCR (qRT-PCR) reaction that was performed in triplicate using TaqMan Gene Expression Assays (Applied Biosystems) on the Applied Biosystems 7900HT Fast Real-time PCR System using fast mode. Amplification of human β-actin mRNA was used as an endogenous control to standardize the amount of sample added to the reaction. The comparative cycle threshold (CT) method was used to analyze the data by generating relative values of the amount of target cDNA (Applied Biosystems). CT represents the number of cycles for the amplification of target to reach a fixed threshold and correlates with the amount of starting material present. To obtain relative values, the following arithmetic formula was used: 2-ΔCT, where ΔCT = difference between the threshold cycles of the target and an endogenous reference.
Flow cytometry analysis
CD reactivity of luminal (CD10 and CD13), basal (CD44 and CD49f (Trop2)), and stem cells (CD117 and CD133) was detected using fluorophore-conjugated CD antibodies (Biolegend). Briefly, trypsinized cells, prepared as a single cell suspension, were resuspended in 50 μl aliquots of 0.1% bovine serum albumin-Hanks’ balanced salt solution (BSA-HBSS). Fluorophore-conjugated CD antibodies were added to the cell suspensions (0.1 µg of anti-CD10, CD13, CD117 and CD133 and 0.05 µg of anti-CD44 and CD49f antibody were added) and incubated for 1 hr at room temperature in the dark. Isotype-specific fluorochromated antibodies were used as negative controls to delineate the negative cell population. The reaction was stopped by the addition of 1 ml of 0.1% BSA-HBSS. The cells were centrifuged and fixed in 0.35 ml of 2% paraformaldehyde. Flow analysis was done by a FACScan (Becton Dickinson, Mountain View, CA) machine fitted with a 488 nm laser. Events that registered outside this trace were scored as positive, and 10,000 events were collected for each sample. The percentage of positive events was determined.
Oganoid cultures
Organoid cultures were set up according to previous protocols [26–28]. Briefly, 120 µl of BD Matrigel® (BD Biosciences, San Jose, CA, Cat# 356230) were added to the bottom of a well 6 well plate (Falcon, Durham, NC, Cat # 353046) and incubate gel at 37 oC for approximately 10 min. Next, a second layer of 1.2 ml matrigel containing 50,000 cell suspension was added on top and allow gel at 37 oC for additional 15-20 min. Then, 2ml F medium with 10 µM Y-27632 (Georgetown Medium) or Clevers’ medium [26–28] will add on the surface. The medium was changed every two days. Organoids were fixed overnight with 4% paraformaldehyde at room temperature and the following day with 70% ethanol. Fixed organoids were embedded in paraffin for sectioning at 5um before probed with antibodies for immunohistochemistry or immunofluorescence. All samples were stained for H&E and the following antibodies were used for immunostaining: 1:125 dilution of rabbit AR (N20) (Santa Cruz; sc-816) and mouse p63 (Santa Cruz; sc25268) and 1:800 dilution of mouse CK 18 (Cell Signaling, 4548P). Images of growing organoids were acquired using EVOS FL Cell Imaging System (Invitrogen) for microscopic imaging.
Agarose assay
To assay anchorage-independent growth, 1 ml of 0.3% agarose containing 1 × 103 cells was layered over 1 ml of 0.6% agarose in each well of 6-well plate. A sterile 3% agarose stock solution was prepared in D-PBS and diluted to the required concentrations by mixing with conditioned medium as previously described. Cultures were overlaid with 0.5 ml of this medium and further additions were made, as necessary, to prevent desiccation for a period of 3–4 weeks.
Tumorigenicity in SCID mice
To determine tumorigenicity, 1x107 cells in 0.2 ml of matrigel were injected subcutaneously into the mid-dorsal intracapular regions of adult male SCID mice. The mice were observed for up to 6 months for tumor development, as previously described [16]. To confirm the tumorigenic capacity of the lines, the following protocol was also used. Normal and tumor cell CRs, grown in the presence of feeder cells and Y-27632 (5 µM) cells, were subsequently plated in conditioned medium and seeded in low attachment plates. Cells were allowed to grow as spheres for 5-days. The media was changed on day 3. Cells were harvested and a single cell suspension was prepared by trysinization of the spheres. 100,000 cells were injected in nude athymic mice. Tumor mass was measured using verneir calipers. Mice were observed for up to one month for tumor development.
Cytogenetic analysis
Chromosome counts, ploidy distribution, and Giemsa (G)-banded karyotypes were prepared by standard protocol as described previously [60].
Exome sequencing data analysis
DNAs from GUMC-29 and -30 were extracted and exome-sequencing was performed with Complete Genomics platform. Sequence reads were processed with a pipeline consisting of the following elements: (1) base calls generated in real-time on the Agilent SureSelect system; (2) Perl scripts developed in-house to produce demultiplexed fastq files by lane and index sequence; (3) demultiplexed BAM files aligned to a human reference (hg19) using BWA (Burrows-Wheeler Aligner). Read-pairs not mapping within +2 standard deviations of the average library size (*125+15 bp for exomes) are removed. All aligned read data were subjected to the following steps: (1) duplicate removal” was performed, (i.e., the removal of reads with duplicate start positions; Picard MarkDuplicates); (2) indel realignment was performed (GATK IndelRealigner) resulting in improved base placement and lower false variant calls; (3) base qualities were recalibrated (GATK Table Recalibration).
Variant detection and genotyping were performed using the UnifiedGenotyper tool from GATK (v3.5). Variant data for each sample were formatted (variant call format) as ‘‘raw’’ calls that contain individual genotype data for one or multiple samples, and flagged using the filtration walker (GATK) to mark sites that are of lower quality/false positives, e.g., low quality scores (<= 50), allelic imbalance (>= 0.75), long homopolymer runs (> 3) and/or low quality by depth (QD< 20).
ACKNOWLEDGMENTS
We thank the Georgetown-Lombardi Nontherapeutic Subject Registry and the Histology and Tissue Shared Resource, the Georgetown-Lombardi Genomics and Epigenomics Shared Resource and the Georgetown-Lombardi Flow Cytometry Shared Resource.
CONFLICTS OF INTEREST
Georgetown University has been awarded a patent by the United States Patent Office (9,279,106) for conditional cell reprogramming. This technology has been licensed exclusively to a new biotechnology company, Propagenix, for commercialization. Georgetown University and the inventors (X.L., R.S.) receive payments and potential royalties from Propagenix.
GRANT SUPPORT
This work was partially supported by NIH R33CA177466 (Schlegel), NIH R21CA180524 (Liu), NIH P30 CA051008 (Weiner), NIH/NCAT TL1-TR001431 (Sandberg, Albanese), DOD-PC140268 (Albanese), and DOD W81XWH-13-1-0327 (Albanese), and internal grant support from Center for Cell Reprogramming at GUMC.
REFERENCES
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015; 65: 5-29. doi: 10.3322/caac.21254.
2. Yu C, Yao Z, Jiang Y, Keller ET. Prostate cancer stem cell biology. Minerva Urol Nefrol. 2012; 64: 19-33.
3. Feldman BJ, Feldman D. The development of androgen-independent prostate cancer. Nat Rev Cancer. 2001; 1: 34-45.
4. Peehl DM. Primary cell cultures as models of prostate cancer development. Endocr Relat Cancer. 2005; 12: 19-47. doi: 10.1677/erc.1.00795.
5. Williams SA, Anderson WC, Santaguida MT, Dylla SJ. Patient-derived xenografts, the cancer stem cell paradigm, and cancer pathobiology in the 21st century. Lab Invest. 2013; 93: 970-82. doi: 10.1038/labinvest.2013.92.
6. Tentler JJ, Tan AC, Weekes CD, Jimeno A, Leong S, Pitts TM, Arcaroli JJ, Messersmith WA, Eckhardt SG. Patient-derived tumour xenografts as models for oncology drug development. Nat Rev Clin Oncol. 2012; 9: 338-50. doi: 10.1038/nrclinonc.2012.61.
7. Jin K, Teng L, Shen Y, He K, Xu Z, Li G. Patient-derived human tumour tissue xenografts in immunodeficient mice: a systematic review. Clin Transl Oncol. 2010; 12: 473-80. doi: 10.1007/s12094-010-0540-6.
8. Shen MM, Abate-Shen C. Molecular genetics of prostate cancer: new prospects for old challenges. Genes & Development. 2010; 24: 1967-2000. doi: 10.1101/gad.1965810.
9. Lawson DA, Witte ON. Stem cells in prostate cancer initiation and progression. J Clin Invest. 2007; 117: 2044-50.
10. Lawson DA, Xin L, Lukacs RU, Cheng D, Witte ON. Isolation and functional characterization of murine prostate stem cells. Proceedings of the National Academy of Sciences. 2007; 104: 181-6. doi: 10.1073/pnas.0609684104.
11. Goldstein AS, Lawson DA, Cheng D, Sun W, Garraway IP, Witte ON. Trop2 identifies a subpopulation of murine and human prostate basal cells with stem cell characteristics. Proceedings of the National Academy of Sciences. 2008; 105: 20882-7. doi: 10.1073/pnas.0811411106.
12. Goldstein AS, Huang J, Guo C, Garraway IP, Witte ON. Identification of a cell of origin for human prostate cancer. Science. 2010; 329: 568-71. doi: 10.1126/science.1189992.
13. Park JW, Lee JK, Phillips JW, Huang P, Cheng D, Huang J, Witte ON. Prostate epithelial cell of origin determines cancer differentiation state in an organoid transformation assay. Proc Natl Acad Sci U S A. 2016; 113: 4482-7.
14. Stoyanova T, Cooper AR, Drake JM, Liu X, Armstrong AJ, Pienta KJ, Zhang H, Kohn DB, Huang J, Witte ON, Goldstein AS. Prostate cancer originating in basal cells progresses to adenocarcinoma propagated by luminal-like cells. Proc Natl Acad Sci U S A. 2013; 110: 20111-6.
15. Goldstein AS, Drake JM, Burnes DL, Finley DS, Zhang H, Reiter RE, Huang J, Witte ON. Purification and direct transformation of epithelial progenitor cells from primary human prostate. Nat Protoc. 2011; 6: 656-67.
16. Liu X, Ory V, Chapman S, Yuan H, Albanese C, Kallakury B, Timofeeva OA, Nealon C, Dakic A, Simic V, Haddad BR, Rhim JS, Dritschilo A, et al. ROCK inhibitor and feeder cells induce the conditional reprogramming of epithelial cells. Am J Pathol. 2012; 180: 599-607. doi: 10.1016/j.ajpath.2011.10.036.
17. Liu X, Krawczyk E, Suprynowicz FA, Palechor-Ceron N, Yuan H, Dakic A, Simic V, Zheng YL, Sripadhan P, Chen C, Lu J, Hou TW, Choudhury S, et al. Conditional reprogramming and long-term expansion of normal and tumor cells from human biospecimens. Nat Protoc. 2016. doi: 10.1038/nprot.2016.174.
18. Boj SF, Hwang CI, Baker LA, Chio, II, Engle DD, Corbo V, Jager M, Ponz-Sarvise M, Tiriac H, Spector MS, Gracanin A, Oni T, Yu KH, et al. Organoid models of human and mouse ductal pancreatic cancer. Cell. 2015; 160: 324-38. doi: 10.1016/j.cell.2014.12.021.
19. Huch M, Bonfanti P, Boj SF, Sato T, Loomans CJ, van de Wetering M, Sojoodi M, Li VS, Schuijers J, Gracanin A, Ringnalda F, Begthel H, Hamer K, et al. Unlimited in vitro expansion of adult bi-potent pancreas progenitors through the Lgr5/R-spondin axis. Embo J. 2013; 32: 2708-21.
20. Huch M, Boj SF, Clevers H. Lgr5(+) liver stem cells, hepatic organoids and regenerative medicine. Regen Med. 2013; 8: 385-7.
21. Huch M, Dorrell C, Boj SF, van Es JH, Li VS, van de Wetering M, Sato T, Hamer K, Sasaki N, Finegold MJ, Haft A, Vries RG, Grompe M, et al. In vitro expansion of single Lgr5+ liver stem cells induced by Wnt-driven regeneration. Nature. 2013; 494: 247-50.
22. Zhang HC, Kuo CJ. Personalizing pancreatic cancer organoids with hPSCs. Nat Med. 2015; 21: 1249-51.
23. Huang JY, Sweeney EG, Sigal M, Zhang HC, Remington SJ, Cantrell MA, Kuo CJ, Guillemin K, Amieva MR. Chemodetection and Destruction of Host Urea Allows Helicobacter pylori to Locate the Epithelium. Cell Host Microbe. 2015; 18: 147-56.
24. Salahudeen AA, Kuo CJ. Toward recreating colon cancer in human organoids. Nat Med. 2015; 21: 215-6.
25. Ootani A, Li X, Sangiorgi E, Ho QT, Ueno H, Toda S, Sugihara H, Fujimoto K, Weissman IL, Capecchi MR, Kuo CJ. Sustained in vitro intestinal epithelial culture within a Wnt-dependent stem cell niche. Nat Med. 2009; 15: 701-6.
26. Karthaus WR, Iaquinta PJ, Drost J, Gracanin A, van Boxtel R, Wongvipat J, Dowling CM, Gao D, Begthel H, Sachs N, Vries RG, Cuppen E, Chen Y, et al. Identification of multipotent luminal progenitor cells in human prostate organoid cultures. Cell. 2015; 159: 163-75.
27. Gao D, Vela I, Sboner A, Iaquinta PJ, Karthaus WR, Gopalan A, Dowling C, Wanjala JN, Undvall EA, Arora VK, Wongvipat J, Kossai M, Ramazanoglu S, et al. Organoid cultures derived from patients with advanced prostate cancer. Cell. 2015; 159: 176-87.
28. Drost J, Karthaus WR, Gao D, Driehuis E, Sawyers CL, Chen Y, Clevers H. Organoid culture systems for prostate epithelial and cancer tissue. Nat Protoc. 2016; 11: 347-58.
29. Yuan H, Myers S, Wang J, Zhou D, Woo JA, Kallakury B, Ju A, Bazylewicz M, Carter YM, Albanese C, Grant N, Shad A, Dritschilo A, et al. Use of reprogrammed cells to identify therapy for respiratory papillomatosis. N Engl J Med. 2012; 367: 1220-7. doi: 10.1056/NEJMoa1203055.
30. Crystal AS, Shaw AT, Sequist LV, Friboulet L, Niederst MJ, Lockerman EL, Frias RL, Gainor JF, Amzallag A, Greninger P, Lee D, Kalsy A, Gomez-Caraballo M, et al. Patient-derived models of acquired resistance can identify effective drug combinations for cancer. Science. 2014; 346: 1480-6. doi: 10.1126/science.1254721.
31. Ringer L, Sirajuddin P, Tricoli L, Waye S, Choudhry MU, Parasido E, Sivakumar A, Heckler M, Naeem A, Abdelgawad I, Liu X, Feldman AS, Lee RJ, et al. The induction of the p53 tumor suppressor protein bridges the apoptotic and autophagic signaling pathways to regulate cell death in prostate cancer cells. Oncotarget. 2014; 5: 10678-91. doi: 10.18632/oncotarget.2528.
32. Pollock CB, McDonough S, Wang VS, Lee H, Ringer L, Li X, Prandi C, Lee RJ, Feldman AS, Koltai H, Kapulnik Y, Rodriguez OC, Schlegel R, et al. Strigolactone analogues induce apoptosis through activation of p38 and the stress response pathway in cancer cell lines and in conditionally reprogrammed primary prostate cancer cells. Oncotarget. 2014; 5: 1683-98. doi: 10.18632/oncotarget.1849.
33. Saeed K, Rahkama V, Eldfors S, Bychkov D, Mpindi JP, Yadav B, Paavolainen L, Aittokallio T, Heckman C, Wennerberg K, Peehl DM, Horvath P, Mirtti T, et al. Comprehensive Drug Testing of Patient-derived Conditionally Reprogrammed Cells from Castration-resistant Prostate Cancer. Eur Urol. 2016. doi: 10.1016/j.eururo.2016.04.019.
34. Chu HW, Rios C, Huang C, Wesolowska-Andersen A, Burchard EG, O’Connor BP, Fingerlin TE, Nichols D, Reynolds SD, Seibold MA. CRISPR-Cas9-mediated gene knockout in primary human airway epithelial cells reveals a proinflammatory role for MUC18. Gene Ther. 2015; 22: 822-9. doi: 10.1038/gt.2015.53.
35. Reynolds SD, Rios C, Wesolowska-Andersen A, Zhuang Y, Pinter M, Happoldt C, Hill CL, Lallier SW, Cosgrove GP, Solomon GM, Nichols DP, Seibold MA. Airway Progenitor Clone Formation is Enhanced by Y-27632-dependent Changes in the Transcriptome. Am J Respir Cell Mol Biol. 2016.
36. Walters BJ, Diao S, Zheng F, Walters BJ, Layman WS, Zuo J. Pseudo-immortalization of postnatal cochlear progenitor cells yields a scalable cell line capable of transcriptionally regulating mature hair cell genes. Sci Rep. 2015; 5: 17792. doi: 10.1038/srep17792.
37. Brown DD, Dabbs DJ, Lee AV, McGuire KP, Ahrendt GM, Bhargava R, Davidson NE, Brufsky AM, Johnson RR, Oesterreich S, McAuliffe PF. Developing in vitro models of human ductal carcinoma in situ from primary tissue explants. Breast Cancer Res Treat. 2015; 153: 311-21. doi: 10.1007/s10549-015-3551-8.
38. Kasper S. Stem cells: The root of prostate cancer? Journal of Cellular Physiology. 2008; 216: 332.
39. Kurita T, Medina RT, Mills AA, Cunha GR. Role of p63 and basal cells in the prostate. Development. 2004; 131: 4955-64. doi: 10.1242/dev.01384.
40. Lawson DA, Zong Y, Memarzadeh S, Xin L, Huang J, Witte ON. Basal epithelial stem cells are efficient targets for prostate cancer initiation. Proc Natl Acad Sci U S A. 2010; 107: 2610-5. doi: 10.1073/pnas.0913873107.
41. Tran CP, Lin C, Yamashiro J, Reiter RE. Prostate stem cell antigen is a marker of late intermediate prostate epithelial cells. Molecular Cancer Research. 2002; 1: 113-21.
42. Uzgare AR, Xu Y, Isaacs JT. In vitro culturing and characteristics of transit amplifying epithelial cells from human prostate tissue. J Cell Biochem. 2004; 91: 196-205. doi: 10.1002/jcb.10764.
43. Suprynowicz FA, Upadhyay G, Krawczyk E, Kramer SC, Hebert JD, Liu X, Yuan H, Cheluvaraju C, Clapp PW, Boucher RC Jr, Kamonjoh CM, Randell SH, Schlegel R. Conditionally reprogrammed cells represent a stem-like state of adult epithelial cells. Proc Natl Acad Sci U S A. 2012; 109: 20035-40. doi: 10.1073/pnas.1213241109.
44. Schwarze SR, Shi Y, Fu VX, Watson PA, Jarrard DF. Role of cyclin-dependent kinase inhibitors in the growth arrest at senescence in human prostate epithelial and uroepithelial cells. Oncogene. 2001; 20: 8184-92.
45. Kokontis JM, Lin HP, Jiang SS, Lin CY, Fukuchi J, Hiipakka RA, Chung CJ, Chan TM, Liao S, Chang CH, Chuu CP. Androgen suppresses the proliferation of androgen receptor-positive castration-resistant prostate cancer cells via inhibition of Cdk2, CyclinA, and Skp2. PLoS One. 2014; 9: e109170.
46. Zhou W, Gui M, Zhu M, Long Z, Huang L, Zhou J, He L, Zhong K. Nicotinamide -methyltransferase is overexpressed in prostate cancer and correlates with prolonged progression-free and overall survival times. Oncol Lett. 2014; 8: 1175-80.
47. Nagpal ML, Davis J, Lin T. Overexpression of CXCL10 in human prostate LNCaP cells activates its receptor (CXCR3) expression and inhibits cell proliferation. Biochim Biophys Acta. 2006; 1762: 811-8.
48. Wright GW, Simon RM. A random variance model for detection of differential gene expression in small microarray experiments. Bioinformatics. 2003; 19: 2448-55.
49. Panaccione A, Chang MT, Carbone BE, Guo Y, Moskaluk CA, Virk RK, Chiriboga L, Prasad ML, Judson B, Mehra S, Yarbrough WG, Ivanov SV. NOTCH1 and SOX10 are Essential for Proliferation and Radiation Resistance of Cancer Stem-Like Cells in Adenoid Cystic Carcinoma. Clin Cancer Res. 2016; 22: 2083-95. doi: 10.1158/1078-0432.CCR-15-2208.
50. Beglyarova N, Banina E, Zhou Y, Mukhamadeeva R, Andrianov G, Bobrov E, Lysenko E, Skobeleva N, Gabitova L, Restifo D, Pressman M, Serebriiskii IG, Hoffman JP, et al. Screening of conditionally reprogrammed patient-derived carcinoma cells identifies ERCC3-MYC interactions as a target in pancreatic cancer. Clin Cancer Res. 2016. doi: 10.1158/1078-0432.CCR-16-0149.
51. Suetens A, Moreels M, Quintens R, Soors E, Buset J, Chiriotti S, Tabury K, Gregoire V, Baatout S. Dose- and time-dependent gene expression alterations in prostate and colon cancer cells after in vitro exposure to carbon ion and X-irradiation. J Radiat Res. 2015; 56: 11-21.
52. Stacey SN, Kehr B, Gudmundsson J, Zink F, Jonasdottir A, Gudjonsson SA, Sigurdsson A, Halldorsson BV, Agnarsson BA, Benediktsdottir KR, Aben KK, Vermeulen SH, Cremers RG, et al. Insertion of an SVA-E retrotransposon into the CASP8 gene is associated with protection against prostate cancer. Hum Mol Genet. 2016; 25: 1008-18.
53. Marhold M, Tomasich E, El-Gazzar A, Heller G, Spittler A, Horvat R, Krainer M, Horak P. HIF1alpha Regulates mTOR Signaling and Viability of Prostate Cancer Stem Cells. Mol Cancer Res. 2015; 13: 556-64.
54. Mirzoeva S, Franzen CA, Pelling JC. Apigenin inhibits TGF-beta-induced VEGF expression in human prostate carcinoma cells via a Smad2/3- and Src-dependent mechanism. Mol Carcinog. 2014; 53: 598-609.
55. Kato T, Kawai K, Egami Y, Kakehi Y, Araki N. Rac1-dependent lamellipodial motility in prostate cancer PC-3 cells revealed by optogenetic control of Rac1 activity. PLoS One. 2014; 9: e97749.
56. Baca SC, Prandi D, Lawrence MS, Mosquera JM, Romanel A, Drier Y, Park K, Kitabayashi N, MacDonald TY, Ghandi M, Van Allen E, Kryukov GV, Sboner A, et al. Punctuated evolution of prostate cancer genomes. Cell. 2013; 153: 666-77. doi: 10.1016/j.cell.2013.03.021.
57. Miki J, Furusato B, Li H, Gu Y, Takahashi H, Egawa S, Sesterhenn IA, McLeod DG, Srivastava S, Rhim JS. Identification of Putative Stem Cell Markers, CD133 and CXCR4, in hTERT-Immortalized Primary Nonmalignant and Malignant Tumor-Derived Human Prostate Epithelial Cell Lines and in Prostate Cancer Specimens. Cancer Research. 2007; 67: 3153-61. doi: 10.1158/0008-5472.can-06-4429.
58. Dennis G Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003; 4: P3.
59. Timofeeva OA, Zhang X, Ressom HW, Varghese RS, Kallakury BV, Wang K, Ji Y, Cheema A, Jung M, Brown ML, Rhim JS, Dritschilo A. Enhanced expression of SOS1 is detected in prostate cancer epithelial cells from African-American men. Int J Oncol. 2009; 35: 751-60.
60. S. VB, A B. (1995). Human Chromosomes: Principles and Techniques (2nd edition): McGraw-Hill, Inc).
61. Palechor-Ceron N, Suprynowicz FA, Upadhyay G, Dakic A, Minas T, Simic V, Johnson M, Albanese C, Schlegel R, Liu X. Radiation induces diffusible feeder cell factor(s) that cooperate with ROCK inhibitor to conditionally reprogram and immortalize epithelial cells. Am J Pathol. 2013; 183: 1862-70. doi: 10.1016/j.ajpath.2013.08.009.