
INTRODUCTION
Under normal physiological conditions, the major function of PD-1 is to inhibit effector T-cell activity and enhance the function and development of Tregs, which inhibit T-cell responses and prevent overstimulation of immune responses in peripheral tissues [1, 2]. The PD-1 pathway can protect the host against autoimmunity [3].
PD-1 pathway plays a key role in the regulation of antifungal and virus immunity [4]. PD-1 knockout (Pdcd1−/−) mice can lead to tissue sensitive to infection or late onset autoimmune disease with variable incidence depending on the background strain carrying the PD1 null animal [5-7]. Whereas, blockade of PD-1 enhances both proliferation of memory B cells and expansion of virus-specific CD8 T cells during chronic simian immunodeficiency virus (SIV) infection in macaques [8]. In persistently infected mice lacking CD4 T-cell help, blockade of the PD-1/PD-L1 inhibitory pathway had a beneficial effect on the ‘helpless’ CD8 T cells, restoring their ability to undergo proliferation, secrete cytokines, kill infected cells, and decrease the viral load [9].
Although tumor cells express numerous antigens, tumor evades T-cell responses and host immunity through negative regulators of the immune system, such as programmed death-1 (PD-1), programmed cell death-ligand-1 (PD-L1)/programmed cell death-ligand-2 (PD-L2), cytotoxic T lymphocyte antigen 4 (CTLA-4), T-cell immunoglobulin mucin 3 (TIM3), 2B4, the B and T lymphocyte attenuator (BTLA), and lymphocyte-activation gene 3 (LAG3) [10-12]. Among these regulators, CTLA-4 is a type 1 transmembrane glycoprotein mainly expressed on activated T cells. CTLA-4 inhibits T-cell function through intracellular signaling regulation via T-cell receptor (TCR) and CD28 in tumors [12, 13]. LAG3 (CD223) is a type I membrane glycoprotein of the immunogloblin (Ig) superfamily expressed in several different cell types, such as plasmacytoid dendritic cells (DCs), B cells, natural killer T cells, γ and δ T cells, exhausted CD8+ T cells, and regulatory T cells (Tregs). Association of LAG3 with PD-1 inhibits signaling passway in T-cell [12, 14]. TIM3 is a transmembrane molecule associated with CD8+ T-cell dysfunction and exhaustion. TIM3 is overexpressed on Tregs in tumor microenvironment. Tregs is related to ovarian tumor size. Blockade of TIM3 restores the inhibitory functions of tumor-infiltrating Tregs [15]. PD-1 and PD-L1/PD-L2 are identified as immune checkpoints that inhibit effector T-cell activity [1, 16].
PD-L1 is overrepresented in the presence of tumor and promotes immune evasion and growth of tumor by suppressing T-cell response [17]. PD-1/PD-L1 plays critical roles in cancer immunology, and blocking antibodies against this receptor provide benefits in clinical trials, with the first of this class recently approved by the Food and Drug Administration (FDA) to treat patients with refractory malignancies [16]. Recently, blockade of PD-1/PD-L1 has been found to treat effectively cancer by enhancing immunity. Several studies on Abs blockade of the PD-1 receptor (nivolumab, MK3475, or combination of nivolumab with the anti-CTLA4 checkpoint inhibitor ipilimumab) have improved survival profiles and acquired high response rates in several solid tumors [18-22]. In melanoma refractory to targeted therapy, pembrolizumab which is a humanized monoclonal IgG4-kappa isotype antibody against PD-1 induced overall response rates (ORRs) of 21%-34%. Among the patients with refractory non-small cell lung cancer (NSCLC), pembrolizumab induced ORRs of 19%-25%. On the basis of these results, pembrolizumab was approved by the USA FDA to treat advanced melanoma and NSCLC [23].
The function of PD-1 in peripheral tolerance and anti-tumor immune response is well established. Moreover, blockade of the PD-1 pathway has achieved good effect on restraining tumor. However, the exact mechanism of dysregulation of PD-1 and its ligands is still unknown. In addition, the manner of PD-1 ligation exerting its effects on specific signaling targets and how these altered signaling events affect T-cell function are yet to be completely understood.
PD-1 AND THE REGULATION OF PD-1 EXPRESSION
PD-1 (also called CD279) was first isolated from 2B4.11 (a murine T-cell hybridoma) and interleukin-3 (IL-3)-deprived LyD9 (a murine hematopoietic progenitor cell line) by using subtractive hybridization technique [24]. PD-1 is encoded by the Pdcd1, which is located on chromosome 2 (2q37) [25-27]. PD-1 is one of the member of B7/CD28 family [28, 29]. PD-1 is also a 288 amino acid (55 kDa) type I transmembrane protein of the immune globulin superfamily, comprising an extracellular N-terminal IgV-like domain, a transmembrane domain, and a cytoplasmic tail [24, 26, 30]. There are two tyrosine residues in the cytoplasmic tail of PD-1; the N-terminal of which is involved in a sequence defined as the immunoreceptor tyrosine-based inhibitory motif (ITIM, I/L/VXYXXL/V); the C-terminal tyrosine is engaged in a sequence defined as immunoreceptor tyrosine-based switch motif (ITSM, TxYxxL) [24, 25, 31, 32]. The amino acid sequence around the C-terminal tyrosine (TEYATIVF) of PD-1 is well conserved between mouse and human and is related to SHP-1 and SHP-2. Whereas, the N-terminal tyrosine of PD-1 does not associate with either SHP-1 or SHP-2 [33].
PD-1 can promote the development, immunity evasion, and prognosis of several kinds of solid tumor, such as NSCLC, melanoma, breast cancer (BC), and renal cell carcinoma (RCC) [34]. Thompson’s research showed that PD-1 was expressed in 56% of nephrectomy specimens of patients with RCC. PD-1 is also expressed in the T cells rather than in RCC tumor cells. Furthermore, the expression of PD-1 was associated with tumor stage, the presence of necrosis or sarcomatoid differentiation, and poor 5 year survival rate [35]. In classical Hodgkin lymphoma (cHL) and mediastinal large B-cell lymphoma, the extended PD-1 ligand/9p24.1 amplification region contains the Janus kinase 2 (JAK2) locus. JAK2 amplification promoted protein expression and activity, specifically inducing PD-1 ligand transcription and enhancing sensitivity to JAK2 inhibition. Therefore, PD-1 ligand/9p24.1 amplification is a disease-specific structural alteration that increases both the gene dosage of PD-1 ligands and their induction by JAK2, defining the PD-1 pathway and JAK2 as complementary rational therapeutic targets [36]. Programmed death 1 expression in the peritumoral microenvironment is an independent prognostic factor for Overall Survival (OS) of patient of cHL and is related to poor prognosis in cHL [37]. Similarly, epithelial-originated malignancy patients with PD-1 positive expression on TILs exhibited significantly shorter OS than the PD-1 negative group [38].
PD-1 is inducibly expressed on activated immune cell types including CD4+ T cells, CD8+ T cells, B cells, natural killer T cells, activated monocytes, DCs, macrophages [10, 28, 29, 39]. Moreover, PD-1 is selectively upregulated in T cells because of persistent exposure to antigens; thus, the expression of PD-1 in T cells is one of the makers of exhausted T cells [40-42]. A few mechanisms are involved in PD-1 expression regulation [43, 44]. Two upstream conserved regulatory regions of PD-1 gene termed as conserved regions B and C (CR-B and CR-C) exist. CR-B and CR-C are hypersensitive to DNase I and are important for PD-1 expression. CR-C was reported to contain a nuclear factor of activated T cells (NFAT) site, which is important for the transcriptional expression of Pdcd1, whereas, the role of CR-B is yet to be known [45]. In CD4+ and CD8+ T cells, the transcriptional activator nuclear factor of activated T cells c1 (NFATc1; also known as NFAT2) binding to CR-C and c-Fos binding to a site located in CR-B enhance the expression of PD-1 after TCR stimulation during the initial phases of Ag recognition [46, 47]. Xiao et al. identified that tumor-infiltrating T cells significantly upregulated the expression of the activator protein 1 (AP-1) subunit c-Fos. C-Fos (AP-1) directly binds to CR-B in the Pdcd1 (gene encoding PD-1) proximal promoter which increases PD-1 expression and enhances antitumor T cell function and restrained tumor growth [48, 49]. Bally et al. identified that NF-ΚB p65 binds to a region which located upstream of PD-1 gene in CR-C and enhances PD-1 expression following stimulation of macrophages with lipopolysaccharide (LPS) [50].
The 55 kDa src kinase-associated protein (SKAP55) and the adhesion and degranulation promoting adaptor protein (ADAP) are located at the killing synapses between CD8+ CTLs and tumor cells [51, 52]. Specifically, ADAP binds to SKAP55 and stabilizes its expression at protein level. Most importantly, ADAP-SKAP55 enhances PD-1 expression in a Fyn-, Ca2+-, and NFATc1 manner. ADAP-SKAP55 module enhances both total and activated NFATc1, which enhances PD-1 expression by binding to the promoter of PD-1. The ADAP-SKAP55-PD-1 pathway represents a “self-control” mechanism to control T-cell activation and adhesion precisely [46].
Multiple cytokines, such as the common γ-chain family (IL-2, IL-7, IL-15, and IL-21) and type I IFNs (IFN- and IFN-β) can also upregulate PD-1 expression [53]. However, with the exception of IFN-α inducing responses from an interferon-stimulated regulatory element (ISRE) located in CR-C, no direct effect of cytokine-induced factors regulating Pdcd1 gene expression has been shown [49]. Kato et al. demonstrated that the concentration of IL-6 was in a high level, which is correlated to poor growth of cytomegalovirus (CMV)-specific T cells and high PD-1 expression on CMV specific T cells, and that disruption of IL-6 or the IL-6 receptor (IL-6R) interaction recovered CMV-specific T-cell growth. IL-6 and IL-12 induce the signal transducer and activator of transcription (STAT) activity STAT3 and STAT4, respectively, via the JAK family of proteins. STAT activity could change the chromatin structure of Pdcd1 and increase the PD-1 expression in splenic CD8 T cells. The NFATc1/STAT regulatory regions interact with the promoter region of the Pdcd1 gene and increase PD-1 expression following cytokine stimulation. Austin et al. found that Pdcd1 was regulated by distal elements, which is a non-biased approach employed across the murine Pdcd1 locus. Their group also found four novel distal regulatory regions. Two of these elements is located on the side of CCCTC-binding factor (CTCF). The third element, located upstream of CR-C, bound NFATc1 and STAT3 or STAT4 in response to TCR and IL-6 or IL-12 signaling, respectively. The final region, located close to the downstream CTCF site also bound NFATc1 and STAT3 or STAT4. Each of the novel NFAT/STAT elements interacts with the Pdcd1 promoter region and the chromatin structure of each regulatory region is altered in response to T-cell activation and cytokine stimulation in CD8 T cells, demonstrating that NFAT/STAT elements is associated with PD-1 expression [49, 54]. Vascular endothelial growth factor-A (VEGF-A) promotes PD-1 expression and other inhibitory checkpoints, which are involved in exhaustion of vascular endothelial growth factor receptor (VEGFR) expressing CD8+ T cells in vitro. Voron et al. identified that blockade of VEGF-A-VEGFR was sufficient to decrease PD-1 expression in intratumoral CD8+ T cells. Sunitinib, a multitarget tyrosine kinase inhibitor (TKI) that inhibits VEGFR1, R2, R3, platelet-derived growth factor receptors, and stem cell factor receptor, has been shown to suppress PD-1 expression at the mRNA level in tumor-infiltrating T cells [43, 55]. In addition, endogenous transforming growth factor-β (TGF-β) is also involved partially in PD-1 expression through TCR activation in T cells [56]. The main signaling pathways of PD-1 transcriptional regulation are shown in Figure 1.
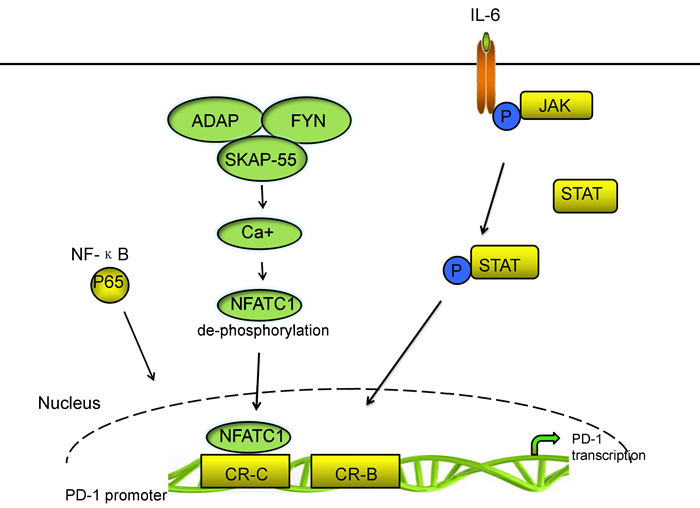
Figure 1: The main signal pathways of PD-1 transcriptional regulation.
By contrast, some transcriptional repressors inhibit PD-1 expression [47]. T-bet binds directly to Pdcd1 within a region of ~500 bp upstream of the transcriptional start site, near or within the CR-B region. This condition directly represses Pdcd1 transcription. When the antigen is persisted, T-bet expression is decreased in T cells; thus PD-1 expression is increased and functions of T cells are inhibited [45]. Meanwhile, the repressive effect of T-bet on PD-1 expression is not enhanced when T-bet is overexpressed. This condition indicates that other factors may also participate in inhibiting PD-1 expression. The B lymphocyte-induced maturation protein 1 (Blimp-1), which is encoded by the prdm1 gene, represses PD-1 gene expression through a feed-forward repressive circuit. On one hand, Blimp-1 binds to a site between CR-B and CR-C of the PD-1 gene to inhibit directly PD-1 gene transcription. On the other hand, Blimp-1 inhibits the expression of NFATc1 and displaces it from CR-C, leading to downregulation of PD-1 expression [47].
PD-1 LIGANDS AND THE REGULATION OF THEIR EXPRESSION
PD-1 is involved in immune tolerance by suppressing activated immune cells via interaction with its ligands. Two known ligands of PD-1 are PD-L1 and PD-L2 [3, 29, 57]. B7-H1 was originally named as the first gene homolog of B7 molecules, and B7-H1 was renamed as PD-L1 after it has been identified as the first ligand of the receptor PD-1 (CD279) in the murine system [58]. PD-L1 gene is located in chromosome 9p24 [36, 59]. PD-L1 is a 290-amino-acid transmembrane glycoprotein [58, 60]. The second known counter-receptor of PD-1, called B7-DC or PD-L2, is also a member of the B7 family [58]. Hino et al. indicated that the degree of PD-L1 expression was correlated to the vertical growth of primary tumors in melanoma. Furthermore, multivariate analysis demonstrated that the survival rate of the PD-L1 high-expression patients was remarkably lower than that of the low-expression patients with stage II melanoma, which indicated that PD-L1 expression was an independent, poor prognostic factor for malignant melanoma [61]. PD-L2 may lead to local cytokine production that is beneficial to the tumor cells [62]. PD-L1 and PD-L2 play different roles in immune regulatory process although both are ligands of PD-1 [63]. PD-L1 inhibits T-cell function in peripheral tissues, whereas PD-L2 suppresses immune T-cell activation in lymphoid organs. PD-L2 inhibits type 2 T-helper (Th-2) lymphocytes, but its role is yet to be fully understood [53, 64].
PD-L1 is expressed on immune cells, including T cells, B cells, DCs, and macrophages [3, 57]. PD-L1 is overrepresented on several types of solid tumor cells, such as glioblastoma multiforme, NSCLC, and some hematologic malignancies [34]. Unlike PD-L1, which is expressed broadly, the expression of PD-L2/B7-DC is limited. PD-L2 is expressed mainly on antigen-presenting cells (APCs), including macrophages and myeloid DCs, and non-hematopoietic tissues, such as the lung [62, 63]. PD-L1 and PD-L2 are expressed on Respiratory tract epithelial cells (BEAS-2B cells). Moreover, the expression of respiratory tract epithelial cells is upregulated by respiratory tract virus infection or treatment with interferon-γ (IFN-γ) and IL-4. PD-L1 was moderately expressed, and PD-L2 was weakly expressed in unstimulated NCI-H292, BEAS-2B, and A549 cells [65]. Similarly, Kan-o et al. identified that polyinosinic-polycytidylic acid (poly IC) upregulates the expression of B7-H1 via activation of the NF-κB. Poly IC increases the generation of reactive oxygen species, which enhances the activation of PI3Kδ and NF-κB. In addition, poly IC-induced upregulation of B7-H1 was observably suppressed by a pan-PI3K inhibitor and partially by an inhibitor or a small interfering (si)RNA for PI3Kδ in BEAS-2B cells [66].
Both PD-L1 mRNA and protein can be upregulated by cytokines produced by infiltrating immune cells, such as IFN-γ, IL-4, IL-10, growth cell stem factors, bacterial LPS, and VEGF [10, 26, 44, 59, 67]. Several pathways exist that IFN-γ increases PD-L1 expression. Abiko et al. found that IFN-γ secreted by CD8+ lymphocytes upregulates PD-L1 in ovarian cancer cells and promotes progression of ovarian cancer. In mouse models, suppressing IFNGR1 (IFN-γ receptor 1) remarkably reduced the level of PD-L1 expression in tumor cells. By contrast, the injection of IFN-γ into subcutaneous tumors increased PD-L1 expression and promoted tumor growth [68]. Moreover, IFN-γ or toll-like receptor (TLR) stimulation upregulated PD-L1 expression in blast cells from patients with acute myeloid leukemia through MEK/ERK- and MyD88/TRAF6 pathway [69]. Chen et al. demonstrated that protein kinase D isoform 2 (PKD2), which is induced by IFN-γ, is an important regulator of PD-L1 expression in human oral squamous carcinoma cells. Inhibition of PKD2 activation not only suppresses PD-L1 expression and enhances an anti-tumor effect but also reduces drug resistance during chemotherapy [69]. By contrast, natural killer (NK) cell activation and secretion of IFN-γ significantly enhanced PD-L1 expression by activating JAK1, JAK2, and STAT1 in tumor cells. Inhibition of JAK pathway activation abrogates increased PD-L1 expression, which enhanced sensibility of tumor cells to NK cell activity [70]. Maine et al. identified that the concentrations of IL-10 and TGF-β in ascites were higher in patients with malignant ovarian tumors than with benign/border-line ovarian tumors. Both IL-10 and TGF-β can increase PD-L1 in monocytes in vitro. Blocking IL-10 with a neutralizing antibody reduced PD-L1 expression [71].
PD-L1 can be upregulated not only by some inflammatory cytokines but also by constitutive oncogene pathway activation [72]. In gene level, oncogenic signaling pathways in tumor cells, such as IFN-γ/JAK2/IFN, ALK/STAT3, PI3K, and MEK/ERK/STAT1 can activate PD-L1 expression [72-74]. Chen et al. demonstrated that the expression of PD-L1 was higher in EGFR-mutant NSCLC cell lines than that in cell lines with wild-type EGFR. Three models of EGFR activation including EGF stimulation, EGFR-19del and EGFR-L858R mutation can increase PD-L1 expression via p-ERK1/2/p-c-Jun and p-AKT/p-S6 pathway [72]. Moreover, exposure to EGFR inhibitor-TKIs can decrease the expression of PD-L1 [73]. Ota’s research has shown that the level of PD-L1 expression in NSCLC cells positive for EML4-ALK is higher than those in wild-type for both EGFR and ALK. EML4-ALK can upregulate PD-L1 expression at the mRNA and protein levels in Ba/F3 via activating PI3K-AKT [74]. The receptor tyrosine kinase c-Met binding to its ligand hepatocyte growth factor can remarkably promote the expression of PD-L1 via activating Ras-PI3K signaling pathway, and this condition can be disrupted following treatment of the cells with pharmacological inhibitors of c-Met [75]. Moreover, Parsa et al. identified that the loss of phosphatase and tensin homolog (PTEN) and the resulting activation of phosphatidylinositol-3-OH kinase (PI-3K) pathway enhanced PD-L1 expression in glioma [72, 76].
In addition, hypoxia-inducible factor-1α (HIF-1α) is also a major regulator of PD-L1 mRNA and protein expression. Hypoxia causes a rapid, dramatic, and selective upregulation of PD-L1 in splenic myeloid-derived suppressor cells (MDSCs), macrophages, DCs, and tumor cells in tumor-bearing mice through HIF-1α. Binding of HIF-1α to a transcriptionally active hypoxia-response elements (HREs), HRE-4 and HRE-1, at two different HRE sites in the PD-L1 proximal promoter in hypoxic MSC-1 cells is comparable with their binding to an established HRE in VEGF, lactate dehydrogenase A (LDHA), and Glut1 genes [77]. Boes et al. identified that TLR3 triggering results in remarkably upregulation of PD-L1 on neuroblastoma cells. In addition, docking protein 3 (DOK3) increases PD-1 ligand expression through abrogating the intensity of calcium signaling at the transcriptional level. DOK3 recruits growth factor receptor-bound protein 2 (Grb2). Together, DOK3 and Grb2 sequester Bruton tyrosine kinase and diminish PLCγ 2 activation and, thereby, attenuate calcium signaling. Calcineurin inhibition study demonstrated that calcium signaling directly negatively regulates PD-1 ligand gene expression [78-82]. Above all, the main signaling pathways of PD-L1 transcriptional regulation are shown in Figure 2.
Despite recent study efforts on PD-L2, the transcriptional regulation of the ligand is yet to be completely clarified [63]. Several inflammatory cytokines, especially IL-4, upregulate the expression of PD-L2 on DCs and macrophages [34, 83, 84].
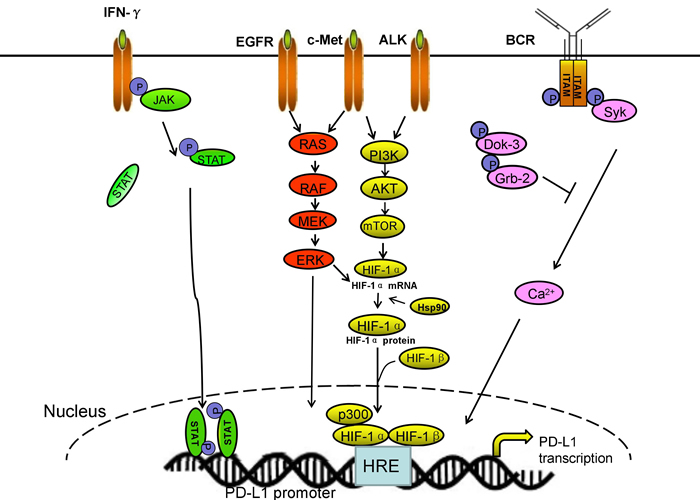
Figure 2: The main signal pathways of PD-L1 transcriptional regulation. Multiple pathways promote PD-L1 expression on transcriptional level.
FUNCTIONAL IMPLICATIONS OF ALTERED BIOCHEMICAL SIGNALING INDUCED BY PD-1
PD-1/PD-L1 axis inhibits T cell response
PD-1/PD-L1 suppresses the effector phase of T-cell response. This condition induces immune tolerance during different phases of T-cell responses, such as regulating the threshold for T-cell activation, inhibiting T-cell proliferation, and promoting apoptosis in activated T cells. The mechanism of PD-1/PD-L1 regulation is correlated with dephosphorylation of signaling molecules belonging to TCR pathway and transmission of inhibitory signal to T cells [31, 85-88]. TCR signaling leads to intracellular Ca2+ flux, which activates multiple signaling pathways required for differentiation and activation of T-cell. PD-1 on T-cell binding to PD-L1 or PD-L2 on APC can activate Lck-mediated phosphorylation of cytoplasmic domain tyrosine and recruitment of SHP-2 to the C-terminal tyrosine of PD-1 cytoplasmic domain [27, 89]. SHP-2 and SHP-1 are two highly related tyrosine phosphatases, which dephosphorylate TCR-proximal signaling molecules, such as protein kinase Cθ, CD3ζ, PI3K and zeta-chain-associated protein kinase (ZAP70), and Syk downstream of B cell antigen receptor (BCR), leading to inhibition of downstream signaling [89, 90]. SHP-2 and SHP-1 are recruited to ITSM of PD-1, leading to inhibition of the PI3K/AKT and mitogen-activated protein kinase signaling pathways downstream of the TCR and blockade of cell-cycle progression in the immune cells [91]. Whereas, PD-1/PD-Ls inhibit Ca2+ flux increasing the number of engaged TCRs required to initiate a Ca2+ flux [92].
PI3K/Akt pathway activation increases Glut1 expression and enhances glucose uptake inducing glycolysis and protein synthesis in T cells [93-95]. After PD-1 recruits the SHP-1 and SHP-2 phosphatase, PI3K/Akt pathway was inhibited. This condition inhibits cell survival proteins, such as B-cell lymphoma-xL (Bcl-xL) which is important for the intrinsic apoptotic pathway; decreases expression of glucose transporters on the plasma membrane and general downregulation of glycolytic enzyme activity, which depresses proliferation of T cells, thereby restraining its survival and decreasing protein synthesis; inhibits CD28-mediated activation and triggers chromatin changes so that the promoter regions of IL-2, TNF-α, and IFN-γ are decreased [28, 96]. Downregulation of IL-2 secretion, driven partially through early growth response protein 2 (EGR2), induces CD8+ T and CD4+ T-cell anergy [97, 98].
Moreover, studies revealed that engagement of PD-1 by PD-L1 inhibited PLC-γ1 and RAS activation and suppressed MEK/Erk MAP pathway [99, 100]. CalDAG is activated downstream of PLC-γ156 promote Ras GRP1 activation. Ras GRP1 promotes exchange of Ras-GDP to Ras-GTP, which suppresses MEK/Erk, suppressing T-cell proliferation. The effect of PD-1 on MEK/Erk and MAP kinases was selective because PD-1 ligation did not inhibit the activation of Jnk and p38 MAP kinases [100, 101]. Engagement of PD-1 and PD-L1 inhibits multiple transcription factors of T cells expression, such as GATA-3 and T-bet, repressing T-cell response [10, 102]. T-bet is a master transcription factor, which can downregulate inhibitory receptors and is essential for differentiation and function of Th1 cells, CD8+ T cells [45, 101, 103]. High level of T-bet maintains exhausted CD8+ T cells and represses inhibitory receptor expression [45]. In addition, the patients with increased level of T-bet+ lymphocytes in tumor of nest and stroma exhibit longer survival time than the low level of such cells [94, 104].
PD-1 can also suppress cell cycle progression of T cell by influencing various regulators of the cell cycle. However, the mechanism is yet to be totally understood. PD-1 signaling prevents cell cycle progression in the G1 phase by increasing the cyclin-dependent kinase (Cdk) inhibitors p27 kip1 and p15 INK4B and repressing the Cdk-activating phosphatase Cdc25A expression. PD-1 inhibits Skp2 by inhibiting PIK3/Akt and Ras/MEK/ERK signaling leading to increase of p27kip1 and inhibition of Cdk2. Cdk2 restraint decreases phosphorylation of Rb and fails to phosphorylate the checkpoint inhibitor Smad3, which inhibits cell cycle progression from the G1 to the S phase in the Cdk2-specific site enhancing transcriptional activity of Smad3 and upregulating p15INK4B expression and restraining the Cdk-activating phosphatase Cdc25A [105, 106].
PD-1/PD-L1 inhibits tumor-infiltrating lymphocytes (TIL) and increases immuno-suppressive Treg function
Engagement of PD-1 and PD-L1 restrains the proliferation, survival, and effector function of CD8+ CTL and promotes apoptosis of TILs [10, 107]. High level of PD-1 expression along with other inhibitory receptors inhibits functions of TILs and decreases its quantity in tumor microenvironment [35, 83, 108]. In addition, TILs increase expression of PD-L1, tryptophan-catabolizing enzyme indoleamine-2,3-dioxygenase (IDO), and FoxP3+ Tregs in the melanoma tumor microenvironment. This condition indicates that TILs are involved in immune-intrinsic negative feedback loop. IDO is in charge of the dissimilation of tryptophan, and influences immune reactions in several situations. PD-1/PD-L1 interaction increases the level of IDO, which exhausts T cells of essential tryptophan and suppresses its metabolites, leading to inhibition of T-cell activation and increasing the number of regulatory T cell [109, 110].
PD-1 pathway activation not only downregulates effector T-cell function but also increases immuno-suppressive Treg function [63]. PD-L1 expression on non-hematopoietic and hematopoietic cells accelerates Treg development and improves Treg function in immune organs and autoimmune attacked tissues [3, 111]. PD-1−/− conventional CD4+ T cells demonstrated a remarkably diminished tendency toward differentiation into peripherally induced Treg (pTreg) cells, which showed that PD-1 is critical for the extrathymic differentiation of pTreg cells in vivo [112]. Previous studies suggested that the number of circulating Tregs of lung cancer patients was nearly twofold compared with healthy controls and the expression levels of PD-1 on Tregs were higher in lung cancer samples than in controls [113]. These results suggest that the PD-1/PD-L1 pathway plays a role in Treg induction and is associated with impaired adaptive immunity. In the tumor microenvironment, PD-1 expressed on Tregs accelerates CD4+ T cells differentiating into Foxp3+ Tregs under the circumstances of CD3 and TGF-β. Foxp3 is a critical transcription factor of Tregs, which suppress Th1 responses [94]. Foxp3+ Tregs is a highly immunosuppressive subset of CD4+ T cells that is critical in s uppressing proliferation and cytokine production of other T cells, inhibiting tumor-specific immune responses and maintaining peripheral immune tolerance in cancer patients [114]. Meanwhile, Treg cells express constitutive high levels of PD-1, which enhance Treg functional response or proliferation and inhibit T cells responses [115, 116].
ANTIBODY BLOCKADE OF PD-1/PD-L1 ON THE TREATMENT OF TUMORS
Efficacy of inhibition of the PD-1 pathway
Blocking PD-1 passway successfully improves T-cell responses in vitro and promotes tumor regression in vivo in animals [117, 118]. In vitro, antibody blockade of PD-1/PD-L1 enhances antitumor immune responses by decreasing the number and/or the suppressive activity of regulatory T cells and by rescuing of the activity of effector T cells in tissues and the tumor microenvironment. In addition, PD-1/PD-L1 antibody blockade decreases the percentages of the highly immunosuppressive MDSC population. Likewise, blockade of PD-1 in B cells may also enhance activity of natural killer cells and increase antigen-specific antibody production via PD-1 positive (PD-1+) B cells [63, 119]. Several studies on syngeneic mouse tumor models demonstrated that the blockade of PD-1/PD-L1 enhances antitumor activity. The level of effector CD4+ T and CD8+ T cells, B cells, and myeloid-derived suppressor cells increased in tumor in mice with PD-1 blockade injected with B16 melanoma cells; T cell proliferation and cytokine production were also enhanced, and tumor sites recruited more effector cells [120, 121]. Blockade of PD-1/PD-L1 has significant influences on different CD4+ T-cell subsets. PD-1 blockade enhanced production of IFN, IL-2, TNF-α, IL-6, and IL-17 and the reduced production of the Th2 cytokines IL-5 and IL-13 [122, 123]. This condition reveals that PD-1 blockade may promote antitumor activity through tipping the Th1/Th2 balance and through stimulation of Th17 cells.
PD-1/PD-L1 blockade demonstrates suppression of tumor growth and less metastases. In the mouse model of bladder cancer, the antibody blockade of PD-1 can increase the number of circulating tumor-specific CD107a-expressing CD8+ T cells and activated (CD25+ FoxP3-) CD4+ splenocytes, as well as significantly reduces tumor size [124]. Monoclonal antibodies blockade of PD-1/PD-L1 has revealed great effect outcomes for a subset of patients with cancer, especially in PD-L1 positive tumors, such as melanoma, hepatocellular carcinoma, lung, kidney, and esophageal cancers, as well as hematological malignancies [19, 112]. The expression of PD-L1 in tumor cells is related to the response of PD-1/PD-L1 inhibitors and may be proposed as a potentially valuable predictive marker for the responsiveness of various cancers, including malignant melanoma, NSCLC, and RCC to PD-L1 or PD-1 blocking antibodies [125].
Clinical trials of mAbs to PD-1
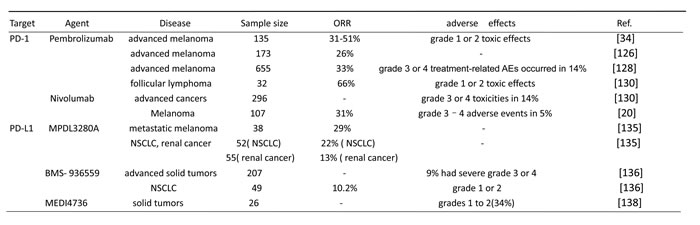
Pembrolizumab is a highly selective, humanized monoclonal IgG4-kappa isotype antibody against PD-1, which binds to the PD-1 receptor on T cells and prevents PD-1 binding to its ligands PD-L1 and PD-L2 [126]. Pembrolizumab is the first PD-1 checkpoint inhibitor for advanced melanoma approved by FDA after the CTLA-4 inhibitor-ipilimumab [127]. Pembrolizumab has remarkable anti-tumor activity and treatment-related toxicity is acceptable. A total of 135 patients with advanced melanoma were in phase 1 study of pembrolizumab. Approximately 38%-52% of patients treated with doses ranging from 2 mg/kg every 3 weeks to 10 mg/kg every 2 weeks showed long-lasting objective responses. In addition, 81% of patients survived for at least 1 year from the beginning of treatment. Grade 1 or 2 adverse events were shown in the majority of patients. 13% of patients have shown grade 3 or 4 adverse events. This result indicated that patients with advanced melanoma treated with pembrolizumab result in a high rate of sustained tumor regression [34]. Robert et al. assessed the clinical effect of pembrolizumab. 173 patients with advanced melanoma received pembrolizumab treatment. The follow-up time was 8 months and ORR was 26%. This result suggested that pembrolizumab is an effective treatment option for patients with ipilimumab-refractory advanced melanoma, wherein few effective treatment options are available [126]. Another trail showed that treatment of advanced melanoma patients with pembrolizumab acquired an overall objective response rate of 33%, 12 month progression-free survival rate of 35%, and median overall survival of 23 months; grade 3 or 4 treatment-related adverse events occurred in 14% of patients with advanced melanoma [128].
Nivolumab is also a MAb of PD-1, which has shown positive therapeutic activity and an acceptable safety profile in treating tumors [19]. In December 2014, the U.S. FDA granted an accelerated approval to nivolumab to treat patients with unresectable or metastatic melanoma and disease progression following ipilimumab and if patients are B-Raf proto-oncogene, serine/threonine kinase (BRAF) V600 mutation positive, which is a BRAF inhibitor [129]. Treatment with nivolumab was first reported in 2012 (sponsored by BMS). Overall response rates and median survival were 28% and 24 months, respectively. The 1 and 2 year survival rates were 62% and 43%, respectively [19, 89]. A multi-dose phase I dose-escalation trial extended the above findings. In this study, 296 patients with the same advanced cancers were given nivolumab at doses from 0.1 mg kg−1 to 10 mg kg−1 every 2 weeks for up to 2 years. Objective responses were observed in 18.4% patients with NSCLC, 27.6% patients with melanoma, and 27.2% patients with RCC. No tumor responses were observed in patients with castrate-resistant prostate cancer (CRPC) or carcinoma of colon and rectum (CRC). Treatment-related adverse events were fatigue, anorexia, nausea, rash, and diarrhea. Grade 3 or 4 toxicities were reported in 14% of patients and evident at all dose levels without obvious dose dependency [130]. A study of 107 patients with melanoma were treated with nivolumab between 2008 and 2012 revealed an overall survival of 16.8 months. The 1 and 2 year survival rates were 62% and 43%, respectively. Objective responses were observed in 31% of patients. The appearance of irAE was 54%, but grades 3 and 4 adverse events were only seen in five patients (5%) [20].
Pidilizumab is a humanized IgG-1 kappa recombinant mAb. It was developed from a murine version, mCT-011 or BAT, that was generated with the immunization of Balb/c mice with membranes of a human B-cell lymphoma cell line [131]. In a Phase I study, 17 patients with hematologic malignancies were treated with escalating doses of pidilizumab ( 0.2 to 6.0 mg/kg). Of the 17 treated patients no clear toxicity reaction was observed during therapy. 33% patients acquired clinical benefit and one acquired complete remission. The study showed the antibody to be safe and well tolerated in this patient population [132]. In an International Phase II Trial, 66 patients with diffuse large B-cell lymphoma (DLBCL) were treated. Treatment-related adverse events was mild. At 16 months after the first treatment, progression-free survival (PFS) was 0.72 (90% CI, 0.60 to 0.82). Among the 24 high-risk patients, PFS was 0.70 (90% CI, 0.51 to 0.82). The study suggested an on-target in vivo effect of pidilizumab [133].
AMP-224 is the first recombinant B7-DC-Fc fusion protein. In a PhaseI trial, patients with advanced solid tumors received low dose cyclophosphamide (CTX) on Day 0, followed by AMP-224 (IV infusion, 0.3 to 30 mg/kg) on Days 1 and 15 of each 28-day cycle. Infusion reactions were observed at higher doses (86% at the 10 mg/kg dose). No drug-related inflammatory adverse events were identified contrary to PD-1 blocking antibodies [17].
Clinical trials of mAbs to PD-L1
MPDL3280A is a humanized IgG4 anti-PD-L1-specific mAb, which is most effective in patients where the immunity response is suppressed by PD-L1 [134]. In a phase I MPDL3280A trial, 38 patients with metastatic melanoma exhibited an ORR of 29% with a 24 week Progression-Free-Survival (PFS) of 43%. This agent was expanded in 52 patients with NSCLC and 55 patients with renal cancer. ORR were 22% and 13%, respectively. Breakthrough designations have also been granted for the clinical development of nivolumab for resistant Hodgkin’s lymphoma and MPDL2380a in advanced bladder cancer [135].
BMS-936559 is also a PD-L1-specific IgG4 mAb, which inhibits the binding of PD-L1 with PD-1 and CD80. A Phase I trial showed good curative effect of BMS-936559. Up to 207 patients with solid tumor were treated with BMS-936559 for 12 weeks (median duration of therapy). Approximately 9% of patients had toxic effects of grades 3 or 4. Complete or partial response were showed in 17 patients [136]. A phase II trial has shown activity in NSCLC patients. Notably, 5 of 49 NSCLC patients had an objective response, and the response lasted for ≥24 weeks in 3 patients. In this study, the adverse reaction belonged to grades 1 or 2, including rash, hypothyroidism, and hepatitis [137].
MEDI4736 is a fully-human anti-PD-L1 antibody, which has a triple mutation in its Fc domain to avoid antibody-dependent cell-mediated cytotoxicity [127]. In a phase I trial, MEDI4736 was administered every 2 or 3 weeks in a 3 + 3 dose escalation in 26 patients with solid tumors. Grade 1 and 2 adverse events were appeared in 34%. Diarrhea, fatigue, rash, and vomiting are the mainly side effects [138]. Their research indicated that MEDI4736 was a promising agent to inhibit malignant processes.
Anti-PD-1 versus anti-CTLA-4 agents
The anti-CTLA-4 and anti-PD-1 treatments are correlated with clinical benefits. However, cancer patients trested with anti-PD-1 agents acquire better PFS and ORR comparied to anti-CTLA-4 treatment. Subgroup analyses demonstrated significant PFS (RR: 0.92 vs. 0.74; P < 0.00001) and ORR (RR: 0.95 vs. 0.76; P = 0.0004) improvement with anti-PD-1 treatment compared with anti-CTLA-4 when each was compared with the control treatments [139]. Similarly, treatment with pembrolizumab was better tolerated and demonstrated superior PFS compared with chemotherapy in ipilimumab-refractory melanoma patients enrolled in phase 2 KEYNOTE-002 trial [128]. Moreover, no clinically meaningful differences were noted between pembrolizumab doses. In the randomized phase 3 KEYNOTE-006 trial, pembrolizumab (10 mg/kg every 2 and 3 weeks) had fewer toxicities and significantly improved PFS, OS, and ORR compared with ipilimumab [128]. The ORR of nivolumab treatment in ipilimumab-refractory patients was lower compared with ipilimumab-naive patients, while the ORRs of the two groups were higher than the control group. Subgroup analyses revealed that the survival benefit was significantly high with anti-PD-1 treatment regardless of previous response to ipilimumab treatment, thereby suggesting that nivolumab or pembrolizumab is a good choice as the first-line treatment [139]. Similarly, the CheckMate 067 trial demonstrated that combined treatment with ipilimumab and nivolumab has better ORR compared with nivolumab monotherapy, especially in PD-L1-positive patients [139, 140]. Collectively, a certain patient population may selectively respond to anti-PD-1 treatment and benefit from the combination treatment with anti-CTLA-4 agents and anti-PD-1 agents.
Table 1: Results of trail using anti-PD-1 or anti-PD-L1 agents.
CONCLUSIONs
The co-inhibitory factor PD-1 binds to its ligands, PD-L1 or PD-L2, to transmit inhibitory signals in T cells and anti-apoptotic signals in tumor cells. Thus, PD-1 binding is characterized as one of the major mechanisms of tumor immune escape. Furthermore, blockade of the PD-1/PD-L1 interaction may help restore anti-tumor immunity by several ways, such as the increase of TILs that restrain Treg function and increase the cytokine secretion. Some recent clinical trials on the antibody blockade of PD-1 and PD-L1 also demonstrated effective response. Despite several previous studies on the important role of PD-1 in inhibiting T-cell activation, the biochemical signaling effects of PD-1 are yet to be fully understood. Therefore, further studies on PD-1 are needed. When the checkpoint PD-1 is better understood, cancer immunotherapy will be more effective.
Conflicts of Interest
The authors report no conflicts of interest.
Funding
This work was supported by Tianjin Natural Science Foundation (No.14JCYBJC27100); National Key Technology R&D Program (2015BALL12B12) and the National Science Foundation of China (No.81472471, 81272221).
References
1. Shindo Y, Yoshimura K, Kuramasu A, Watanabe Y, Ito H, Kondo T, Oga A, Ito H, Yoshino S, Hazama S, Tamada K, Yagita H, Oka M. Combination immunotherapy with 4-1BB activation and PD-1 blockade enhances antitumor efficacy in a mouse model of subcutaneous tumor. Anticancer Res. 2015; 35:129-136.
2. Ritprajak P1, Azuma M. Intrinsic and extrinsic control of expression of the immunoregulatory molecule PD-L1 in epithelial cells and squamous cell carcinoma. Oral Oncol. 2015; 51: 221-228. doi: 10.1016/j.oraloncology.2014.11.014
3. Francisco LM, Sage PT, Sharpe AH. The PD-1 pathway in tolerance and autoimmunity. Immunol Rev. 2010; 236: 219-242. doi: 10.1111/j.1600-065X.2010.00923.x.
4. Lázár-Molnár E, Gácser A, Freeman GJ, Almo SC, Nathenson SG, Nosanchuk JD. The PD-1/PD-L costimulatory pathway critically affects host resistance to the pathogenic fungus Histoplasma capsulatum. Proc Natl Acad Sci U S A. 2008; 105: 2658-2663. doi: 10.1073/pnas.0711918105.
5. Ellestad KK, Thangavelu G, Ewen CL, Boon L, Anderson CC. PD-1 is not required for natural or peripherally induced regulatory T cells: Severe autoimmunity despite normal production of regulatory T cells. Eur J Immunol. 2014; 44: 3560-3572. doi: 10.1002/eji.201444688.
6. Nishimura H, Nose M, Hiai H, Minato N, Honjo T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity. 1999; 11: 141-151.
7. Nishimura H, Okazaki T, Tanaka Y, Nakatani K, Hara M, Matsumori A, Sasayama S, Mizoguchi A, Hiai H, Minato N, Honjo T. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science. 2001; 291: 319-322.
8. Velu V, Titanji K, Zhu B, Husain S, Pladevega A, Lai L, Vanderford TH, Chennareddi L, Silvestri G, Freeman GJ, Ahmed R, Amara RR. Enhancing SIV-specific immunity in vivo by PD-1 blockade. Nature. 2009; 458: 206-210. doi: 10.1038/nature07662.
9. Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, Freeman GJ, Ahmed R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006; 439: 682-687.
10. Ji M, Liu Y, Li Q, Li XD, Zhao WQ, Zhang H, Zhang X, Jiang JT, Wu CP. PD-1/PD-L1 pathway in non-small-cell lung cancer and its relation with EGFR mutation. J Transl Med. 2015; 13: 5. doi: 10.1186/s12967-014-0373-0.
11. Freeman-Keller M, Weber JS. Anti-programmed death receptor 1 immunotherapy in melanoma: rationale, evidence and clinical potential. Ther Adv Med Oncol. 2015; 7: 12-21. doi: 10.1177/1758834014551747.
12. Huang RY, Eppolito C, Lele S, Shrikant P, Matsuzaki J, Odunsi K. LAG3 and PD1 co-inhibitory molecules collaborate to limit CD8+ T cell signaling and dampen antitumor immunity in a murine ovarian cancer model. Oncotarget. 2015; 6: 27359-27377. doi: 10.18632/oncotarget.4751.
13. Wang SD, Li HY, Li BH, Xie T, Zhu T, Sun LL, Ren HY, Ye ZM. The role of CTLA-4 and PD-1 in anti-tumor immune response and their potential efficacy against osteosarcoma. Int Immunopharmacol. 2016; 38: 81-89. doi: 10.1016/j.intimp.2016.05.016.
14. Okamura T, Fujio K, Sumitomo S, Yamamoto K. Roles of LAG3 and EGR2 in regulatory T cells. Ann Rheum Dis. 2012; 71: i96-100. doi: 10.1136/annrheumdis-2011-200588.
15. Bu M, Shen Y, Seeger WL, An S, Qi R, Sanderson JA, Cai Y. Ovarian carcinoma-infiltrating regulatory T cells were more potent suppressors of CD8(+) T cell inflammation than their peripheral counterparts, a function dependent on TIM3 expression. Tumour Biol. 2016; 37: 3949-3956. doi: 10.1007/s13277-015-4237-x.
16. Pedoeem A, Azoulay-Alfaguter I, Strazza M, Silverman GJ, Mor A. Programmed death-1 pathway in cancer and autoimmunity. Clin Immunol. 2014; 153: 145-152. doi: 10.1016/j.clim.2014.04.010.
17. Henick BS, Herbst RS, Goldberg SB. The PD-1 pathway as a therapeutic target to overcome immune escape mechanisms in cancer. Expert Opin Ther Targets. 2014; 12: 1407-1420. doi: 10.1517/14728222.2014.955794.
18. Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, Wolchok JD, Hersey P, Joseph RW, Weber JS, Dronca R, Gangadhar TC, Patnaik A, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med. 2013; 369: 134-144. doi: 10.1056/NEJMoa1305133.
19. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, Leming PD, Spigel DR, Antonia SJ, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012; 366: 2443-2454. doi: 10.1056/NEJMoa1200690.
20. Topalian SL, Sznol M, McDermott DF, Kluger HM, Carvajal RD, Sharfman WH, Brahmer JR, Lawrence DP, Atkins MB, Powderly JD, Leming PD, Lipson EJ, Puzanov I, et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol. 2014; 32: 1020-1030. doi: 10.1200/JCO.2013.53.0105.
21. Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, Segal NH, Ariyan CE, Gordon RA, Reed K, Burke MM, Caldwell A, Kronenberg SA, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013; 369: 122-133. doi: 10.1056/NEJMoa1302369.
22. Rajan A, Gulley JL. Gulley, Nivolumab (anti-PD-1, BMS-936558, ONO-4538) in patients with advanced non-small cell lung cancer. Transl Lung Cancer Res. 2014; 3: 403-405. doi: 10.3978/j.issn.2218-6751.
23. Kwok G, Yau TC, Chiu JW, Tse E, Kwong YL. Pembrolizumab (Keytruda). Hum Vaccin Immunother. 2016.
24. Ishida Y, Agata Y, Shibahara K, Honjo T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992; 11: 3887-3895.
25. Shinohara T, Taniwaki M, Ishida Y, Kawaichi M, Honjo T. Structure and chromosomal localization of the human PD-1 gene (PDCD1). Genomics. 1994; 23: 704-706.
26. Riley JL. PD-1 signaling in primary T cells. Immunol Rev. 2009; 229: 114-125. doi: 10.1111/j.1600-065X.2009.00767.x.
27. McPherson RC, Konkel JE, Prendergast CT, Thomson JP, Ottaviano R, Leech MD, Kay O, Zandee SE, Sweenie CH, Wraith DC, Meehan RR, Drake AJ, Anderton SM. Epigenetic modification of the PD-1 (Pdcd1) promoter in effector CD4(+) T cells tolerized by peptide immunotherapy. Elife. 2014; 3. doi: 10.7554/eLife.03416.
28. Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. 2008; 26: 677-704. doi: 10.1146/annurev.immunol.26.021607.090331.
29. Liu C, Jiang J, Gao L, Hu X, Wang F, Shen Y, Yu G, Zhao Z, Zhang X. A Promoter Region Polymorphism in PDCD-1 Gene Is Associated with Risk of Rheumatoid Arthritis in the Han Chinese Population of Southeastern China. Int J Genomics. 2014; 2014: 247637. doi: 10.1155/2014/247637.
30. Zhang X, Schwartz JC, Guo X, Bhatia S, Cao E, Lorenz M, Cammer M, Chen L, Zhang ZY, Edidin MA, Nathenson SG, Almo SC. Structural and functional analysis of the costimulatory receptor programmed death-1. Immunity. 2004; 20: 337-347.
31. Okazaki T, Maeda A, Nishimura H, Kurosaki T, Honjo T. PD-1 immunoreceptor inhibits B cell receptor-mediated signaling by recruiting src homology 2-domain-containing tyrosine phosphatase 2 to phosphotyrosine. Proc Natl Acad Sci U S A. 2001; 98: 13866-13871.
32. Unkeless JC, Jin J. Inhibitory receptors, ITIM sequences and phosphatases. Curr Opin Immunol. 1997; 9: 338-343.
33. Starr R, Willson TA, Viney EM, Murray LJ, Rayner JR, Jenkins BJ, Gonda TJ, Alexander WS, Metcalf D, Nicola NA, Hilton DJ. A family of cytokine-inducible inhibitors of signalling. Nature. 1997; 387: 917-921.
34. Ohaegbulam KC, Assal A, Lazar-Molnar E, Yao Y, Zang X. Human cancer immunotherapy with antibodies to the PD-1 and PD-L1 pathway. Trends Mol Med. 2015; 21: 24-33. doi: 10.1016/j.molmed.2014.10.009.
35. Thompson RH, Dong H, Lohse CM, Leibovich BC, Blute ML, Cheville JC, Kwon ED. PD-1 is expressed by tumor-infiltrating immune cells and is associated with poor outcome for patients with renal cell carcinoma. Clin Cancer Res. 2007; 13: 1757-1761.
36. Green MR, Monti S, Rodig SJ, Juszczynski P, Currie T, O’Donnell E, Chapuy B, Takeyama K, Neuberg D, Golub TR, Kutok JL, Shipp MA. Integrative analysis reveals selective 9p24.1 amplification, increased PD-1 ligand expression, and further induction via JAK2 in nodular sclerosing Hodgkin lymphoma and primary mediastinal large B-cell lymphoma. Blood. 2010; 116: 3268-3277. doi: 10.1182/blood-2010-05-282780.
37. Koh YW, Jeon YK, Yoon DH, Suh C, Huh J. Programmed death 1 expression in the peritumoral microenvironment is associated with a poorer prognosis in classical Hodgkin lymphoma. Tumour Biol. 2016; 37: 7507-7514. doi: 10.1007/s13277-015-4622-5.
38. Zhang Y, Kang S, Shen J, He J, Jiang L, Wang W, Guo Z, Peng G, Chen G, He J, Liang W. Prognostic significance of programmed cell death 1 (PD-1) or PD-1 ligand 1 (PD-L1) Expression in epithelial-originated cancer: a meta-analysis. Medicine (Baltimore). 2015; 94: e515. doi: 10.1097/MD.0000000000000515.
39. Agata Y, Kawasaki A, Nishimura H, Ishida Y, Tsubata T, Yagita H, Honjo T. Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int Immunol. 1996; 8: 765-772.
40. Zou W, Chen L. Inhibitory B7-family molecules in the tumour microenvironment. Nat Rev Immunol. 2008; 8: 467-477. doi: 10.1038/nri2326.
41. Ahmadzadeh M, Johnson LA, Heemskerk B, Wunderlich JR, Dudley ME, White DE, Rosenberg SA. Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood. 2009; 114: 1537-1544. doi: 10.1182/blood-2008-12-195792.
42. Matsuzaki J, Gnjatic S, Mhawech-Fauceglia P, Beck A, Miller A, Tsuji T, Eppolito C, Qian F, Lele S, Shrikant P, Old LJ, Odunsi K. Tumor-infiltrating NY-ESO-1-specific CD8+ T cells are negatively regulated by LAG-3 and PD-1 in human ovarian cancer. Proc Natl Acad Sci U S A. 2010;107: 7875-7880. doi: 10.1073/pnas.1003345107.
43. Voron T, Colussi O, Marcheteau E, Pernot S, Nizard M, Pointet AL, Latreche S, Bergaya S, Benhamouda N, Tanchot C, Stockmann C, Combe P, Berger A, et al. VEGF-A modulates expression of inhibitory checkpoints on CD8+ T cells in tumors. J Exp Med. 2015; 212: 139-148. doi: 10.1084/jem.20140559.
44. Saito H, Kuroda H, Matsunaga T, Osaki T, Ikeguchi M. Increased PD-1 expression on CD4+ and CD8+ T cells is involved in immune evasion in gastric cancer. J Surg Oncol. 2013; 107: 517-522. doi: 10.1002/jso.23281.
45. Kao C, Oestreich KJ, Paley MA, Crawford A, Angelosanto JM, Ali MA, Intlekofer AM, Boss JM, Reiner SL, Weinmann AS, Wherry EJ. Transcription factor T-bet represses expression of the inhibitory receptor PD-1 and sustains virus-specific CD8+ T cell responses during chronic infection. Nat Immunol. 2011; 12: 663-671. doi: 10.1038/ni.2046.
46. Li C, Li W, Xiao J, Jiao S, Teng F, Xue S, Zhang C, Sheng C, Leng Q, Rudd CE, Wei B, Wang H. ADAP and SKAP55 deficiency suppresses PD-1 expression in CD8+ cytotoxic T lymphocytes for enhanced anti-tumor immunotherapy. EMBO Mol Med. 2015; 7: 754-769. doi: 10.15252/emmm.201404578.
47. Lu P, Youngblood BA, Austin JW, Mohammed AU, Butler R, Ahmed R, Boss JM. Blimp-1 represses CD8 T cell expression of PD-1 using a feed-forward transcriptional circuit during acute viral infection. J Exp Med. 2014; 211: 515-527. doi: 10.1084/jem.20130208.
48. Xiao G, Deng A, Liu H, Ge G, Liu X. Activator protein 1 suppresses antitumor T-cell function via the induction of programmed death 1. Proc Natl Acad Sci U S A. 2012. 109: 15419-15424.
49. Austin JW, Lu P, Majumder P, Ahmed R, Boss JM. STAT3, STAT4, NFATc1, and CTCF regulate PD-1 through multiple novel regulatory regions in murine T cells. J Immunol. 2014; 192: 4876-4886. doi: 10.4049/jimmunol.1302750.
50. Bally AP, Lu P, Tang Y, Austin JW, Scharer CD, Ahmed R, Boss JM. NF-kappaB regulates PD-1 expression in macrophages. J Immunol. 2015; 194: 4545-4554. doi: 10.4049/jimmunol.1402550.
51. Liu J, Kang H, Raab M, da Silva AJ, Kraeft SK, Rudd CE. FYB (FYN binding protein) serves as a binding partner for lymphoid protein and FYN kinase substrate SKAP55 and a SKAP55-related protein in T cells. Proc Natl Acad Sci U S A. 1998; 95: 8779-8784.
52. Marie-Cardine A, Hendricks-Taylor LR, Boerth NJ, Zhao H, Schraven B, Koretzky GA. Molecular interaction between the Fyn-associated protein SKAP55 and the SLP-76-associated phosphoprotein SLAP-130. J Biol Chem. 1998; 40: 25789-25795.
53. Massari F, Santoni M, Ciccarese C, Santini D, Alfieri S, Martignoni G, Brunelli M, Piva F, Berardi R, Montironi R, Porta C, Cascinu S, Tortora G. PD-1 blockade therapy in renal cell carcinoma: current studies and future promises. Cancer Treat Rev. 2015; 41: 114-121. doi: 10.1016/j.ctrv.2014.12.013.
54. O’Shea JJ, Murray PJ. Murray, Cytokine signaling modules in inflammatory responses. Immunity. 2008;28: 477-487. doi: 10.1016/j.immuni.2008.03.002.
55. Ozao-Choy J, Ma G, Kao J, Wang GX, Meseck M, Sung M, Schwartz M, Divino CM, Pan PY, Chen SH. The novel role of tyrosine kinase inhibitor in the reversal of immune suppression and modulation of tumor microenvironment for immune-based cancer therapies. Cancer Res. 2009; 69: 2514-2522. doi: 10.1158/0008-5472.CAN-08-4709.
56. Rekik R, Belhadj Hmida N, Ben Hmid A, Zamali I, Kammoun N, Ben Ahmed M. PD-1 induction through TCR activation is partially regulated by endogenous TGF-beta. Cell Mol Immunol. 2015; 12: 648-649. doi: 10.1038/cmi.2014.104.
57. Parry RV, Chemnitz JM, Frauwirth KA, Lanfranco AR, Braunstein I, Kobayashi SV, Linsley PS, Thompson CB, Riley JL. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol. 2005; 25: 9543-9553.
58. Sanmamed MF, Chen L. Inducible expression of B7-H1 (PD-L1) and its selective role in tumor site immune modulation. Cancer J. 2014; 20: 256-261. doi: 10.1097/PPO.0000000000000061.
59. Bellmunt J, Mullane SA, Werner L, Fay AP, Callea M, Leow JJ, Taplin ME, Choueiri TK, Hodi FS, Freeman GJ, Signoretti S. Association of PD-L1 expression on tumor-infiltrating mononuclear cells and overall survival in patients with urothelial carcinoma. Ann Oncol. 2015; 26: 812-817. doi: 10.1093/annonc/mdv009.
60. Dong H, Zhu G, Tamada K, Chen L. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat Med. 1999; 5: 1365-1369.
61. Hino R, Kabashima K, Kato Y, Yagi H, Nakamura M, Honjo T, Okazaki T, Tokura Y. Tumor cell expression of programmed cell death-1 ligand 1 is a prognostic factor for malignant melanoma. Cancer. 2010; 116: 1757-1766. doi: 10.1002/cncr.24899.
62. Riella LV, Paterson AM, Sharpe AH, Chandraker A. Role of the PD-1 pathway in the immune response. Am J Transplant. 2012; 12: 2575-2587. doi: 10.1111/j.1600-6143.2012.04224.x.
63. Momtaz P, Postow MA. Immunologic checkpoints in cancer therapy: focus on the programmed death-1 (PD-1) receptor pathway. Pharmgenomics Pers Med. 2014; 7: 357-365. doi: 10.2147/PGPM.S53163. eCollection 2014.
64. Topalian SL, Drake CG, Pardoll DM. Targeting the PD-1/B7-H1(PD-L1) pathway to activate anti-tumor immunity. Curr Opin Immunol. 2012; 24: 207-212. doi: 10.1016/j.coi.2011.12.009.
65. Stanciu LA, Bellettato CM, Laza-Stanca V, Coyle AJ, Papi A, Johnston SL. Expression of programmed death-1 ligand (PD-L) 1, PD-L2, B7-H3, and inducible costimulator ligand on human respiratory tract epithelial cells and regulation by respiratory syncytial virus and type 1 and 2 cytokines. J Infect Dis. 2006; 193: 404-412.
66. Kan-o K, Matsumoto K, Asai-Tajiri Y, Fukuyama S, Hamano S, Seki N, Nakanishi Y, Inoue H. PI3K-delta mediates double-stranded RNA-induced upregulation of B7-H1 in BEAS-2B airway epithelial cells. Biochem Biophys Res Commun. 2013; 435: 195-201. doi: 10.1016/j.bbrc.2013.04.082.
67. Chen J, Feng Y, Lu L, Wang H, Dai L, Li Y, Zhang P. Interferon-gamma-induced PD-L1 surface expression on human oral squamous carcinoma via PKD2 signal pathway. Immunobiology. 2012; 217: 385-393. doi: 10.1016/j.imbio.2011.10.016.
68. Abiko K, Matsumura N, Hamanishi J, Horikawa N, Murakami R, Yamaguchi K, Yoshioka Y, Baba T, Konishi I, Mandai M. IFN-gamma from lymphocytes induces PD-L1 expression and promotes progression of ovarian cancer. Br J Cancer. 2015; 112: 1501-1509. doi: 10.1038/bjc.2015.101.
69. Krönig H, Kremmler L, Haller B, Englert C, Peschel C, Andreesen R, Blank CU. Interferon-induced programmed death-ligand 1 (PD-L1/B7-H1) expression increases on human acute myeloid leukemia blast cells during treatment. Eur J Haematol. 2014; 92: 195-203. doi: 10.1111/ejh.12228.
70. Bellucci R, Martin A, Bommarito D, Wang K, Hansen SH, Freeman GJ, Ritz J. Interferon-gamma-induced activation of JAK1 and JAK2 suppresses tumor cell susceptibility to NK cells through upregulation of PD-L1 expression. Oncoimmunology. 2015; 4: e1008824.
71. Maine CJ, Aziz NH, Chatterjee J, Hayford C, Brewig N, Whilding L, George AJ, Ghaem-Maghami S. Programmed death ligand-1 over-expression correlates with malignancy and contributes to immune regulation in ovarian cancer. Cancer Immunol Immunother. 2014; 63: 215-224. doi: 10.1007/s00262-013-1503-x.
72. Chen N, Fang W, Zhan J, Hong S, Tang Y, Kang S, Zhang Y, He X, Zhou T, Qin T, Huang Y, Yi X, Zhang L. Upregulation of PD-L1 by EGFR Activation Mediates the Immune Escape in EGFR-Driven NSCLC: Implication for Optional Immune Targeted Therapy for NSCLC Patients with EGFR Mutation. J Thorac Oncol. 2015; 10: 910-923. doi: 10.1097/JTO.0000000000000500.
73. Akbay EA, Koyama S, Carretero J, Altabef A, Tchaicha JH, Christensen CL, Mikse OR, Cherniack AD, Beauchamp EM, Pugh TJ, Wilkerson MD, Fecci PE, Butaney M, et al. Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors. Cancer Discov. 2013; 3: 1355-1363. doi: 10.1158/2159-8290.CD-13-0310.
74. Ota K, Azuma K, Kawahara A, Hattori S, Iwama E, Tanizaki J, Harada T, Matsumoto K, Takayama K, Takamori S, Kage M, Hoshino T, Nakanishi Y, et al. Induction of PD-L1 expression by the EML4-ALK oncoprotein and downstream signaling pathways in non-small cell lung cancer. Clin Cancer Res. 2015; 21: 4014-4021. doi: 10.1158/1078-0432.CCR-15-0016.
75. Balan M, Mier y Teran E, Waaga-Gasser AM, Gasser M, Choueiri TK, Freeman G, Pal S. Novel roles of c-Met in the survival of renal cancer cells through the regulation of HO-1 and PD-L1 expression. J Biol Chem. 2015; 290: 8110-8120. doi: 10.1074/jbc.M114.612689.
76. Parsa AT, Waldron JS, Panner A, Crane CA, Parney IF, Barry JJ, Cachola KE, Murray JC, Tihan T, Jensen MC, Mischel PS, Stokoe D, Pieper RO. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat Med. 2007; 13: 84-88.
77. Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, Bronte V, Chouaib S. PD-L1 is a novel direct target of HIF-1alpha, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J Exp Med. 2014; 211: 781-790. doi: 10.1084/jem.20131916.
78. Scharenberg AM, Humphries LA, Rawlings DJ. Calcium signalling and cell-fate choice in B cells. Nat Rev Immunol. 2007; 7: 778-789.
79. Mashima R, Hishida Y, Tezuka T, Yamanashi Y. The roles of Dok family adapters in immunoreceptor signaling. Immunol Rev. 2009; 232: 273-285. doi: 10.1111/j.1600-065X.2009.00844.x.
80. Ng CH, Xu S, Lam KP. Dok-3 plays a nonredundant role in negative regulation of B-cell activation. Blood. 2007; 110: 259-266.
81. Stork B, Neumann K, Goldbeck I, Alers S, Kähne T, Naumann M, Engelke M, Wienands J. Subcellular localization of Grb2 by the adaptor protein Dok-3 restricts the intensity of Ca2+ signaling in B cells. EMBO J. 2007; 26: 1140-1149.
82. Ou X, Xu S, Li YF1, Lam KP. Adaptor protein DOK3 promotes plasma cell differentiation by regulating the expression of programmed cell death 1 ligands. Proc Natl Acad Sci U S A. 2014; 111: 11431-11436. doi: 10.1073/pnas.1400539111.
83. Wang C, Thudium KB, Han M, Wang XT, Huang H, Feingersh D, Garcia C, Wu Y, Kuhne M, Srinivasan M, Singh S, Wong S, Garner N, et al. In vitro characterization of the anti-PD-1 antibody nivolumab, BMS-936558, and in vivo toxicology in non-human primates. Cancer Immunol Res. 2014; 2: 846-856. doi: 10.1158/2326-6066.CIR-14-0040.
84. Xiao Y, Yu S, Zhu B, Bedoret D, Bu X, Francisco LM, Hua P, Duke-Cohan JS, Umetsu DT, Sharpe AH, DeKruyff RH, Freeman GJ. RGMb is a novel binding partner for PD-L2 and its engagement with PD-L2 promotes respiratory tolerance. J Exp Med. 2014; 211: 943-959. doi: 10.1084/jem.20130790.
85. Chikuma S, Terawaki S, Hayashi T, Nabeshima R, Yoshida T, Shibayama S, Okazaki T, Honjo T. PD-1-mediated suppression of IL-2 production induces CD8+ T cell anergy in vivo. J Immunol. 2009; 182: 6682-6689. doi: 10.4049/jimmunol.0900080.
86. Bishop KD, Harris JE, Mordes JP, Greiner DL, Rossini AA, Czech MP, Phillips NE. Depletion of the programmed death-1 receptor completely reverses established clonal anergy in CD4(+) T lymphocytes via an interleukin-2-dependent mechanism. Cell Immunol. 2009; 256: 86-91. doi: 10.1016/j.cellimm.2009.01.008.
87. Dai S, Jia R, Zhang X, Fang Q, Huang L. The PD-1/PD-Ls pathway and autoimmune diseases. Cell Immunol. 2014; 290: 72-79. doi: 10.1016/j.cellimm.2014.05.006.
88. Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, Fitz LJ, Malenkovich N, Okazaki T, Byrne MC, Horton HF, Fouser L, Carter L, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000; 192: 1027-1034.
89. Hersey P, Gowrishankar K. Pembrolizumab joins the anti-PD-1 armamentarium in the treatment of melanoma. Future Oncol. 2015; 11: 133-140. doi: 10.2217/fon.14.205.
90. Zhao S, Li F, Leak RK, Chen J, Hu X. Regulation of neuroinflammation through programed death-1/programed death ligand signaling in neurological disorders. Front Cell Neurosci. 2014; 8: 271. doi: 10.3389/fncel.2014.00271.
91. Atefi M, Avramis E, Lassen A, Wong DJ, Robert L, Foulad D, Cerniglia M, Titz B, Chodon T, Graeber TG, Comin-Anduix B, Ribas A. Effects of MAPK and PI3K pathways on PD-L1 expression in melanoma. Clin Cancer Res. 2014; 20: 3446-3457. doi: 10.1158/1078-0432.CCR-13-2797.
92. Wei F, Zhong S, Ma Z, Kong H, Medvec A, Ahmed R, Freeman GJ, Krogsgaard M, Riley JL. Strength of PD-1 signaling differentially affects T-cell effector functions. Proc Natl Acad Sci U S A. 2013; 110: E2480-2489. doi: 10.1073/pnas.1305394110.
93. Bachegowda LS, Barta SK. Genetic and molecular targets in lymphoma: implications for prognosis and treatment. Future Oncol. 2014; 10: 2509-2528. doi: 10.2217/fon.14.112.
94. Lu B, Chen L, Liu L, Zhu Y, Wu C, Jiang J, Zhang X. T-cell-mediated tumor immune surveillance and expression of B7 co-inhibitory molecules in cancers of the upper gastrointestinal tract. Immunol Res. 2011; 50: 269-275. doi: 10.1007/s12026-011-8227-9.
95. Gianchecchi E, Delfino DV, Fierabracci A. Recent insights into the role of the PD-1/PD-L1 pathway in immunological tolerance and autoimmunity. Autoimmun Rev. 2013; 12: 1091-1100. doi: 10.1016/j.autrev.2013.05.003.
96. Bryan LJ, Gordon LI. Blocking tumor escape in hematologic malignancies: the anti-PD-1 strategy. Blood Rev. 2015; 29: 25-32. doi: 10.1016/j.blre.2014.09.004.
97. Jandus C, Bioley G, Speiser DE, Romero P. Selective accumulation of differentiated FOXP3(+) CD4 (+) T cells in metastatic tumor lesions from melanoma patients compared to peripheral blood. Cancer Immunol Immunother. 2008; 57: 1795-1805. doi: 10.1007/s00262-008-0507-4.
98. Zheng Y, Zha Y, Driessens G, Locke F, Gajewski TF. Transcriptional regulator early growth response gene 2 (Egr2) is required for T cell anergy in vitro and in vivo. J Exp Med. 2012; 209: 2157-2163. doi: 10.1084/jem.20120342.
99. Qian N, Gao L, Dong L, Liu H, Fu J, Meng D, Gao X, Zhang J, Gao Y3, Song H. Construction, expression, purification, and characterization of a dual-targeting PD-1/VEGF-A fusion protein (P-V). Protein Expr Purif. 2015; 109: 1-6. doi: 10.1016/j.pep.2015.01.004.
100. Boussiotis, V.A., P. Chatterjee, and L. Li, Biochemical signaling of PD-1 on T cells and its functional implications. Cancer J, 2014. 20: 265-271. doi: 10.1097/PPO.0000000000000059.
101. Li J, Jie HB, Lei Y, Gildener-Leapman N, Trivedi S, Green T, Kane LP, Ferris RL. PD-1/SHP-2 inhibits Tc1/Th1 phenotypic responses and the activation of T cells in the tumor microenvironment. Cancer Res. 2015; 75: 508-518. doi: 10.1158/0008-5472.CAN-14-1215.
102. Nurieva R, Thomas S, Nguyen T, Martin-Orozco N, Wang Y, Kaja MK, Yu XZ, Dong C. T-cell tolerance or function is determined by combinatorial costimulatory signals. EMBO J. 2006; 25: 2623-2633.
103. Stolarczyk E, Lord GM, Howard JK. The immune cell transcription factor T-bet: A novel metabolic regulator. Adipocyte. 2014; 3: 58-62. doi: 10.4161/adip.26220.
104. Chen LJ, Sun J, Wu HY, Zhou SM, Tan Y, Tan M, Shan BE, Lu BF, Zhang XG. B7-H4 expression associates with cancer progression and predicts patient’s survival in human esophageal squamous cell carcinoma. Cancer Immunol Immunother. 2011; 60: 1047-1055. doi: 10.1007/s00262-011-1017-3.
105. Patsoukis N, Sari D, Boussiotis VA. PD-1 inhibits T cell proliferation by upregulating p27 and p15 and suppressing Cdc25A. Cell Cycle. 2012; 11: 4305-4309. doi: 10.4161/cc.22135.
106. Patsoukis N, Brown J, Petkova V, Liu F, Li L, Boussiotis VA. Selective effects of PD-1 on Akt and Ras pathways regulate molecular components of the cell cycle and inhibit T cell proliferation. Sci Signal. 2012; 5: ra46. doi: 10.1126/scisignal.2002796.
107. Kim JW, Nam KH, Ahn SH, Park do J, Kim HH, Kim SH, Chang H, Lee JO, Kim YJ, Lee HS, Kim JH, Bang SM, Lee JS, et al. Prognostic implications of immunosuppressive protein expression in tumors as well as immune cell infiltration within the tumor microenvironment in gastric cancer. Gastric Cancer. 2016; 19: 42-52. doi: 10.1007/s10120-014-0440-5.
108. Blank C, Kuball J, Voelkl S, Wiendl H, Becker B, Walter B, Majdic O, Gajewski TF, Theobald M, Andreesen R, Mackensen A. Blockade of PD-L1 (B7-H1) augments human tumor-specific T cell responses in vitro. Int J Cancer. 2006; 119: 317-327.
109. Schlößer HA, Theurich S, Shimabukuro-Vornhagen A, Holtick U, Stippel DL, von Bergwelt-Baildon M. Overcoming tumor-mediated immunosuppression. Immunotherapy., 2014; 6: 973-988. doi: 10.2217/imt.14.58.
110. Godin-Ethier J, Hanafi LA, Piccirillo CA, Lapointe R. Indoleamine 2,3-dioxygenase expression in human cancers: clinical and immunologic perspectives. Clin Cancer Res. 2011; 17: 6985-6991. doi: 10.1158/1078-0432.CCR-11-1331.
111. Francisco LM, Salinas VH, Brown KE, Vanguri VK, Freeman GJ, Kuchroo VK, Sharpe AH. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med. 2009; 206: 3015-3029. doi: 10.1084/jem.20090847.
112. Chen X, Fosco D, Kline DE, Meng L, Nishi S, Savage PA, Kline J. PD-1 regulates extrathymic regulatory T-cell differentiation. Eur J Immunol. 2014; 44: 2603-2616. doi: 10.1002/eji.201344423.
113. Zhong A, Pan X, Shi M. Expression of PD-1 by CD4(+)CD25(+)CD127(low) Treg cells in the peripheral blood of lung cancer patients. Onco Targets Ther. 2015; 8: 1831-1833. doi: 10.2147/OTT.S90538.
114. Ghebeh H, Barhoush E, Tulbah A, Elkum N, Al-Tweigeri T, Dermime S. FOXP3+ Tregs and B7-H1+/PD-1+ T lymphocytes co-infiltrate the tumor tissues of high-risk breast cancer patients: Implication for immunotherapy. BMC Cancer. 2008; 8: 57. doi: 10.1186/1471-2407-8-57.
115. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012; 12: 252-264. doi: 10.1038/nrc3239.
116. Tykodi SS. PD-1 as an emerging therapeutic target in renal cell carcinoma: current evidence. Onco Targets Ther. 2014; 7: 1349-1359. doi: 10.2147/OTT.S48443.
117. Chauvin JM, Pagliano O, Fourcade J, Sun Z, Wang H, Sander C, Kirkwood JM, Chen TH, Maurer M, Korman AJ, Zarour HM. TIGIT and PD-1 impair tumor antigen-specific CD8(+) T cells in melanoma patients. J Clin Invest. 2015; 125: 2046-2058. doi: 10.1172/JCI80445.
118. Fourcade J, Sun Z, Benallaoua M, Guillaume P, Luescher IF, Sander C, Kirkwood JM, Kuchroo V, Zarour HM. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen-specific CD8+ T cell dysfunction in melanoma patients. J Exp Med. 2010; 207: 2175-2186. doi: 10.1084/jem.20100637.
119. Zheng W, Xiao H, Liu H, Zhou Y. Expression of programmed death 1 is correlated with progression of osteosarcoma. APMIS. 2015; 123: 102-107. doi: 10.1111/apm.12311.
120. van Dam LS, de Zwart VM, Meyer-Wentrup FA. The role of programmed cell death-1 (PD-1) and its ligands in pediatric cancer. Pediatr Blood Cancer. 2015; 62: 190-197. doi: 10.1002/pbc.25284.
121. Iwai Y, Terawaki S, Honjo T. PD-1 blockade inhibits hematogenous spread of poorly immunogenic tumor cells by enhanced recruitment of effector T cells. Int Immunol. 2005; 17: 133-144.
122. Shiner EK, Holbrook BC, Alexander-Miller MA. CD4+ T cell subset differentiation and avidity setpoint are dictated by the interplay of cytokine and antigen mediated signals. PLoS One. 2014; 9: e100175. doi: 10.1371/journal.pone.0100175.
123. Dulos J, Carven GJ, van Boxtel SJ, Evers S, Driessen-Engels LJ, Hobo W, Gorecka MA, de Haan AF, Mulders P, Punt CJ, Jacobs JF, Schalken JA, Oosterwijk E, et al. PD-1 blockade augments Th1 and Th17 and suppresses Th2 responses in peripheral blood from patients with prostate and advanced melanoma cancer. J Immunother. 2012; 35: 169-178. doi: 10.1097/CJI.0b013e318247a4e7.
124. Mangsbo SM, Sandin LC, Anger K, Korman AJ, Loskog A, Tötterman TH. Enhanced tumor eradication by combining CTLA-4 or PD-1 blockade with CpG therapy. J Immunother. 2010; 33: 225-235. doi: 10.1097/CJI.0b013e3181c01fcb.
125. Meng X, Huang Z, Teng F, Xing L, Yu J. Predictive biomarkers in PD-1/PD-L1 checkpoint blockade immunotherapy. Cancer Treat Rev. 2015; 41: 868-876. doi: 10.1016/j.ctrv.2015.11.001.
126. Robert C, Ribas A, Wolchok JD, Hodi FS, Hamid O, Kefford R, Weber JS, Joshua AM, Hwu WJ, Gangadhar TC, Patnaik A, Dronca R, Zarour H, et al. Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma: a randomised dose-comparison cohort of a phase 1 trial. Lancet. 2014; 384: 1109-1117. doi: 10.1016/S0140-6736(14)60958-2.
127. Lee SM, Chow LQ. A new addition to the PD-1 checkpoint inhibitors for non-small cell lung cancer-the anti-PDL1 antibody-MEDI4736. Transl Lung Cancer Res. 2014; 3: 408-410. doi: 10.3978/j.issn.2218-6751.2014.11.10.
128. Ribas A, Hamid O, Daud A, Hodi FS, Wolchok JD, Kefford R, Joshua AM, Patnaik A, Hwu WJ, Weber JS, Gangadhar TC, Hersey P, Dronca R, et al. Association of Pembrolizumab With Tumor Response and Survival Among Patients With Advanced Melanoma. JAMA. 2016; 315: 1600-1609. doi: 10.1001/jama.2016.4059.
129. Swaika A, Hammond WA, Joseph RW. Current state of anti-PD-L1 and anti-PD-1 agents in cancer therapy. Mol Immunol. 2015; 67: 4-17. doi: 10.1016/j.molimm.2015.02.009.
130. Philips GK, Atkins M. Therapeutic uses of anti-PD-1 and anti-PD-L1 antibodies. Int Immunol. 2015; 27: 39-46. doi: 10.1093/intimm/dxu095.
131. Hardy B, Yampolski I, Kovjazin R, Galli M, Novogrodsky A. A monoclonal antibody against a human B lymphoblastoid cell line induces tumor regression in mice. Cancer Res. 1994; 54: 5793-5796.
132. Berger R, Rotem-Yehudar R, Slama G, Landes S, Kneller A, Leiba M, Koren-Michowitz M, Shimoni A, Nagler A. Phase I safety and pharmacokinetic study of CT-011, a humanized antibody interacting with PD-1, in patients with advanced hematologic malignancies. Clin Cancer Res. 2008; 14: 3044-3051. doi: 10.1158/1078-0432.CCR-07-4079.
133. Armand P, Nagler A, Weller EA, Devine SM, Avigan DE, Chen YB, Kaminski MS, Holland HK, Winter JN, Mason JR, Fay JW, Rizzieri DA, Hosing CM, et al. Disabling immune tolerance by programmed death-1 blockade with pidilizumab after autologous hematopoietic stem-cell transplantation for diffuse large B-cell lymphoma: results of an international phase II trial. J Clin Oncol. 2013. 31: 4199-4206. doi: 10.1200/JCO.2012.48.3685.
134. Kim JW, Eder JP. Prospects for targeting PD-1 and PD-L1 in various tumor types. Oncology (Williston Park). 2014; 28 3: 15-28.
135. Sunshine J, Taube JM. PD-1/PD-L1 inhibitors. Curr Opin Pharmacol. 2015; 23: 32-38. doi: 10.1016/j.coph.2015.05.011.
136. Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, Pitot HC, Hamid O, Bhatia S, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012; 366: 2455-2465. doi: 10.1056/NEJMoa1200694.
137. Langer CJ. Emerging immunotherapies in the treatment of non-small cell lung cancer (NSCLC): the role of immune checkpoint inhibitors. Am J Clin Oncol. 2015. 38: 422-430. doi: 10.1097/COC.0000000000000059.
138. Zielinski CC. A phase I study of MEDI4736, NNT-PD-L1 antibody in patients with advanced solid tumors. Transl Lung Cancer Res. 2014; 3: 406-407. oi: 10.3978/j.issn.2218-6751.2014.08.07.
139. Yun S, Vincelette ND, Green MR, Wahner Hendrickson AE, Abraham I. Targeting immune checkpoints in unresectable metastatic cutaneous melanoma: a systematic review and meta-analysis of anti-CTLA-4 and anti-PD-1 agents trials. Cancer Med. 2016; 5: 1481-1491. doi: 10.1002/cam4.732.
140. Valsecchi ME. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N Engl J Med. 2015; 373: 1270. doi: 10.1056/NEJMc1509660#SA1.