
INTRODUCTION
Polycystic ovary syndrome (PCOS), as a highly complex endocrine disorder, is usually comprised of phenotypical and heterogeneous reproductive effects [1] as well as metabolic symptoms [2–5]. Thus, there is great genetic heterogeneity and different pathophysiological mechanisms of various PCOS phenotypes. The genetic heterogeneity, combined with the pronounced variability in the diagnostic criteria, makes the genetic study and susceptibility gene identification of PCOS particularly difficult. Although PCOS has been studied for decades, the genetic contributions to this disorder are not fully understood. Furthermore, lack of sufficiently large cohorts also reduces the power to identify specific genes of PCOS [6].
It has been reported that candidate genes associated with PCOS contribute to different biological processes and phenotypes [7]. For example, as the association between obesity and PCOS [8], gene variants affecting fat mass, such as FTO [9], have been found to be involved in PCOS. The effect of common variants in TCF7L2 and KCNJ11 is likely to be mediated by the impairment of insulin secretion from β-cells, as a well-established pathogenic pathway in PCOS [10]. The hyperandrogenemia of PCOS is most commonly characterized by increased testosterone levels, resulting from enhanced ovarian biosynthesis [11]. Several genes (CYP11A, CYP19) involved in steroid biosynthesis pathways are also potential candidate genes for PCOS. However, the candidate genetic markers significantly contributing to the diagnosis and prognosis of PCOS needs to be further explored.
Recently, gene expression profiling with microarray and RNA sequencing has been used to discover gene markers and signaling pathways associated with various complex diseases [12]. Gene expression datasets from PCOS patients and normal controls have been collected for the successful identification of gene expression signatures [13]. However, as the extreme heterogeneity and small sample size among studies, the reproducibility of various studies is very low [14]. If a larger sample size is not attainable, integrated analyses of multi-center collaborative studies become very useful [15].
In this study, to investigate the candidate diagnostic and prognostic genetic markers for PCOS, we performed comprehensive and statistical analyses on 9 case-control studies with 13 gene expression profiling datasets. Finally, we identified 13 genetic markers may be potential molecular factors for the diagnosis and prognosis of PCOS patients. Further functional studies of these candidate genes may improve the understanding and treatment of PCOS diseases.
RESULTS
Characteristics of included studies
Nine well-designed GEO studies (accession numbers: GSE1615, GSE5850, GSE5090, GSE6798, GSE8157, GSE10946, GSE34526, GSE43264 and GSE48301) with PCOS patients and normal controls were included in this analysis [16–22]. Five different microarray platforms (GPL96, GPL 97, GPL570, GPL15362 and GPL6244) were used to generate 13 datasets from the nine case-control studies. Detailed descriptions of each dataset were shown in Table 1. After dataset preparing as shown in Figure 1, expression profiles for a total of 8470 genes in these datasets were extracted for further analysis.
Table 1: Characteristics of the 13 datasets included in the analysis
ID |
GSE Acc |
GPL |
Details |
#Control |
#Case |
Country |
PMID |
#Probe |
#Symbol |
|---|---|---|---|---|---|---|---|---|---|
1 |
GSE1615 |
GPL96/97 |
Theca cell |
4 |
5 |
USA |
15598877 |
44928 |
19923 |
2 |
GSE5090 |
GPL96 |
Omental adipose |
8 |
9 |
Spain |
17062763 |
22283 |
13236 |
3 |
GSE5850 |
GPL570 |
Oocytes MII |
6 |
6 |
USA |
17148555 |
54675 |
22878 |
4 |
GSE6798 |
GPL570 |
Vastus lateralis muscle |
13 |
16 |
Denmark |
17563058 |
54675 |
22878 |
5 |
GSE8157 |
GPL570 |
Vastus lateralis muscle |
13 |
10 |
Denmark |
18560589 |
54675 |
22878 |
6 |
GSE10946_lean |
GPL570 |
Cumulus cells(lean) |
6 |
5 |
Canada |
19141487 |
54675 |
22878 |
7 |
GSE10946_obese |
GPL570 |
Cumulus cells(obese) |
5 |
7 |
Canada |
19141487 |
54675 |
22878 |
8 |
GSE34526 |
GPL570 |
Granulosa cells |
3 |
7 |
India |
22904171 |
54675 |
22878 |
9 |
GSE43264 |
GPL15362 |
Subcutaneous adipose |
7 |
8 |
Ireland |
unpub |
17126 |
16910 |
10 |
GSE48301_eEN |
GPL6244 |
Endothelial cells |
4 |
3 |
USA |
23824412 |
32321 |
19740 |
11 |
GSE48301_eEP |
GPL6244 |
Epithelial cells |
3 |
4 |
USA |
23824412 |
32321 |
19740 |
12 |
GSE48301_eMSC |
GPL6244 |
Mesenchymal cells |
4 |
3 |
USA |
23824412 |
32321 |
19740 |
13 |
GSE48301_eSF |
GPL6244 |
Stromal fibroblasts cells |
4 |
4 |
USA |
23824412 |
32321 |
19740 |
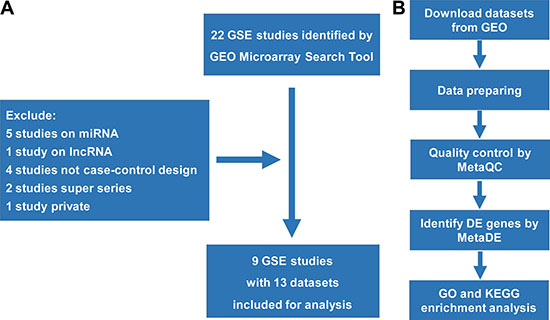
Figure 1: The process of data collection, selection, processing, and analysis. (A) The process of data collection and selection; (B) The process of data processing and further analyses.
Correlation of gene expression in various PCOS profiles
The expressing pattern for each dataset was obtained by the methods of calculating the expression variation score (EVS) [23]. By clustering with Pearson, Spearman and Kendall correlations, the relationships for the 13 datasets were shown in Figure 2A, 2B and 2C, respectively. The coefficience values suggested that two datasets (GSE6798 and GSE8157) from PCOS muscles [20, 21] were extremely consistent with each other and obviously apart from the other 11 datasets. The high consistency may be resulting from the specificity of muscle tissues and the same laboratory. Thus, we integrated these two datasets as Muscle2 and performed further analysis. Among the remained 11 PCOS datasets (Figure 2A–2C), there only three datasets from GSE48301 showed reasonable similarities. As heterogeneity of sampling tissues and low quality of datasets, the quality control should be performed before meta-analysis.
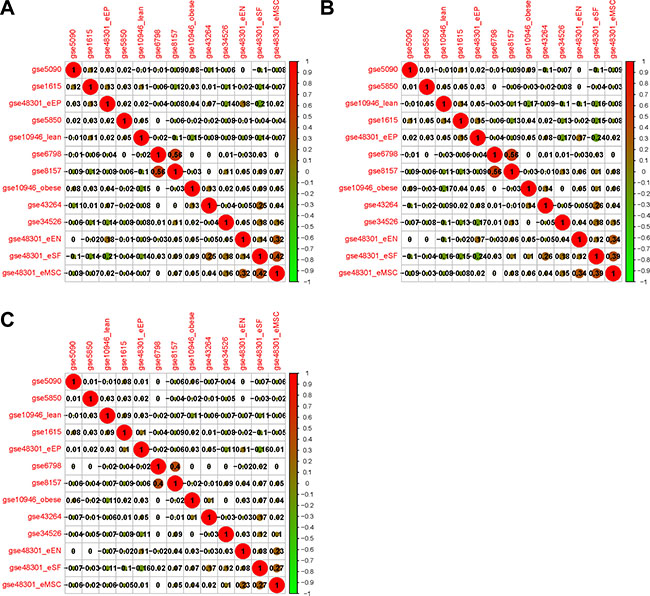
Figure 2: Correlation analyses of the relationships among 13 datasets based on gene expression variation profiles. (A) Clustering with Pearson; (B) Clustering with Spearman; (C) Clustering with Kendall correlation coefficience. The positive and negative correlations between pairs of datasets are shown as red and green respectively, and the size of node index the strength of correlation.
Quality control (QC) assessment in 11 datasets
To identify datasets with high quality and consistency, we carried out the quality assessment for the 11 PCOS datasets by the R package MetaQC [24]. Six QC assessments including homogeneity of coexpression structure, accuracy and consistency of biomarkers detection with or without pathway information were calculated. As shown in Table 2 and Figure 3 with PCA biplots, the top five datasets (GSE43264, GSE48301_eSF, GSE1615, GSE10946_obese and GSE48301_eMSC) performed relatively well in most criteria, while GSE48301_eEP as borderline case. After excluded the bottom five datasets (GSE34526, GSE48301_eEN, GSE5090, GSE5850 and GSE10946_lean) with low quality assessment, there were 6 datasets, named as PCOS6, remained for the following analysis.
Table 2: Results of quality control measures and SMRs for 11 datasets
ID |
Study |
IQC |
EQC |
CQCg |
CQCp |
AQCg |
AQCp |
SMR |
Quality |
|---|---|---|---|---|---|---|---|---|---|
1 |
GSE43264 |
4.02 |
4 |
2.51 |
3.32 |
2.63 |
4.54 |
2.67 |
high |
2 |
GSE48301_eSF |
9.49 |
2.82 |
8.89 |
2.44 |
12.97 |
1.17* |
2.83 |
high |
3 |
GSE1615 |
2.43 |
3 |
1.92* |
2.64 |
0.92* |
2.78 |
4.67 |
high |
4 |
GSE10946_obese |
7.45 |
4 |
0.27* |
2.89 |
0.1* |
0.7* |
5.17 |
high |
5 |
GSE48301_eMSC |
6.1 |
2.62 |
7.12 |
0.59* |
8.35 |
0.49* |
5.33 |
high |
6 |
GSE48301_eEP |
6.25 |
3.1 |
1.88* |
0.23* |
0.46* |
1.16* |
5.5 |
borderline |
7 |
GSE34526 |
1.74* |
4 |
0.35* |
1.36* |
0.14* |
0.05* |
7.17 |
low |
8 |
GSE48301_eEN |
4.17 |
2.34* |
0.46* |
1.06* |
0.63* |
0.08* |
7.17 |
low |
9 |
GSE5090 |
0.98* |
1.68* |
0.51* |
0.77* |
1.86* |
0.05* |
8.33 |
low |
10 |
GSE5850 |
0* |
2.32* |
0.05* |
3.07 |
0.07* |
0.41* |
8.5 |
low |
11 |
GSE10946_lean |
3.15 |
2.8 |
0* |
0.03* |
0.05* |
0.99* |
8.67 |
low |
*P value not significant after Bonferroni correction (i.e. P > 0.05/# of studies).
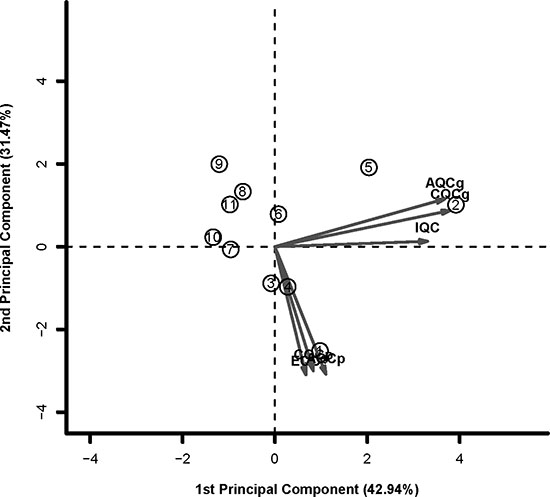
Figure 3: The PCA biplots of QC measures by MetaQC in 11 datasets. The top five datasets (GSE43264, GSE48301_eSF, GSE1615, GSE10946_obese and GSE48301_eMSC) performed relatively well in most criteria. The bottom five datasets (GSE34526, GSE48301_eEN, GSE5090, GSE5850 and GSE10946_lean) were defined as exclusion cases. GSE48301_eEP was defined as borderline case.
Identification of differentially expressed genes (DEG) by MetaDE
Further analyses for both groups of Muscle2 and PCOS6 were performed by the MetaDE package. The datasets in two groups were re-merged and filtered, respectively. From the detection competency curves (Figure 4A, 4B), most methods of meta-analysis were useful to detected common DEGs among different datasets, especially for the Fisher method. Under the criteria of q value less than 0.05, there were 869 and 287 DEGs were identified in Muscle2 and PCOS6, respectively (Supplementary Table S1, S2). The expression patterns of these DEGs were shown in the heatmaps (Figure 4C, 4D), suggesting that the common DEGs were almost consistency in each dataset.
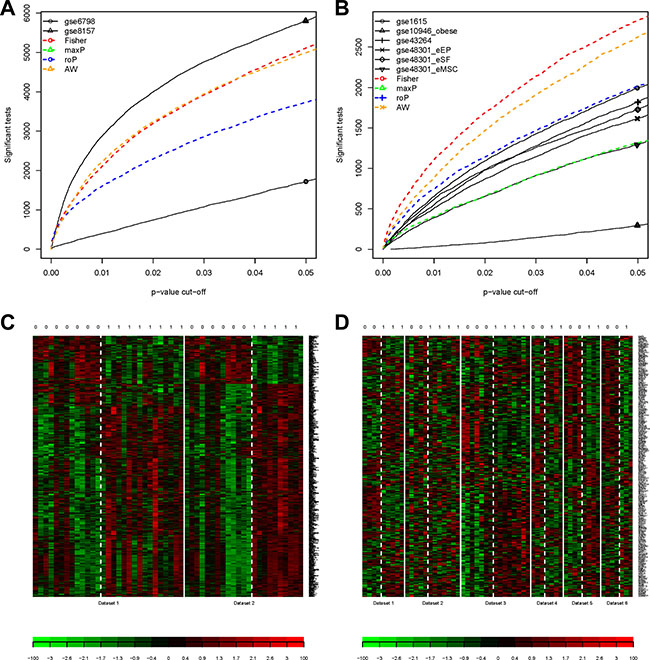
Figure 4: Results of DEGs by metaDE. (A) The detection competency curves for Muscle2; (B) The detection competency curves for PCOS6; (C) The heatmap plot of DEG expression profiles for Muscle2; (D) The heatmap plot of DEG expression profiles for PCOS6. The column with 0 on top stands for normal samples, while 1 stands for PCOS patients.
Functional enrichment and signaling pathway analyses
Functional enrichment analyses were performed with the DAVID web server. We found that the 869 DEGs in Muscle2 datasets were particularly enriched in the muscle system, metabolic process and activity (Figure 5A, Supplementary Table S3), agreed with previous reports [20, 21]. Meanwhile, the 287 DEGs in PCOS6 datasets mainly focused on the response to stimulus from endogenous, hormone and steroid hormone (Figure 5B, Supplementary Table S4).
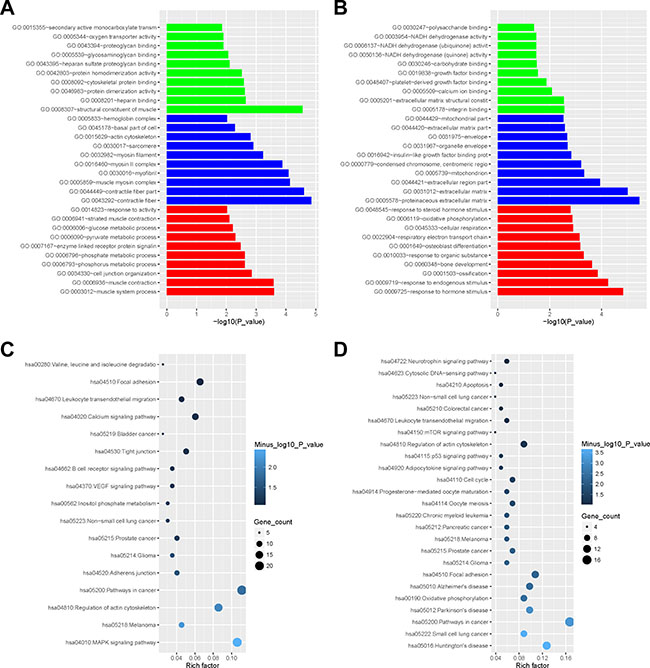
Figure 5: The enrichment analysis of the DEGs by DAVID. (A) The barplot of gene ontology enrichment for Muscle2 (green for molecular function, blue for cellular component, red for biological process); (B) The barplot of gene ontology enrichment for PCOS6; (C) Rich factor plot of pathway enrichment for Muscle2; (D) Rich factor plot of pathway enrichment for PCOS6.
Further signaling pathway analyses showed that the DEGs in Muscle2 almost enriched in several types of cancers, junction pathway, and signaling pathways including B cell receptor, VEGF, Calcium and MAPK (Figure 5C, Supplementary Table S5). Then, the PCOS6 DEGs directly focused on the oocyte meiosis and oocyte maturation, and also enriched in cancer pathway, apoptosis, adhesion, adipocytokine, neurotrophin, mTOR and p53 signaling pathways (Figure 5D, Supplementary Table S6).
Further analyses of common and potential DEGs in PCOS
The DEGs overlapped in Muscle2 and PCOS6 groups were shown in a Venn diagram (Figure 6). Interestingly, there were 13 DEGs (SIAE, S100A8, ICAM1, EIF4E2, RAB32, FN1, MORC4, RGS10, SLC1A1, FGF7, SLC35D2, PDGFRA and APCDD1) identified in both groups. Then, these DEGs were classified as common markers of PCOS (Table 3).
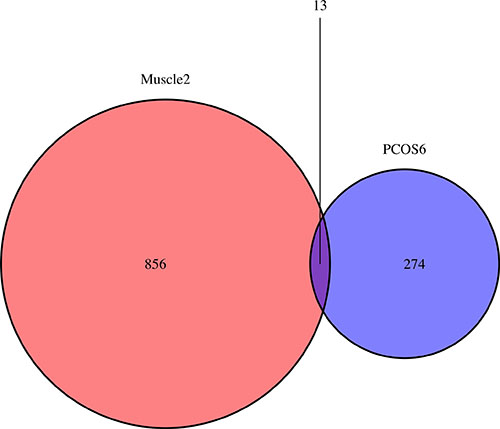
Figure 6: The venn plot for common biomarkers among DEGs of Muscle2 and PCOS6. The 856 DEGs are specific in Muscle2, while 274 DEGs are specific in PCOS6. There are 13 DEGs identified in both groups are SIAE, S100A8, ICAM1, EIF4E2, RAB32, FN1, MORC4, RGS10, SLC1A1, FGF7, SLC35D2, PDGFRA and APCDD1.
Table 3: The 13 common DEGs identified in both Muscle2 and PCOS6
Gene |
Muscle2 |
PCOS6 |
||||
|---|---|---|---|---|---|---|
Meta.stat |
Meta.p_value |
Meta.q_value |
Meta.stat |
Meta.p_value |
Meta.q_value |
|
SIAE |
24.8724 |
7.09E-05 |
0.0094 |
36.5715 |
0.0006 |
0.0383 |
S100A8 |
17.3211 |
0.0022 |
0.0485 |
36.2393 |
0.0006 |
0.0404 |
ICAM1 |
20.0402 |
0.0007 |
0.0290 |
49.7231 |
8.01E-06 |
0.0150 |
EIF4E2 |
32.4194 |
2.34E-06 |
0.0012 |
39.4171 |
0.0002 |
0.0324 |
RAB32 |
18.2467 |
0.0015 |
0.0410 |
40.2864 |
0.0002 |
0.0298 |
FN1 |
22.7958 |
0.0002 |
0.0172 |
43.3634 |
5.93E-05 |
0.0208 |
MORC4 |
21.3159 |
0.0004 |
0.0236 |
34.4672 |
0.0011 |
0.0469 |
RGS10 |
19.1576 |
0.0010 |
0.0343 |
47.2925 |
1.87E-05 |
0.0156 |
SLC1A1 |
18.3162 |
0.0014 |
0.0403 |
38.6278 |
0.0003 |
0.0333 |
FGF7 |
19.3963 |
0.0009 |
0.0328 |
34.6910 |
0.00103 |
0.0460 |
SLC35D2 |
19.9783 |
0.0007 |
0.0292 |
38.0259 |
0.0004 |
0.0347 |
PDGFRA |
18.5395 |
0.0013 |
0.0382 |
38.8920 |
0.0003 |
0.0333 |
APCDD1 |
27.6092 |
1.82E-05 |
0.0040 |
35.3975 |
0.0008 |
0.0430 |
Furthermore, the multivariate Cox proportional hazard analyses of the 13 DEGs among patients with ovarian serous cystadenocarcinoma (TCGA: http://cancergenome.nih.gov) were performed. We found that four genes (SIAE, ICAM1, FN1, and FGF7) were significantly correlated with disease-free survival, while the other three genes (SIAE, FGF7, and PDGFRA) were significantly correlated with overall survival (Table 4). These findings further confirmed that the critical role of these candidate genes in PCOS.
Table 4: Cox proportional hazard analysis of the 13 common DEGs for DFS and OS among patients with ovarian serous cystadenocarcinoma
Variable |
PHA Test* |
HR |
P |
95%CI_L |
95%CI_H |
|---|---|---|---|---|---|
DFS |
|||||
SIAE |
0.7022 |
1.1439 |
0.0354† |
1.0092 |
1.2965 |
ICAM1 |
0.9702 |
1.2007 |
0.0361† |
1.0119 |
1.4247 |
EIF4E2 |
0.7475 |
1.2159 |
0.0707 |
0.9837 |
1.5030 |
RAB32 |
0.1007 |
0.7993 |
0.0586 |
0.6336 |
1.0082 |
FN1 |
0.4617 |
0.7808 |
0.0158† |
0.6387 |
0.9547 |
FGF7 |
0.3707 |
1.4987 |
0.0001† |
1.2204 |
1.8404 |
OS |
|||||
AGE |
0.8117 |
1.0325 |
0.0008† |
1.0134 |
1.0520 |
FGF7 |
0.1797 |
1.4019 |
0.0129† |
1.0741 |
1.8298 |
PDGFRA |
0.2893 |
0.5232 |
0.0052† |
0.3321 |
0.8243 |
Abbreviations: DFS, disease-free survival; OS, overall survival; PHA test, proportional hazards assumption test; HR, hazard ratio. *!PHA test P < 0.05 violates the hazards assumption. †Significant.
DISCUSSION
PCOS is a highly complex endocrine disorder and affected by phenotypically heterogeneous. As its phenotypic heterogeneity and studies with small sample sizes, the power is low to identify specific genes for PCOS. To increase the sample size and make powerful analysis, we performed the integrated and meta-analyses to improve the quality of gene association studies. Then, we identified 13 genetic markers may be potential molecular factors for the diagnosis and prognosis of PCOS patients.
Recently, microarray analysis of gene expression profiles has been widely used to identify genes and biological pathways associated with various complex diseases, including PCOS. However, previous studies have sampled different tissues from PCOS patients. The pathological factors and mechanisms in various tissues of PCOS patients may be similar. In this study, to identify the common PCOS-associated genes in multiple types of tissues, we carried out the integrated analysis and identified 13 common DEGs for PCOS. Taking ICAM-1 for an example, two SNPs of ICAM-1, G241R and K469E, have been found to be risk factors for PCOS [25]. Moreover, ICAM1 K469E is associated with obesity and PCOS, according to serum triglyceride levels [26]. As evidenced by gene ontology and pathway analyses, these DEGs in Muscle2 datasets directly involved in muscle mysion complex, myofibril, muscle system and muscle contraction. The DEGs in PCOS6 datasets mainly focused on oocyte pathways, including oocyte meiosis (PPP2R1B, RPS6KA6, MAD2L1, SGOL1, CAMK2D, IGF1, and ANAPC11) and progesterone-mediated oocyte maturation (RPS6KA6, MAD2L1, IGF1, ANAPC11, PIK3R3, and CCNA2). In-depth functional studies of these candidate genes and signaling pathways may improve the understanding of PCOS.
Along with next generation sequencing (NGS), increasing numbers of studies explored differences in ncRNA and epigenetic modifications (such as DNA methylation) between PCOS patients and normal controls. It has been reported that serum miR-21 is markedly increased in PCOS patients. Through targeting LATS1, miR-21 may promote PCOS progression and act as a novel non-invasive biomarker for the diagnosis of PCOS [27]. lncRNA SRA has been found to be associated with PCOS, and may be an important mediator of adiposity-related processes in individual susceptible to PCOS [28]. The global methylation of peripheral leukocyte DNA has no difference between PCOS patients and controls [29]. The methods for genome-wide DNA methylation analysis at a single base pair resolution are evolving quickly [30]. Therefore, it will be more feasible to carry out genome-wide studies with sufficient samples in different tissue types.
In conclusion, this meta-analysis successfully integrated gene expression datasets for PCOS. This process stabilized the effects of the studies’ extreme heterogeneity and small sample sizes, and uncovered genes and biological pathways associated with PCOS. Finally, we identified 13 common genes among various PCOS tissues, providing novel and potential molecular markers for the diagnosis and prognosis of PCOS diseases. Further functional studies on these genes may improve understanding of the pathological processes of PCOS.
MATERIALS AND METHODS
Collection and inclusion criteria of studies
Using the Gene Expression Omnibus (GEO) Microarray Search Tool (http://gbnci-abcc.ncifcrf.gov/geo/ [31] , we searched the GEO database for publicly available studies from its inception to August 31, 2016, using the following keywords: “Homo sapiens” (organism), “PCOS” or “Polycystic ovary syndrome” (study keywords), and “RNA” (sample type). After a systematic review, 22 GSE studies were collected. The inclusion criteria for studies were as follows: 1) patients diagnosed with PCOS diseases and normal controls; 2) gene expression profiling of mRNA; and 3) sufficient information to perform the analysis. Then, 13 datasets from 9 studies were collected for subsequent analysis (Table 1). Figure 1A provides details of the process of data collection and study selection. The workflow of data processing and analysis is illustrated in Figure 1B.
Dataset preparing
Thirteen gene expression datasets were downloaded from the GEO database, which were completed in five different platforms. Because different probes were used to detect gene expression in different platforms, the number of detectable genes varied across platforms (Table 1). To merge these datasets from different studies, we used three functions in the R package MetaDE: MetaDE.match, MetaDE.merge, and MetaDE.filtering [15]. First, the probe with largest interquartile range (IQR) among all probes annotated to the same gene was selected to represent the expression level of the gene. Second, we merged the gene expression profiles to maintain the commonly profiled genes across the 13 datasets. Finally, either un-expressed (10% genes with small mean intensity) or un-informative genes (10% small standard deviation) were filtered, and the remaining 8470 genes were retained for further analysis.
Statistical analyses
The ind.analysis function in the MetaDE package was used to compare gene expression between PCOS patients and normal controls in each dataset. We used moderated t statistics to evaluate statistical significance.
The transcriptome of each dataset was evaluated with the expression variation score (EVS), introduced by Marina Sirota [23]. In each dataset, the EVS for the gene j is defined as EVSj=-sign(tj)log(pj), where tj is the moderated t statistic for the gene j and pj is the p-value of the test. The scale and sign of the EVS represent the degree of significance and the ‘direction’ of the association, respectively. Then, we used the EVS vector to represent the pattern of the gene expression profile for each dataset. The consistency of the transcriptome tendency was estimated by the correlation coefficients of the EVS vectors between datasets [23].
Quality control
Before meta-analysis, the MetaQC package [24] was used to determine the inclusion/exclusion criteria for meta-analysis. The six quantitative quality control (QC) measures were calculated by MetaQC, and the principal component analysis (PCA) biplots and standardized mean ranks (SMR) were helpful to identify and exclude study with low quality before further meta-analysis.
Meta-analysis
The meta-analysis in each group was completed by MetaDE package. The q-values less than 0.05 for Fisher method, which were subjected to multiple-testing correction with the Benjamini-Hochberg method [32], were the critical to DEGs. The expression patterns in datasets of all DEGs were shown on heatmap plot.
Gene ontology and pathway enrichment analyses
The gene ontology and pathway enrichment analyses of interested gene sets were completed by DAVID web servers (https://david.ncifcrf.gov) [33].
TCGA and survival analysis
A study on ovarian serous cystadenocarcinoma (ov_tcga) with 586 patients was collected form TCGA database (TCGA: http://cancergenome.nih.gov). Based on information of disease-free survival (DFS) and overall survival (OS) for ov patients, we used a multivariate Cox proportional hazards model to determine hazard ratios (HRs) for biomarkers. All analyses were performed with R statistical software (survival and risksetROC packages).
CONFLICTS OF INTEREST
No potential competing interest.
GRANT SUPPORT
This work was supported by grants obtained from the National Natural Science Foundation of China (31471250, 31371510, 31571519, 81600675, 81530042, 31571529, 31210103905, 31571390, 31401126), Ministry of Education Grants IRT_15R51, the Science and Technology Commission of Shanghai Municipality (grant number 15JC1403200), and Fundamental Research Funds for Central Universities (2000219136, 1507219042).
Authors’ contributions
LCQ, WL, JN and WGY designed the study and downloaded and analyzed the data; LCQ, LXQ, YJ, ZXB and WGY drafted the manuscript; and LCQ, HHR, ZSH, LXL and WGY contributed to the interpretation of data and the calculation of results. All authors read and approved the manuscript.
REFERENCES
1. Legro RS. Diagnostic criteria in polycystic ovary syndrome. Semin Reprod Med. 2003; 21:267–75.
2. Obermayer-Pietsch B, Trummer C, Schwetz V, Schweighofer N, Pieber T. Genetics of insulin resistance in polycystic ovary syndrome. Curr Opin Clin Nutr Metab Care. 2015; 18:401–6.
3. Panidis D, Farmakiotis D, Rousso D, Kourtis A, Katsikis I, Krassas G. Obesity, weight loss, and the polycystic ovary syndrome: effect of treatment with diet and orlistat for 24 weeks on insulin resistance and androgen levels. Fertil Steril. 2008; 89:899–906.
4. Legro RS, Kunselman AR, Dodson WC, Dunaif A. Prevalence and predictors of risk for type 2 diabetes mellitus and impaired glucose tolerance in polycystic ovary syndrome: a prospective, controlled study in 254 affected women. J Clin Endocrinol Metab. 1999; 84:165–9.
5. Talbott EO, Guzick DS, Sutton-Tyrrell K, McHugh-Pemu KP, Zborowski JV, Remsberg KE, Kuller LH. Evidence for association between polycystic ovary syndrome and premature carotid atherosclerosis in middle-aged women. Arterioscler Thromb Vasc Biol. 2000; 20:2414–21.
6. Kosova G, Urbanek M. Genetics of the polycystic ovary syndrome. Mol Cell Endocrinol. 2013; 373:29–38.
7. Barber TM, Franks S. Genetics of polycystic ovary syndrome. Front Horm Res. 2013; 40:28–39.
8. Naderpoor N, Shorakae S, Joham A, Boyle J, De Courten B, Teede HJ. Obesity and polycystic ovary syndrome. Minerva Endocrinol. 2015; 40:37–51.
9. Li T, Wu K, You L, Xing X, Wang P, Cui L, Liu H, Cui Y, Bian Y, Ning Y, Zhao H, Tang R, Chen ZJ. Common variant rs9939609 in gene FTO confers risk to polycystic ovary syndrome. PLoS One. 2013; 8:e66250.
10. Rutter GA, Parton LE. The beta-cell in type 2 diabetes and in obesity. Front Horm Res. 2008; 36(118–34.
11. Barber TM, McCarthy MI, Wass JA, Franks S. Obesity and polycystic ovary syndrome. Clin Endocrinol (Oxf). 2006; 65:137–45.
12. Shi X, Shen S, Liu J, Huang J, Zhou Y, Ma S. Similarity of markers identified from cancer gene expression studies: observations from GEO. Brief Bioinform. 2014; 15:671–84.
13. Ben-Shlomo I, Younis JS. Basic research in PCOS: are we reaching new frontiers? Reprod Biomed Online. 2014; 28:669–83.
14. Motulsky HJ. Common misconceptions about data analysis and statistics. Pharmacol Res Perspect. 2015; 3:e00093.
15. Wang X, Kang DD, Shen K, Song C, Lu S, Chang LC, Liao SG, Huo Z, Tang S, Ding Y, Kaminski N, Sibille E, Lin Y, et al. An R package suite for microarray meta-analysis in quality control, differentially expressed gene analysis and pathway enrichment detection. Bioinformatics. 2012; 28:2534–6.
16. Corton M, Botella-Carretero JI, Benguria A, Villuendas G, Zaballos A, San Millan JL, Escobar-Morreale HF, Peral B. Differential gene expression profile in omental adipose tissue in women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2007; 92:328–37.
17. Kaur S, Archer KJ, Devi MG, Kriplani A, Strauss JF 3rd, Singh R. Differential gene expression in granulosa cells from polycystic ovary syndrome patients with and without insulin resistance: identification of susceptibility gene sets through network analysis. J Clin Endocrinol Metab. 2012; 97:E2016–21.
18. Kenigsberg S, Bentov Y, Chalifa-Caspi V, Potashnik G, Ofir R, Birk OS. Gene expression microarray profiles of cumulus cells in lean and overweight-obese polycystic ovary syndrome patients. Mol Hum Reprod. 2009; 15:89–103.
19. Piltonen TT, Chen J, Erikson DW, Spitzer TL, Barragan F, Rabban JT, Huddleston H, Irwin JC, Giudice LC. Mesenchymal stem/progenitors and other endometrial cell types from women with polycystic ovary syndrome (PCOS) display inflammatory and oncogenic potential. J Clin Endocrinol Metab. 2013; 98:3765–75.
20. Skov V, Glintborg D, Knudsen S, Jensen T, Kruse TA, Tan Q, Brusgaard K, Beck-Nielsen H, Hojlund K. Reduced expression of nuclear-encoded genes involved in mitochondrial oxidative metabolism in skeletal muscle of insulin-resistant women with polycystic ovary syndrome. Diabetes. 2007; 56:2349–55.
21. Skov V, Glintborg D, Knudsen S, Tan Q, Jensen T, Kruse TA, Beck-Nielsen H, Hojlund K. Pioglitazone enhances mitochondrial biogenesis and ribosomal protein biosynthesis in skeletal muscle in polycystic ovary syndrome. PLoS One. 2008; 3:e2466.
22. Wood JR, Dumesic DA, Abbott DH, Strauss JF 3rd. Molecular abnormalities in oocytes from women with polycystic ovary syndrome revealed by microarray analysis. J Clin Endocrinol Metab. 2007; 92:705–13.
23. Sirota M, Schaub MA, Batzoglou S, Robinson WH, Butte AJ. Autoimmune disease classification by inverse association with SNP alleles. PLoS Genet. 2009; 5:e1000792.
24. Kang DD, Sibille E, Kaminski N, Tseng GC. MetaQC: objective quality control and inclusion/exclusion criteria for genomic meta-analysis. Nucleic Acids Res. 2012; 40:e15.
25. Vural P, Kanmaz-Ozer M, Dogru-Abbasoglu S, Gedikbasi A, Cil E, Karadag B, Uysal M. Lack of association between intercellular adhesion molecule-1 (ICAM-1) polymorphisms and polycystic ovary syndrome. J Assist Reprod Genet. 2011; 28:869–75.
26. Ojeda-Ojeda M, Martinez-Garcia MA, Alpanes M, Luque-Ramirez M, Escobar-Morreale HF. Association of TLR2 S450S and ICAM1 K469E polymorphisms with polycystic ovary syndrome (PCOS) and obesity. J Reprod Immunol. 2016; 113:9–15.
27. Jiang L, Li W, Wu M, Cao S. Ciculating miRNA-21 as a Biomarker Predicts Polycystic Ovary Syndrome (PCOS) in Patients. Clin Lab. 2015; 61:1009–15.
28. Liu Z, Hao C, Huang X, Zhang N, Bao H, Qu Q. Peripheral blood leukocyte expression level of lncRNA steroid receptor RNA activator (SRA) and its association with polycystic ovary syndrome: a case control study. Gynecol Endocrinol. 2015; 31:363–8.
29. Xu N, Azziz R, Goodarzi MO. Epigenetics in polycystic ovary syndrome: a pilot study of global DNA methylation. Fertil Steril. 2010; 94:781–3 e1.
30. Laird PW. Principles and challenges of genomewide DNA methylation analysis. Nat Rev Genet. 2010; 11:191–203.
31. Zhu Y, Davis S, Stephens R, Meltzer PS, Chen Y. GEOmetadb: powerful alternative search engine for the Gene Expression Omnibus. Bioinformatics. 2008; 24:2798–800.
32. Wang L, Wu LF, Lu X, Mo XB, Tang ZX, Lei SF, Deng FY. Integrated Analyses of Gene Expression Profiles Digs out Common Markers for Rheumatic Diseases. PLoS One. 2015; 10:e0137522.
33. Huang DW, Sherman BT, Tan Q, Kir J, Liu D, Bryant D, Guo Y, Stephens R, Baseler MW, Lane HC, Lempicki RA. DAVID Bioinformatics Resources: expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Res. 2007; 35:W169–75.