
INTRODUCTION
Treatment of metastatic renal cell carcinoma (mRCC) has dramatically changed over the last decade and multiple targeted therapies have replaced IL-2 and IFN-α immunotherapy as the primary treatment option. These new agents, such as Sorafenib, Sunitinib or Temsirolimus, mainly target two pathways, vascular endothelial growth factor (VEGF) signaling and the mammalian target of rapamycin (mTOR) [1, 2, 3, 4, 5, 6]. Although targeted agents have substantially improved patient outcomes, 14.080 deaths due to kidney and renal pelvic cancer are estimated for 2015 in the United States [7], not least because complete response is rare and many tumors eventually develop resistance [8, 9, 10]. Therefore, novel treatment approaches are urgently needed and efforts to identify novel therapeutic targets based on molecular tumor characteristics are necessary.
MET encodes for a receptor tyrosine kinase (RTK) called c-Met or hepatocyte growth factor receptor (HGFR). Cells of epithelial origin widely present HGFR on their surface and overexpression of HGFR is common in carcinomas [11]. The ligand for HGFR is hepatocyte growth factor/scatter factor (HGF/SF) and binding induces recruitment of several signaling effectors. Downstream signaling of HGFR includes activation of MAPK and PI3K-AKT pathway and induction of the vascular endothelial growth factor (VEGF) and in consequence evokes a variety of pleiotropic pro-tumorigenic responses, like cell migration, proliferation and angiogenesis [12].
Mechanisms of MET activation include mutation and amplification. Activating germline MET mutations have been observed in patients with hereditary papillary RCCs (papRCC) and in 13% of sporadic papillary RCC [13]. Moreover, somatic mutations of MET have been reported in head and neck squamous cell carcinomas [14], lung adenocarcinomas [15], and childhood hepatocellular carcinomas [16]. MET amplifications have been reported in colorectal, gastric, esophageal, lung and clear-cell ovarian cancer [17, 18, 19, 20, 21]. High HGFR protein expression is frequently observed in carcinomas with aggressive phenotype and associated with poor prognosis in non-small cell lung, ovarian and colorectal cancer [22, 23, 24]. Besides papillary RCC, MET upregulation has also been observed in clear-cell RCC (ccRCC) and association with poor pathologic features and unfavorable prognosis has been described [25, 26]. However, larger studies addressing HGFR protein levels and genetic alterations are missing.
Therefore, we systematically assessed the molecular status of MET in ccRCC in correlation with clinical features in a large, hospital-based series with long-term follow-up information and show that elevated HGFR expression and MET amplifications are evident in a substantial percentage of metastatic and/or aggressive ccRCCs and emerge de novo in RCC metastasis.
RESULTS
Immunohistochemistry and CISH Analyses
Immunostains and Chromogenic-in-situ-Hybridisation (CISH) were performed on tissue microarrays (TMA) containing tumor and corresponding normal renal tissue from 932 patients with renal cell carcinomas. The study is focused on 763 clear-cell RCC. In total 572 cases could be successfully scored for both, HGFR expression by immunohistochemistry and MET copy number by CISH analysis. The remaining cases were excluded from further analyses either because of insufficient tumor tissue, poor tissue preservation or missing patient information. Figure 1(A-D) depicts immunohistochemical HGFR expression and corresponding MET CISH results. The clinical and pathological features of the study population are summarized in Table 1.
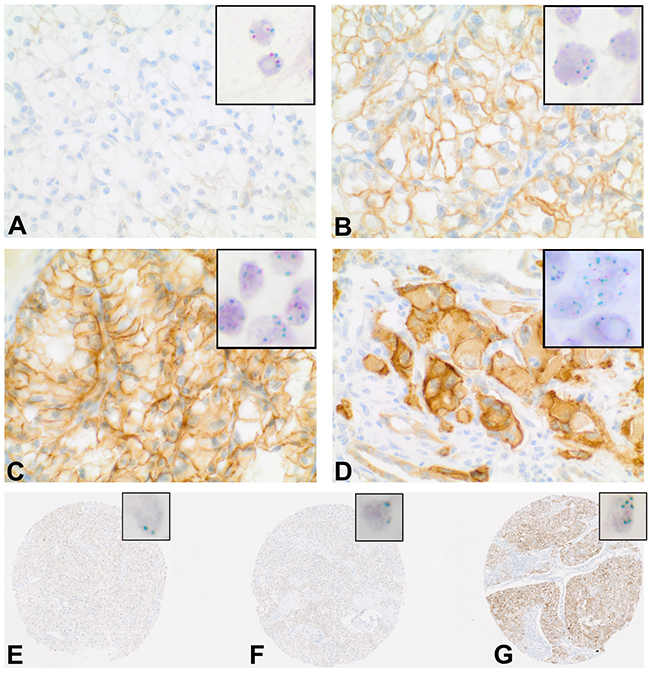
Figure 1: HGFR expression and MET copy number in clear-cell RCC. A-G. Immunohistochemical demonstration of HGFR expression; Insert: corresponding Chromogenic-in-situ-Hybridisation (CISH) with centromeric region of chromosome 7 (red) and MET signals (green) in the nuclei. A-D. HGFR expression and MET copy numbers in primary ccRCC. (A) No elevated protein levels, no copy number increase. (B) Immunoreactive score (IRS) = 8, MET copy number >4. (C) IRS = 12; MET copy number >4. (D) IRS = 12, MET copy number >8. E-G: MET status in different regions of the primary tumor and matching metastasis. E+F. Primary tumor, no elevated protein levels and no copy number increase. (G) Metastasis, IRS = 12, MET copy number >4.
Table 1: Clinicopathological characteristics of the study population
Variable |
n(%) |
Study Population |
572 |
Fuhrman Grade |
|
1 |
163(29) |
2 |
317(55) |
3 |
92(16) |
4 |
2(0) |
Primary tumor |
|
pT1 |
313(55) |
pT2 |
46(8) |
pT3 |
191(33) |
pT4 |
22(4) |
Synchronous distant metastasis |
|
yes |
91(15) |
no |
481(84) |
Local lymphnode metastasis |
|
pN1 |
35(6) |
pN0 |
331(58) |
pNx |
206(36) |
Sex |
|
female |
234(41) |
male |
338(59) |
Age at surgery |
|
>65 |
251(44) |
≤65 |
321(56) |
ECOG |
|
0 |
353(62) |
≥1 |
219(38) |
HGFR was immunohistochemically detected in a total of 83% cases (476/572), 51% (292/572) showed low (defined as immunoreactive score (IRS) < 6) and 32% (184/572) high cytoplasmic positivity (defined as IRS ≥ 6), the remaining 96 cases were completely negative (17%). MET copy number gains could be detected in 310 cases, 47% (272/572) tumors exhibited >2-4 MET copies per nucleus and another 7% (38/572) tumors more than 4 copies per nucleus, the remaining 46% (262/572) cases showed 2 copies per nucleus. Increase of MET/centromer-7 ratio > 2 was only seen in 7 tumors, all being positive by immunohistochemistry; furthermore 37 out of 38 tumors with more than 4 copies per nucleus were positive by HGFR immunohistochemistry (Supplementary Figure S1A+B). In turn, out of 187 tumors with high HGFR protein levels only 5 tumors showed MET/centromer-7 ratio >2, respectively 141 tumors an average MET copy number >2, the remaining tumors (n=46) 2 MET copies/nucleus. However, the protein levels of HGFR and MET copy number showed a weak positive correlation (Spearman’s rank correlation rho=0.377; p < 0.001, Supplementary Figure S1C).
HGFR expression / MET copy numbers and tumor progression
As MET signaling has been reported to be pivotal in development of cancer and metastasis [12] we created another TMA including tissue of 18 primary ccRCCs and corresponding metastasis. Out of 15 primary ccRCCs with low/no HGFR expression, high HGFR protein levels were detected in 6 corresponding metastases. Figure 1(E-G) demonstrates strong increase of HGFR expression in the metastasis of a primarily HGFR-negative ccRCC. CISH analyses revealed that increase of protein levels was accompanied by MET copy number gains in 5 out of these 6 cases with an average of two MET copies per nucleus in the primary tumor and an average of 4.4 MET copies per nucleus in the metastasis. The remaining case showed stable MET copy numbers in primary tumor and metastasis.
Mutation analysis
As increased HGFR protein levels were observed in the absence of MET copy number gains, HGFR overexpression has to be based on other mechanisms than amplification. As mutations that inactivate the Cbl binding site lead to constitutive HGFR expression [27], we checked for MET mutations by targeted next-generation sequencing in 9 tumors with high HGFR protein levels and 2 MET copies/nucleus and 2 tumors with high HGFR protein levels and gains of MET copy numbers. No MET mutations were found using Ion AmpliSeqTM Cancer Hotspot Panel v2 comprising exon 2, 11, 14, 16, and 19 of the MET gene. Besides VHL mutations, we did not identify other recurrent mutations in the examined genes.
Comparison of HGFR expression / MET copy numbers with Clinical and Pathological Features
The proportion of tumors positive for HGFR by immunohistochemistry increased with dedifferentiation (P < 0.001) and occurrence of distant metastasis (P = 0.049) (Figure 2). For example, percentage of tumors with high HGFR expression was 17% in G1 compared to 46% in G3 carcinomas. No consistent association of HGFR expression and tumor extent, lymph node metastasis and patient age was observed.
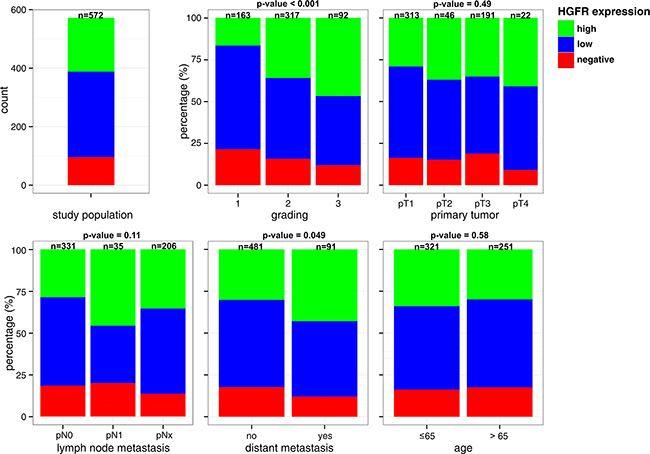
Figure 2: Comparison of HGFR expression with Clinical and Pathological Features.
MET copy number gains were accompanied by dedifferentiation (P < 0.001), higher tumor extent (P < 0.001), positive lymphnode status (P = 0.006), and occurrence of distant metastasis (P = 0.02) (Figure 3). For instance, the proportion of tumors with more than four MET copies/nucleus increased about 10 times in high-grade carcinomas compared to low-grade carcinomas (20% vs 2%). Whereas more than half of T1-carcinomas (54%) showed two MET copy numbers/nucleus, tumors without copy number gains represent only 18% of locally advanced (T4) carcinomas. No consistent association between MET copy numbers and patient age was observed.
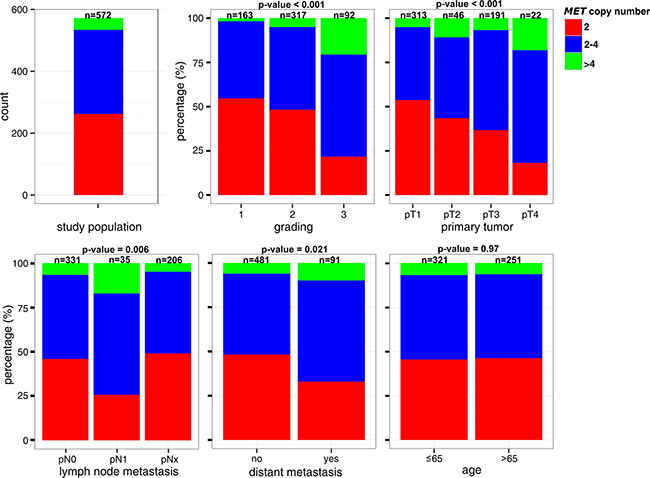
Figure 3: Comparison of MET copy number status with Clinical and Pathological Features.
HGFR expression / MET copy numbers and patient prognosis
The median time of follow-up was 8 years (mean 7.8, min 0.014, max 23.7 years), among the 572 patients, 189 had died from ccRCC by the end of the follow-up.
When tumors were grouped according to HGFR expression, univariate survival analysis revealed a decrease in cancer-specific survival (P = 0.008) in patients affected by tumors with high HGFR expression compared to tumors with low/no HGFR expression. In contrast, no consistent relationship was observed between HGFR expression and time to progression (TTP). When tumors were grouped based on MET copy numbers per nucleus, cases with more than two MET copies exhibited significant shorter cancer-specific survival (P = 0.001) and time to progression (P = 0.046). Kaplan-Meier plots are depicted in Figure 4.
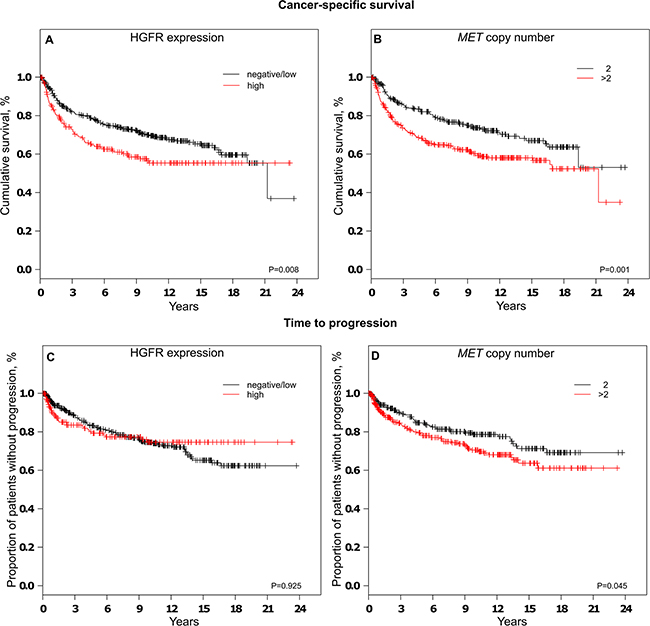
Figure 4: Analysis of cancer-specific survival A+B and time to progression C+D depending on HGFR expression A+C or MET copy number status B+D.
Differences in cancer-specific survival or time to progression did not remain statistically significant after adjustment for established prognostic factors in the multiple regression analysis (Table 2 and Supplementary Table S1).
Table 2: Uni- and multivariate analyses of prognostic factors influencing cancer-specific survival (CSS) in clear-cell RCC
Univariate |
Multivariate |
|||||||
M0+M1 |
M0 |
M1 |
||||||
HR(95% CI) |
P |
HR (95% CI) |
P |
HR(95% CI) |
P |
HR (95% CI) |
P |
|
Grade of malignancy1 |
4.7 (3.45-6.39) |
<0.001 |
2.08 (1.45-2.97) |
<0.001 |
2.58 (1.58-4.22) |
<0.001 |
1.94 (1.14-3.3) |
0.014 |
Primary tumor 2 |
3.9 (2.9-5.24) |
<0.001 |
2.23 (1.59-3.12) |
<0.001 |
2.79 (1.84-4.22) |
<0.001 |
1.3 (0.73-2.34) |
0.36 |
Lymphnode metastasis3 |
5.24 (3.52-7.78) |
<0.001 |
1.65 (1.06-2.57) |
0.026 |
2.61 (1.4-4.85) |
0.002 |
1.41 (0.74-2.68) |
0.29 |
Distant metastasis4 |
11.2 (8.23-15.22) |
<0.001 |
7.0 (4.98-9.84) |
<0.001 |
- |
- |
- |
- |
ECOG5 |
0.5 (0.38-0.67) |
<0.001 |
0.68 (0.51-0.91) |
0.01 |
0.75 (0.5-1.13) |
0.17 |
0.69 (0.43-1.11) |
0.13 |
Age6 |
1.1 (0.82-1.47) |
0.54 |
1.13 (0.83-1.53) |
0.43 |
1.13 (0.76-1.7) |
0.53 |
1.18 (0.71-1.95) |
0.53 |
Sex7 |
0.68 (0.5-0.91) |
0.01 |
0.81 (0.60-1.11) |
0.19 |
0.67 (0.45-1.0) |
0.052 |
0.91 (0.55-1.5) |
0.7 |
HGFR-Expression8 |
1.49 (1.11-2.0) |
0.008 |
1.02 (0.73-1.43) |
0.89 |
1.05 (0.69-1.61) |
0.81 |
0.84 (0.47-1.5) |
0.56 |
MET copy number9 |
1.62 (1.2-2.18) |
0.001 |
1.15 (0.82-1.61) |
0.42 |
0.83 (0.54-1.27) |
0.39 |
1.7 (0.94-3.08) |
0.08 |
1 G3/4 vs G1/G2.
2 pT3/pT4 vs pT1/pT2.
3 pN1/pN2 vs pNx/pN0
4 M1 vs M0.
5 0 vs ≥1
6 > 65 vs ≤ 65
7 Female vs Male
8 high vs negative/low
9 >2 vs 2
HR, hazard ratio; CI, confidence interval. Probability values and hazard ratios considered statistically significant are shown in bold.
DISCUSSION
Despite comprehensive translational research efforts in renal cell carcinoma and availability of targeted therapy options, at present neither prognostic nor predictive biomarkers are established for routine clinical treatment stratification [28]. Independently to the lack of molecular therapy prediction, targeted therapy has demonstrated modest benefit [29]. However, the rarity of cures and intrinsic or acquired resistance demands novel treatment approaches and identification of predictive and prognostic biomarkers. c-Met/hepatocyte growth factor receptor (HGFR) encoded by the MET oncogene is a validated therapeutic target for a number of malignancies and results obtained from clinical trials are encouraging [30].
Our results demonstrate that elevated HGFR protein levels are associated with dedifferentiation and distant metastasis and MET copy number gains with dedifferentiation, tumor extent, lymph node, and distant metastasis. Limitations of this study are the retrospective single-institution design and the use of tissue micro array technique, which enables studies on large collectives, but may disregard tumor heterogeneity. However, our findings are in accordance with previous reports showing that elevated HGFR expression correlates with worse cancer-specific survival [25]. Moreover, we demonstrate that MET copy number gains correlate with unfavorable patient outcome. In addition, a recent study on brain metastasis of ccRCC indicated MET overexpression as an independent prognostic factor for brain metastasis-specific survival [31].
At present, reliable data regarding diagnostic criteria and predictive cut offs for anti-MET treatment are not available for ccRCC, thus further studies have to define applicable parameters. Recently published final results of the phase 3 trial METEOR comparing cabozantinib (an oral inhibitor of tyrosine kinases including MET, VEGFR, and AXL) versus everolimus in patients with advanced renal cell carcinoma [32] do not provide evidence for a significant relationship between MET expression levels and treatment outcomes. Unfortunately, HGFR expression was only investigated in a subset of RCCs and MET copy number status was not determined. Schoffski et al. reported that ALK/ROS/MET inhibitor Crizotinib induced long lasting disease control in metastatic papillary renal cell carcinoma type 1 with MET mutation and long term stable disease in a case with MET amplification. In contrast, among patients with papRCCs devoid of MET amplification or mutation, none achieved a response upon Crizotinib treatment [33].
The MET pathway is associated with breast cancer progression and anti-Met therapies are currently evaluated in breast cancer patients [34, 35, 36]. A phase II study of Foretinib (an oral multi-kinase inhibitor of MET, RON, AXL, TIE-2, and VEGF receptors) in patients with triple-negative breast cancer observed a clinical benefit rate of 46 %. Unfortunately, only 6.7 % of tumors were MET positive by IHC and none of the tumors harbored a MET amplification. Hence, the study was unable to confirm MET status as a predictive biomarker [37].
In non-small cell lung cancer the results are inconsistent [38], however HGFR protein levels have been reported to be predictive for anti-MET treatment in clinical studies [39, 40, 41] whereas these results were not confirmed in a phase III trial (METLung) [42]. In contrast, MET amplification seems to be predictive based on recently reported results of a Crizotinib phase I study [43].
At present it remains unclear, if MET copy number assessment or examination of HGFR protein expression is the approach of choice to identify potential therapy responder. Due to discrepancies in protein levels and copy number status in a subset of tumors, we recommend to perform both tests, in situ hybridization and immunohistochemistry, until reliable data is available.
Our results reveal that MET is deregulated in a subset of ccRCC and MET copy number gains (>4/nucleus) occur in 7% of therapy-naive ccRCC. Importantly, this proportion increases to 20% in patients with high-grade carcinomas. Naturally, tumor spreading and consecutive need for systemic therapy is more likely in patients with high-grade ccRCC. Furthermore, we identified elevated HGFR expression in 6 and MET copy number increase in 5 metastases out of 15 ccRCCs without elevated expression of HGFR or copy number gains of MET in the primary tumor. These findings indicate that the MET pathway may be a promising target especially in ccRCC patients with high grade or metastasized disease and the MET status should be re-evaluated in recurrences or metastasis.
MET is transcriptionally activated by hypoxia and acts as mediator of antiangiogenic therapy resistance in models of solid tumors [44, 45]. Importantly, also in Sunitinib-resistant RCC, increase in MET expression and activation was observed [46]. Therefore, therapeutically targeting of MET may prevent or overcome antiangiogenic therapy resistance in RCCs. This implies that not only therapy-naive ccRCC with primary MET upregulation are in the focus of therapeutic MET inhibition but also Sunitinib-resistant RCCs. Further studies on RCCs with prior antiangiogenic therapy have to investigate if MET amplification is seen in therapy resistant RCCs, resembling a resistance mechanism evident in lung cancers with EGFR activating mutations treated with Gefitinib [47].
Association with clinical and pathological features in primary tumor, MET copy number gains in the context of metastatic spread and initial in-vitro data indicating that MET-signaling acts as mediator of antiangiogenic therapy [46], suggest a biologic relevance of MET signaling in ccRCCs.
Several compounds including small-molecule TKIs, monoclonal antibodies, and anti-HGF compounds are available for selective inhibition of MET signaling [48, 49] or inhibition of MET in combination with vascular endothelial growth factor receptor (VEGFR) [32, 50] and are already tested in preclinical and clinical trials [51], making anti-MET therapy a near-term feasible treatment option.
In conclusion, our findings highlight inhibition of MET signaling pathway as a promising new target for the treatment of ccRCC patients and prognostic significance of the molecular status of MET. Furthermore, HGFR expression and MET copy numbers should not only be assessed in the primary tumor, but also, maybe even more important, in recurrences or metastasis to guide anti-MET therapy in ccRCC patients.
PATIENTS AND METHODS
Patients
Tissue samples from 932 patients with primary renal cell carcinomas treated at the Department of Urology at the University of Heidelberg between 1987 and 2005 were collected. The human tissue samples were provided by the Tumour Tissue Bank of the National Centre for Tumour Diseases Heidelberg after approval by the Ethics Committee of the University of Heidelberg. As described previously, clinical follow-up was available for 911 patients, who were prospectively evaluated every 3 months for the first 2 years after treatment, every 6 months for the next 3 years, and yearly thereafter (chest x-ray or thoracic computed tomographic (CT) scan; abdominal sonography or CT scan or magnetic resonance imaging; serum chemistry). No adjuvant treatment of localized disease was administered. Patients with metastasized disease and with a Karnofsky performance index of ≥80 and no medical contraindications received palliative interferon-alpha– and IL-2–based immunotherapy. No targeted therapeutic approaches were performed [52].
Tissue-Micro-Array
A tissue microarray containing 932 primary tumor and corresponding normal tissue samples of 932 patients was composed. The tumors were graded according to the three-tiered nuclear grading system recommended by the WHO Classification of Tumours 2004 and pathologically staged based on the TNM classification (2009). In total, a set of 19 array blocks was generated, each containing 200 tissue core specimens, representing 50 patients per array. A morphologically representative region was chosen from each of the renal cell carcinomas and two cylindrical core tissue specimens per tumor block measuring 0.6 mm in diameter were punched from these regions and arrayed into the recipient paraffin block. Further details have been described previously [53].
In addition, together with the Tumor Tissue Bank of the National Centre for Tumor Diseases Heidelberg, a second tissue microarray was composed containing tumor tissue of 18 patients with metastasized RCCs. Briefly, representative regions of the primary tumor and the matching metastasis was compiled; three (2x primary tumor, 1x metastasis) cylindrical cores (diameter 1 mm) were punched from the donor blocks and arrayed into the recipient paraffin block using a semiautomatic system (Beecher Instruments, Silver Spring, MD, USA). The origins of metastases were adrenal gland (2x), bone (2x) and lymph node (14x).
Immunohistochemistry
After heat-induced antigen retrieval using the target retrieval solution ULTRA Cell Conditioning (ULTRA CC1; Ventana Medical Systems, Tucson, AZ, USA; 950-224) tissue microarray slides were stained with an anti-total c-MET (SP44) rabbit monoclonal primary antibody (Ventana Medical Systems; 790-443). Staining was performed using an automated staining system BenchMark ULTRA (Ventana Medical Systems) in accordance with the manufacturer’s instructions, the following solutions were used: OptiView DAB IHC Detection Kit (760-700), Hematoxylin I (790-2208), Bluing Reagent (760-2037). The arrays were independently scored by two pathologists (S.M.-G. and W.R.) blinded to tissue annotations and patient outcomes. For the immunohistochemical semiquantitative assessment of HGFR expression, the product of the scores of staining intensity and quantity of immunoreactive tumor cells was calculated based on the following scoring system: the intensity ranged from 0 = negative, 1 = low, 2 = medium to 3 = high; the quantity comprised 0 = no expression, 1 < 10% of positive cells, 2 = positivity in 10% to 50%, 3 = positivity in 51% to 80%, and 4 = positivity in more than 80%. The final immunoreactive score (IRS) score (ranging from 0 to 12) is obtained by multiplication of the intensity score and the quantity score.
Chromogenic in situ hybridization (CISH)
The commercial available Probe (ZytoDot 2C SPEC MET/CEN 7 Probe, ZytoVision, Bremerhaven, Germany; C-3057-400) a mixture of a Dinitrophenyl-labeled CEN 7 probe specific for the alpha satellite centromeric region of chromosome 7 (D7Z1) and a Digoxigenin-labeled probe specific for the MET gene at 7q31 has been used according to the manufacturer’s instructions.
Mutation analysis
Mutation analysis was performed as described in detail previously [54]. Briefly, for library preparation, the multiplex PCR-based Ion Torrent AmpliSeqTM technology (Life Technologies) with the Cancer HotSpot Panel v2 (IonTorrent / Thermo Fisher Scientific, Waltham, USA) was used. Amplicon library preparation was performed with the Ion AmpliSeq Library Kit v2.0 using approximately 10 ng of DNA. Briefly, the DNA was mixed with the primer pool, containing all primers for generating the 207 amplicons and the AmpliSeq HiFi Master Mix and transferred to a PCR cycler (BioRad, Munich, Germany). After the end of the PCR reaction, primer end sequences were partially digested using FuPa reagent, followed by the ligation of barcoded sequencing adapters (Ion Xpress Barcode Adapters, Life Technologies). The final library was purified using AMPure XP magnetic beads (Beckman Coulter, Krefeld, Germany) and quantified using qPCR (Ion Library Quantitation Kit, Thermo Fisher Scientific, Waltham, USA) on a StepOne qPCR machine (Thermo Fisher Scientific, Waltham, USA). The individual libraries were diluted to a final concentration of 100 pM and eight to ten libraries were pooled and processed to library amplification on Ion Spheres using Ion PGMTM Template OT2 200 Kit. Unenriched libraries were quality-controlled using Ion Sphere quality control measurement on a QuBit instrument. After library enrichment (Ion OneTouch ES), the library was processed for sequencing using the Ion Torrent 200 bp sequencing v2 chemistry and the barcoded libraries were loaded onto a chip. Our way of pooling eight samples on a 318 chip resulted in a mean coverage of 3000 fold per amplicon.
Variant calling and annotation
Data analysis was performed using the Ion Torrent Suite Software (version 4.4). After base calling, the reads were aligned against the human genome (hg19) using the TMAP algorithm within the Torrent Suite. Variant calling was performed with the variant caller plugin within the Torrent Suite Software and the IonReporter package using a corresponding bed-file containing the coordinates of the amplified regions. Only variants with an allele frequency > 5% and minimum coverage > 100 reads were taken into account. Variant annotation was performed using Annovar [55]. Annotations included information about nucleotide and amino acid changes of RefSeq annotated genes, COSMIC and dbSNP entries as well as detection of possible splice site mutations. For data interpretation and validation, the aligned reads were visualized using the IGV browser (Broad Institute) [56].
Statistical methods
Survival was calculated from the date of nephrectomy to two different events: cancer-specific survival (CSS, event: tumor-related death, survival time was censored for patients who did not experience the investigated event) and time to progression (TTP, event: recurrence, metastasis, deaths before progression were censored). Association between survival times and HGFR expression / MET copy number increase was first assessed by log-rank tests and represented using Kaplan-Meier plots. In order to account for the influence of established prognostic factors, hazard ratios (HRs) and 95% confidence intervals (CIs) were adjusted for patient gender and age, tumor extent, lymph node metastasis, distant metastasis, grade of malignancy, and ECOG Performance Status in a multiple Cox proportional hazard regression. Data were analysed using the R software package (https://cran.r-project.org/). For count data, Fisher’s exact test (two-sided) was used. Spearman’s rank correlation was estimated to quantify the relationship between MET copy numbers and HGFR expression. Probability values <0.05 were considered to indicate a statistically significant result.
ACKNOWLEDGMENTS
We thank David Jansen and Angelika Bönisch for excellent technical assistance, Hildegard Jakobi for excellent help with the patient data and the Tissue Bank of the National Centre for Tumour Diseases, Heidelberg.
CONFLICTS OF INTEREST
Humphrey Gardner is an employee of AstraZeneca.
REFERENCES
1. Escudier B, Eisen T, Stadler WM, Szczylik C, Oudard S, Siebels M, Negrier S, Chevreau C, Solska E, Desai AA, Rolland F, Demkow T, Hutson TE, et al. Sorafenib in advanced clear-cell renal-cell carcinoma. N Engl J Med. 2007; 356: 125–134.
2. Motzer RJ, Escudier B, Oudard S, Hutson TE, Porta C, Bracarda S, Grünwald V, Thompson JA, Figlin RA, Hollaender N, Urbanowitz G, Berg WJ, Kay A, et al. Efficacy of everolimus in advanced renal cell carcinoma: a double-blind, randomised, placebo-controlled phase III trial. Lancet. 2008; 372: 449–456.
3. Rini BI, Escudier B, Tomczak P, Kaprin A, Szczylik C, Hutson TE, Michaelson MD, Gorbunova VA, Gore ME, Rusakov IG, Negrier S, Ou YC, Castellano D, et al. Comparative effectiveness of axitinib versus sorafenib in advanced renal cell carcinoma (AXIS): a randomised phase 3 trial. Lancet. 2011; 378: 1931–1939.
4. Rini BI, Halabi S, Rosenberg JE, Stadler WM, Vaena DA, Ou SS, Archer L, Atkins JN, Picus J, Czaykowski P, Dutcher J, Small EJ. Bevacizumab plus interferon alfa compared with interferon alfa monotherapy in patients with metastatic renal cell carcinoma: CALGB 90206. J Clin Oncol 2010; 28: 2137–2143.
5. Sternberg CN, Davis ID, Mardiak J, Szczylik C, Lee E, Wagstaff J, Barrios CH, Salman P, Gladkov OA, Kavina A, Zarbá JJ, Chen M, McCann L, et al. Pazopanib in locally advanced or metastatic renal cell carcinoma: results of a randomized phase III trial. J Clin Oncol. 2010; 28: 1061–1068.
6. Hudes G, Carducci M, Tomczak P, Dutcher J, Figlin R, Kapoor A, Staroslawska E, Sosman J, McDermott D, Bodrogi I, Kovacevic Z, Lesovoy V, Schmidt-Wolf IG, et al. Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N Engl J Med. 2007; 356: 2271–2281.
7. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015; 65: 5–29.
8. Albiges L, Oudard S, Negrier S, Caty A, Gravis G, Joly F, Duclos B, Geoffrois L, Rolland F, Guillot A, Laguerre B, Legouffe E, Kohser F, et al. Complete remission with tyrosine kinase inhibitors in renal cell carcinoma. J Clin Oncol. 2012; 30: 482–487.
9. Motzer RJ, Hutson TE, Tomczak P, Michaelson MD, Bukowski RM, Oudard S, Negrier S, Szczylik C, Pili R, Bjarnason GA, Garcia-del-Muro X, Sosman JA, Solska E, et al. Overall survival and updated results for sunitinib compared with interferon alfa in patients with metastatic renal cell carcinoma. J Clin Oncol. 2009; 27: 3584–3590.
10. Rasmussen N, Rathmell WK. Looking beyond inhibition of VEGF/mTOR: emerging targets for renal cell carcinoma drug development. Curr Clin Pharmacol. 2011; 6: 199–206.
11. Petrini I. Biology of MET: a double life between normal tissue repair and tumor progression. Ann Transl Med. 2015; 3: 82.
12. Birchmeier C, Birchmeier W, Gherardi E, Vande Woude GF. Met, metastasis, motility and more Nat Rev Mol Cell Biol. 2003; 4: 915–925.
13. Schmidt L, Duh FM, Chen F, Kishida T, Glenn G, Choyke P, Scherer SW, Zhuang Z, Lubensky I, Dean M, Allikmets R, Chidambaram A, Bergerheim UR, et al. Germline and somatic mutations in the tyrosine kinase domain of the MET proto-oncogene in papillary renal carcinomas. Nat Genet. 1997; 16: 68–73.
14. Di Renzo MF, Olivero M, Martone T, Maffe A, Maggiora P, Stefani AD, Valente G, Giordano S, Cortesina G, Comoglio PM. Somatic mutations of the MET oncogene are selected during metastatic spread of human HNSC carcinomas. Oncogene. 2000; 19: 1547–1555.
15. The Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014; 511: 543–550.
16. Park WS, Dong SM, Kim SY, Na EY, Shin MS, Pi JH, Kim BJ, Bae JH, Hong YK, Lee KS, Lee SH, Yoo NJ, Jang JJ, Pack S, et al. Somatic mutations in the kinase domain of the Met/hepatocyte growth factor receptor gene in childhood hepatocellular carcinomas. Cancer Res. 1999; 59: 307–310.
17. Zeng ZS, Weiser MR, Kuntz E, Chen CT, Khan SA, Forslund A, Nash GM, Gimbel M, Yamaguchi Y, Culliford AT 4th, D’Alessio M, Barany F, Paty PB. c-Met gene amplification is associated with advanced stage colorectal cancer and liver metastases. Cancer Lett. 2008; 265: 258–269.
18. Nakajima M, Sawada H, Yamada Y, Watanabe A, Tatsumi M, Yamashita J, Matsuda M, Sakaguchi T, Hirao T, Nakano H. The prognostic significance of amplification and overexpression of c-met and c-erb B-2 in human gastric carcinomas. Cancer. 1999; 85: 1894–1902.
19. Miller CT, Lin L, Casper AM, Lim J, Thomas DG, Orringer MB, Chang AC, Chambers AF, Giordano TJ, Glover TW, Beer DG. Genomic amplification of MET with boundaries within fragile site FRA7G and upregulation of MET pathways in esophageal adenocarcinoma. Oncogene. 2006; 25: 409–418.
20. Schildhaus HU, Schultheis AM, Rüschoff J, Binot E, Merkelbach-Bruse S,Fassunke J, Schulte W, Ko YD, Schlesinger A, Bos M, Gardizi M, Engel-Riedel W,Brockmann M, et al. MET amplification status in therapy-naive adeno- and squamous cell carcinomas of the lung. Clin Cancer Res. 2015; 21: 907–915.
21. Yamamoto S, Tsuda H, Miyai K, Takano M, Tamai S, Matsubara O. Gene amplification and protein overexpression of MET are common events in ovarian clear-cell adenocarcinoma: their roles in tumor progression and prognostication of the patient. Mod Pathol. 2011; 24: 1146–1155.
22. Benedettini E, Sholl LM, Peyton M, Reilly J, Ware C, Davis L, Vena N, Bailey D, Yeap BY, Fiorentino M, Ligon AH, Pan BS, Richon V, et al. Met activation in non-small cell lung cancer is associated with de novo resistance to EGFR inhibitors and the development of brain metastasis. Am J Pathol. 2010; 177: 415–423.
23. Sawada K, Radjabi AR, Shinomiya N, Kistner E, Kenny H, Becker AR, Turkyilmaz MA, Salgia R, Yamada SD, Vande Woude GF, Tretiakova MS, Lengyel E. c-Met overexpression is a prognostic factor in ovarian cancer and an effective target for inhibition of peritoneal dissemination and invasion. Cancer Res. 2007; 67: 1670–1679.
24. Lee CT, Chow NH, Su PF, Lin SC, Lin PC, Lee JC. The prognostic significance of RON and MET receptor coexpression in patients with colorectal cancer. Dis Colon Rectum. 2008; 51: 1268–1274.
25. Gibney GT, Aziz SA, Camp RL, Conrad P, Schwartz BE, Chen CR, Kelly WK, Kluger HM. c-Met is a prognostic marker and potential therapeutic target in clear cell renal cell carcinoma. Ann Oncol. 2013; 24: 343–349.
26. Miyata Y, Kanetake H, Kanda S. Presence of phosphorylated hepatocyte growth factor receptor/c-Met is associated with tumor progression and survival in patients with conventional renal cell carcinoma. Clin Cancer Res. 2006; 12: 4876–4881.
27. Peschard P, Fournier TM, Lamorte L, Naujokas MA, Band H, Langdon WY, Park M. Mutation of the c-Cbl TKB domain binding site on the Met receptor tyrosine kinase converts it into a transforming protein. Mol Cell. 2001; 8: 995–1004.
28. Maroto P, Rini B. Molecular biomarkers in advanced renal cell carcinoma. Clin Cancer Res. 2014; 20: 2060–2071.
29. Escudier B, Albiges L, Sonpavde G. Optimal management of metastatic renal cell carcinoma: current status. Drugs. 2013; 73: 427–438.
30. Vigna E, Comoglio PM. Targeting the oncogenic Met receptor by antibodies and gene therapy. Oncogene. 2015; 34: 1883–1889.
31. Schiefer AI, Mesteri I, Berghoff AS, Haitel A, Schmidinger M, Preusser M, et al. Evaluation of tyrosine kinase receptors in brain metastases of clear cell renal cell carcinoma reveals cMet as a negative prognostic factor. Histopathology. 2015;67:799-805.
32. Choueiri TK, Escudier B, Powles T, Tannir NM, Mainwaring PN, Rini BI, et al. Cabozantinib versus everolimus in advanced renal cell carcinoma (METEOR): final results from a randomised, open-label, phase 3 trial. Lancet Oncol. 2016;17:917-927.
33. Schoffski P, Wozniak A, Escudier B, Rutkowski P, Anthoney A, Bauer S, et al. Crizotinib achieves objective responses and long-lasting disease control in patients (pts) with metastatic papillary renal cell carcinoma type 1 (PRCC1) with somatic MET mutations. EORTC phase II trial 90101 “CREATE”. [abstract]. Cancer Res. 2016;76: Abstract nr CT006.
34. Jardim DL, Tang C, Gagliato Dde M, Falchook GS, Hess K, et al. Analysis of 1,115 patients tested for MET amplification and therapy response in the MD Anderson Phase I Clinic. Clin Cancer Res. 2014;20:6336-6345.
35. Ho-Yen CM, Green AR, Rakha EA, Brentnall AR, Ellis IO, Kermorgant S, et al. C-Met in invasive breast cancer: is there a relationship with the basal-like subtype? Cancer. 2014;120:163-171.
36. Ho-Yen CM, Jones JL, Kermorgant S. The clinical and functional significance of c-Met in breast cancer: a review. Breast Cancer Res. 2015;17:52.
37. Rayson D, Lupichuk S, Potvin K, Dent S, Shenkier T, Dhesy-Thind S, et al. Canadian Cancer Trials Group IND197: a phase II study of foretinib in patients with estrogen receptor, progesterone receptor, and human epidermal growth factor receptor 2-negative recurrent or metastatic breast cancer. Breast Cancer Res Treat. 2016;157:109-116.
38. Finocchiaro G, Toschi L, Gianoncelli L, Baretti M, Santoro A. Prognostic and predictive value of MET deregulation in non-small cell lung cancer. Ann Transl Med. 2015; 3: 83
39. Spigel DR, Ervin TJ, Ramlau RA, Daniel DB, Goldschmidt JH Jr, Blumenschein GR Jr, Krzakowski MJ, Robinet G, Godbert B, Barlesi F, Govindan R, Patel T, Orlov SV. Randomized phase II trial of Onartuzumab in combination with erlotinib in patients with advanced non-small-cell lung cancer. J Clin Oncol. 2013; 31: 4105–4114.
40. Koeppen H, Yu W, Zha J, Pandita A, Penuel E, Rangell L, Raja R, Mohan S, Patel R, Desai R, Fu L, Do A, Parab V, et al. Biomarker analyses from a placebo-controlled phase II study evaluating erlotinib onartuzumab in advanced non-small cell lung cancer: MET expression levels are predictive of patient benefit. Clin Cancer Res. 2014; 20: 4488–4498.
41. Scagliotti G, von Pawel J, Novello S, Ramlau R, Favaretto A, Barlesi F, Akerley W, Orlov S, Santoro A, Spigel D, Hirsh V, Shepherd FA, Sequist LV, et al. Phase III Multinational, Randomized, Double-Blind, Placebo-Controlled Study of Tivantinib (ARQ 197) Plus Erlotinib Versus Erlotinib Alone in Previously Treated Patients With Locally Advanced or Metastatic Nonsquamous Non-Small-Cell Lung Cancer. J Clin Oncol. 2015; 33: 2667–2674.
42. Perol M. Negative results of METLung study: an opportunity to better understand the role of MET pathway in advanced NSCLC. Transl Lung Cancer Res. 2014; 3: 392–394.
43. Camidge DR, Ou SI, Shapiro G, Otterson GA, Villaruz LC, Villalona-Calero MA, Iafrate AJ, Varella-Garcia M, Dacic S, Cardarella S, Zhao W, Tye L, Stephenson P, et al. Efficacy and safety of crizotinib in patients with advanced c-MET-amplified non-small cell lung cancer (NSCLC). J Clin Oncol 2014; 32: abstr 8001.
44. Shojaei F, Lee JH, Simmons BH, Wong A, Esparza CO, Plumlee PA, Feng J, Stewart AE, Hu-Lowe DD, Christensen JG. HGF/c-Met Acts as an Alternative Angiogenic Pathway in Sunitinib-Resistant Tumors. Cancer Res. 2010; 70: 10090–10100.
45. Jahangiri A, De Lay M, Miller LM, Carbonell WS, Hu YL, Lu K, Tom MW, Paquette J, Tokuyasu TA, Tsao S, Marshall R, Perry A, Bjorgan KM, et al. Gene Expression Profile Identifies Tyrosine Kinase c-Met as a Targetable Mediator of Antiangiogenic Therapy Resistance. Clin Cancer Res. 2013; 19: 1773–1783.
46. Ciamporcero E, Miles KM, Adelaiye R, Ramakrishnan S, Shen L, Ku S, Pizzimenti S, Sennino B, Barrera G, Pili R. Combination strategy targeting VEGF and HGF/c-met in human renal cell carcinoma models. Mol Cancer Ther. 2015; 14: 101–110.
47. Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen J, Kosaka T, Holmes AJ, Rogers AM, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007; 316: 1039–1043.
48. Schuller AG, Barry ER, Jones RD, Henry RE, Frigault MM, Beran G, Linsenmayer D, Hattersley M, Smith A, Wilson J, Cairo S, Déas O, Nicolle D, et al. The MET inhibitor AZD6094 (Savolitinib, HMPL-504) induces regression in papillary renal cell carcinoma patient derived xenograft models. Clin Cancer Res. 2015. 21: 2811-2819.
49. Santoro A, Rimassa L, Borbath I, Daniele B, Salvagni S, Van Laethem JL, Van Vlierberghe H, Trojan J, Kolligs FT, Weiss A, Miles S, Gasbarrini A, Lencioni M, et al. Tivantinib for second-line treatment of advanced hepatocellular carcinoma: a randomised, placebo-controlled phase 2 study. Lancet Oncol. 2013; 14: 55–63.
50. Nakagawa T, Tohyama O, Yamaguchi A, Matsushima T, Takahashi K, Funasaka S, Shirotori S, Asada M, Obaishi H.. E7050: a dual c-Met and VEGFR-2 tyrosine kinase inhibitor promotes tumor regression and prolongs survival in mouse xenograft models. Cancer Sci. 2010; 101: 210–215.
51. Choueiri TK, Escudier B, Powles T, Mainwaring PN, Rini BI, Donskov F, Hammers H, Hutson TE, Lee JL, Peltola K, Roth BJ, Bjarnason GA, Géczi L, et al. Cabozantinib versus Everolimus in Advanced Renal-Cell Carcinoma. N Engl J Med. 2015; 373: 1814–1823.
52. Macher-Goeppinger S, Roth W, Wagener N, Hohenfellner M, Penzel R, Haferkamp A, Schirmacher P, Aulmann S. Molecular heterogeneity of TFE3 activation in renal cell carcinomas. Mod Pathol. 2012; 25: 308-15.
53. Macher-Goeppinger S, Aulmann S, Wagener N, Funke B, Tagscherer KE, Haferkamp A, Hohenfellner M, Kim S, Autschbach F, Schirmacher P, Roth W. Decoy receptor 3 is a prognostic factor in renal cell cancer. Neoplasia. 2008; 10: 1049–1056.
54. Endris V, Penzel R, Warth A, Muckenhuber A, Schirmacher P, Stenzinger A, Weichert W. Molecular diagnostic profiling of lung cancer specimens with a semiconductor-based massive parallel sequencing approach: feasibility, costs, and performance compared with conventional sequencing. J Mol Diagn. 2013; 15: 765–775.
55. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010; 38: e164.
56. Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. Integrative genomics viewer. Nat Biotechnol. 2011; 29: 24–26.