
INTRODUCTION
Prostate cancer is one of the most prevalent malignancies among men worldwide. Although the 5-year survival rate is > 90% in patients with localized disease, the rate is < 30% in patients who develop distant metastases, typically after the primary tumor becomes insensitive to androgen-deprivation therapy [1]. Castration-resistant prostate cancer (CRPC) has a high tendency to metastasize to bone, leading to various skeletal-related events, decreased bone strength, spinal cord compression fractures, debilitating pain, and bone marrow failure [2, 3]. Treatment of metastatic CRPC (mCRPC) includes the use of bisphosphonates and the anti-RANKL antibody denosumab, which act on the bone environment and reduce the incidence of skeletal-related events [4–6]. However, both treatments fail to prevent metastasis and do not improve either progression-free or overall survival [7].
Radiotherapy is another option for treating mCRPC. Targeted therapeutic radioisotopes are preferred over external beam radiation therapy (EBRT) for patients with diffuse, widespread bone involvement because they spare normal tissue from unnecessary irradiation [8, 9]. Radium-223 dichloride (Xofigo®; 223Ra) is the first of a new class of alpha-emitting radiopharmaceuticals to be approved by the U.S. Food and Drug Administration (FDA) for the treatment of bone metastases in mCRPC. Due to its chemical similarity to calcium, 223Ra forms complexes with the bone mineral hydroxyapatite at areas of increased osteoblastic activity, such as bone metastases [10–13], and has demonstrated an overall survival benefit in patients receiving intravenous treatment. The beta-emitting radionuclides previously approved by the FDA, strontium-89 (89Sr) and samarium-153 (153Sm), provide palliation for patients with multifocal bone metastases, but have failed to extend overall survival when given as monotherapies [9, 10].
The primary advantage of treating metastases with alpha-emitting rather than beta-emitting radionuclides is the potential to deliver a greater dose of radiation in a highly localized manner [14, 15]. Alpha particles are heavily charged, composed as they are of a helium nucleus with 2 positive charges, while beta particles are much smaller and take the form of either electrons or positrons. Furthermore, the path lengths of 89Sr and 153Sm are millimeters in both soft tissue and bone, while the range of alpha particles emitted from 223Ra is < 100 μm, or approximately 10 cell diameters in length [10, 15]. This short range of activity limits damage to surrounding normal tissue, leading to improved safety profiles compared to beta-emitting therapies. In particular, 223Ra delivered to bone surfaces has been reported to spare the bone marrow compartment and minimize the myelosuppressive effects observed with beta emitters [14, 16]. On the other hand, the linear energy transfer of alpha particles is about 100–1000 times greater than the average linear energy transfer of beta particles [13]. This ultimately leads to a relatively higher frequency of complex DNA double-strand breaks that are more difficult to repair than those created by beta emitters [9, 13, 14]. Still, phase III clinical studies in mCRPC have demonstrated that 223Ra is able to improve patient survival with limited toxicity, while having positive effects on changes in serum alkaline phosphatase and prostate-specific antigen (PSA) biomarker levels. In addition, the improvement in survival appears to be independent of prior exposure to both docetaxel and bisphosphonates [17].
Although the cytotoxic effects of 223Ra are well studied, it is currently unknown if this alpha emitter can act synergistically with immunotherapy strategies to enhance the killing of tumor cells that survive this treatment modality. Some tumor cells survive sublethal radiation due to dosing constraints dictated by the need to minimize toxicity to the patient, which often translates into dose heterogeneity within a given tumor mass. In addition to initiating a diverse immune response through antigen cascade, radiation therapy can also alter the phenotype of the surviving tumor cells in ways that render them more susceptible to cytotoxic T lymphocyte (CTL)-mediated attack. This process, known as immunogenic modulation, largely involves the regulation of molecules involved in antigen presentation, which can be critical for effective T-cell recognition [18–21]. Such events have been extensively characterized in a variety of human carcinoma cell lines following EBRT and proton radiation therapy in vitro [22]. We have also demonstrated clinically the immunomodulatory effects of EBRT, as it elicits a greater tumor antigen-specific CD8 T-cell response and a consequent reduction in tumor burden in combination with vaccine than with either modality alone.
In this study we explored the ability of 223Ra to induce immunogenic modulation and enhance CTL-mediated lysis of human prostate, breast, and lung carcinoma cell lines, each harboring a different p53, triple-negative, or K-Ras mutational status, respectively. While 223Ra is already FDA-approved for the treatment of mCRPC, it may also have clinical benefit against breast and lung cancers, as they also frequently metastasize to bone [23, 24].
RESULTS
Increasing doses of 223Ra inhibited cell proliferation with minimal effect on viability
The goal of this study was to analyze the immunomodulatory effects of alpha-emitting 223Ra on a variety of tumor types, and to determine whether those phenotypic changes enhanced CTL-mediated lysis of the surviving tumor-cell population. Therefore, to establish a nonlethal dose for subsequent experiments, we needs to understand how 223Ra affects both cell viability and proliferation over a range of doses in vitro. Over a 96-hour period, we exposed prostate (LNCaP and PC3), breast (MDA-MB-231 and ZR75-1), and lung (H441 and H1703) carcinoma cells (1 × 106 cells) to 0, 2, 4, 10, and 40 Gy of 223Ra, a range that includes and exceeds clinically relevant doses. We strategically chose 2 prostate, breast, and lung tumor lines that were genotypically distinct based on their p53, triple-negative, or K-Ras mutational status, respectively (Table 1).
Table 1: Human carcinoma cell lines of diverse origin and phenotype treated with 223Ra alpha radiation
Tumor Class |
Cell Line |
Notable Characteristic |
|---|---|---|
Breast |
MDA-MB-231 |
Triple Negative (ER–/PR–/Her2neu–) |
ZR75-1 |
Triple Positive (ER+/PR+/Her2neu+) |
|
Prostate |
LNCaP |
P53 Wild type |
PC3 |
P53 Null |
|
Lung |
H1703 |
Ras Wild Type |
H441 |
Ras Mutant (G12V) |
Overall, we found that the number of cells harvested did not significantly change after exposure to 2 and 4 Gy of 223Ra (Figure 1). The greatest effect on cell proliferation in the MDA-MB-231, LNCaP, PC3, H441, and H1703 tumor-cell lines was observed after the radiation dose was increased from 4 to 10 Gy. Surprisingly, only minor changes were observed in the total cell number when the amount of radiation was increased to 40 Gy of 223Ra. Cell viabilities (italicized in Figure 1) following each treatment were normalized to that of their respective mock-irradiated (0 Gy) controls. Cell viability was not less than 96% following 4 Gy of 223Ra in any of the tumor-cell lines; viability was at least 86% after 10 Gy of 223Ra in 5/6 cell lines. H441 had a viability of 76% after 10 Gy of alpha radiation. As was the case with the total cell number, viability underwent only minor changes when the dose was increased to 40 Gy. Unlike the other tumor cell lines, ZR75-1 was largely unaffected by 223Ra, as demonstrated by the slight reduction in proliferation rate and the lack of change in viability. Although the relative biological effectiveness values of 223Ra and EBRT are 20 and 1, respectively, both treatments had comparable effects on tumor-cell proliferation and viability [15].
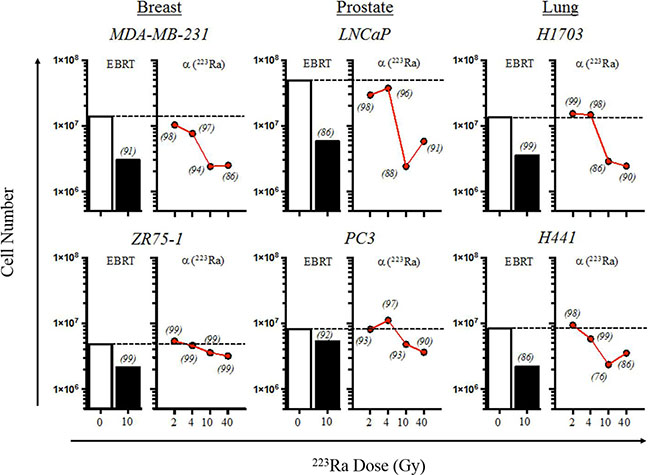
Figure 1: 223Ra alpha radiation inhibits tumor proliferation, with minimal effects on cell viability. Human prostate, breast, and lung carcinoma cells were mock-irradiated (0 Gy, open bars) or treated with either 10 Gy of photon radiation (EBRT, closed bars) or 2–40 Gy of 223Ra (red circles). EBRT-treated cells were irradiated and then cultured for an additional 96 h. Alpha-irradiated cells were cultured with an appropriate amount of 223Ra so that the total cumulative dose was 2, 4, 10, or 40 Gy after 96 h. Cells were harvested, counted, and stained with AO/PI viability dye. Cell viabilities for each treatment, normalized to that of the mock-irradiated samples, are italicized. Data are representative of 2–3 independent experiments.
Exposure of human carcinoma cells to sublethal doses of 223Ra significantly increased sensitivity to antigen-specific CTL lysis
After establishing that 223Ra doses up to 40 Gy did not induce substantial tumor-cell death, we next examined whether alpha radiation affected the susceptibility of surviving tumor cells to CTL-mediated lysis. For this analysis, we focused primarily on the clinically relevant 4- and 10-Gy doses of 223Ra. Breast, prostate, and lung carcinoma cells were first exposed to 223Ra for 96 hours, then co-cultured with CD8+ effector T cells specific for carcinoembryonic antigen (CEA; HLA-A2-restricted), mucin-1 (MUC-1; HLA-A2-restricted), and brachyury (HLA-A2/A24-restricted) epitopes. As shown in Figure 2A, treatment with either dose of 223Ra increased the sensitivity of the breast, prostate, and lung carcinoma cell lines to CTL-mediated lysis targeting the CEA, MUC-1, and brachyury tumor antigens. We observed a significant increase in T cell-mediated killing of 2/2 breast, 1/1 prostate, and 2/2 lung tumor cell lines by CEA- and MUC-1-specific CTLs after both 4 and 10 Gy of radiation. However, no consistent dose-dependent effects were observed, as only 1/5 and 2/5 cell lines were killed to a greater degree with the CEA- and MUC-1-specific CTLs, respectively, at the higher dose of 223Ra. We also observed a significant increase in T cell-mediated killing of 1/2 breast, 2/2 prostate, and 2/2 lung carcinoma cell lines by brachyury-specific CTLs in response to 4 Gy of 223Ra. An increase in tumor-cell lysis occurred in all 6 cell lines following 10 Gy of radiation. Only 3/6 cell lines were more sensitive to brachyury-specific CTL-mediated lysis when the 223Ra dose was increased from 4 to 10 Gy. Interestingly, only the lung carcinoma cell lines (H1703 and H441) demonstrated a propensity to be killed at a greater level by each of the 3 antigen-specific CTLs at the higher dose of 223Ra.
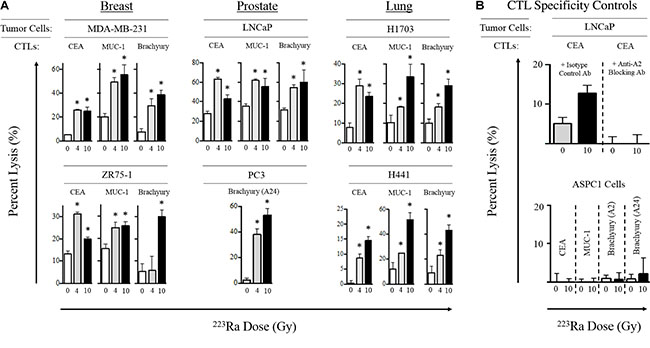
Figure 2: Sublethal exposure to 223Ra increases CTL lysis of breast, prostate, and lung carcinoma cells in vitro. (A) Tumor cells were either left untreated (0 Gy, white bars) or exposed to 4 Gy (grey bars) or 10 Gy (black bars) of 223Ra over a 96-h incubation period, then used as targets in an overnight CTL lysis assay. CEA-, MUC-1-, and brachyury-specific CD8+ T cells were used as effectors at an E:T ratio of 30:1. (B) CTL HLA restriction was verified by incubating tumor cells with anti-HLA-A2 blocking mAb (top) or by performing the killing assay with CEA+HLA-A2– AsPC-1 cells as target controls (bottom). Experiments were repeated 1–3 times with similar results. *denotes statistical significance relative to untreated cells (P < 0.05).
We conducted 2 different CTL specificity control experiments to verify that the CTL killing was major histocompatibility complex class I (MHC-I)-restricted. The addition of an anti-HLA-A2 blocking antibody during the killing assay abolished CEA-specific CTL-mediated lysis of LNCaP tumor cells (HLA-A2+) exposed to 10 Gy 223Ra (Figure 2B). Moreover, exposure of the HLA-A2/A24– AsPC-1 carcinoma cell line to 10 Gy of 223Ra over 96 hours did not result in any significant lysis by any of the CEA, MUC-1, or brachyury CTLs used in this study (Figure 2B, bottom panel). Taken together, these results indicate that sublethal doses of 223Ra enhance HLA-restricted, antigen-specific, CTL-mediated lysis of various human carcinomas, and that such killing can be achieved targeting a broad repertoire of tumor-associated antigens.
Exposure of human carcinoma cells to 223Ra significantly increased expression of histocompatibility leukocyte antigens (HLA)
CTL killing requires interaction between the T-cell receptor and CD8-restricted epitopes presented by MHC-I molecules on the surface of tumor cells. We examined changes in HLA-A, B, and C (HLA-ABC) expression by immunofluorescence in each cell line following a 96-hour exposure to 4 and 10 Gy of 223Ra. A quantitative image analysis (Figure 3A) showed at least 2-fold increases in HLA-ABC for each cell line at either the 4- or 10-Gy dose relative to mock-irradiated controls. 223Ra induced dose-dependent upregulation of HLA-ABC in all prostate and lung tumor cell lines and in the MDA-MB-231 breast tumor cell line. At 10 Gy 223Ra, HLA-ABC expression increased 7.02-fold and 3.80-fold in LNCaP and PC3 prostate carcinoma lines, respectively. HLA-ABC also increased 3.32-fold and 1.71-fold in MDA-MB-231 and ZR-75-1 breast carcinoma cells, respectively, and 3.58-fold and 2.80-fold in H441 and H1703 lung tumor cell lines, respectively. Figure 3B presents a series of representative HLA-ABC immunofluorescence images at 0, 4, and 10 Gy 223Ra for each tumor type.
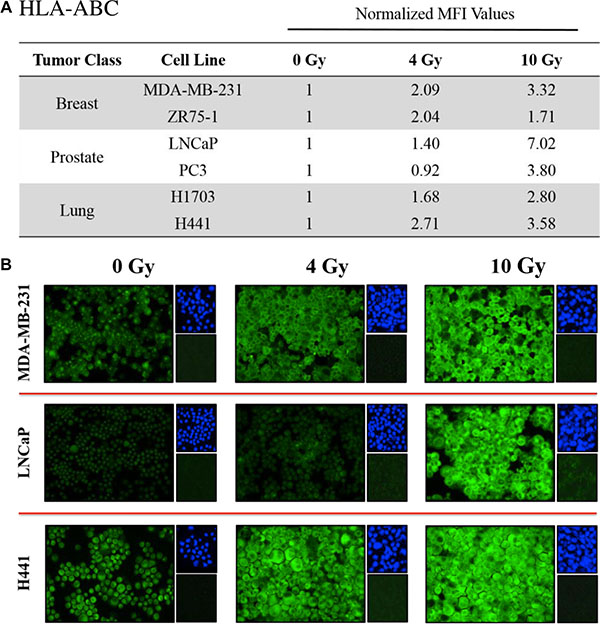
Figure 3: Sublethal doses of 223Ra significantly increase HLA expression in various tumor types. Human prostate, breast, and lung carcinoma cells were mock-irradiated (0 Gy) or exposed to a cumulative dose of 223Ra totaling 4 or 10 Gy over a 96-h incubation period. Changes in HLA-ABC expression were determined by immunofluorescence imaging post-irradiation. (A) HLA-ABC expression was quantified using ImageJ software. The data presented are mean fluorescence intensity (MFI) values normalized to their respective mock-irradiated controls. (B) HLA-ABC immunofluorescence stains (green, 20× magnification). Upper right panels: 4′6-diamidino-2-phenylindole nuclear stain (blue). Lower right panels: isotype control. Data are representative of 2–3 independent experiments.
Exposure of human carcinoma cells to 223Ra significantly increased expression of calreticulin
Calreticulin is a chaperone protein of the antigen-processing machinery (APM) that helps guide peptide loading into MHC-I molecules. Due to its potential role in modulating CTL sensitivity, we studied the effects of 223Ra on calreticulin expression by immunofluorescence. Similar to the results obtained for HLA-ABC, calreticulin was markedly upregulated upon exposure to 223Ra. We observed elevated levels of total calreticulin protein in all of the tested cell lines in response to 4 Gy of 223Ra, and the amount of calreticulin further increased after exposure to 10 Gy (Figure 4A). At 10 Gy 223Ra, calreticulin expression increased 5.5-fold and 2.5-fold in LNCaP and PC3 prostate carcinoma cells, respectively, compared to untreated controls. Calreticulin also increased 3.48-fold and 3.07-fold in MDA-MB-231 and ZR-75-1 breast carcinoma cells, respectively, and 4.27-fold and 4.28-fold in the lung tumor cell lines, respectively (Figure 4B).
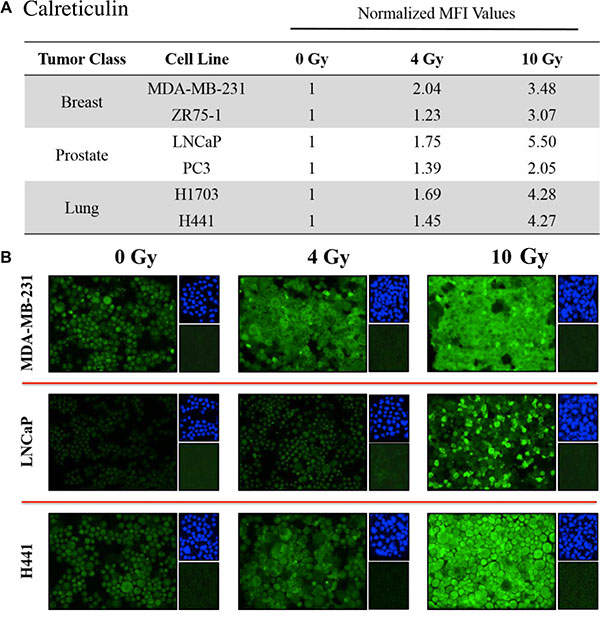
Figure 4: Tumor cells exposed to 223Ra have increased expression of calreticulin. Immunofluorescence imaging was used to analyze calreticulin upregulation in human prostate, breast, and lung cancer cells following exposure to 223Ra. (A) Calreticulin expression was quantified using ImageJ software. The data presented are MFI values normalized to their respective mock-irradiated controls. (B) Calreticulin immunofluorescence stains following a cumulative dose of either 4 or 10 Gy radiation over a 96-h treatment period (green, 20× magnification). Upper right panels: 4′6-diamidino-2-phenylindole nuclear stain (blue). Lower right panels: isotype control. Data are representative of 2–3 independent experiments.
223Ra induced the ER stress response and surface translocation of calreticulin in tumor cells
Radiation-induced immunogenic modulation of calreticulin in carcinoma cells is mediated by the endoplasmic reticulum (ER) stress response, as previously reported [25]. Based on the observed effects of 223Ra on calreticulin protein expression, we next examined whether this alpha emitter is similarly capable of activating the ER stress response in tumors. We stably transduced LNCaP prostate tumor cells with an ER stress response element that drives firefly luciferase expression, and then either mock-irradiated the cells or treated them with a cumulative dose of 10 Gy 223Ra over 96 hours. 223Ra triggered the ER stress response by 72 hours (P < 0.001) after the start of treatment, and the cells maintained this activated state at 96 hours (P < 0.001), at which time we performed CTL killing assays (Figure 5A).
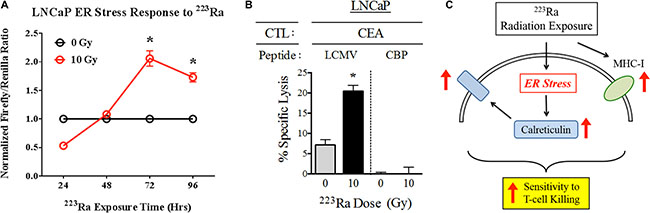
Figure 5: 223Ra activates the ER stress response in LNCaP cells. (A) LNCaP cells stably transduced with a firefly luciferase ER stress reporter element were either mock-irradiated (0 Gy) or exposed to a total dose of 10 Gy 223Ra over a 96-h incubation period. At the indicated time points, firefly and Renilla luciferase activities were determined using a Dual Luciferase Reporter Assay System. The results are shown as the ratio of firefly luciferase activity to Renilla luciferase control. (B) Functional role of cell-surface calreticulin on CTL-mediated lysis. LNCaP cells were either mock-irradiated or exposed to 10 Gy 223Ra, then co-cultured with CEA-specific CD8+ T cells in the presence of calreticulin blocking peptide (CBP, right) or lymphocytic chriomeningitis virus control peptide (LCMV, left). (C) Schematic illustrating the immunomodulatory effects of 223Ra on tumor cells. *denotes statistical significance relative to untreated cells (P < 0.05).
One of the functional consequences of ER stress induction in tumor cells is translocation of calreticulin to the cell surface, where it enhances target killing by enhancing tumor-cell/T-cell recognition. We analyzed this immunomodulatory effect by investigating how an exogenous calreticulin blocking peptide, designed to inhibit the interaction between calreticulin and its receptor on T cells, would affect CTL killing following 223Ra exposure. As expected, 10 Gy of 223Ra induced greater CEA-specific CTL killing of LNCaP cells in the presence of a control lymphocytic choriomeningitis virus peptide LCM. However, the increase in CTL-mediated lysis was completely abrogated by the addition of calreticulin blocking peptide, demonstrating the importance of surface calreticulin in T-cell killing of tumor cells upon exposure to 223Ra. This result led us to develop a working model (Figure 5C) of immunogenic modulation of tumor cells following 223Ra therapy. Here, we show that while 223Ra increases expression of MHC-I, it can also induce ER stress in tumor cells and subsequently lead to upregulation and surface translocation of calreticulin. These phenotypic changes ultimately enhance tumor sensitivity to CTL- mediated lysis.
DISCUSSION
The primary goal of radionuclide therapy is to reduce tumor burden through direct killing of tumor cells. Unfortunately, dose delivery constraints designed to limit toxicity allow for the survival of tumor cells that are not exposed to a lethal dose of radiation. Radiolabeled antibodies also are likely to be delivered in non-lytic doses because poor vascularization of solid tumor tissue hampers the infiltration of these agents [13]. However, radiotherapies delivered in sublethal doses can still be therapeutic anticancer modalities if they can take advantage of other immune-activating agents as part of a combination regimen [26, 27]. This may be achieved by inducing a spectrum of phenotypic changes in the surviving tumor cells that collectively augment their susceptibility to antigen-specific T cell-mediated destruction, a process known as immunogenic modulation. The changes that occur primarily involve multiple components of the APM, such as immunoproteosome subunits, peptide transporters, and protein chaperones, all of which contribute to enhanced presentation of tumor antigens for CTL recognition [19, 20, 25, 28].
223Ra is currently prescribed for patients with mCRPC at a dose of 55 kBq/kg of body weight (or 48 μCi/kg) [16]. For a 70-kg patient, assuming an absorbed radiation dose of 4.262 μCi/rad to osteogenic cells, this translates to approximately 4 Gy of 223Ra. In our studies, we exposed a variety of cell lines representative of tumors that frequently metastasize to bone, notably prostate, breast, and lung (Table 1), to 223Ra over 96 hours. Although cumulative doses of 4 and 10 Gy were found to be sublethal (Figure 1), they were still able to enhance T cell-mediated lysis of each tumor cell line by CTLs specific for MUC-1, brachyury, and CEA tumor antigens (Figure 2). The dose effect on CTL killing (Figure 2) did not entirely reflect the dose effect on surface MHC-1 (Figure 3). We have previously noted that radiation has several effects on antigen processing machinery independent of MHC-1 surface expression [20]. Doses as low as 2 Gy have been shown to increase tumor sensitivity to CTL killing. It would be challenging to target a given tumor exposure dose with 223Ra due to its uptake being dependent on osteogenic bone turnover rate. Given these observations, treating patients with the approved dose of 223Ra would be predicted to modulate tumor phenotype, resulting in enhanced T-cell killing.
In addition to greater CTL sensitivity, each tumor-cell line displayed some of the cardinal signs of immunogenic modulation following 223Ra therapy [20], such as upregulation of HLA-ABC (Figure 3) and calreticulin (Figure 4), both of which are important for effective antigen presentation. T cell-mediated killing relies on recognition of specific CD8-restricted epitopes associated with MHC-I molecules on the surface of tumor cells. However, proper loading of MHC-I molecules is guided in part by calreticulin, a critical chaperone protein that resides in the ER membrane. Calreticulin is a component of the APM essential for efficient antigen presentation, folding of newly synthesized glycoproteins, and enrichment of endogenous peptides in the ER. Increased calreticulin expression has been observed in various tumor-cell lines following in vitro and in vivo treatment with sublethal EBRT and proton radiation, and is believed to play a critical role in enhancing antigen-specific CTL recognition of tumor cells [22, 25].
Previous studies with EBRT and proton radiation therapy have shown that translocation of calreticulin to the cell surface is as important as its upregulation during immunogenic modulation [22]. Surface calreticulin has the ability to bind CTLs and enhance tumor-cell lysis, possibly due to more stable and/or prolonged tumor cell/T cell interaction. In previous studies, inhibiting this interaction through the use of a calreticulin blocking peptide significantly reduced the sensitivity of irradiated tumor cells to CTL-mediated lysis [18, 22]. Here, we observed the same effect after treatment with 223Ra (Figure 5). The presence of calreticulin blocking peptide during the CTL killing assay completely abrogated any increase in tumor-cell lysis that occurred following radiation treatment. This finding demonstrates that treatment with 223Ra induces calreticulin translocation, and that enhanced CTL killing is dependent on interaction with surface calreticulin.
Calreticulin surface exposure and ER stress have previously been linked in immunogenic modulation studies of EBRT, which led us to investigate whether 223Ra also induces ER stress in tumor cells. In response to EBRT, ER stress initiates the unfolded protein response, a protective cellular mechanism that attempts to restore ER homeostasis by arresting protein translation and upregulating multiple APM components and other immune-related proteins [25]. One of the immunologically relevant changes that occur during the ER stress response is calreticulin surface expression [20, 25]. Using an LNCaP cell line transduced with an ER stress reporter element, we discovered that ER stress is induced within the first 72 hours after 223Ra treatment (Figure 5). It is likely that ER stress is the main driver of calreticulin translocation and is thus responsible for increased tumor sensitivity to CTL lysis. However, future mechanistic studies incorporating the ER stress inducer thapsigargin will provide better insight into ER stress’s role in the immunomodulatory effects of 223Ra [25].
223Ra’s ability to drive immunogenic modulation and enhance CTL lysis in a variety of tumor-cell lines suggests that it has broad applicability in cancer therapy. Here, we show that 223Ra has the potential to treat a variety of human carcinomas of distinct origin and genotype. Sensitivity to CTL lysis was enhanced in prostate, breast, and lung tumor cells regardless of their p53, triple-negative, or K-Ras mutational status, respectively (Table 1), suggesting that 223Ra may be used to effectively treat bone metastases arising from each of these tumor types. Moreover, with the development of novel radionuclide-labeled antibodies, alpha particle-emitting agents such as 223Ra may be targeted to primary and metastatic tumor lesions not only in bone, but also in soft tissue sites throughout the body that generally exhibit poor uptake of bone-seeking radionuclides [15, 29–31].
Various studies are currently exploring the use of 223Ra in combination therapies, primarily for the treatment of mCRPC. 223Ra is under clinical evaluation in combination with immunomodulatory chemotherapy drugs (docetaxel), androgen-deprivation therapies (enzalutamide and abiraterone), and osteoclast-targeted agents, including bisphosphonates (zoledronic acid) and anti-RANKL antibodies (denosumab) [2, 4, 18, 32, 33]. The studies presented here support the use of therapeutic cancer vaccines that can generate antigen-specific T-cell responses and exploit the immunostimulatory environment created by 223Ra radiotherapy to achieve robust and effective antitumor responses [34–36]. Further studies are required to determine optimal timing for the use of 223Ra in combination with immunotherapies.
Such an approach with alpha-emitting 223Ra is based on preclinical studies demonstrating that beta-emitting 153Sm is able to induce immunogenic modulation and render tumor cells more amenable to immune-mediated killing [37]. A recent phase II study by our group demonstrated the clinical benefit of combining PSA-TRICOM, a poxviral-based cancer vaccine, with 153Sm in late-stage mCRPC [38]. PSA-TRICOM is designed to activate PSA-specific T cells and slow tumor growth by expanding the antigen repertoire of T cells through epitope spreading. The combination of 153Sm and PSA-TRICOM increased progression-free survival, enhanced the number of CD4+ and CD8+ PSA-specific T cells expressing type I cytokines, and decreased PSA and soluble CD40L levels in patient sera compared with 153Sm alone [38]. Our studies with both 153Sm and 223Ra show that, compared to radiotherapy with beta-emitters, radiotherapy with alpha-emitters safely delivers much higher energy to tumor tissue in a more localized manner, providing a rationale for investigating the combination of 223Ra and PSA-TRICOM for the treatment of mCRPC.
MATERIALS AND METHODS
Tumor-cell lines
Cell lines used in these studies were from tumors capable of metastasizing to bone. Cells of human breast (MDA-MB-231 and ZR-75-1), prostate (LNCaP and PC3), pancreatic (AsPC-1), and lung carcinomas (NCI-H1703 and NCI-H441) were obtained from American Type Culture Collection (Manassas, VA). Cells were maintained at 37°C/5% CO2 in cell culture medium according to the supplier’s specifications, supplemented with 10% FBS, 100 μg/mL streptomycin, and 100 units/mL penicillin.
Tumor irradiation
Tumor cells were treated with the alpha particle-emitting radiopharmaceutical radium-223 dichloride (Xofigo®; 223Ra), obtained from Bayer Pharmaceuticals (Whippany, NJ) under a Material Cooperative Research and Development Agreement. Each cell line was cultured in normal growth medium supplemented with a volume of 223Ra that would deliver cumulative radiation doses totaling 2, 4, 10, or 40 Gy by the end of a 96-h treatment period. The amount of 223Ra added to each flask was mathematically derived by assuming a cell culture mass of 10 g and by applying the 223Ra dose constant of 4.2 × 10−12 Gy kg/Bq/sec [14]. After the 96-h incubation period, the irradiated culture medium was removed from the flasks and cells were harvested for subsequent analysis. Tumor cells in suspension were also irradiated with a single 10-Gy dose of EBRT (photons, Cs-137 source, Gammacell-40; AECL/Nordion, Ottawa, ONT) and then returned to culture for an additional 96 h.
CD8+ CTLs
CTLs specific for CEA, MUC-1, and brachyury were used to study the susceptibility of cancer cells to CD8+ T cell-mediated killing following 223Ra irradiation. The CEA-specific CTLs were HLA-A2-restricted and recognized the epitope YLSGANLNL (CAP-1) [39, 40]. The MUC-1-specific CTLs were HLA-A2-restricted and recognized the epitope ALWGQDVTSV [41, 42]. The brachyury-specific CTLs were either HLA-A2- or HLA-A24-restricted and recognized the epitopes WLLPGTSTL (T-p2A) and KYQNEEITAL, respectively [43].
Cytotoxicity assays
Tumor cells were either mock-irradiated (0 Gy) or exposed to 4 or 10 Gy of 223Ra for 96 h. After treatment, cells were trypsinized and used as targets in a standard CD8+ T-cell cytotoxicity assay. Tumor cells were metabolically labeled with 111In-oxyquinoline (Medi-Physics, Arlington Heights, IL) and then co-incubated in 96-well round-bottom plates overnight at 37°C/5%CO2 with the CEA, MUC-1, and brachyury HLA-restricted CTL cell lines at an effector:target ratio of 30:1. Supernatant fluids were harvested and measured on a gamma counter to quantify the amount of 111In release. CTL killing was calculated according to the following formula: % specific lysis = (experimental release – spontaneous release) / (maximum release – spontaneous release) × 100. Spontaneous and maximum releases were determined by incubating the tumor cells with either regular culture medium or upon lysis with 2% Triton X-100 (Sigma-Aldrich, St. Louis, MO), respectively. CTL specificity control experiments were performed in the presence of anti-HLA-A2 or isotype control IgG2b antibodies (Bio-Rad Laboratories, Hercules, CA) at a concentration of 20 μg/mL. Negative control cytotoxicity assays were also performed with the AsPC-1 cell line (HLA-A2–, HLA-A24–/CEA+, brachyury+, MUC-1+) to confirm HLA specificity. For indicated experiments, the CTL assay was performed in the presence of calreticulin blocking peptide (MBL International, Woburn, MA) or lymphocytic choriomeningitis virus peptide NP118−132 (CPC Scientific, Sunnyvale, CA).
Immunofluorescence
After a 96-h exposure to 223Ra, expression of calreticulin and HLA-ABC were analyzed by immunofluorescence. A total of 1 × 105 cells were deposited onto microscope slides using cytocentrifugation (Shandon Cytospin; Thermo Fisher Scientific, Waltham, MA), and then fixed with 3% methanol-free paraformaldehyde (Electron Microscopy Sciences, Hatfield, PA) for 20 min. After permeabilization with 0.05% Triton X-100, the tumor cells were stained with mouse monoclonal antibodies specific for calreticulin (Abcam, Cambridge, UK) or HLA-ABC (BD Biosciences, San Jose, CA) at a 1:100 dilution, followed by an anti-mouse Alexa Fluor-488 conjugated secondary antibody (Thermo Fisher Scientific). The cells were then counterstained with 4′6-diamidino-2-phenylindole and examined by fluorescence microscopy (Leica Microsystems, Wetzlar, Germany). Relative calreticulin and HLA-ABC expression levels were calculated using ImageJ software by normalizing the intensity values to their respective mock-irradiated (0 Gy) controls.
Luciferase ER stress reporter assay
LNCaP prostate tumor cells were stably transduced to express firefly luciferase under control of the ER stress transcriptional response element, as previously described [44]. As an internal control, these cells were also transduced to constitutively express Renilla luciferase (Thermo Fisher Scientific) under control of the cytomegalovirus promoter. Transduced cells were selected in medium containing 1 μg/mL puromycin (Life Technologies, Carlsbad, CA), and single-cell clones were either mock-irradiated (0 Gy) or exposed to 10 Gy 223Ra over a 96-h incubation period. Luciferase activity was quantified using the Dual-Luciferase Reporter Assay System (Promega, Madison, WI) at multiple time points after the start of treatment to monitor the progressive onset of the ER stress response pathway.
Statistical analysis
Statistical analyses were performed using 2-tailed Student’s t-tests with a 95% confidence interval. All analyses were conducted using Prism Graphpad 6.0f software, and probability values of P < 0.05 were considered significant.
ACKNOWLEDGMENTS
The authors thank Dr. Jeffrey Schlom for his helpful suggestions. The authors also thank Marion Taylor and Michelle Padget for their excellent technical expertise and Bonnie L. Casey for editorial assistance in the preparation of this manuscript.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
FUNDING
This research was supported by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, National Institutes of Health.
REFERENCES
1. Brawley OW. Trends in prostate cancer in the United States. Journal of the National Cancer Institute Monographs. 2012; 2012:152–156.
2. Body JJ, Casimiro S, Costa L. Targeting bone metastases in prostate cancer: improving clinical outcome. Nature reviews Urology. 2015; 12:340–356.
3. Ahmadzadehfar H, Eppard E, Kurpig S, Fimmers R, Yordanova A, Schlenkhoff CD, Gartner F, Rogenhofer S, Essler M. Therapeutic response and side effects of repeated radioligand therapy with 177Lu-PSMA-DKFZ-617 of castrate-resistant metastatic prostate cancer. Oncotarget. 2016; 7:12477–12488. doi: 10.18632/oncotarget.7245.
4. Gartrell BA, Coleman R, Efstathiou E, Fizazi K, Logothetis CJ, Smith MR, Sonpavde G, Sartor O, Saad F. Metastatic Prostate Cancer and the Bone: Significance and Therapeutic Options. European urology. 2015; 68:850–858.
5. Coleman R. Treatment of Metastatic Bone Disease and the Emerging Role of Radium-223. Seminars in nuclear medicine. 2016; 46:99–104.
6. Saad F, Markus R, Goessl C. Targeting the receptor activator of nuclear factor-kappaB (RANK) ligand in prostate cancer bone metastases. BJU international. 2008; 101:1071–1075.
7. Abou DS, Ulmert D, Doucet M, Hobbs RF, Riddle RC, Thorek DL. Whole-Body and Microenvironmental Localization of Radium-223 in Naive and Mouse Models of Prostate Cancer Metastasis. Journal of the National Cancer Institute. 2015 Dec 18; 108.
8. Goyal J, Antonarakis ES. Bone-targeting radiopharmaceuticals for the treatment of prostate cancer with bone metastases. Cancer letters. 2012; 323:135–146.
9. Sgouros G. Alpha-particles for targeted therapy. Advanced drug delivery reviews. 2008; 60:1402–1406.
10. Lewington VJ. Bone-seeking radionuclides for therapy. Journal of nuclear medicine. 2005; 46 Suppl 1:38S-47S.
11. Harrison MR, Wong TZ, Armstrong AJ, George DJ. Radium-223 chloride: a potential new treatment for castration-resistant prostate cancer patients with metastatic bone disease. Cancer management and research. 2013; 5:1–14.
12. Nilsson S, Larsen RH, Fossa SD, Balteskard L, Borch KW, Westlin JE, Salberg G, Bruland OS. First clinical experience with alpha-emitting radium-223 in the treatment of skeletal metastases. Clinical cancer research. 2005; 11:4451–4459.
13. Sartor O, Maalouf B, CR H, Macklis R. Targeted use of alpha particles: current status in cancer therapeutics. J Nucl Med Radiat Ther. 2012; 3:136.
14. Bruland OS, Nilsson S, Fisher DR, Larsen RH. High-linear energy transfer irradiation targeted to skeletal metastases by the alpha-emitter 223Ra: adjuvant or alternative to conventional modalities? Clinical cancer research. 2006; 12:6250s-6257s.
15. Elgqvist J, Frost S, Pouget JP, Albertsson P. The potential and hurdles of targeted alpha therapy - clinical trials and beyond. Frontiers in oncology. 2014; 3:324.
16. Coleman R, Aksnes AK, Naume B, Garcia C, Jerusalem G, Piccart M, Vobecky N, Thuresson M, Flamen P. A phase IIa, nonrandomized study of radium-223 dichloride in advanced breast cancer patients with bone-dominant disease. Breast cancer research and treatment. 2014; 145:411–418.
17. Parker CC. The role of bisphosphonates in the treatment of prostate cancer. BJU international. 2005; 95:935–938.
18. Hodge JW, Garnett CT, Farsaci B, Palena C, Tsang KY, Ferrone S, Gameiro SR. Chemotherapy-induced immunogenic modulation of tumor cells enhances killing by cytotoxic T lymphocytes and is distinct from immunogenic cell death. International journal of cancer. 2013; 133:624–636.
19. Hodge JW, Ardiani A, Farsaci B, Kwilas AR, Gameiro SR. The tipping point for combination therapy: cancer vaccines with radiation, chemotherapy, or targeted small molecule inhibitors. Seminars in oncology. 2012; 39:323–339.
20. Gameiro SR, Ardiani A, Kwilas A, Hodge JW. Radiation-induced survival responses promote immunogenic modulation to enhance immunotherapy in combinatorial regimens. Oncoimmunology. 2014; 3:e28643.
21. Garnett CT, Palena C, Chakraborty M, Tsang KY, Schlom J, Hodge JW. Sublethal irradiation of human tumor cells modulates phenotype resulting in enhanced killing by cytotoxic T lymphocytes. Cancer research. 2004; 64:7985–7994.
22. Gameiro SR, Malamas AS, Bernstein MB, Tsang KY, Vassantachart A, Sahoo N, Tailor R, Pidikiti R, Guha CP, Hahn SM, Krishnan S, Hodge JW. Tumor Cells Surviving Exposure to Proton or Photon Radiation Share a Common Immunogenic Modulation Signature, Rendering Them More Sensitive to T Cell-Mediated Killing. International journal of radiation oncology, biology, physics. 2016; 95:120–130.
23. Hobbs RF, Song H, Watchman CJ, Bolch WE, Aksnes AK, Ramdahl T, Flux GD, Sgouros G. A bone marrow toxicity model for Ra alpha-emitter radiopharmaceutical therapy. Physics in medicine and biology. 2012; 57:3207–3222.
24. Takalkar A, Paryani B, Adams S, Subbiah V. Radium-223 dichloride therapy in breast cancer with osseous metastases. BMJ case reports. 2015.
25. Gameiro SR, Jammeh ML, Wattenberg MM, Tsang KY, Ferrone S, Hodge JW. Radiation-induced immunogenic modulation of tumor enhances antigen processing and calreticulin exposure, resulting in enhanced T-cell killing. Oncotarget. 2014; 5:403–416. doi: 10.18632/oncotarget.1719.
26. Twyman-Saint Victor C, Rech AJ, Maity A, Rengan R, Pauken KE, Stelekati E, Benci JL, Xu B, Dada H, Odorizzi PM, Herati RS, Mansfield KD, Patsch D, et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature. 2015; 520:373–377.
27. Derer A, Frey B, Fietkau R, Gaipl US. Immune-modulating properties of ionizing radiation: rationale for the treatment of cancer by combination radiotherapy and immune checkpoint inhibitors. Cancer immunology, immunotherapy: CII. 2016; 65:779–786.
28. Hodge JW, Kwilas A, Ardiani A, Gameiro SR. Attacking malignant cells that survive therapy: Exploiting immunogenic modulation. Oncoimmunology. 2013; 2:e26937.
29. Baidoo KE, Yong K, Brechbiel MW. Molecular pathways: targeted alpha-particle radiation therapy. Clinical cancer research. 2013; 19:530–537.
30. Kim YS, Brechbiel MW. An overview of targeted alpha therapy. Tumour biology. 2012; 33:573–590.
31. Hagemann UB, Wickstroem K, Wang E, Shea AO, Sponheim K, Karlsson J, Bjerke RM, Ryan OB, Cuthbertson AS. In Vitro and In Vivo Efficacy of a Novel CD33-Targeted Thorium-227 Conjugate for the Treatment of Acute Myeloid Leukemia. Molecular cancer therapeutics. 2016
32. Ardiani A, Gameiro SR, Kwilas AR, Donahue RN, Hodge JW. Androgen deprivation therapy sensitizes prostate cancer cells to T-cell killing through androgen receptor dependent modulation of the apoptotic pathway. Oncotarget. 2014; 5:9335–9348. doi: 10.18632/oncotarget.2429.
33. Saad F, Carles J, Gillessen S, Heidenreich A, Heinrich D, Gratt J, Levy J, Miller K, Nilsson S, Petrenciuc O, Tucci M, Wirth M, Federhofer J, et al. Radium-223 and concomitant therapies in patients with metastatic castration-resistant prostate cancer: an international, early access, open-label, single-arm phase 3b trial. The Lancet Oncology. 2016; 17:1306–1316.
34. Kudo-Saito C, Schlom J, Camphausen K, Coleman CN, Hodge JW. The requirement of multimodal therapy (vaccine, local tumor radiation, and reduction of suppressor cells) to eliminate established tumors. Clinical cancer research. 2005; 11:4533–4544.
35. Chakraborty M, Abrams SI, Coleman CN, Camphausen K, Schlom J, Hodge JW. External beam radiation of tumors alters phenotype of tumor cells to render them susceptible to vaccine-mediated T-cell killing. Cancer research. 2004; 64:4328–4337.
36. Chakraborty M, Gelbard A, Carrasquillo JA, Yu S, Mamede M, Paik CH, Camphausen K, Schlom J, Hodge JW. Use of radiolabeled monoclonal antibody to enhance vaccine-mediated antitumor effects. Cancer immunology, immunotherapy: CII. 2008; 57:1173–1183.
37. Chakraborty M, Wansley EK, Carrasquillo JA, Yu S, Paik CH, Camphausen K, Becker M, Goeckler W, Schlom J, Hodge JW. Use of Samarium-153-EDTMP to modulate phenotype of tumor cells and enhance T-cell-mediated killing, Clinical cancer research. 2008; 14:4316–4325.
38. Heery CR, Madan RA, Stein MN, Stadler WM, DiPaola RS, Rauckhorst M, Steinberg SM, Marte JL, Chen CC, Grenga I, Donahue RN, Jochems C, Dahut WL, et al. Samarium-153-EDTMP (Quadramet(R)) with or without vaccine in metastatic castration-resistant prostate cancer: A randomized Phase 2 trial. Oncotarget. 2016; 7:69014–23. doi: 10.18632/oncotarget.10883.
39. Tsang KY, Zhu M, Nieroda CA, Correale P, Zaremba S, Hamilton JM, Cole D, Lam C, Schlom J. Phenotypic stability of a cytotoxic T-cell line directed against an immunodominant epitope of human carcinoembryonic antigen. Clinical cancer research. 1997; 3:2439–2449.
40. Tsang KY, Zaremba S, Nieroda CA, Zhu MZ, Hamilton JM, Schlom J. Generation of human cytotoxic T cells specific for human carcinoembryonic antigen epitopes from patients immunized with recombinant vaccinia-CEA vaccine. Journal of the National Cancer Institute. 1995; 87:982–990.
41. Tsang KY, Palena C, Gulley J, Arlen P, Schlom J. A human cytotoxic T-lymphocyte epitope and its agonist epitope from the nonvariable number of tandem repeat sequence of MUC-1. Clinical cancer research. 2004; 10:2139–2149.
42. Jochems C, Tucker JA, Vergati M, Boyerinas B, Gulley JL, Schlom J, Tsang KY. Identification and characterization of agonist epitopes of the MUC1-C oncoprotein. Cancer immunology, immunotherapy : CII. 2014; 63:161–174.
43. Tucker JA, Boyerinas B, Fallon J, Greiner JW, Palena C, Rodell TC, Schlom J, Tsang KY. Identification and characterization of a cytotoxic T-lymphocyte agonist epitope of brachyury, a transcription factor involved in epithelial to mesenchymal transition and metastasis. Cancer immunology, immunotherapy: CII. 2014; 63:1307–1317.
44. Gameiro SR, Malamas AS, Tsang KY, Ferrone S, Hodge JW. Inhibitors of histone deacetylase 1 reverse the immune evasion phenotype to enhance T-cell mediated lysis of prostate and breast carcinoma cells. Oncotarget. 2016; 7:7390–7402. doi: 10.18632/oncotarget.7180.