
INTRODUCTION
Children with B-cell precursor acute lymphoblastic leukemia (BCP-ALL) with the BCR-ABL1 fusion gene form a small patient group with a poor prognosis, which has been substantially improved in recent treatment protocols with the addition of imatinib and other tyrosine kinase inhibitors [1–2]. Approximately 15% of cases of BCP-ALL are characterized by a gene expression signature similar to that of BCR-ABL1-positive disease and an unfavorable prognosis [3–4]. This BCR-ABL1-like subtype is found in approximately 50% of so-called B-other cases, which are BCP-ALL cases negative for the sentinel cytogenetic lesions BCR-ABL1, ETV6-RUNX1, TCF3-PBX1, rearrangement of MLL, or high hyperdiploidy (51-65 chromosomes). BCR-ABL1-like BCP-ALL shows a high frequency of B-cell development gene aberrations, especially IKZF1 deletions [3–5], and tyrosine kinase activating lesions [6–7].
Tyrosine kinase activating lesions are diverse and include tyrosine kinase fusion proteins, cytokine receptor overexpression, or activating point mutations in genes encoding kinases, cytokine receptors, or signaling molecules (recently reviewed in [8]). In our study, we focus on tyrosine kinase fusion genes because they are most similar to the well-known BCR-ABL1 fusion gene and expected to be clonal leukemia drivers. Detection of tyrosine kinase fusions could guide targeted treatment with tyrosine kinase inhibitors and improve outcome in a similar way as currently for BCR-ABL1-positive patients.
Tyrosine kinases known to be involved in fusions include the ABL class kinases ABL1, ABL2, PDGFRB and CSF1R as well as the JAK class kinase JAK2. Each of these kinases has been detected in in-frame fusions with multiple 5’ partner genes resulting in the expression of a constitutively active, oncogenic kinase. The number of patients without sentinel chromosomal abnormalities and the diversity of novel tyrosine kinase fusions poses a challenge to routine fusion gene detection. Previous studies have described the discovery and oncogenic potential of ABL and JAK class fusion genes and the sensitivity of patients’ cells or in vitro cell models to tyrosine kinase inhibition [6–7]. Selection of patients for fusion detection in these studies was based on gene expression profiling indicating Philadelphia-like ALL [6–7]. A large Japanese study screened all B-other cases by transcriptome analysis or multiplexed RT-PCR for 15 fusions and found both methods similarly sensitive [9]. Other studies describe systematic screens in B-other cases by FISH or RT-PCR to detect specific tyrosine kinase fusions [10–11].
We aimed at the detection of recently identified tyrosine kinase fusion genes in 153 B-other cases of a population-based cohort of 574 Dutch/German pediatric BCP-ALL patients at initial diagnosis to address the frequency of tyrosine kinase fusions, their clinical response characteristics, and associated genetic lesions in B-cell development genes.
RESULTS
Tyrosine kinase fusions are restricted to BCR-ABL1-like subtype
We identified 14 ABL/JAK class tyrosine kinase activating fusion genes among 77 BCR-ABL1-like cases (18%), and none among 76 non-BCR-ABL1-like B-other cases (Table 1; Supplementary Table 1; Supplementary Figure 1). We found nine tyrosine kinase fusions predictive for activated ABL signaling: 4 EBF1-PDGFRB, 2 SSBP1-CSF1R, and one each of ZMIZ1-ABL1, FOXP1-ABL1, RCSD1-ABL2. Five tyrosine kinase fusions are predictive for activated JAK signaling: 3 PAX5-JAK2, one each of BCR-JAK2 and TERF2-JAK2. The exons included in the fusion transcripts were the same as described previously [6, 12–13], except for RCSD1 exon 3-ABL2 exon 5 (Supplementary Figure 2), and TERF2 exon 10-JAK2 exon 19 with a deletion of the last 11 coding nucleotides of TERF2 exon 10 (Supplementary Figure 3). All detected fusion transcripts encode in-frame fusion proteins as evaluated using ProteinPaint [14].
Table 1: Frequency of identified tyrosine kinase fusion genes
Marker |
BCR-ABL1-like (n=77) |
Remaining B-other (n=76) |
|---|---|---|
ABL1/ABL2 fusion |
3.9% |
0% |
ZMIZ1-ABL1 |
1 |
|
FOXP1-ABL1 |
1 |
|
RCSD1-ABL2 |
1 |
|
PDGFRB fusion |
5.2% |
0% |
EBF1-PDGFRB |
4 |
|
CSF1R fusion |
2.6% |
0% |
SSBP2-CSF1R |
2 |
|
JAK2 fusion |
6.5% |
0% |
PAX5-JAK2 |
3 |
|
BCR-JAK2 |
1 |
|
TERF2-JAK2 |
1 |
|
CRLF2 high expression* |
15.6% |
15.8% |
PAR1 deletion** |
10.5% |
10.7% |
*Expression of Affymetrix U133 Plus 2.0 probe set 208303_s_at above the 90th percentile of the total BCP-ALL group, as described previously [5].
**Deletion of IL3RA and CSF2RA and retention of CRLF2 as determined by MLPA.
Table 2 summarizes the genomic analyses of the tyrosine kinase fusion cases. All PDGFRB, CSF1R and ABL1 fusions showed aberrant FISH patterns with the appropriate break apart FISH probes (Table 2, Supplementary Figure 4). In addition, both ABL1 fusions showed add(9)(q34) in their karyotypes (ABL1 is located on 9q34), together with add(10)(q21) in the ZMIZ1-ABL1 case (ZMIZ1 is located on 10q22), and with del(3)(p13) in the FOXP1-ABL1 case (FOXP1 is located on 3p13). Two EBF1-PDGFRB cases showed an interstitial 5q deletion on array-CGH, one case arose by balanced t(5;5) translocation, and one case showed a more complex copy number alteration, with loss of EBF1 exon 16 and gain of a genomic region encompassing EBF1 exons 7-9 (Table 2; Supplementary Figure 5A-B). EBF1 exon 16 deletion was confirmed by MLPA in the two interstitial 5q deletion cases and the complex case (Table 2). Genomic breaks were also detected in SSBP2-CSF1R, BCR2-JAK2 and TERF2-JAK2 fusions (Table 2, Supplementary Figure 5C-F). Interestingly, one SSBP2-CSF1R fusion probably arose by chromothripsis of chromosome 5 (Supplementary Figure 5D). Finally, when we ordered the gene expression levels of the involved tyrosine kinases for the 153 B-other BCP-ALL cases, the expression in the fusion cases ranked in the top (median percentile 2.6%, range 0.7-14%; Table 2).
Table 2: Molecular characteristics of the identified tyrosine kinase fusion genes
Case |
Tyrosine kinase fusion |
Simplified karyotype |
Array-CGHa |
EBF1 exon 16b |
FISHc |
Expr rankd |
Exons fused |
Validation |
|---|---|---|---|---|---|---|---|---|
R32 |
EBF1-PDGFRB |
ND |
deletion chr5: 149,494,702-158,058,047 |
0.64 |
PDGFRB split |
1% |
e15-e11 |
RT-PCR |
A288 |
EBF1-PDGFRB |
46,XY,t(2;12)(q23~24; q12)[20]/46,XY[14] |
diploid |
ND |
PDGFRB split |
5% |
e14-e11 |
RT-PCR |
A472 |
EBF1-PDGFRB |
46,XY[20] |
deletion chr5: 149,494,702-158,030,723; |
0.62 |
PDGFRB split |
4% |
e15-e11 |
RT-PCR |
A428 |
EBF1-PDGFRB |
46,XY[20].ish 7(cep7,D7S522)x1,i(7) |
gain chr5: 158,222,469-158,428,865 |
0.54 |
PDGFRB split |
6% |
e15-e11 |
RT-PCR |
A123 |
SSBP2-CSF1R |
ND |
small deletions typical of chromothripsis |
1.0 |
PDGFRB split |
1% |
e16-e12 |
RT-PCR |
A526 |
SSBP2-CSF1R |
46,XY[10] |
gain chr5: 80,740,416-149,403,322 |
1.1 |
PDGFRB split |
3% |
e16-e12 |
RT-PCR |
A91 |
ZMIZ1-ABL1 |
47,XY,add(9)(q34), add(10)(q21),mar[9] |
diploid |
1.1 |
ABL1 split |
2% |
e18-e2 |
RT-PCR |
A26 |
FOXP1-ABL1 |
46,XY,del(3)(p13), add(9)(q34),i(22)(q10), inc[6] |
diploid |
1.0 |
ABL1 split |
14% |
e27-e4 |
TLA; PCR |
A530 |
RCSD1-ABL2 |
46,XY[4]/42~ 46,XY,inc[3] |
ND |
0.71 |
ND |
1% |
e3-e5 |
RT-PCR; TLA |
A31 |
PAX5-JAK2 |
46,XX,del(1)(q3?2q4?4), inc[2]/46,XX[18] |
diploid |
0.94 |
ND |
1% |
e5-e19 |
RT-PCR |
A286 |
PAX5-JAK2 |
46,XX[17] |
diploid |
0.81 |
ND |
6% |
e5-e19 |
RT-PCR |
A204 |
PAX5-JAK2 |
ND |
diploid |
0.96 |
ND |
3% |
e5-e19 |
RT-PCR |
A214 |
TERF2-JAK2 |
46,XY[20] |
gain chr16: 67,958,182-88,690,630; |
0.95 |
ND |
3% |
e10-e19 |
RT-PCR; TLA |
A216 |
BCR-JAK2 |
ND |
deletion chr9: 194,193-5,068,342; gain chr9: 5,080,443-5,647,733; |
0.91 |
ND |
1% |
e1-e17 |
RT-PCR; TLA |
aGenome positions in build hg18, copy number changes shown for the genes involved in the fusion; see also Supplementary Figure 5.
bEBF1 exon 16 copy number ratio determined by MLPA. A value below 0.75 is called as a deletion.
cFISH break apart probes from Cytocell were used. The centromeric PDGFRB probe overlaps with CSF1R and can be used to detect breakpoints in CSF1R as well as in PDGFRB.
dExpr rank indicates the percentile in rank of the tyrosine kinase gene expression among 153 B-other BCP-ALL cases on Affymetrix U133 Plus 2 arrays.
ND, not determined.
CRLF2 high expression and PAR1 deletions are common in both BCR-ABL1-like and B-other cases
High gene expression levels of the cytokine receptor CRLF2 were found in 16% of BCR-ABL1-like cases, none of which overlapped with ABL/JAK class tyrosine kinase fusion cases, and also in 16% of non-BCR-ABL1-like B-other cases. About 50% of CRLF2-high cases carried a JAK2 mutation [15]. The frequency of CRFL2 high expression cases with a PAR1 deletion, JAK2 mutation or both was similarly high in BCR-ABL1-like (9/12 cases) and B-other cases (11/12 cases (Figure 1). CRLF2 expression had no prognostic value in this Dutch/German BCP-ALL cohort as described previously [5].
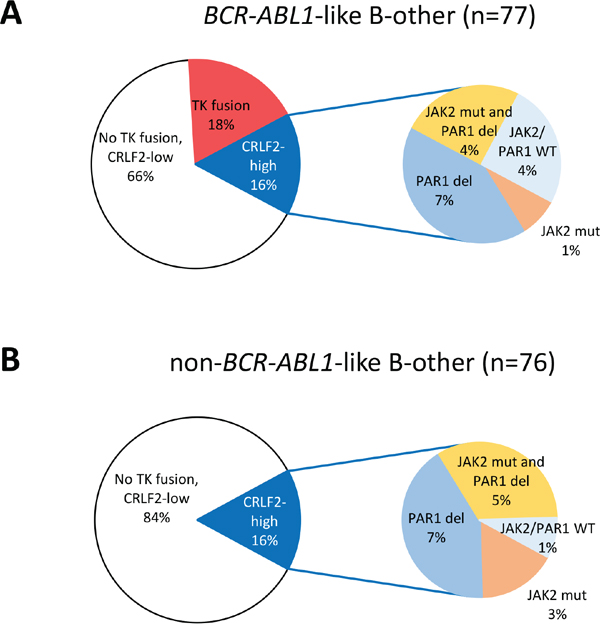
Figure 1: Distribution of tyrosine kinase fusions and CRLF2 high expression cases. Pie diagrams showing the percentages of tyrosine kinase (TK) fusion cases and CRLF2 high expression among A. 77 BCR-ABL1-like B-other cases, and B. 76 non-BCR-ABL1-like B-other cases. Within the CRLF2 high expression cases, a sub-distribution of cases with JAK2 mutation and/or PAR1 deletion is shown. In the non-BCR-ABL1-like B-other cases, 2 PAR1-deleted cases were not tested for JAK2 mutations.
Tyrosine kinase fusion cases are enriched for IKZF1 deletion variants
Next, we compared the frequency of B-cell development gene lesions between the tyrosine kinase fusion-positive BCR-ABL1-like cases, the fusion-negative BCR-ABL1-like cases, and the non-BCR-ABL1-like B-other cases. While IKZF1 deletions were common both in fusion-positive and fusion-negative BCR-ABL1-like cases (64% vs. 40%; p=0.14), IKZF1 deletions other than exon 4-7 or full deletion, including deletion of exons 1, 1-2, 1-3, 2-3, 2-7, 2-8, 4-8, 5 and 7-8, occurred more frequently in the fusion-positive compared with the fusion-negative BCR-ABL1-like cases (43% vs. 15%; p=0.03; Table 3). Except for deletions in EBF1, enriched in EBF1-PDGFRB fusion cases due to interstitial 5q deletion, none of the other genes detected by P335 MLPA (PAX5, ETV6, RB1, BTG1, CDKN2A/B, PAR1) showed an aberrant deletion frequency in tyrosine kinase fusion cases compared with non-BCR-ABL1-like B-other cases (Table 2). In the fusion-positive cases, CDKN2A/B deletion was less frequent than in the fusion-negative BCR-ABL1-like cases (14% vs. 63%, p=0.002; Table 3 ). Two large genomic lesions previously described to occur in BCR-ABL1-like BCP-ALL, dic(9;20) and intrachromosomal amplification of chromosome 21 [3, 16], were mutually exclusive with the identified tyrosine kinase fusions (Table 3).
Table 3: Clinical and molecular features of ABL/JAK class tyrosine kinase fusions
TK fusion-positive |
TK fusion-negative |
Fisher P |
non-BCR-ABL1-like B-other |
Fisher P |
||
|---|---|---|---|---|---|---|
Clinical features |
||||||
male |
11/14 (79%) |
28/63 (44%) |
0.036 |
44/76 (58%) |
0.23 |
|
age ≥ 10 years |
7/14 (50%) |
18/63 (29%) |
0.2 |
25/76 (33%) |
0.24 |
|
WBC ≥ 50x109/L |
6/14 (43%) |
32/63 (51%) |
0.77 |
23/76 (30%) |
0.37 |
|
NCI high riska |
11/14 (79%) |
43/63 (68%) |
0.53 |
42/76 (55%) |
0.14 |
|
MRD TP1b |
high |
7/11 (64%) |
17/36 (47%) |
0.093 |
10/45 (22%) |
0.001 |
MRD TP2b |
high |
4/7 (57%) |
4/30 (13%) |
0.037 |
1/34 (3%) |
0.002 |
Risk arm MR or HR |
12/14 (86%) |
52/62 (84%) |
1 |
47/74 (64%) |
0.13 |
|
Prednisone poor responsec |
2/7 (29%) |
3/31 (10%) |
0.22 |
3/31 (10%) |
0.22 |
|
No CR after inductiond |
5/12 (42%) |
10/58 (17%) |
0.11 |
5/73 (7%) |
0.004 |
|
Molecular features |
||||||
IKZF1 deletion |
Total |
9/14 (64%) |
25/62 (40%) |
0.14 |
13/73 (18%) |
0.0009 |
Commone |
3/14 (21%) |
16/62 (26%) |
1 |
5/73 (7%) |
0.11 |
|
Otherf |
6/14 (43%) |
9/62 (15%) |
0.026 |
8/73 (11%) |
0.009 |
|
EBF1 deletion |
4/14 (29%) |
9/62 (15%) |
0.24 |
3/73 (4%) |
0.012 |
|
EBF1 single exon 16 del |
4/14 (29%) |
0/62 (0%) |
0.0008 |
0/73 (0%) |
0.0005 |
|
PAX5 deletion/amp |
3/14 (21%) |
32/62 (52%) |
0.072 |
26/73 (36%) |
0.37 |
|
ETV6 deletion |
2/14 (14%) |
9/62 (15%) |
1 |
18/75 (24%) |
0.73 |
|
BTG1 deletion |
2/14 (14%) |
1/62 (2%) |
0.085 |
6/74 (8%) |
0.61 |
|
CDKN2A/B deletion |
2/14 (14%) |
39/62 (63%) |
0.002 |
29/75 (39%) |
0.13 |
|
RB1 deletion |
1/14 (7%) |
6/62 (10%) |
1 |
5/74 (7%) |
1 |
|
CRLF2 high expression |
0/14 (0%) |
12/63 (19%) |
0.11 |
12/76 (16%) |
0.2 |
|
PAR1 deletion |
0/14 (0%) |
8/62 (13%) |
0.34 |
8/75 (11%) |
0.35 |
|
dic(9;20) |
0/14 (0%) |
15/57 (26%) |
0.031 |
2/74 (3%) |
1 |
|
iAMP21 |
0/14 (0%) |
10/57 (18%) |
0.19 |
2/74 (3%) |
1 |
|
aNCI-Rome high risk is defined by white blood cell count (WBC) ≥_50x109/L and/or age ≥ 10 years.
bMinimal residual disease PCR high (≥10-3), intermediate (≥10-4 and <10-3), low (<10-4); TP1, after the first induction course of chemotherapy, DCOG ALL-10 protocol day 33, ALL-9 protocol day 42, COALL day 28; TP2, before consolidation, day 79.
cPrednisone response on day 8 ≥1.0x109 blasts/L (DCOG ALL-8/10 protocols).
dComplete remission (CR) after induction (day 33 in DCOG ALL-9/10, day 42 in ALL-8) is defined on morphological grounds by the presence of <5% leukemic blasts and regenerating hematopoiesis.
eCommon IKZF1 deletions are defined as full deletion (exons 1-8) and deletion of exons 4-7.
fOther IKZF1 deletions included deletion of exons 1, 1-2, 1-3, 2-3, 2-7, 2-8, 4-8, 5 and 7-8.
Tyrosine kinase fusion cases show poor treatment response to induction therapy and high minimal residual disease levels
Finally, we evaluated clinical response characteristics. Of 12 tyrosine kinase fusion-positive cases with evaluable data, 5 patients did not achieve complete morphological remission at the end induction therapy; 4 of them were late responders, and one was a non-responder resulting in early death (Figure 2A). This non/late response rate at the end of induction therapy was higher in the fusion-positive cases compared with non-BCR-ABL1-like B-other (42% vs. 7%, p=0.004) and also higher, albeit not statistically significant, compared with fusion-negative BCR-ABL1-like cases (42% vs. 17%, p=0.11; Table 3). Furthermore, fusion-positive cases were characterized by higher levels of minimal residual disease (MRD) compared with non-BCR-ABL1-like B-other cases at end of induction therapy (p=0.001) and before consolidation (p=0.002) and also, although less significant, compared with fusion-negative BCR-ABL1-like cases (p=0.09, p=0.04, respectively; Table 3). The 8-year cumulative incidence of relapse (CIR) in the BCR-ABL1-like group with tyrosine kinase fusions (35% ± 16%) was comparable with the remaining BCR-ABL1-like group (35% ± 6%), and worse than the non-BCR-ABL1-like B-other group (17% ± 5%; Gray p-value 0.066; Figure 2B). Of the three relapsed fusion-positive cases, one died of relapse and two were alive at end of follow-up (6-12 months). Remarkably, the percentage of males was higher in fusion-positive than fusion-negative BCR-ABL1-like cases (79% vs. 44%, p=0.04; Table 3).
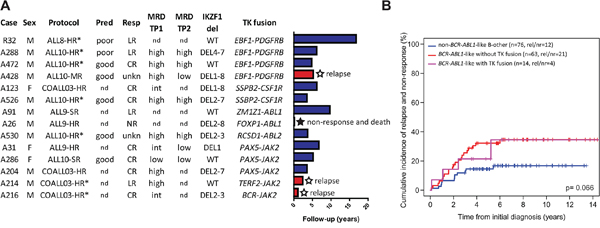
Figure 2: BCR-ABL1-like tyrosine kinase fusion cases. A. Clinical characteristics and follow-up for the tyrosine kinase fusion cases. Barplot representing years from diagnosis to event or censoring. Treatment protocol and arm, prednison window response (Pred), morphological response after induction therapy (Resp), minimal residual disease (MRD), and deletion status of IKZF1 are shown. MRD monitoring by PCR was performed for research purposes in ALL-9 and COALL03 and for MRD-guided risk stratification in ALL-10. WT indicates no IKZF1 deletion. For definition of Pred, Resp, and MRD see footnotes Table 3. Response ‘unkn’ indicates patients with low cellularity bone marrow which could not be evaluated. * Indicates patients who received bone marrow transplant, for which HR-treated cases were eligible. B. Cumulative incidence of relapse and non-response curves for tyrosine kinase fusion positive BCR-ABL1-like cases, fusion-negative BCR-ABL1-like cases, and non-BCR-ABL1-like B-other cases. Relapse and non-response were considered as events, death as competing event. Cumulative incidence probabilities were estimated using a competing risk model, equality was tested with the Gray test.
DISCUSSION
We identified ABL/JAK class tyrosine kinase fusion genes in 14/153 (9%) of DCOG/COALL B-other cases and these fusions were restricted to the BCR-ABL1-like B-other subgroup. Given that these 153 cases were derived from a population-based selection of BCP-ALL [5] and that these fusions do not co-occur with other driver leukemic fusions (BCR-ABL1, rearranged MLL, TCF3-PBX1, ETV6-RUNX1), ABL/JAK class tyrosine kinase fusions cases are estimated to represent approximately 2.5% of pediatric BCP-ALL, making this heterogeneous subgroup similar in size to the BCR-ABL1-positive cytogenetic subtype. Although the number of cases evaluated in our study is modest, the frequency of tyrosine kinase fusion cases is similar to those reported in other cohorts: 3% ABL/JAK class fusions in a high-risk pediatric US cohort [6] and 5% ABL/JAK class fusions among Japanese pediatric B-other cases [9].
The most comprehensive detection of novel tyrosine kinase fusion genes requires transcriptome or whole genome sequencing. These costly methods have not been applied to complete cohorts but rather to subsets of cases, for example cases selected based on Philadelphia-like gene expression or absence of sentinel chromosomal abnormalities, mostly in retrospective studies [6, 9]. More targeted approaches to detect recently identified tyrosine kinase fusions include RT-PCR [9], capture-targeted sequencing [17] or targeted locus amplification (TLA) and sequencing [18]. We combined genomic information from copy number, gene expression, targeted RT-PCR, FISH, and TLA to detect tyrosine kinase fusion genes in a representative cohort of 153 BCP-ALL cases without sentinel chromosomal aberrations that were taken from a population-based cohort of 574 cases at initial diagnosis of BCP-ALL. We found tyrosine kinase fusions exclusively in the BCR-ABL1-like B-other group, and found them non-overlapping with two chromosomal abnormalities that were described earlier to occur in BCR-ABL1-like cases, iAMP21 and dic(9;20) [3]. Moreover, while CRLF2 high expression cases are present both in BCR-ABL1-like and non-BCR-ABL1-like B-others, we found that tyrosine kinase fusions and high CRLF2 expression were mutually exclusive. The current study did not aim at discovering novel TK fusions, therefore there is a small probability that a sample with a novel tyrosine kinase gene is included in the tyrosine kinase fusion negative group.
Our finding that tyrosine kinase fusions are only found in patients with BCR-ABL1-like features, and our earlier finding that tyrosine kinase fusion cases are identified by both the St. Jude and Erasmus MC BCR-ABL1-like expression signatures [16], means that a pre-screening method based on gene expression, such as the US-developed low density array, is suitable to narrow down the patient population to be screened for tyrosine kinase fusions. An alternative approach screens patients selected by poor early clinical response for tyrosine kinase fusions [10].
We found that ABL/JAK kinase fusion cases were characterized by poor initial response to induction treatment, high MRD levels, and a higher relapse rate compared with non-BCR-ABL1-like B-other ALL cases but a comparable relapse rate as BCR-ABL1-like ALL without tyrosine kinase fusions. In correspondence, a recent Japanese study showed that the event-free survival of tyrosine kinase-activating fusion cases (albeit including CRLF2 rearrangements) was unfavorable compared with fusion-negative BCP-ALL [9], Schwab et al. described that EBF1-PDGFRB-positive patients were MRD positive at the end of induction [10], and Roberts et al. described MRD positivity and inferior survival in the total group of Philadelphia-like cases [6]. Recent protocols using MRD-based risk-directed therapy suggest that initial poor treatment response in tyrosine kinase fusion-positive cases or the total group of Philadelphia-like cases can be overcome with intensive chemotherapy, leading to durable remission [10, 19]. Ex vivo leukemic patients’ cells with JAK and ABL class fusions were shown to be sensitive to tyrosine kinase inhibitors [6–7, 15]. Promising early clinical results suggests that ABL class tyrosine kinase fusion patients respond well to ABL class inhibitors imatinib or dasatinib [6, 10, 20–21]. Together with our data, these reports stress the importance of prospective detection of tyrosine kinase fusions and incorporation of tyrosine kinase inhibitors in ALL treatment protocols to improve outcome.
MATERIALS AND METHODS
Patient samples
This study comprised 574 children with newly diagnosed BCP-ALL enrolled in consecutive Dutch Childhood Oncology Group trials (DCOG ALL-8, ALL-9 and ALL-10) [5, 22] and German Cooperative ALL trials (COALL 06-97 and 07-03) [23–24]. These patient cohorts were described and analyzed together previously [3, 5]. Written informed consent was obtained from parents or guardians and institutional review boards approved the use of excess of diagnostic material for research purposes. These studies were conducted in accordance with the Declaration of Helsinki. Mononuclear cells were collected using Lymphoprep sucrose gradient centrifugation (Nycomed Pharma, Oslo, Norway) from bone marrow aspirates and peripheral blood samples obtained prior to treatment. Where needed, mononuclear cells were enriched to >90% leukemic cells by depleting normal cells using anti-CD marker coated magnetic beads (Dynal, Oslo, Norway) as described previously [25]. DNA and total RNA were isolated using TRIzol (Invitrogen Life Technologies, Breda, the Netherlands) and quantified using a Nanodrop ND1000 (Nanodrop, Wilmington, DE). From this cohort, we focused on B-other patients, defined as patients without the sentinel chromosomal abnormalities BCR-ABL1, MLL rearrangements, ETV6-RUNX1, TCF3-PBX1, and high hyperdiploidy. Among the B-other cases, BCR-ABL1-like cases identified by hierarchical clustering of gene expression data were described previously in our cohort [5]. For 153 out of 204 B-other/BCR-ABL1-like cases in the cohort, tyrosine kinase fusion testing was performed (see flow chart in Supplementary Figure 1).
Tyrosine kinase fusion detection
Detection of tyrosine kinase fusion genes was performed by RT-PCR followed by Sanger sequencing for seven fusion transcripts EBF1-PDGFRB, PAX5-JAK2, NUP214-ABL1, RANBP2-ABL1, ETV6-ABL1, RCSD1-ABL1 and STRN3-JAK2 described by Roberts et al. [7]. In addition, we performed a more extensive RT-PCR panel [6] enabling detection of 30-39 fusion genes (depending on availability of material). For RT-PCR primers see Supplementary Table 2. We used targeted locus amplification for 21 cases to detect fusion genes involving ABL1, PDGFRB, CSF1R, ABL2, TYK2, and JAK2 (TLA, Cergentis, Utrecht, the Netherlands) [18, 26]. We used break apart FISH with PDGFRB/CSF1R and ABL1 probes (Cytocell) to confirm fusions. The methods applied to each case depended on the type and amount of available patient material and are indicated in Supplementary Table 3 and summarized in Supplementary Figure 1. For the comparison of tyrosine kinase fusion positive versus negative patients, we included 153 samples that were tested at least by the 7-fusion RT-PCR panel. For expression rank analysis (Table 2), the Affymetrix microarray gene expression values for each tyrosine kinase gene were ranked from high to low for the 153 samples. Then the percentile rank of the fusion-positive case among the 153 samples was calculated.
Genome-wide DNA copy number arrays (array-CGH)
Copy number analysis was performed using Agilent SurePrint G3 Hmn 4x180K arrays (Agilent Technologies, Amstelveen, the Netherlands) co-hybridized with 1 μg patient DNA labeled with ULS-Cy5 and 1 μg reference genomic DNA male pool (G147A, Promega, Leiden, the Netherlands) labeled with ULS-Cy3 (Agilent Genomic DNA ULS Labeling Kit). Copy number microarray data were normalized using median log ratio in the CGHcall [27] version 2.14.0, centralized using CGHnormaliter [28] version 1.8.0, and segmented and called using CGHcall default settings (-1 for loss, 0 for diploid, 1 for gain and 2 for amplification) in R version 2.14.1.
Multiplex ligation-dependent probe amplification
The SALSA P335 ALL-IKZF1 (a3) and the SALSA P202 Multiplex Ligation-dependent Probe Amplification (MLPA) assays (MRC-Holland) were used to identify or confirm genomic lesions on the following genes: IKZF1, CDKN2A, CDKN2B, ETV6, PAX5, RB1, BTG1 and EBF1 as described previously [5, 29]. In short, 125 ng of genomic DNA was used to generate DNA fragments with incorporated FAM nucleotides according to the manufacturer’s protocol. The amplified fragments were quantified using an ABI-3130 genetic analyzer (Applied Biosystems, Carlsbad, CA). Peak intensities were normalized to the manufacturer’s control probes and to a synthetic control reference generated from five normal DNA samples in the same MLPA run. A peak ratio lower than 0.75 was considered a deletion, a ratio between 0.75 and 1.3 was considered normal copy number, a ratio higher than 1.3 was considered a gain in copy number.
Targeted locus amplification
Targeted Locus Amplification (TLA) combined with deep-sequencing was used to detect fusion genes and sequence mutations in regions up to 100 kb around a pre-selected primer pair by crosslinking of physically proximal genomic sequences as described before [26]. Briefly, DNA and protein in 10-15 million viable leukemic blast cells were crosslinked in a 2% formaldehyde solution. Cells were lysed and DNA was digested with NlaIII, followed by ligation, de-crosslinking and DNA purification. DNA molecules were trimmed with NspI and ligated at a concentration of 5 ng/μl to promote intramolecular ligation to DNA fragments of approximately 2 kb. These chimeric fragments were PCR amplified, sonicated and adaptor-ligated for paired-end high-throughput Illumina sequencing. A total of 31 primer sets targeting 19 recurrently affected genes were designed and multiplexed, including the genes involved in the classical cytogenetic subtypes MLL, RUNX1, TCF3, and IKZF1, the tyrosine kinase genes ABL1, ABL2, PDGFRB, CSF1R, JAK1, JAK2, JAK3, FLT3, and TYK2, and the cytokine signaling genes CRLF2, EPOR, IL7R, TSLP, SH2B3, and IL2RB [17].
Fluorescent in-situ hybridization (FISH)
FISH was performed on interface nuclei using break apart probes (Cytocell, Cambridge, UK) for PDGFRB and ABL1. The FISH probes for PDGFRB overlap with the neighboring CSF1R locus. At least 100 interphase nuclei were evaluated.
Reverse transcriptase PCR (RT-PCR)
cDNA was synthesized from 1 μg total or copy RNA using M-MLV reverse transcriptase and combined oligo-dT and pdN6 priming in 20 μl (Promega, Madison, WI). PCR was performed on 2.5 μl cDNA using Taq polymerase, MgCl2 and buffer from Applied Biosystems (Bleiswijk, Netherlands). For primer sequences see Supplementary Table 2, for RT-PCR results per sample see Supplementary Table 3.
Association with clinical outcome
Cumulative incidence of relapse (CIR) was estimated using a competing risks model. We considered relapse as event, and death as competing event. To test for equality of CIRs, Gray’s test has been applied. The CIR probability (pCIR) with standard error was reported. Outcome analyses were performed in R 3.0.1, using the packages cmprsk version 2.2-6 [30], mstate version 0.2.6 [31], and survival version 2.37-4 [32].
ACKNOWLEDGMENTS
We thank Oksana Montecchini and Alice Poropat for technical assistance.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
GRANT SUPPORT
This work was supported by the VICI program grant 016.126.612 from Netherlands Organisation for Scientific Research (NWO), the Foundation Pediatric Oncology Center Rotterdam (SKOCR), the Dutch Cancer Society grants EMCR 2007-3718 and AMC 2008-4265, the KiKa Foundation (Kika-132 grant) and the European Union’s Seventh Framework Program FP7/2007-2013 ENCCA grant HEALTH-F2-2011-261474.
Author contributions
JMB, EMPS, JRMM, JJB, HBB, AB, VHJvdV, CEvdS and RPK designed and performed experiments, and analyzed and interpreted data. RP, HAdGK and GE provided patients’ characteristics and clinical outcome data. RP and MLdB conceptualized the study, and interpreted data. JMB and MLdB drafted the manuscript. The manuscript was revised and approved by all authors.
REFERENCES
1. Biondi A, Schrappe M, De Lorenzo P, Castor A, Lucchini G, Gandemer V, Pieters R, Stary J, Escherich G, Campbell M, Li CK, Vora A, Arico M, et al. Imatinib after induction for treatment of children and adolescents with Philadelphia-chromosome-positive acute lymphoblastic leukaemia (EsPhALL): a randomised, open-label, intergroup study. Lancet Oncol. 2012; 13:936-945.
2. Schultz KR, Carroll A, Heerema NA, Bowman WP, Aledo A, Slayton WB, Sather H, Devidas M, Zheng HW, Davies SM, Gaynon PS, Trigg M, Rutledge R, et al. Long-term follow-up of imatinib in pediatric Philadelphia chromosome-positive acute lymphoblastic leukemia: Children's Oncology Group study AALL0031. Leukemia. 2014; 28:1467-1471.
3. Den Boer ML, van Slegtenhorst M, De Menezes RX, Cheok MH, Buijs-Gladdines JG, Peters ST, Van Zutven LJ, Beverloo HB, Van der Spek PJ, Escherich G, Horstmann MA, Janka-Schaub GE, Kamps WA, et al. A subtype of childhood acute lymphoblastic leukaemia with poor treatment outcome: a genome-wide classification study. Lancet Oncol. 2009; 10:125-134.
4. Mullighan CG, Su X, Zhang J, Radtke I, Phillips LA, Miller CB, Ma J, Liu W, Cheng C, Schulman BA, Harvey RC, Chen IM, Clifford RJ, et al. Deletion of IKZF1 and prognosis in acute lymphoblastic leukemia. N Engl J Med. 2009; 360:470-480.
5. Van der Veer A, Waanders E, Pieters R, Willemse ME, Van Reijmersdal SV, Russel LJ, Harrison CJ, Evans WE, Van der Velden VHJ, Hoogerbrugge PM, Van Leeuwen F, Escherich G, Horstmann MA, et al. Independent prognostic value of BCR-ABL1-like signature and IKZF1 deletion, but not high CRLF2 expression, in children with B-cell precursor ALL. Blood. 2013; 122:2622-2629.
6. Roberts KG, Li Y, Payne-Turner D, Harvey RC, Yang YL, Pei D, McCastlain K, Ding L, Lu C, Song G, Ma J, Becksfort J, Rusch M, et al. Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. N Engl J Med. 2014; 371:1005-1015.
7. Roberts KG, Morin RD, Zhang J, Hirst M, Zhao Y, Su X, Chen SC, Payne-Turner D, Churchman ML, Harvey RC, Chen X, Kasap C, Yan C, et al. Genetic alterations activating kinase and cytokine receptor signaling in high-risk acute lymphoblastic leukemia. Cancer Cell. 2012; 22:153-166.
8. Izraeli S. Beyond Philadelphia: 'Ph-like' B cell precursor acute lymphoblastic leukemias - diagnostic challenges and therapeutic promises. Curr Opin Hematol. 2014; 21:289-296.
9. Imamura T, Kiyokawa N, Kato M, Imai C, Okamoto Y, Yano M, Ohki K, Yamashita Y, Kodama Y, Saito A, Mori M, Ishimaru S, Deguchi T, et al. Characterization of pediatric Philadelphia-negative B-cell precursor acute lymphoblastic leukemia with kinase fusions in Japan. Blood Cancer J. 2016; 6:e419.
10. Schwab C, Ryan SL, Chilton L, Elliott A, Murray J, Richardson S, Wragg C, Moppett J, Cummins M, Tunstall O, Parker CA, Saha V, Goulden N, et al. EBF1-PDGFRB fusion in pediatric B-cell precursor acute lymphoblastic leukemia (BCP-ALL): genetic profile and clinical implications. Blood. 2016; 127:2214-2218.
11. Zaliova M, Moorman AV, Cazzaniga G, Stanulla M, Harvey RC, Roberts KG, Heatley SL, Loh ML, Konopleva M, Chen IM, Zimmermannova O, Schwab C, Smith O, et al. Characterization of leukemias with ETV6-ABL1 fusion. Haematologica. 2016; 101:1082-1093.
12. Soler G, Radford-Weiss I, Ben-Abdelali R, Mahlaoui N, Ponceau JF, Macintyre EA, Vekemans M, Bernard OA, Romana SP. Fusion of ZMIZ1 to ABL1 in a B-cell acute lymphoblastic leukaemia with a t(9;10)(q34;q22.3) translocation. Leukemia. 2008; 22:1278-1280.
13. Ernst T, Score J, Deininger M, Hidalgo-Curtis C, Lackie P, Ershler WB, Goldman JM, Cross NC, Grand F. Identification of FOXP1 and SNX2 as novel ABL1 fusion partners in acute lymphoblastic leukaemia. Br J Haematol. 2011; 153:43-46.
14. Zhou X, Edmonson MN, Wilkinson MR, Patel A, Wu G, Liu Y, Li Y, Zhang Z, Rusch MC, Parker M, Becksfort J, Downing JR, Zhang J. Exploring genomic alteration in pediatric cancer using ProteinPaint. Nature Genetics. 2015; 48:4-6.
15. Steeghs EMP, Jerchel IS, de Goffau-Nobel W, Hoogkamer AQ, Boer JM, Boeree A, van de Ven C, Koudijs MJ, Cuppen E, De Groot-Kruseman HA, Horstmann MA, Pieters R, Den Boer ML. JAK2 aberrations in childhood precursor B cell acute lymphoblastic leukemia. Submitted.
16. Boer JM, Marchante JRM, Evans WE, Horstmann MA, Escherich G, Pieters R, Den Boer ML. BCR-ABL1-Like Cases in Pediatric Acute Lymphoblastic Leukemia: a Comparison Between DCOG/Erasmus MC and COG/St. Jude Signatures. Haematologica 2015; 100:e354-357.
17. Escherich G, Zur Stadt U, Alawi M, Horstmann M. Rapid capture targeted next generation sequencing (NGS) for detection of genomic kinase- and cytokine receptor rearrangements in B-precursor acute lymphoblastic leukemia. Annual Meeting Abstracts Blood. 2015; 126:2609.
18. Kuiper RP, Van Reijmersdal SV, Simonis M, Yu J, Sonneveld E, Scheijen B, Boer JM, Boeree A, Klous P, Hoogerbrugge P, Yilmaz M, Van Leeuwen FN, Den Boer ML, et al. Targeted locus amplification & next generation sequencing for the detection of recurrent and novel gene fusions for improved treatment decisions in pediatric acute lymphoblastic leukemia. Annual Meeting Abstracts Blood. 2015; 126:696.
19. Roberts KG, Pei D, Campana D, Payne-Turner D, Li Y, Cheng C, Sandlund JT, Jeha S, Easton J, Becksfort J, Zhang J, Coustan-Smith E, Raimondi SC, et al. Outcomes of Children With BCR-ABL1-Like Acute Lymphoblastic Leukemia Treated With Risk-Directed Therapy Based on the Levels of Minimal Residual Disease. Journal of Clinical Oncology. 2014; 32:3012-3020.
20. Weston BW, Hayden MA, Roberts KG, Bowyer S, Hsu J, Fedoriw G, Rao KW, Mullighan CG. Tyrosine Kinase Inhibitor Therapy Induces Remission in a Patient With Refractory EBF1-PDGFRB-Positive Acute Lymphoblastic Leukemia. Journal of Clinical Oncology. 2013; 31:e413-416.
21. Lengline E, Beldjord K, Dombret H, Soulier J, Boissel N, Clappier E. Successful tyrosine kinase inhibitor therapy in a refractory B-cell precursor acute lymphoblastic leukemia with EBF1-PDGFRB fusion. Haematologica. 2013; 98:e146-148.
22. Kamps WA, van der Pal-de Bruin KM, Veerman AJ, Fiocco M, Bierings M, Pieters R. Long-term results of Dutch Childhood Oncology Group studies for children with acute lymphoblastic leukemia from 1984 to 2004. Leukemia. 2010; 24:309-319.
23. Escherich G, Horstmann MA, Zimmermann M, Janka-Schaub GE, Grp CS. Cooperative study group for childhood acute lymphoblastic leukaemia (COALL): long-term results of trials 82,85,89,92 and 97. Leukemia. 2010; 24:298-308.
24. Escherich G, Zimmermann M, Janka-Schaub G. Doxorubicin or daunorubicin given upfront in a therapeutic window are equally effective in children with newly diagnosed acute lymphoblastic leukemia. A randomized comparison in trial CoALL 07-03. Pediatr Blood Cancer. 2013; 60:254-257.
25. Den Boer ML, Harms DO, Pieters R, Kazemier KM, Göbel U, Körholz D, Graubner U, Haas RJ, Jorch NJ, Spaar HJ, Kaspers GJL, Kamps WA, Van der Does-Van den Berg A, et al. Patient stratification based on prednisolone-vincristine-asparaginase resistance profiles in children with acute lymphoblastic leukemia. J Clin Oncol. 2003; 21:3262-3268.
26. de Vree PJ, de Wit E, Yilmaz M, van de Heijning M, Klous P, Verstegen MJ, Wan Y, Teunissen H, Krijger PH, Geeven G, Eijk PP, Sie D, Ylstra B, et al. Targeted sequencing by proximity ligation for comprehensive variant detection and local haplotyping. Nat Biotechnol. 2014; 32:1019-1025.
27. van de Wiel MA, Kim KI, Vosse SJ, van Wieringen WN, Wilting SM, Ylstra B. CGHcall: calling aberrations for array CGH tumor profiles. Bioinformatics. 2007; 23:892-894.
28. van Houte BP, Binsl TW, Hettling H, Pirovano W, Heringa J. CGHnormaliter: an iterative strategy to enhance normalization of array CGH data with imbalanced aberrations. BMC Genomics. 2009; 10:401.
29. Boer JM, Koenders JE, van der Holt B, Exalto C, Sanders MA, Cornelissen JJ, Valk PJ, den Boer ML, Rijneveld AW. Expression profiling of adult acute lymphoblastic leukemia identifies a BCR-ABL1-like subgroup characterized by high non-response and relapse rates. Haematologica. 2015; 100:e261-264.
30. Gray RJ. cmprsk: Subdistribution Analysis of Competing Risks. R package version 22-6 2013; http://CRAN.R-project.org/package=cmprsk.
31. De Wreede LC, Fiocco M, Putter H. mstate: An R Package for the Analysis of Competing Risks and Multi-State Models. J Stat Softw. 2011; 38:1-30.
32. Therneau T. A Package for Survival Analysis in S. R package version 236-12. 2012.