
INTRODUCTION
Ovarian cancer is the eighth most common cause of cancer death in Taiwanese women [1]. Approximately 238,700 new cases are diagnosed each year worldwide, which are responsible for 151,900 deaths [2]. The 5-year survival rates of ovarian cancer are stage-dependent and range between 27% and 92% [3]. Common adverse prognostic factors include advanced stages and disease recurrence [4].
Mutations of the BRCA1/2 genes occur in approximately 20% of high-grade ovarian serous carcinoma [5] and are associated with better survival outcomes [6–10]. Inactivating BRCA1/2 mutations portend an increased risk of malignant transformation because of their ability to impair homologous recombination-dependent DNA repair [11]. Conversely, inactivation of homologous recombination renders BRCA1/2 mutant tumors sensitive to platinum [6, 8, 10, 12] and poly (ADP-ribose) polymerase (PARP) inhibitors (PARPi) [13]. Unfortunately, the identification of patients who could benefit from PARPi remains challenging.
The United States (US) Food and Drug Administration (FDA) approved the use of the PARPi olaparib when BRCA1/2 germline mutations are present [14, 15]. However, the US FDA did not approve olaparib for patients who carry somatic BRCA1/2 mutations only (approximately one third of all BRCA1/2 mutant patients) [5, 6]. In contrast, the European Medicines Agency (EMA) approved olaparib for ovarian cancer patients who have either germline or somatic BRCA1/2 mutations [13, 16]. Besides such regulatory discrepancies, not all patients with pathogenic BRCA mutations successfully respond to PARPi. A potential explanation lies in the fact that BRCA mutations require a loss of heterozygosity (LOH) to cause biallelic BRCA inactivation [17]. Although the loss of a single BRCA allele is sufficient per se to induce genomic instability and drive malignant transformation [18, 19], cells with monoallelic BRCA inactivation might not be sensitive to PARPi [20]. Unfortunately, the occurrence of biallelic inactivation of BRCA1/2 has not been specifically analyzed in most of the available studies. In addition, few data on somatic and germline BRCA1/2 mutations have been published in patients with Asian descent, and for Taiwanese patients, even the prevalence of germline mutations is only insufficiently investigated [21]. Notably, clear cell carcinomas are underrepresented in most studies because of their low incidence in North America and Europe (1−12% of all cases) compared to Asia (13−25%) [22–24]. BRCA1/2 mutations have been reported in clear cell carcinomas [8, 25, 26] and endometrioid tumors [6, 8, 27–29], but their clinical significance requires further scrutiny.
Although Sanger sequencing is traditionally used for identifying BRCA mutations in clinical samples [30], next generation sequencing (NGS) has recently allowed obtaining a complete coverage of all exonic regions. This is essential since BRCA mutations differ among patients of different ethnicity [31]. Somatic BRCA1/2 mutations can be successfully identified by NGS in formalin-fixed paraffin-embedded (FFPE) samples, but biallelic inactivation has not been thus far investigated [29].
Using NGS (covering all of the BRCA1/2 exons as well as the exon-intron junctions) of FFPE specimens obtained from 99 Taiwanese patients with ovarian cancer, we analyzed 1) germline and somatic BRCA1/2 mutations and 2) the occurrence of biallelic BRCA1/2 inactivation.
RESULTS
Analytic workflow
We included 99 patients with ovarian cancer who were unselected for their family history of malignancies. The histological subtypes included serous (n = 46), endometrioid (n = 24), and clear cell (n = 29) carcinomas. Supplementary Figure 1 shows the workflow for the identification of BRCA1/2 variants identified in the current study.
Detection of BRCA1 and BRCA2 variants by NGS and Sanger sequencing
NGS was used for BRCA1/2 sequencing of FFPE tumor samples (average sequencing depth: >5700×; mean uniformity: 91.1%). Forty-four nonsynonymous and splice variants were identified in 36 patients. Of them, 13 were pathogenic variants (Table 1), 23 VUS (Supplementary Table S1), and eight benign variants. NGS-identified variants with an allele frequency >10% were confirmed by Sanger sequencing of tumor samples. To distinguish between germline and somatic mutations, Sanger sequencing was also performed for normal tissues (for all pathogenic variants; 13/13 and for all VUS with available control samples; 22/23). In addition to Sanger sequencing, NGS was performed in normal control samples when a sufficient amount of DNA was available (for pathogenic variants; 10/13 and for VUS; 18/23). A 100% concordance rate between the results of NGS and Sanger sequencing in tumor samples was observed.
Table 1: BRCA variant description and clinical characteristics for patients with pathogenic BRCA1/2 mutations
ID |
Germline (G)/ Somatic (S) |
Gene |
Change for nucleotides (nt) and amino acids (aa)# |
Variant classification (ARUP/ BIC/ BRCA Share/ ClinVar/ LOVD) |
Type§ |
Age (y) |
FIGO stage/grade |
FH¶ |
|
|---|---|---|---|---|---|---|---|---|---|
nt |
aa |
||||||||
Recurrent variants (n = 2) |
|||||||||
1 |
G |
BRCA2 |
c.5164_5165 delAG |
p.S1722fs |
path/ path/ NA/ path/ path |
Ser |
50 |
IV/ 3 |
No |
2 |
G |
BRCA2 |
c.5164_5165 delAG |
p.S1722fs |
path/ path/ NA/ path/ path |
Ser |
57 |
I/ 3 |
No |
Unique variants (n = 11) |
|||||||||
3 |
G |
BRCA1 |
c.2188G>T |
p.E730* |
path/ path/ NA/ path/ NA |
Ser |
53 |
IV/ 3 |
NA |
4 |
G |
BRCA1 |
c.2387C>T |
p.T796I |
NA/ VUS/ NA/ VUS/ path |
Ser |
42 |
III/ 3 |
No |
5 |
G |
BRCA1 |
c.5332+1G>A |
p.Œ_splice |
path/ VUS/ path/ path/ path |
Ser |
48 |
III/ 3 |
No |
6 |
G |
BRCA1 |
c.3858_3861 delTGAG |
p.S1286fs |
path/ path/ NA/ path/ path |
Ser |
56 |
III/ 3 |
NA |
6 |
S |
BRCA2 |
c.8488-1G>A |
p.Œ_splice |
path/ NA/ NA/ VUS/ path |
Ser |
56 |
III /3 |
NA |
7 |
G |
BRCA2 |
c.2339C>G |
p.S780* |
NA/ NA/ path/ NA/ NA |
Ser |
71 |
II/ 3 |
No |
8 |
G |
BRCA2 |
c.773_774 delAA |
p.E260fs |
NA/ NA/ NA/ NA/ NA |
Ser |
69 |
II/ 3 |
NA |
9 |
S |
BRCA1 |
c.726delT |
p.S242fs |
NA/ NA/ NA/ NA/ NA |
Ser |
78 |
III/ 3 |
NA |
10 |
S |
BRCA1 |
c.2964delA |
p.F989fs |
NA/ NA/ NA/ NA/ NA |
Ser |
63 |
III/ 3 |
No |
11 |
S |
BRCA1 |
c.5211_5212 delAG |
p.G1738fs |
NA/ NA/ NA/ NA/ NA |
Ser |
49 |
III/ 3 |
No |
12 |
S |
BRCA2 |
c.4351delG |
p.D1451fs |
NA/ NA/ NA/ NA/ NA |
Ser |
65 |
III/ 3 |
No |
#HGVSp - the Human Genome Variation Society (HGVS) protein sequence name. The Annotation is based on the BRCA1 transcript ENSG00000012048 (NM_007294) and the BRCA2 transcriptENSG00000139618 (NM_000059). §Histological subtype; ser = Serous. ¶ Family history (FH) refers to a positive history of breast and/or ovarian cancer in the first- and second-degree relatives.
Cases in bold letter are novel mutations.
Databases: BIC (Breast Cancer Information Core, http://research.nhgri.nih.gov/bic/), ClinVar (http://www.ncbi.nlm.nih.gov/clinvar/) database, LOVD (Leiden Open Variation Database, http://www.lovd.nl/3.0/home), ARUP (http://arup.utah.edu/database/BRCA/) database and BRCA Share (http://www.umd.be/BRCA1/) database
Abbreviations: NA, not applicable; path, pathogenic; VUS, variant of uncertain significance; y, years.
Patient characteristics and distribution of BRCA1/2 mutations in this cohort
The distribution of pathogenic and potentially pathogenic BRCA1 and BRCA2 variants according to tumor histological subtypes is depicted in Figure 1. Table 2 summarizes the patient characteristics according to the BRCA mutation status.
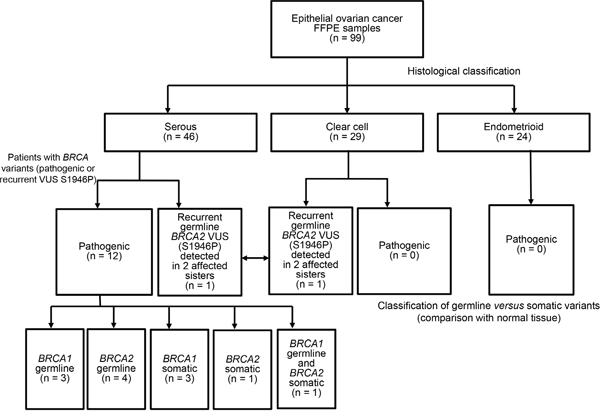
Figure 1: BRCA1/2 genetic variants identified in the study cohort. Distribution of pathogenic BRCA1/2 mutations according to different histological subtypes. The recurrent BRCA2 VUS p.S1946P was deemed to be pathogenic owing to its occurrence in two sisters with ovarian cancer (one with clear cell carcinoma and the other with serous carcinoma).
Table 2: Clinical characteristics of the study patients according to the BRCA mutation status
Entire cohort |
BRCA-positive patients |
BRCA-negative patients |
P |
||
|---|---|---|---|---|---|
n (%) |
n (%) |
n (%) |
|||
99 (100) |
12 (12.1) |
87 (87.9) |
|||
Histology |
0.001 |
||||
Serous carcinoma |
46 (46.5) |
12 (26.1) |
34 (73.9) |
||
Endometrioid carcinoma |
24 (24.2) |
0 (0) |
24 (100) |
||
Clear cell carcinoma |
29 (29.3) |
0 (0) |
29 (100) |
||
Age, years |
0.069 |
||||
Median |
52 |
56 |
52 |
||
Range |
23−83 |
42−78 |
23−83 |
||
Mean ± SD |
52.9 ± 11.1 |
58.4 ± 10.8 |
52.2 ± 11.0 |
||
FIGO stage |
0.228 |
||||
I, II |
42 (42.4)* |
3 (7.1)# |
39 (92.9)# |
||
III, IV |
57 (57.6)* |
9 (15.8)§ |
48 (84.2)§ |
||
Grade※ |
0.579 |
||||
1 |
5 (7.1)¶ |
0 (0) ǂ |
5 (100) ǂ |
||
2, 3 |
65 (92.9)¶ |
12 (18.5)ψ |
53 (81.5)ψ |
Patients with pathogenic BRCA1/2 mutations were considered as BRCA-positive, whereas the remaining patients were regarded as BRCA-negative. Comparisons between BRCA-positive and BRCA-negative patients were performed with Fisher’s exact tests, χ2 tests, or Student’s t-tests, as appropriate.
※Clear cell carcinomas were not graded.
*Percentage calculated on the entire cohort (n = 99).
#Percentage calculated on patients with FIGO stages I and II (n = 42).
§ Percentage calculated on patients with FIGO stages III and IV (n = 57).
¶Percentage calculated on patients whose tumors were graded (n = 70).
ǂPercentage calculated on patients with grade 1 tumors (n = 5).
ψPercentage calculated on patients with grade 2–3 tumors (n = 65).
Twelve patients carried pathogenic BRCA1/2 mutations. Specifically, BRCA1/2 germline mutations were identified in seven patients (BRCA1, n = 3; BRCA2, n = 4), BRCA1/2 somatic mutations in four patients (BRCA1, n = 3; BRCA2, n = 1), whereas one patient carried both a BRCA1 germline mutation and a BRCA2 somatic mutation. All of the 12 BRCA-positive patients had a diagnosis of serous ovarian carcinomas (P = 0.001). The frequencies of BRCA1/2 pathogenic mutations in the entire cohort and the subgroup of patients with serous carcinoma were 12.1% (12/99) and 26.1% (12/46), respectively. Somatic and germline BRCA1/2 mutation rates in serous ovarian cancer are 4/46 (8.7%) and 8/46 (17%), respectively. The mean age at onset for patients with and without pathogenic BRCA1/2 variants was 56 years and 52 years, respectively (P = 0.069). Carriers of BRCA germline mutations tended to have a lower median age at diagnosis (55 years) compared with women bearing only somatic mutations (64 years). In the entire study cohort, 57.6% of patients (n = 57) had advanced disease (FIGO stage III–IV). Moreover, 92.9% of patients (n = 65) with serous or endometrioid carcinomas had high-grade malignancies. Of them, 12 patients harbored pathogenic BRCA1/2 variants. Tumor grade is not routinely determined in clear cell carcinoma [32]. Notably, we observed a recurrent BRCA2 variant of uncertain significance (VUS) in two sisters (one with clear cell carcinoma and the other with serous carcinoma; Figure 1).
Pathogenic variants in BRCA1 and BRCA2
Thirteen pathogenic BRCA1/2 variants were identified in 12 patients (Table 1). Two patients carried the same variant (BRCA2 p.S1722fs), whereas one patient (subject 6) had two pathogenic variants (one in BRCA1 and one in BRCA2). Of the 13 pathogenic variants, eight were frameshift mutations, two nonsense mutations, one missense mutation, and two splice donor variants. Seven mutations were detected in BRCA1 and five in BRCA2. Five novel frameshift mutations were identified, including four somatic variants (BRCA1 p.S242fs, BRCA1 p.F989fs, BRCA1 p.G1738fs, and BRCA2 p.D1451fs) and a germline variant (BRCA2 p.E260fs; Figure 2 and Supplementary Figure S2). There were seven pathogenic BRCA1/2 germline mutations and five pathogenic somatic mutations (Figure 2).
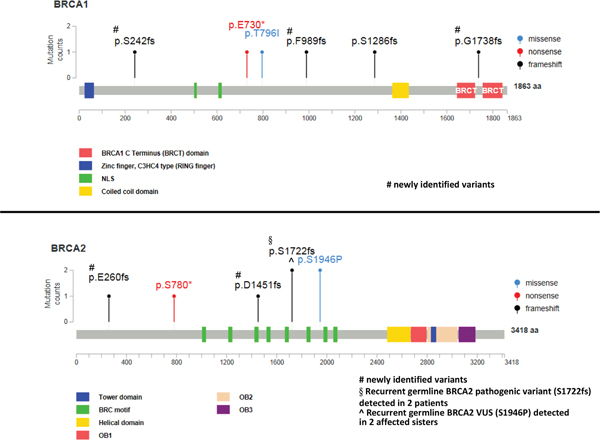
Figure 2: Pathogenic BRCA1/2 variants identified in the study cohort according to their amino acid position. The recurrent BRCA2 VUS p.S1946P—deemed to be pathogenic owing to its occurrence in two sisters with ovarian cancer—is included. However, splice site mutations are not displayed.
VUS and benign variants in BRCA1 and BRCA2
Of the 99 patients, 19.2 % (n = 19) had BRCA1/2 VUS, all of them being missense variants (Supplementary Table S1). The histological subtypes in the 19 patients with BRCA1/2 VUS included serous (n = 10; 52.6%), endometrial (n = 5; 26.3%), and clear cell (n = 4; 21.2%) carcinomas. One patient with serous ovarian cancer (subject 7) carried an additional pathogenic variant. A total of 23 VUS were detected, with two patients harboring two VUS (subject 22 and 26) and one patient harboring three VUS (subject 14). One of the three VUS identified in this patient was also evident in another study participant (subject 13). Fifteen (63.6%) and seven (30.4%) VUS were germline and somatic, respectively. One BRCA2 variant was not classifiable because DNA from non-tumor control tissues was not available. Four benign variants were identified in eight patients, i.e., BRCA1 variants p.V1247I and p.M1628T, BRCA2 variant p.I1929V (identified in four patients), and p.R2842H (identified in two patients).
Nine of the detected VUS have not been previously described. Of note, six novel VUS (three in BRCA1 and three in BRCA2) are predicted to be pathogenic by at least one of Grantham/ SIFT/ Polyphen and not present in 997 healthy volunteers (Supplementary Table S1), suggesting their pathogenic potential.
Identification of a potentially pathogenic BRCA VUS
Three patients carrying a VUS had a family history of breast or ovarian cancer in the absence of any additional pathogenic BRCA variant. One patient (subject 24) carried a variant predicted to be pathogenic according to the Grantham and SIFT scores but classified as benign by PolyPhen. The remaining two patients were two sisters (one with clear cell carcinoma and the other with serous carcinoma) who carried the BRCA2 VUS p.S1946P (predicted to be pathogenic by Grantham but classified as benign by SIFT and PolyPhen). The sister with serous carcinoma carried a BRCA2 LOH (Table 3), consistent with the two-hit hypothesis of tumor suppressor gene inactivation. The variant is rare in healthy subjects of the Taiwanese population (Supplementary Table S1). To shed more light on the potential pathogenic role of the BRCA2 VUS p.S1946P, the family history of cancer was analyzed (Figure 3). Among the patients’ four siblings, no cases of cancer were evident. The analysis of germline variants in the patient siblings revealed that two of the patients’ sisters were BRCA2 p.S1946P carriers, whereas the other sister and the brother were not. From the paternal side, the only known case of malignancy was a lung cancer in a paternal cousin. However, the patients’ mother had pancreatic cancer and their maternal uncle (as well as his son) had colorectal cancer. Although hereditary cancer was plausible, no samples were available from the three family members to investigate the presence of the BRCA2 VUS p.S1946P. However, the tumor samples of the two ovarian cancer patients were analyzed for the presence of pathogenic variants in the coding regions of the 29 genes involved in DNA repair (ATM, ATR, BLM, BRIP1, CHEK1, CHEK2, ERCC1, FANCA, FANCC, FANCD2, FANCF, MLH1, MRE11A, MSH2, MSH6, PALB2, PER1, PMS1, PMS2, PRKDC, PTEN, RAD50, RECQL4, SMUG1, TP53, WRN, XPA, XPC, and XRCC2). However, no shared pathogenic variants were evident (data not shown). Notably, the BRCA2 VUS p.S1946P is located nearby the BRC motif (Figure 2) which is essential for BRCA2-mediated recombination repair. We thus speculate that the serine-to-proline amino substitution may potentially exert pathogenic effects via an altered BRC conformation.
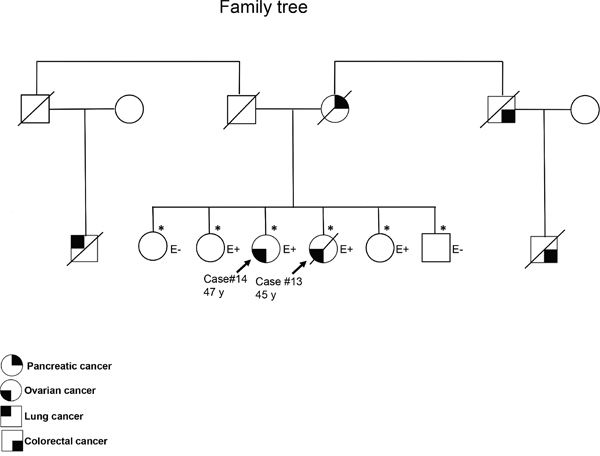
Figure 3: Family tree of the two sisters with ovarian cancer harboring the BRCA2 VUS p.S1946P. The asterisks denote subjects who have been tested for the BRCA2 VUS p.S1946P. Subjects with and without the variant of interest are reported as E+ and E-, respectively.
Table 3: Clinical feasibility of PARP inhibitors in patients harboring pathogenic BRCA1/2 mutations and in two sisters carrying the BRCA2 VUS p.S1946P
ID |
Variant classification |
Gene |
Amino acid change# |
Variant frequency (%) |
Germline (G)/ somatic (S) |
LOH/ biallelic inactivation |
Eligibility for PARPi (USFDA label) |
Eligibility for PARPi (EMA label) |
|---|---|---|---|---|---|---|---|---|
1 |
pathogenic |
BRCA2 |
p.S1722fs |
78 |
G |
Yes |
Yes |
Yes |
2 |
pathogenic |
BRCA2 |
p.S1722fs |
74 |
G |
Yes |
Yes |
Yes |
3 |
pathogenic |
BRCA1 |
p.E730* |
77 |
G |
Yes |
Yes |
Yes |
4ψ |
pathogenic |
BRCA1 |
p.T796I |
53 |
G |
Unknown§ |
Yes |
Yes |
5 |
pathogenic |
BRCA1 |
p.Œ_splice |
78 |
G |
Yes |
Yes |
Yes |
6 |
pathogenic |
BRCA1 |
p.S1286fs |
62 |
G |
Yes |
Yes |
Yes |
7 |
pathogenic |
BRCA2 |
p.S780* |
80 |
G |
Yes |
Yes |
Yes |
8 |
pathogenic |
BRCA2 |
p.E260fs |
75 |
G |
Yes |
Yes |
Yes |
9 |
pathogenic |
BRCA1 |
p.S242fs |
79 |
S |
Yes |
No |
Yes |
10 |
pathogenic |
BRCA1 |
p.F989fs |
59 |
S |
Yes |
No |
Yes |
11 |
pathogenic |
BRCA1 |
p.G1738fs |
68 |
S |
Yes |
No |
Yes |
12 |
pathogenic |
BRCA2 |
p.D1451fs |
43 |
S |
Yes |
No |
Yes |
13 |
VUS |
BRCA2 |
p.S1946P |
75 |
G |
Yes |
Yes¶ |
Yes¶ |
14 |
VUS |
BRCA2 |
p.S1946P |
48 |
G |
No |
Yes¶ |
Yes¶ |
Patients eligible for PARPi (Yes/Unknown/No) |
10/0/4 |
14/0/0 |
||||||
ψPatient 4 harbored an additional somatic pathogenic BRCA2 variant characterized by a low frequency (6%). #HGVSp - the Human Genome Variation Society (HGVS) protein sequence name. The Annotation is based on the BRCA1 transcript ENSG00000012048 (NM_007294) and the BRCA2 transcript ENSG00000139618 (NM_000059). §The SNP analysis did not allow a conclusion about the presence of an LOH. ¶Based on the assumption that the VUS p.S1946P can be classified as pathogenic.
Abbreviations: EMA, European Medicines Agency; LOH, loss of heterozygosity;PARPi, poly (ADP-ribose) polymerase inhibitors; USFDA, United States Food and Drug Administration; VUS, variant of uncertain significance.
Biallelic BRCA1/2 inactivation and its therapeutic implications
We hypothesized that the presence of a biallelic BRCA inactivation (regardless of the germline or somatic origin of the investigated variant) may identify patients who benefit from PARPi. The presence of LOH was tested in 14 patients, of whom 12 had pathogenic BRCA mutations (Table 3) and the remaining two were the sisters harboring the BRCA2 germline variant p.S1946P. One patient carried both a germline and a somatic BRCA mutation (subject 6), but only the germline mutation was tested. A biallelic inactivation of the mutated BRCA gene by LOH was detected in 12 patients (85.7%). Importantly, LOH was identified in all of the four patients who carried somatic pathogenic BRCA mutations. Of the 10 patients with germline BRCA mutations, eight had a LOH, whereas no LOH could be detected in two patients. However, in one case the tumor purity was very low, which is insufficient for LOH detection. According to the US FDA, the use of PARPi in our patients would be limited to cases with BRCA germline mutations (n = 10), whereas all patients were eligible for PARPi under the EMA label.
DISCUSSION
Herein we reported five novel pathogenic BRCA1/2 mutations (Table 1) and six novel VUS with pathogenic potential (Supplementary Table S1) in patients with ovarian cancer. These results expand our knowledge on the occurrence of both germline and somatic BRCA1/2 mutations in Taiwanese patients with different histological subtypes of ovarian cancer. Screening of BRCA mutations in patients with ovarian cancer may have implications for allocating patients to PARPi.
We also confirmed the findings of a recent report regarding the feasibility of using FFPE tumor samples for NGS analysis of alterations in the BRCA1/2 genes [29]. To our knowledge, only one BRCA1 germline stop mutation has been previously identified by single-strand conformation polymorphism in a sample of 68 Taiwanese patients with ovarian cancer [21]. By using NGS, we were able to identify deleterious BRCA1/2 mutations in 12.1% of our patients, with germline mutations being evident in 8.1% of the samples. A similar prevalence of germline mutations has been reported in Chinese patients (6.9%) [33], whereas a higher frequency has been observed in Japanese (12.6%) [28] and Western cohorts (13.2−15.3%) [8, 34, 35]. A Korean study found BRCA1/2 mutations in 13.5% of patients who had a negative family history [36]. The prevalence of pathogenic germline mutations identified in our patients with serous tumors (17.4%) was in accordance with that of Japanese (16.2%) [28].
The combined prevalence of BRCA1/2 germline and somatic mutations in patients with ovarian cancer differs according to the detection method used (Table 4). However, the reported frequency of BRCA1/2 mutations in patients with high-grade serous carcinoma has been shown to vary between 19 and 30% [5, 6, 27, 29], which is in line with the results of our study (26.1%). Similarly, the observed frequency of somatic mutations in our cohort (33%) is in line with the published literature (16−40%) [5, 6, 27, 29, 37]. The BRCA mutation rate in endometrioid carcinoma of the ovary is ≤ 10% [8, 28, 33, 34]. Albeit less investigated, a similarly low frequency has been reported for clear cell carcinomas [25, 26, 28, 34], a feature that may explain their generally poor response to platinum-based chemotherapy [38]. Here, we were unable to identify pathogenic BRCA1/2 mutations in patients with endometrioid or clear cell carcinomas. However, we observed a potentially deleterious BRCA2 germline variant (BRCA2 p.S1946P) in two sisters with ovarian cancer (one with clear cell carcinoma and the other with serous carcinoma). The patients had a positive family history of malignancies (including pancreatic and colorectal cancer) known to be associated with BRCA mutations [39, 40]. We initially reasoned that the malignancies of the two patients could have been caused by the Lynch syndrome [41] or mutations in TP53 or other DNA repair genes [42–44]. However, both sisters did not show any shared pathogenic variant in 29 DNA repair genes (including TP53, MLH1, MSH2, MSH6, PMS2 and genes involved in homologous recombination). Moreover, the change from serine to proline may affect protein conformational changes locally or globally and change protein functions [45]. pS1946P is located in exon 11 where the BRC motif binds to RAD51 and belongs to the ovarian cancer cluster region (OCCR). Mutations in this region have been shown to confer a higher risk for ovarian cancer [46, 47]. Taken together, our data suggest that the VUS BRCA2 p.S1946P may be pathogenic.
Table 4: Prevalence of germline and somatic BRCA1/2 mutations in ovarian cancer
Authors |
Samples |
Somatic plus germline % (n/N) |
Somatic only % (n/N) |
Analytical method |
|---|---|---|---|---|
High-grade serous ovarian carcinomas |
||||
Mafficini, 2016 [29] |
FFPE |
28 (13/47) |
23 (3/13) |
NGS (Ion Torrent) |
McAlpine, 2012 [27] |
Fresh |
30 (31/103) |
16 (5/31) |
Illumina exome sequencing, DHPLC (part), MLPA |
TCGA, 2011 [5] |
Fresh |
22 (70/316) |
27 (19/70) |
NGS |
Hennessy, 2010 [6] |
Fresh |
19 (44/235) |
39 (11/28) |
Agilent high-density tiling array |
Current study |
FFPE |
26 (12/46) |
33 (4/12) |
NGS (Ion Torrent) |
High-grade serous and non-serous ovarian carcinomas |
||||
Hilton, 2002 [37] |
Fresh |
33 (30/92) |
40 (12/30) |
PTT |
Current study |
FFPE |
12 (12/99) |
33 (4/12) |
NGS (Ion Torrent) |
Abbreviations: FFPE, formalin-fixed paraffin-embedded; DHPLC, denaturing high-performance liquid chromatography; MLPA, multiplex ligation-dependent probe amplification; NGS, next-generation sequencing; PTT, protein truncation test; TCGA, The Cancer Genome Atlas Research Network.
We then analyzed the eligibility for PARPi of the 14 patients who carried either pathogenic BRCA1/2 mutations or the BRCA2 VUS p.S1946P. Only 10 patients (71.4%) met the FDA criteria for use of PARPi (owing to the presence of germline mutations), whereas all of them should have been eligible according to the EMA guidelines. Biallelic BRCA1/2 inactivation was also considered, because tumor cells with monoallelic BRCA inactivation are unresponsive to PARPi [20]. In our study, BRCA1/2 inactivation was identified in most – but not all – patients with pathogenic or potentially deleterious BRCA variants. However, an accurate estimation of the proportion of patients carrying monoallelic BRCA1/2 inactivation was unfeasible because of the small sample size.
There were several limitations in our study. First, we did not analyze whether BRCA1/2 genes were inactivated by epigenetic silencing [5]. Second, other genes associated with homologous recombination deficiency in patients with ovarian cancer patients were not analyzed [5, 10]. Consequently, we cannot exclude that the proportion of patients eligible for treatment with PARP inhibitors may have been underestimated. Third, the sample size of this study is small especially in the group of serous carcinoma. However, we provide the mutation rates in the understudied East Asian region, where we showed similar mutation rates to those reported in Caucasians and other ethnic subgroups. Fourth, the family history of each studied case was only retrieved from charts. Of note, none of the germline BRCA1/2 had a family history of breast and ovarian cancer. Careful cancer screening on the family members of any detected pathogenic germline BRCA1/2 mutation should be advised.
In conclusion, BRCA1/2 mutations are common in Taiwanese patients with serous ovarian carcinoma. Several novel BRCA1/2 mutations reported herein warrant further validation for their pathogenic potentials. The identification of BRCA1/2 somatic mutations may have implications for guiding therapeutic decisions in patients with ovarian cancer.
MATERIALS AND METHODS
Patients
The study protocol was approved by the Institutional Review Board of the Chang Gung Memorial Hospital (IRB No. 104-6241B). Ninety-nine patients with ovarian cancer were included. The histological variants included serous (n = 46), endometrioid (n = 24), and clear cell (n = 29) carcinomas. The occurrence of breast and ovarian cancer in first- and second-degree relatives was investigated.
Samples and DNA extraction
We used FFPE samples obtained by surgical removal of the primary tumors. Normal FFPE samples were used as paired controls for specimens carrying genetic variants. Genomic DNA was isolated from two 10 μm-thick FFPE sections using the QIAamp DNA FFPE Tissue Kit (Qiagen, Hilden, Germany). We obtained blood samples from the family members of the two sisters harboring the BRCA2 VUS p.S1946P. Genomic DNA was extracted from blood white cells using the QIAamp DNA Blood Midi Kit (Qiagen). The concentration and integrity of the purified DNA were checked using the Quanti-iT dsDNA HS Assay (Invitrogen, Carlsbad, CA, USA) and a Fragment Analyzer (Advanced Analytical Technologies, Ankeny, IA, USA), respectively.
BRCA1/2 sequencing and data processing
Genomic DNA (40 ng) was amplified using 250 primer pairs (GeneRead DNASeq Targeted panels v2, Qiagen) to target all of the exonic regions as well as the intronic regions within 20-bp of a splicing junction. Amplicons were ligated to a barcode adaptor using the Ion Xpress™ Plus Fragment Library Kit (Life Technologies, Carlsbad, CA, USA). The barcoded library was then enriched by emulsion PCR using OneTouch2 and OneTouch ES instruments (Life Technologies) following the Ion Torrent protocol provided by Life Technologies. The library’s quantity and quality were examined on a fragment analyzer (Advanced Analytical Technologies) and a Qubit fluorometer (Invitrogen). The enriched library was sequenced using the Ion Personal Genome Machine (PGM) with an Ion 318 chip (Life Technologies) following the manufacturer’s instructions. The mean sequencing depth for FFPE tumor samples was >5700×, with a mean uniformity of 91.1%. Variants with a frequency >10% were confirmed by Sanger sequencing of tumor DNA. Germline DNA was analyzed by NGS or Sanger sequencing. Germline DNA from the siblings of the two sisters carrying the BRCA2 VUS p.S1946P was also examined by Sanger sequencing.
Analysis of genetic variants
Raw sequence data were mapped to the human reference genome (hg19) using The Torrent Suite Server (v. 4.2). Variant calling was performed with the Torrent Suite Variant Caller plug-in (v. 4.2) and the Variant Effect Predictor (VEP) was used for annotation. Variants with a read count <25 and a variant frequency <5% were not analyzed further. Common single nucleotide polymorphisms (SNPs) were identified using the dbSNP (release 138), 1000 Genome (phase 1 data), and 5000 Exome data sets. Previously reported BRCA1/2 mutations were identified and classified with the BIC (Breast Cancer Information Core, http://research.nhgri.nih.gov/bic/), ClinVar (http://www.ncbi.nlm.nih.gov/clinvar/), LOVD (Leiden Open Variation Database, http://www.lovd.nl/3.0/home), ARUP (http://arup.utah.edu/database/BRCA/), and BRCA Share (http://www.umd.be/BRCA1/) data sets. A variant classified as “likely benign” or “likely pathogenic” by the ClinVar was considered as VUS. Variants were classified as pathogenic if 1) they were labeled as such in any of the data sets used for the study or 2) they were frameshift or stop mutations. Variants were considered as benign if they were unequivocally classified as such (i.e., without a concurrent classification either as VUS or pathogenic within the same database) in the consulted data sets. All of the previously unidentified variants that were not clearly benign or pathogenic were regarded as VUS. A pathogenic variant was considered as novel if it was absent in the abovementioned data sets as well as in the COSMIC database (version 70, http://cancer.sanger.ac.uk/cosmic). SIFT (http://sift.jcvi.org/), PolyPhen2 (http://genetics.bwh.harvard.edu/pph2/index.shtml), and Grantham (http://asia.ensembl.org/info/genome/variation/predicted_data.html) were used to predict the functional impact of the detected variants (Supplementary Table S2). Variants classified as “probably damaging” in PolyPhen2 were considered as pathogenic, whereas “possibly damaging” variants were classified as VUS. SNP data of 997 healthy subjects of the population-based project in Taiwan were downloaded for comparison (https://taiwanview.twbiobank.org.tw/index).
In line with previous methodology [48], LOH was determined by analyzing the frequency of the patient’s SNPs within the mutated BRCA gene using the ADTEx tool. This method may also detect large genomic rearrangements.
Statistical analysis
Intergroup differences were analyzed using Fisher’s exact tests, χ2 tests, or Student’s t-tests, as appropriate. All calculations were performed using the IBM SPSS software package (version 17.0; IBM, Armonk, NY, USA). P values < 0.05 (two-tailed) were considered statistically significant.
ACKNOWLEDGMENTS
We are grateful to Chiao-Yun Lin, Yen-Jung Lu, and Jung-Erh Yang for their technical assistance.
CONFLICTS OF INTEREST
Shu-Jen Chen, Nina Lapke, and Chien-Hung Chen are employees of ACT Genomics, Co. Ltd.
GRANT SUPPORT
This study was supported in part by the Chang Gung Medical Foundation (grants CMRPG3E0391/2/3 and CMRPG3B0063 to A. Chao, CMRPG3c0941/2/3 to T. H. Wang).
REFERENCES
1. Department of Health, the Executive Yuan. Cancer registration annual report in Taiwan. 2014.
2. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015; 65:87-108.
3. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015; 65:5-29.
4. Modesitt SC, Jazaeri AA. Recurrent epithelial ovarian cancer: pharmacotherapy and novel therapeutics. Expert Opin Pharmacother. 2007; 8:2293-2305.
5. Integrated genomic analyses of ovarian carcinoma. Nature. 2011; 474:609-615.
6. Hennessy BT, Timms KM, Carey MS, Gutin A, Meyer LA, Flake DD, 2nd, Abkevich V, Potter J, Pruss D, Glenn P, Li Y, Li J et al. Somatic mutations in BRCA1 and BRCA2 could expand the number of patients that benefit from poly (ADP ribose) polymerase inhibitors in ovarian cancer. J Clin Oncol. 2010; 28:3570-3576.
7. Yang D, Khan S, Sun Y, Hess K, Shmulevich I, Sood AK, Zhang W. Association of BRCA1 and BRCA2 mutations with survival, chemotherapy sensitivity, and gene mutator phenotype in patients with ovarian cancer. JAMA. 2011; 306:1557-1565.
8. Alsop K, Fereday S, Meldrum C, deFazio A, Emmanuel C, George J, Dobrovic A, Birrer MJ, Webb PM, Stewart C, Friedlander M, Fox S et al. BRCA mutation frequency and patterns of treatment response in BRCA mutation-positive women with ovarian cancer: a report from the Australian Ovarian Cancer Study Group. J Clin Oncol. 2012; 30:2654-2663.
9. Bolton KL, Chenevix-Trench G, Goh C, Sadetzki S, Ramus SJ, Karlan BY, Lambrechts D, Despierre E, Barrowdale D, McGuffog L, Healey S, Easton DF et al. Association between BRCA1 and BRCA2 mutations and survival in women with invasive epithelial ovarian cancer. JAMA. 2012; 307:382-390.
10. Pennington KP, Walsh T, Harrell MI, Lee MK, Pennil CC, Rendi MH, Thornton A, Norquist BM, Casadei S, Nord AS, Agnew KJ, Pritchard CC et al. Germline and somatic mutations in homologous recombination genes predict platinum response and survival in ovarian, fallopian tube, and peritoneal carcinomas. Clin Cancer Res. 2014; 20:764-775.
11. Nielsen FC, van Overeem Hansen T, Sorensen CS. Hereditary breast and ovarian cancer: new genes in confined pathways. Nat Rev Cancer. 2016.
12. Dann RB, DeLoia JA, Timms KM, Zorn KK, Potter J, Flake DD, 2nd, Lanchbury JS, Krivak TC. BRCA1/2 mutations and expression: response to platinum chemotherapy in patients with advanced stage epithelial ovarian cancer. Gynecol Oncol. 2012; 125:677-682.
13. Ledermann J, Harter P, Gourley C, Friedlander M, Vergote I, Rustin G, Scott CL, Meier W, Shapira-Frommer R, Safra T, Matei D, Fielding A et al. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: a preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol. 2014; 15:852-861.
14. Kaufman B, Shapira-Frommer R, Schmutzler RK, Audeh MW, Friedlander M, Balmana J, Mitchell G, Fried G, Stemmer SM, Hubert A, Rosengarten O, Steiner M et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J Clin Oncol. 2015; 33:244-250.
15. Domchek SM, Aghajanian C, Shapira-Frommer R, Schmutzler RK, Audeh MW, Friedlander M, Balmana J, Mitchell G, Fried G, Stemmer SM, Hubert A, Rosengarten O et al. Efficacy and safety of olaparib monotherapy in germline BRCA1/2 mutation carriers with advanced ovarian cancer and three or more lines of prior therapy. Gynecol Oncol. 2016; 140:199-203.
16. Ledermann J, Harter P, Gourley C, Friedlander M, Vergote I, Rustin G, Scott C, Meier W, Shapira-Frommer R, Safra T, Matei D, Macpherson E et al. Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer. N Engl J Med. 2012; 366:1382-1392.
17. Maxwell KN, Sloover DD, Wubbenhorst B, Wenz B, Lunceford N, Emery L, D'Andrea K, Daber RD, Feldman MD, Domchek SM, Nathanson KL. Abstract 2990: Evidence for diverse mechanisms of tumorigenesis in breast and ovarian tumors of BRCA1/2 carriers. Can Res. 2015; 75.
18. Sedic M, Skibinski A, Brown N, Gallardo M, Mulligan P, Martinez P, Keller PJ, Glover E, Richardson AL, Cowan J, Toland AE, Ravichandran K et al. Haploinsufficiency for BRCA1 leads to cell-type-specific genomic instability and premature senescence. Nat Commun. 2015; 6:7505.
19. Cousineau I, Belmaaza A. BRCA1 haploinsufficiency, but not heterozygosity for a BRCA1-truncating mutation, deregulates homologous recombination. Cell Cycle. 2007; 6:962-971.
20. Konishi H, Mohseni M, Tamaki A, Garay JP, Croessmann S, Karnan S, Ota A, Wong HY, Konishi Y, Karakas B, Tahir K, Abukhdeir AM et al. Mutation of a single allele of the cancer susceptibility gene BRCA1 leads to genomic instability in human breast epithelial cells. Proc Natl Acad Sci U S A. 2011; 108:17773-17778.
21. Wang PH, Shyong WY, Li YF, Lee HH, Tsai WY, Chao HT, Wu CY, Tsai YC, Yuan CC. BRCA1 mutations in Taiwanese with epithelial ovarian carcinoma and sporadic primary serous peritoneal carcinoma. Jpn J Clin Oncol. 2000; 30:343-348.
22. Chao A, Lai CH, Tsai CL, Hsueh S, Hsueh C, Lin CY, Chou HH, Lin YJ, Chen HW, Chang TC, Wang TH. Tumor stress-induced phosphoprotein1 (STIP1) as a prognostic biomarker in ovarian cancer. PLoS One. 2013; 8:e57084.
23. del Carmen MG, Birrer M, Schorge JO. Clear cell carcinoma of the ovary: a review of the literature. Gynecol Oncol. 2012; 126:481-490.
24. Chiang YC, Chen CA, Chiang CJ, Hsu TH, Lin MC, You SL, Cheng WF, Lai MS. Trends in incidence and survival outcome of epithelial ovarian cancer: 30-year national population-based registry in Taiwan. J Gynecol Oncol. 2013; 24:342-351.
25. Lakhani SR, Manek S, Penault-Llorca F, Flanagan A, Arnout L, Merrett S, McGuffog L, Steele D, Devilee P, Klijn JG, Meijers-Heijboer H, Radice P et al. Pathology of ovarian cancers in BRCA1 and BRCA2 carriers. Clin Cancer Res. 2004; 10:2473-2481.
26. Goodheart MJ, Rose SL, Hattermann-Zogg M, Smith BJ, De Young BR, Buller RE. BRCA2 alteration is important in clear cell carcinoma of the ovary. Clin Genet. 2009; 76:161-167.
27. McAlpine JN, Porter H, Kobel M, Nelson BH, Prentice LM, Kalloger SE, Senz J, Milne K, Ding J, Shah SP, Huntsman DG, Gilks CB. BRCA1 and BRCA2 mutations correlate with TP53 abnormalities and presence of immune cell infiltrates in ovarian high-grade serous carcinoma. Mod Pathol. 2012; 25:740-750.
28. Sakamoto I, Hirotsu Y, Nakagomi H, Ouchi H, Ikegami A, Teramoto K, Amemiya K, Mochizuki H, Omata M. BRCA1 and BRCA2 mutations in Japanese patients with ovarian, fallopian tube, and primary peritoneal cancer. Cancer. 2016; 122:84-90.
29. Mafficini A, Simbolo M, Parisi A, Rusev B, Luchini C, Cataldo I, Piazzola E, Sperandio N, Turri G, Franchi M, Tortora G, Bovo C et al. BRCA somatic and germline mutation detection in paraffin embedded ovarian cancers by next-generation sequencing. Oncotarget. 2016; 7:1076-1083. doi: 10.18632/oncotarget.6834.
30. Frank TS, Deffenbaugh AM, Reid JE, Hulick M, Ward BE, Lingenfelter B, Gumpper KL, Scholl T, Tavtigian SV, Pruss DR, Critchfield GC. Clinical characteristics of individuals with germline mutations in BRCA1 and BRCA2: analysis of 10,000 individuals. J Clin Oncol. 2002; 20:1480-1490.
31. Kim YC, Zhao L, Zhang H, Huang Y, Cui J, Xiao F, Downs B, Wang SM. Prevalence and spectrum of BRCA germline variants in mainland Chinese familial breast and ovarian cancer patients. Oncotarget. 2016; 7:9600-9612. doi: 10.18632/oncotarget.7144.
32. Kurman RJ, Carcangiu, M.L., Herrington, C.S., Young, R.H., 4th edn: International Agency for Research on Cancer; 2014.
33. Hasmad HN, Lai KN, Wen WX, Park DJ, Nguyen-Dumont T, Kang PC, Thirthagiri E, Ma'som M, Lim BK, Southey M, Woo YL, Teo SH. Evaluation of germline BRCA1 and BRCA2 mutations in a multi-ethnic Asian cohort of ovarian cancer patients. Gynecol Oncol. 2016; 141:318-322.
34. Pal T, Permuth-Wey J, Betts JA, Krischer JP, Fiorica J, Arango H, LaPolla J, Hoffman M, Martino MA, Wakeley K, Wilbanks G, Nicosia S et al. BRCA1 and BRCA2 mutations account for a large proportion of ovarian carcinoma cases. Cancer. 2005; 104:2807-2816.
35. Risch HA, McLaughlin JR, Cole DE, Rosen B, Bradley L, Fan I, Tang J, Li S, Zhang S, Shaw PA, Narod SA. Population BRCA1 and BRCA2 mutation frequencies and cancer penetrances: a kin-cohort study in Ontario, Canada. J Natl Cancer Inst. 2006; 98:1694-1706.
36. Choi MC, Heo JH, Jang JH, Jung SG, Park H, Joo WD, Lee C, Lee JH, Lee JM, Hwang YY, Kim SJ. Germline Mutations of BRCA1 and BRCA2 in Korean Ovarian Cancer Patients: Finding Founder Mutations. Int J Gynecol Cancer. 2015; 25:1386-1391.
37. Hilton JL, Geisler JP, Rathe JA, Hattermann-Zogg MA, DeYoung B, Buller RE. Inactivation of BRCA1 and BRCA2 in ovarian cancer. J Natl Cancer Inst. 2002; 94:1396-1406.
38. Sugiyama T, Kamura T, Kigawa J, Terakawa N, Kikuchi Y, Kita T, Suzuki M, Sato I, Taguchi K. Clinical characteristics of clear cell carcinoma of the ovary: a distinct histologic type with poor prognosis and resistance to platinum-based chemotherapy. Cancer. 2000; 88:2584-2589.
39. Couch FJ, Johnson MR, Rabe KG, Brune K, de Andrade M, Goggins M, Rothenmund H, Gallinger S, Klein A, Petersen GM, Hruban RH. The prevalence of BRCA2 mutations in familial pancreatic cancer. Cancer Epidemiol Biomarkers Prev. 2007; 16:342-346.
40. Phelan CM, Iqbal J, Lynch HT, Lubinski J, Gronwald J, Moller P, Ghadirian P, Foulkes WD, Armel S, Eisen A, Neuhausen SL, Senter L et al. Incidence of colorectal cancer in BRCA1 and BRCA2 mutation carriers: results from a follow-up study. Br J Cancer. 2014; 110:530-534.
41. Hampel H, Frankel WL, Martin E, Arnold M, Khanduja K, Kuebler P, Nakagawa H, Sotamaa K, Prior TW, Westman J, Panescu J, Fix D et al. Screening for the Lynch syndrome (hereditary nonpolyposis colorectal cancer). N Engl J Med. 2005; 352:1851-1860.
42. Ruijs MW, Verhoef S, Rookus MA, Pruntel R, van der Hout AH, Hogervorst FB, Kluijt I, Sijmons RH, Aalfs CM, Wagner A, Ausems MG, Hoogerbrugge N et al. TP53 germline mutation testing in 180 families suspected of Li-Fraumeni syndrome: mutation detection rate and relative frequency of cancers in different familial phenotypes. J Med Genet. 2010; 47:421-428.
43. Hirasawa A, Masuda K, Akahane T, Ueki A, Yokota M, Tsuruta T, Nomura H, Kataoka F, Tominaga E, Banno K, Makita K, Susumu N et al. Family history and BRCA1/BRCA2 status among Japanese ovarian cancer patients and occult cancer in a BRCA1 mutant case. Jpn J Clin Oncol. 2014; 44:49-56.
44. Norquist BM, Harrell MI, Brady MF, Walsh T, Lee MK, Gulsuner S, Bernards SS, Casadei S, Yi Q, Burger RA, Chan JK, Davidson SA et al. Inherited Mutations in Women With Ovarian Carcinoma. JAMA Oncol. 2015:1-9.
45. Williamson MP. The structure and function of proline-rich regions in proteins. Biochem J. 1994; 297:249-260.
46. Rebbeck TR, Mitra N, Wan F, Sinilnikova OM, Healey S, McGuffog L, Mazoyer S, Chenevix-Trench G, Easton DF, Antoniou AC, Nathanson KL, Consortium C et al. Association of type and location of BRCA1 and BRCA2 mutations with risk of breast and ovarian cancer. JAMA. 2015; 313:1347-1361.
47. Narod SA. Modifiers of risk of hereditary breast and ovarian cancer. Nat Rev Cancer. 2002; 2:113-123.
48. Amarasinghe KC, Li J, Hunter SM, Ryland GL, Cowin PA, Campbell IG, Halgamuge SK. Inferring copy number and genotype in tumour exome data. BMC Genomics. 2014; 15:732.