
INTRODUCTION
The breakthrough of the anticancer properties of cisplatin cis-[Pt(NH3)2Cl2] around 1965, promoted very much attention in the area of metal-based anticancer agents [1]. The anticancer effects observed for cisplatin suggested that platinum and non-platinum metal-based compounds might be as valuable as organic anticancer drugs [2, 3]. Cisplatin, and a few related platinum complexes, such as carboplatin and oxaliplatin are along with the most commonly used anticancer agents [4]. The extensive clinical success of platinum compounds has improved the synthesis of other platinum and non-platinum metallodrugs that might demonstrate different cytotoxic properties and characterized by a different prototype of anticancer specificities, active against cisplatin-resistant tumor cells and with an encouraging toxicological profile. Thus, in the span of three to four decades, a variety of metal compounds were investigated as potential anticancer agents based on several non-platinum metals such as ruthenium, palladium, titanium, gold and copper [2–10].
It is known that Au(III) is iso-electronic with d8 system and iso-structural with Pt(II). Due to the structural similarity, the square planar gold(III) complexes have been qualified as appropriate candidates for the potential anti-cancer activity evaluation [3, 11–14].
Since the beginning of 21th century, a series of new gold(III) complexes turned out to be stable under physiological conditions and also exhibited appreciable in vitro and in vivo anticancer activity [12, 13, 15–18]. Parish et al. [19] described the synthesis of stable gold(III) complex using damp (2-(Dimethylamino)methyl)phenyl) as uni-negative bidentate ligand as an analogy in the structural features of cisplatin as neutral coordination complex in the mid of 1990s. The acceptable solution stability of these gold(III) compounds permitted to put into practice extensive both in vitro and in in vivo pharmacological testing. Therefore, several other series of cytotoxic gold(III) compounds were synthesized in other laboratories around the world, including the gold(III) dithiocarbamates [20] and the gold(III) porphyrins [21, 22].
Multidentate ligands such as polyamines, cyclam, terpyridine and phenanthroline were preferentially used in order to augment the stability of the gold(III) center and target gold(III) complexes with cationic coordination spheres [Au(en2)]3+, [Au(dien)Cl]2+, [Au(cyclam)]3+, [Au(phen)Cl2]+ and [Au(terpy)Cl]2+ were characterized. The investigation of their solution behavior was carried out through a variety of physical and chemical methods and demonstrated their stability [23, 24].
There are many examples of gold(III) complexes linked to dithiocarbamate, polyamine, polypyridine and their derivative ligands [2, 3]. In particular, dithiocarbamate ligands were combined with gold(III) complexes to inhibit the interaction of the metal center with thiol-containing renal enzymes and avoid cisplatin-induced nephrotoxicity [16].
In this connection, the idea of coupling the well-known anticancer properties of some metal ions with a potential chemo-protective function of dithiocarbamates was turned out to be an effective strategy. In fact, some gold(III) dithiocarbamate compounds were demonstrated to be cytotoxic, to inhibit both the proteasome and the thioredoxin reductase activity in human breast and prostate cancer cell cultures as well as in tumor xenografts [2, 3, 15, 17, 18, 25, 26].
Some other gold(III) complexes of bipyridine were synthesized. Akhmadullina et al. described the synthesis of [(bipy)AuCl2]BF4 and [(phen)AuCl2]BF4 [27]. Casini et al. prepared and characterized square planar gold(III) complexes that contain functionalized bipyridine ligands of general formula [Au(N^N)Cl2][PF6] [where N^N = 2,2′-bipyridine, 4,4′-dimethyl-2,2′-bipyridine, 4,4′-dimethoxy-2,2′-bipyridine and 4,4′-diamino-2,2′-bipyridine] [28].
Ogawa et al. [29] recently elaborated the introduction of a Au(III) ion into a mesogenic core, [M(Bdt)(Cnbpy)]+ (Bdt = 1,2-benzenedithiolato and Cnbpy = 4,4′-di-alkyl-2,2′-bipyridine (n = 13 (4,4′-di-tridecyl-2,2′-bipyridine (C13bpy)) and 8,10 (4,4′-di-(3-octyltridecyl)-2,2′-bipyridine (C8,10bpy), leading to the formation of ionic molecular assemblies in crystalline and mesophases. Successive syntheses of precursor complexes, [AuCl2(Cnbpy)]PF6 (n = 13 (1) and 8,10 (2)), followed by the target complexes, [Au(Bdt)(Cnbpy)]PF6 (n = 13 (3) and 8,10 (4)), were achieved [29].
In order to improve the antitumor properties and reduce toxic side-effects of the previously reported gold(III) analogues, we designed new gold(III) complexes [Au(BPY)(S2CNR2)]Cl2 (BPY = 2,2′-bipyridine, 5,5′-dimethyl-2,2′-bipyridine, or 6,6′-dimethyl-2,2′-bipyridine and R = Methyl, Ethyl, or benzyl in S2CNR2 is Dialkyl/aryldithiocarbammate) with nitrogen and sulfur donor ligands. Here, we report synthesis, chemical characterization, and in vitro anticancer activity of these new gold(III) complexes.
RESULTS
Characterization by FT-IR
Gold(III) mixed ligands complexes 1–9 were identified via the presence of the stretching bands around 3030 and 2925 cm-1 for ν(C–H) the aromatic (phenyl and 2,2′-dipyridyl) and ν(C–H) saturated aliphatic methyl and ethyl groups of coordinated ligands respectively. All gold(III) compounds exhibited characteristic absorbance peaks primarily for ν(C–N) and ν(C–S). The infrared region 1480–1550 cm-1 is mostly related with the R2N–CSS ‘thioureide’ band in the IR spectra of dithiocarbamate compounds which defines the carbon-nitrogen bond order between a single bond at 1250–1350 cm-1 and a double bond at 1640–1690 cm-1 [30]. The thioureide band, ν(C–N) was detected at 1470–1490 cm-1 in compounds 1–9 respectively. These frequency absorption bands lie in between those associated with single C–N and double C = N bonds, hence the partial double bond character of ‘thioureide’ bond was confirmed for all gold(III) compounds [31]. The presence of strong absorption band in the range of 1470–1550 in FTIR spectra clearly indicates the formation dithiocarbamato gold(III) compounds [32, 33]. Similarly, C = S stretching with medium intensity around 1070 and 970 cm-1 for compounds 1–9 is an additional evidence of the formation of mixed ligands compounds. These absorption bands are comparable to free sodium salt of diethyldithiocarbamate ligands [31].
Characterization by NMR
The 1H and 13C NMR chemical shifts of all nine compounds are given in synthesis part of experimental section and small downfield and upfield shifts for proton(s) of the coordinated dimethyl dithiocarbamate, diethyl dithiocarbamate and dibenzyldithiocarbamate have been seen in gold(III) compounds 1–9 in comparison to free dialkyl/diaryldithiocarbamate ligands [24, 34, 35]. The 13C NMR spectra of compounds 4, 7 and 1 showed seven and six resonances respectively; which confirmed the coordination of dimethyldithiocarmato and bipyridyl ligands with Au(III) ion (given in synthesis part of experimental section). The 13C NMR spectra of compounds 5, 8 and 2 showed one additional chemical shift comparative to compounds 4, 7 and 1 spectra as expected. Whereas, multiple chemical shifts are observed in the 13C NMR spectra of compounds 3, 6 and 9 due to presence of dibenzyl and pyridyl functional groups. There is an up-field chemical shift of NC = S carbon of coordinated dialkyl/diaryldithiocarbamate ligands with respect to free dialkyl/diaryldithiocarbamate ligands for all gold(III) compounds. The 13C chemical shifts of NC = S carbon of bonded dimethyl thiocarbamate, diethyl thiocarbamate and dibenzyl thiocarbamate are observed in the range 186–200 ppm in our synthesized compounds 1–9 [24, 34, 35].
Mass spectrometry characterization
Positive-ion mode ESI mass spectra for electrosprayed aqueous-methanol solutions of compounds 1, 2 and 3 are presented in Supplementary Figure S1. The mass spectra profile of compounds 1, 2 and 3 present a common peak ion at 157 m/z corresponding to the protoned 2,2′-bipyridine moiety ligand (a) or its sodium adduct (b) at 179 m/z as reported for compound 3. The ESI-MS spectra of compound 1 presents an intense peak ion at 437 m/z assigned to a positive ion where Au(III) is coordinated by two dimethyl dithiocarbamate moieties. Compound 2 presents instead an intense peak at 493 m/z corresponding to the coordination of two diethyl dithiocarbamate moieties and analogously the ESI-MS scan for compound 3 presents a signal at 741 m/z associated to the coordination of two dibenzyldithiocarbamate moieties. Compounds 4, 5 and 6 in solution present the same ESI-MS behavior of compounds 1, 2 and 3 with the differences that in all ESI-MS spectra scan of compounds 4, 5 and 6 present a common positive Ion at 185 m/z that can be assigned to the free protoned dimethyl-2,2-bipyridine ligand (g) (Supplementary Figure S2). The structure of the dithiocarbamate derivatives c, d and f is shown in Supplementary Figure S3.
Stability determination and interactions with lysozyme
Electronic spectra were recorded on each complex freshly prepared in DMSO solution at room temperature. Then, their electronic spectra were monitored over 24 hours at 37oC. The resulting UV-Vis absorption spectra of compound 1, 2 and 3 are shown in Supplementary Figure S4A–S4C. The UV spectra of the compounds confirm the stability under physiological temperature.
The interaction between compounds 1–3 and the lysozyme in solution have been probed by studying the change in their electrochemical profiles in presence of different concentration of protein (Supplementary Figures S5, S6 and S7). Both CV and SWV demonstrated that in the presence of lysozyme all compounds 1–3 showed a substantial decrease in the peak current as well as a clear peak potential shift (Supplementary Figures S5, S6 and S7). The significant change of their redox state due to their interaction with lysozyme suggests a significative chemical interaction of such gold compounds and proteins [36]. This chemical aspect deserves high attention because it could be involved in the mechanism of action of these innovative molecules as anticancer drugs.
Cytotoxic effects
Gold(III)complexes (compounds 1–9) (Figure 1) were tested for in vitro cytotoxicity toward human Hodgkin lymphoma (L-540), androgen-resistant prostate cancer (PC3), breast cancer (MCF-7) and ovarian adenocarcinoma cisplatin-sensitive (A2780) and -resistant (A2780cis) cell lines. For comparison purposes, cisplatin activity was also evaluated.
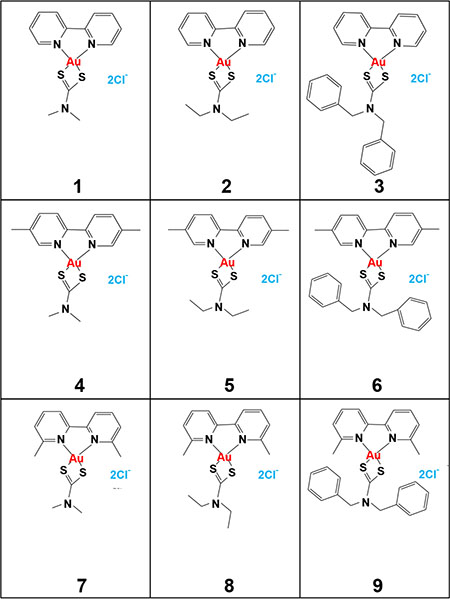
Figure 1: Chemical structure of the nine new synthesized gold(III) complexes (compound 1–9).
In L-540 cells gold(III) complexes showed an IC50 higher or similar to that of cisplatin (Table 1). Compounds 3, 6 and 9, all characterized by the addition of two benzoic groups (Figure 1, right panel), were less active than cisplatin in all cell lines tested showing higher or similar IC50 values (Tables 1 and 2). The other gold(III) complexes (compounds 1, 2, 4, 5, 7, 8) (Figure 1) were generally more active than the reference drug cisplatin with an IC50 ranging from 0.27 to 0.47 μM in MCF-7 cells, from 0.42 to 1.6 μM in PC3 cells (Table 1), from 0.18 to 0.73 μM in A2780 cells (Table 2) and from 0.17 to 0.45 μM in its cisplatin-resistant clone A2780cis (Table 2). Compound 1 showed the lowest IC50 values in the entire panel of the investigated tumor cell lines (Tables 1 and 2) and its representative dose-response curves are shown in Figure 2A. It was proved about 60-fold and 80-fold more active than cisplatin in inhibiting cell proliferation of cisplatin-resistant ovarian adenocarcinoma A2780cis cells (Table 2) and breast cancer MCF-7 cells (Table 1), respectively. With reference to Table 1, the IC50 values for complex 1, 2, 4, 5, 7 and 8 are comparable. Using three representative gold(III) complexes (compounds 1, 2 and 3) with different cytotoxic activity, we demonstrated the intracellular gold uptake by L-540 cells. Consistently with the cytotoxic effects, the gold uptake of both compound 1 and 2 was greater than that of compound 3 (Supplementary Figure S8A).
Table 1: Growth inhibition by gold(III) compounds in MCF-7, PC3 and L-540 cells
Compound |
IC50 (μM) |
||
|---|---|---|---|
MCF-7 |
PC3 |
L-540 |
|
Cisplatin |
22.2 ± 0.20 |
3.30 ± 0.30 |
2.50 ± 0.10 |
1 |
0.27 ± 0.02 |
0.42 ± 0.04 |
2.68 ± 0.24 |
2 |
0.27 ± 0.02 |
0.70 ± 0.06 |
2.80 ± 0.25 |
3 |
23.0 ± 2.07 |
11.2 ± 1.00 |
12.0 ± 1.08 |
4 |
0.28 ± 0.02 |
0.59 ± 0.05 |
2.70 ± 0.22 |
5 |
0.29 ± 0.03 |
0.86 ± 0.08 |
2.90 ± 0.26 |
6 |
18.0 ± 1.62 |
11.6 ± 1.04 |
6.50 ± 0.06 |
7 |
0.35 ± 0.03 |
0.77 ± 0.07 |
3.60 ± 0.32 |
8 |
0.47 ± 0.04 |
1.60 ± 0.14 |
6.20 ± 0.56 |
9 |
31.0 ± 2.79 |
11.5 ± 1.03 |
35.75 ± 3.22 |
Cell lines were exposed to increasing concentrations of gold(III) compounds and Cisplatin included as reference drug. After 72 h exposure the number of viable cells was evaluated by MTT and MTS assay. Results represent the mean ± SEM of three replicate wells from three independent experiments. IC50 values were calculated using the Calcusyn software.
Table 2: Growth inhibition by gold(III) compounds in ovarian cancer cell lines A2780 and cisplatin-resistant A2780cis
Compound |
IC50 (μM) |
Fold resistance |
|
|---|---|---|---|
A2780 |
A2780cis |
||
Cisplatin |
1.5 ± 0.1 |
10.4 ± 0.9 |
6.93 |
1 |
0.18 ± 0.02 |
0.17 ± 0.01 |
0.94 |
2 |
0.24 ± 0.02 |
0.18 ± 0.01 |
0.58 |
3 |
4.66 ± 0.13 |
14.3 ± 1.12 |
3.07 |
4 |
0.28 ± 0.03 |
0.17 ± 0.01 |
0.94 |
5 |
0.59 ± 0.05 |
0.23 ± 0.02 |
0.39 |
6 |
6.35 ± 0.06 |
15.7 ± 1.41 |
2.47 |
7 |
0.27 ± 0.02 |
0.23 ± 0.02 |
0.85 |
8 |
0.73 ± 0.06 |
0.45±0.03 |
0.62 |
9 |
6.7 ± 0.6 |
16.0 ± 1.44 |
2.39 |
Cell lines were exposed to increasing concentrations of gold(III) compounds and Cisplatin included as reference drug. After 72 h exposure viable cells were evaluated by MTT assay. Results represent the mean ± SEM of three replicate wells from three independent experiments. IC50 values were calculated using the Calcusyn software and compared with cisplatin. Compounds in which the ratio A2780cis IC50/A2780 IC50 resulted < 1 are reported as bold numbers.
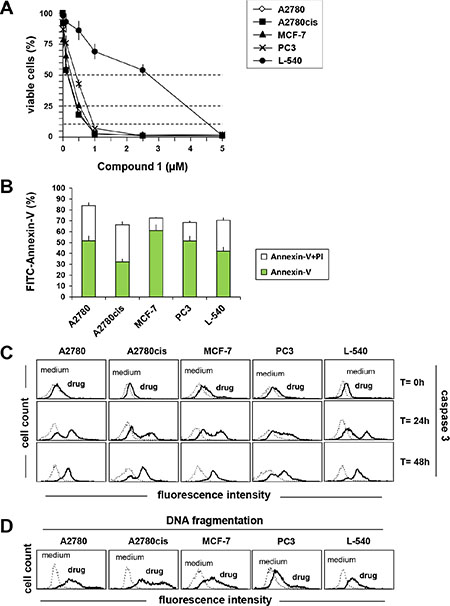
Figure 2: Compound 1 induces apoptosis in A2780, A2780cis, MCF-7, PC3, L-540 cells. (A) Cells were exposed to increasing concentrations of compound 1. After 72 h the viable cell number was evaluated by MTT or MTS (B) FACS analysis of cells after 72 h incubation with compound 1 (IC75) and double stained with Annexin V/FITC and PI. (C) analysis of caspase 3 activation using FLICA reagent by flow cytometry after treating cells with compound 1 (IC75) for 0, 24 and 48 h. (D) DNA fragmentation (Apo-Direct) was assessed by flow cytometry after treatment for 72 h with compound 1 (IC75). Dotted lines indicate background fluorescence of cells. The x- and y-axes indicate the logarithms of the relative fluorescence intensity and relative cell number, respectively. FACS histograms are representative of one of three different experiments. Results represent the mean ± SEM of three independent experiments.
Induction of apoptosis
There are several mechanisms involved in the anticancer activity of Gold(I) and (III) complexes including the induction of apoptosis [15, 17, 37]. Therefore, we evaluated Annexin-V/PI double staining, caspase 3 activation and DNA fragmentation (Figure 2) in tumor cells treated with the most active gold(III) complex (compound 1). As shown in Figure 2B, compound 1 induced apoptosis in the cell lines tested. Treatment with compound 1 resulted in a substantial phosphatidylserine exposure (Annexin-V) together with an high percentage of cells permeable to PI staining, thus supporting apoptosis as a major mechanism of cell death (Figure 2B). Consistently, treatment with compound 1 (IC75) induced a time-dependent activation of caspase 3 (Figure 2C). Apoptosis induction was confirmed by DNA fragmentation (Figure 2D) with Apo-Direct analysis. Compound 1 was active at the same concentration in both cisplatin-sensitive (A2780) and -resistant (A2780cis) [38] ovarian cancer cells, and resulted more active in tumor cells than in normal human Mesenchymal stromal cells (MSCs) (Supplementary Figure S8B) (IC50 = 0.96 μM). The final DMSO concentration was un-effective (data not shown).
Inhibition of mitochondrial functions and Reactive oxygen species (ROS) accumulation
Treatment with Compound 1 promoted mitochondrial membrane permeabilization (Figure 3A) and cytochrome-c (Cyt-c) release (Figure 3B) from the mitochondria and consistently induced activation of caspase 9 (Figure 3C), suggesting that the anticancer activity involves the mitochondrial intrinsic apoptotic pathway. Compound 1 (IC75) increased mitochondrial ROS levels, and especially in breast cancer MCF-7 cells (Figure 4A). In order to establish a possible relationship between ROS overproduction and decreased viability, tumor cells were incubated with the ROS scavenger NAC (Figure 4A and 4B). NAC blocked ROS production (Figure 4A) and almost completely neutralized the anti-proliferative effect of compound 1 (Figure 4B), suggesting that the cytotoxic/pro-apoptotic activity is due to ROS generation. Cell cycle progression was only slightly affected by compound 1 (data not shown).
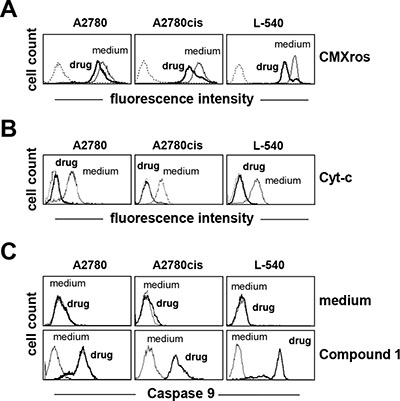
Figure 3: Compound 1 induces the mitochondrial intrinsic apoptotic pathway. (A) mitochondrial membrane permeabilization (CMXRos), and (B) Cytochrome-c (Cyt-c) release were assessed by flow cytometry after treatment for 24 h with compound 1(IC75). (C) analysis of caspase 9 activation using FLICA reagent by flow cytometry after treating cells with compound 1 (IC75) for 24 h. Dotted lines indicate background fluorescence of cells. The x- and y-axes indicate the logarithms of the relative fluorescence intensity and relative cell number, respectively. FACS histograms are representative of one of three different experiment
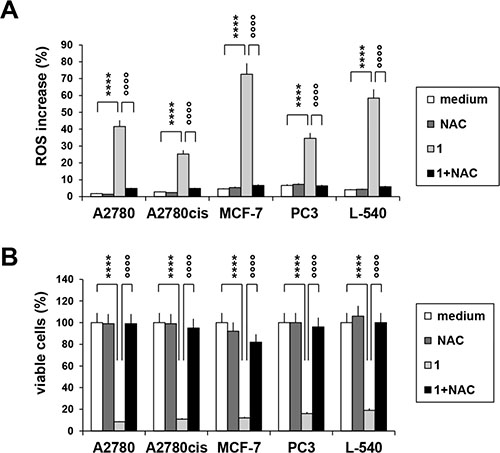
Figure 4: ROS accumulation after treatment with compound 1. (A) cells were treated for 72 h with compound 1 (IC75) in the presence or in the absence of the antioxidant NAC (5 mM), then stained with MitoSox reagent and analyzed by flow cytometry. Histograms represent the percentage of ROS positive cells. (B) percentage of cell viability upon treatment with compound 1 (IC75) and NAC (5 mM) for 72 h. Values represent the mean ± SEM of three different experiments. *p < 0.05; **p < 0.01; ****p < 0.0001, compound 1 vs. control; °°°°p < 0.0001, compound 1 vs. compound 1 + NAC.
p53 status and Gold(III) complexes cytotoxicity
As response to chemotherapy can be influenced by the status of the tumor suppressor TP53 gene, four p53 wild-type (WT-p53), four p53 mutant (Mut-p53) and one p53 deleted (Null-p53) were utilized to determine whether the new gold(III) complexes have p53 dependence.
Compounds (3, 4, 6, 7 and 9) showed an IC50 ranging between 0.16–4.1 μM. Representative dose-response curves in A549 cells are shown in Figure 5A, which indicate that compounds 4 and 7 have greater potencies than compounds 3, 6 and 9 also in lung cancer cells. Dose-response curves were used to obtain IC50 values in the entire NSCLC panel, and are shown in Figure 5B–5F. These IC50 values indeed demonstrate that compound 4 (IC50, 0.12–0.31 μM) and 7 (IC50, 0.14–0.29 μM) have greater potencies (at least 10-fold) than compound 3 (2.4–4.1 μM), 6 (1.5–2.8 μM) and 9 (2.1–3.9 μM) in all NSCLC cell lines. It is clear that structural differences regulate the potency of the molecules. Moreover, these results indicate that the IC50 values in WT- p53 models were similar to those in Mut- and Null-p53 cell lines. To confirm that sensitivity to gold(III) complexes was not dependent on p53 status, we utilized WT-p53 A2780 ovarian cancer cells in which p53 was knocked out using the CRISPR/Cas9 technology (CRISPR p53 KO). The corresponding control cells were transfected with a control plasmid and retained WT-p53 status. Results from the MTT assay revealed there were no significant differences in IC50 for representative compounds 3 (CRISPR Control, 0.87 ± 0.11 μM vs. CRISPR p53 KO, 1.13 ± 0.39 μM ) and 4 (CRISPR Control, 0.087 ± 0.003 μM vs. CRISPR p53 KO, 0.083 ± 0.003 μM) (Figure 6).
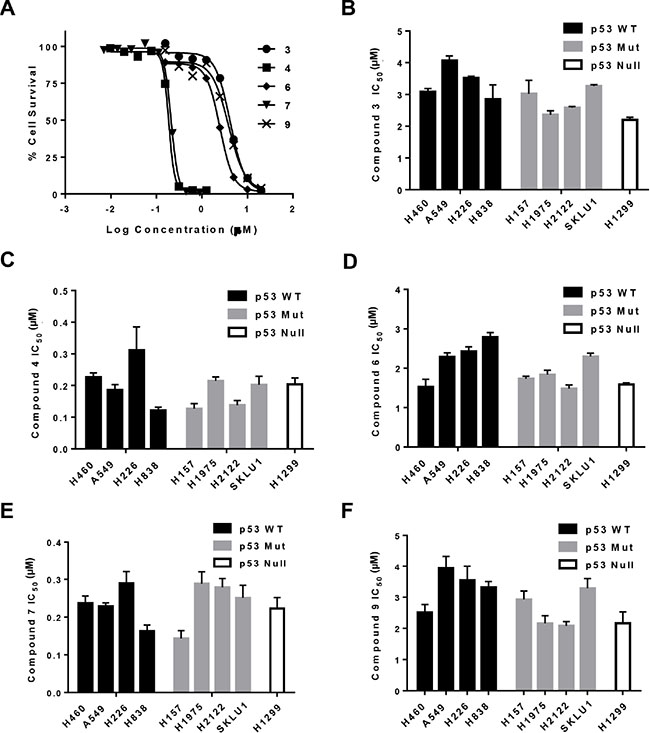
Figure 5: Cytotoxicity of gold complexes in a panel of NSCLC cell lines. Determination of IC50 for compounds 3, 4, 6, 7 and 9 was by MTT assay. (A) Representative sigmoidal dose-response curves for A549 cells obtained 3 days after initiating exposure to the gold complex. (B–F), IC50 values to the indicated gold compounds for NSCLC cells grouped by p53 status. Data are presented as mean +/− SEM of three independent experiments.
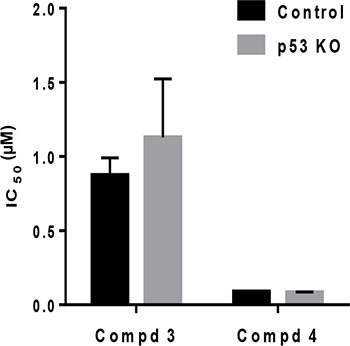
Figure 6: Cytotoxicity of gold complexes is independent of p53. The IC50 values for compound 3 and 4 were obtained in an A2780 clone following transfection with either CRISPR control (Control) or CRISPR p53-sgRNA plasmid (p53 KO). Data are presented as mean +/− SEM of three independent experiments.
Thus, the potential of the gold complexes to circumvent this resistance was investigated. For this purpose the IC50 and fold resistance (A2780cis/A2780) values for selected gold complexes were determined using the sensitive A2780 cancer cell line and the corresponding cisplatin-resistant A2780cis and 2780CP-16 [39] cell lines. While the A2780cis (resistant) cells showed an IC50 for cisplatin about 7-fold higher than A2780 (sensitive) cells, the IC50 of the most active compounds (1, 2, 4, 5, 7, 8) resulted similar or lower in A2780cis respect to A2780 (fold resistance or resistant factor less than 1) (Table 2). Similar results were obtained also in the 20-fold cisplatin-resistant 2780CP cells (data not shown), indicating that gold complexes under investigation in this study have the ability to overcome cisplatin-resistance to a substantial extent.
DISCUSSION
The design of more active and less toxic antitumor agents is a major goal to develop new-targeted therapeutic strategies. Dithiocarbammate gold(III) complexes have recently gained increasing attention due to their strong cytotoxic effects associated with low nephrotoxicity and their capability to overcome cisplatin-resistance [15, 16].
In this study we described the synthesis and evaluated the anticancer activity of new bipyridine gold(III) dithiocarbamate-containing complexes. They demonstrated a potent cytotoxic activity in ovarian, lung, breast, prostate cancer and Hodgkin lymphoma cells, induced apoptosis and Reactive Oxygen species. They were active in cisplatin-resistant ovarian carcinoma cells and independently of p53 status in both lung and ovarian cancer cell lines and less toxic in non-cancer human Mesenchymal stromal cells.
These new gold(III) complexes have dithiocarbamate ligands containing methyl (compounds 1, 4, 7) and ethyl (compounds 2, 5, 8) chains. The less active complexes (3, 6 and 9) have dithiocarbamate ligands containing dibenzyl substitution. The aliphatic containing dithiocarbamate ligands showed a higher anticancer activity respect to the benzyl dithiocarbamate complexes, summarized as Me ~ Et >> benzyl dithiocarbamate containing complexes. The different in vitro cytotoxic effects could be attributed to the small side chain of Methyl and Ethyl chains compared to the large dibenzyl group. However, even if our results indicate the consistent difference in potency (IC50) between different compounds, higher potencies are not an indicator of success at the clinical level, and this is well demonstrated by the ~10-fold difference in potency between cisplatin and the equal successful carboplatin [40]. Thus, other factors have to be taken into consideration to assess the clinical potential of less active new gold complexes, such as in vivo toxicity.
Cisplatin resistance is a critical factor that limits its clinical utility [1, 41]. The most active compounds (1, 2, 4, 5, 7, 8) were able to overcome the resistance to the reference drug cisplatin in ovarian and breast cancer cells, ruling out the occurrence of any cross-resistance phenomena. A fundamental mechanism of cisplatin resistance is the failure of this drug to activate p53, even in tumor cells primed for wild-type p53 function [42, 43]. Therefore, the identification of new drugs that do not depend for their anticancer activity on wild-type p53 status is an important goal since both NSCLC and ovarian cancer patients with p53 mutation have shorter overall survival as well as shorter time to progression [44, 45]. We found that the anticancer activity of the new gold(III) complexes was independent of p53 status: it was similar in WT-p53 models, in Mut- and Null-p53 cell lines and in WT-A2780 cells in which p53 was knocked out.
According to the above reported experimental results, complex 1 exerted a potent cytotoxic effect by inducing apoptosis, as assessed by Annexin-V, activation of caspase 3 and DNA fragmentation. Moreover, it induces mitochondrial membrane depolarization, cytochrome-c release and caspase 9 activation, indicating that its anticancer activity is exerted through the mitochondrial intrinsic apoptotic pathway. It induced ROS overproduction and growth inhibition, which was blocked by pre-treatment with the ROS scavenger NAC. However, we cannot exclude that thiol-containing antioxidant NAC could block the effects of compound 1 by binding with the active site of compound 1, as demonstrated for other gold complexes [46].
We found that the investigated compounds are able to interact with the small protein lysozymes. Moreover, the ESI-MS experiments indicated that in aqueous-methanol solution the compounds fast rearrange to generate stable derivatives where the gold(III) was coordinated by the four sulfur atom from two dithiocarbamate moieties. Indeed, we cannot exclude that such dithiocarbamate derivatives may have a role in the biological activity of the gold complexes. Further deep studies are however needed to better establish which chemical species is effectively responsible for the cellular cytotoxicity.
MATERIALS AND METHODS
Reagents
Disodium hydrogen phosphate, sodium dihydrogen phosphate, hen egg white lysozyme, and ethanol were obtained from Sigma-Aldrich. During the experimental work, the double distilled water was used for electrochemical measurements and acquired from Lab based Water Still Aquatron A 4000 D unit. Compounds (1–9) were dissolved (10 mM) in DMSO and stored at −80°C in volumes of 500 μL or aliquoted in volumes of 10 μL and stored at −20°C (used once without refreezing). They were then diluted in RPMI medium immediately before use. The culture medium with the same amount of drug-free DMSO was used as negative control in all experiments.
Synthesis of gold(III) complexes
Sodium tetrachloroaurate(III) dihydrate, Sodium dimethyldithiocarbamate monohydrate, sodium diethyldithiocarbamate trihydarte, sodium dibenzyldithiocarbamate hydrate, 2,2′-Bipyridine, 5,5′-dimethyl-2,2′-bipyridine and 6,6′-dimethyl-2,2′-bipyridine were purchased from Sigma-Aldrich Co. St. Louis, Missouri United States. All solvents including ethanol, dichloromethane were purchased from Merck Darmstadt, Germany and used without further purification. All reactions were carried out under ambient conditions.
Elemental analyses of gold(III) complexes (compounds 1–9) were performed on Perkin Elmer Series 11 (CHNS/O), Analyzer 2400. The solid state FTIR spectra of free ligands and their corresponding gold(I) complexes were recorded on a Perkin–Elmer FTIR 180 spectrophotometer or NICOLET 6700 FTIR using Potassium bromide (KBr) pellets over the range 4000–400 cm-1. 1H and 13C NMR spectra were recorded on a LAMBDA 500 spectrophotometer operating at 500.01, 125.65 and 200.0 MHz respectively, corresponding to a magnetic field of 11.74 T. Tetramethylsilane (TMS) was used as an internal standard for 1H and 13C. The 13C NMR spectra were obtained with 1H broadband decoupling, and the spectral conditions were: 32 k data points, 0.967 s acquisition time, 1.00 s pulse delay and 45 pulse angle.
[Au(BPY)(DMTC)]Cl2
The compound 1 was synthesized by stepwise synthesis; Na[AuCl4]. 2H2O, 0.5 mM (200 mg) and 2,2′-Bipyridine 0.5 mM (78 mg) were added simultaneously in 20 mL of ethanol and mixture was stirred for 3 h at room temperature. The sodium dimethyl dithiocarbamate dihydrate 0.5 mM (72 mg) in 10 mL distilled water was added slowly in the pale yellow turbid solution obtained of first step. The reaction mixture was stirred for an additional 1 h at room temperature. The final product appeared as light yellow precipitate in solution. The precipitate was collected by filtration, washed with fresh distilled water (3 × 10 mL) and dried at room temperature under vacuum. Yield: 83.09% (219.98 mg). FT-IR (KBr, υmax, cm−1): 3570 (m), 3045 (w), 2926 (w); 1586 (m), 1482 (s), 1242 (m), 1159 (w), 1039 (m), 993 (m), 967 (w), 761 (s), 565 (m), 439 (m). 1H NMR (500 MHz, DMSO-d6): δ = 3.36 (6H, 2 × CH3), 7.55, 8.06, 8.44 and 8.73 (2H, 2 × CH, BPY). 13C NMR (125.1 MHz, DMSO-d6): δ = 40.29 (CH3), 121.65, 125.19, 139.20 and 148.50 (2,2′-BPY), 189.87 (NC = S). Anal. calc. for C13H14Cl2N3S2Au (544.27): C, 28.69; H, 2.59; N, 7.72; S, 11.78%. Found: C, 28.55; H, 2.51; N, 7.75; S, 11.80%.
[Au(BPY)(DETC)]Cl2
Compound 2 was synthesized according to the procedure as mention above for complex (1). The solution of sodium diethyldithiocarbamate trihydrate 1.0 mM (226 mg) in 10 mL of distilled water was added slowly to the reaction mixture of first step as described above for complex (1) and was stirred for 1 h at room temperature. The final product of compound 2 appeared as yellow precipitate in the reaction medium. The product was collected by filtration, washed with fresh distilled water (3 × 10 mL) and dried at room temperature under vacuum for 24 h. The product appeared as yellow crystalline powder. Yield: 86.87% (166.87 mg). FT-IR (KBr, υmax, cm−1): 3568 (m), 3048 (w), 2923 (w); 1583 (m), 1485 (s), 1247 (m), 1155 (w), 1038 (m), 988 (m), 958 (w), 768 (s), 555 (m), 435 (m). 1H NMR (500 MHz, DMSO-d6): δ = 1.25 (6H, 2 × CH3), 3.76 (4H, 2 × CH2), 7.60, 8.11, 8.49 and 8.76 (2H, 2 × CH, BPY). 13C NMR (125.1 MHz, DMSO-d6): δ = 12.07 (CH3), 46.57 (CH2), 121.63, 125.23, 139.23 and 148.36 (2,2′-BPY), 193.88 (NC = S). Anal. calc. for C15H18Cl2N3S2Au (572.33): C, 31.48; H, 3.17; N, 7.34; S, 11.21%. Found: C, 31.37; H, 3.08; N, 7.19; S, 11.15%.
[Au(BPY)(DBTC)]Cl2
Compound 3 was synthesized according to the procedure as mention above for compounds 1 and 2. The solution of sodium dibenzyldithiocarbamate hydrate 0.5 mM (148 mg) in 10 mL distilled water was added slowly in the reaction mixture of Na[AuCl4].2H2O, 0.5 mM (200 mg) and 2,2′-Bipyridine 0.5 mM (78 mg) as described above for complex (1) and stirred at room temperature for 1 h. The final product appeared as brown lumps in solution. The solid product was collected by filteration, washed with fresh distilled water (3 × 10 mL) and dried at room temperature under vacuum for 24 h. Yield: 85.55% (226.13 mg). FT-IR (KBr, υmax, cm−1): 3587 (m), 3143 (w), 2925 (w); 1552 (s), 1481 (m), 1224 (m), 1120 (w), 1078 (m), 981 (m), 916 (w), 551 (m), 473 (m). 1H NMR (500 MHz, DMSO-d6): δ = 5.03 (4H, 2 × CH2), 7.38 (10H, 2 × C6H5), 7.47, 7.96, 8.39 and 8.69 (2H, 2 × CH, BPY). 13C NMR (125.1 MHz, DMSO-d6): δ = 55.18 (CH2), 120.67, 124.43, 137.73 and 149.08 (2,2′-BPY), 128.24–132.50 and 154.64 (C6H5), 191.76 (NC = S). Anal. calc. for C25H22Cl2N3S2Au (696.46): C, 43.11; H, 3.18; N, 6.03; S, 9.21%. Found: C, 43.33; H, 3.15; N, 6.20; S, 9.33%.
[Au(5,5′-Me2BPY)(DMTC)]Cl2
The compound 4 was synthesized by following the same procedure as described for compound 1 with minor modifications. The Na[AuCl4].2H2O, 0.5 mM (200 mg) and 5,5′-dimethyl-2,2′-dipyridyl 0.5 mM (92 mg) were added simultaneously in 20 mL of ethanol and mixture was stirred for 3 h at room temperature. The sodium dimethyldithiocarbamate dihydrate 0.5 mM (72 mg) in 10 mL distilled water was added slowly in the bright yellow turbid solution of above reaction mixture. The reaction mixture was stirred for 1 h at room temperature. The final product appeared as pale yellow precipitates in solution. The precipitates were collected by filtration, washed with fresh distilled water (3 × 10 mL) and dried at room temperature under vacuum. Yield: 80.09% (187.27 mg). FT-IR (KBr, υmax, cm−1): 3373 (m), 3037 (w), 2922 (w), 1555 (s), 1477 (s), 1272 (m), 1155 (m), 1028 (m), 981 (m), 896 (w), 567 (s), 468 (m). 1H NMR (500 MHz, DMSO-d6): δ = 2.37 (6H, 2 × CH3), 3.35 (6H, 2 × CH3), 7.85, 8.31 and 8.53 (2H, 2 × CH, BPY). 13C NMR (125.1 MHz, DMSO-d6): δ = 17.78 (CH3), 40.29 (CH3), 121.28, 135.33, 140.27 and 147.80 (2,2′-BPY), 193.87 (NC = S). Anal. calc. for C15H18Cl2N3S2Au (572.33): C, 31.48; H, 3.17; N, 7.34; S, 11.21%. Found: C, 31.50; H, 3.08; N, 7.25; S, 11.17%.
[Au(5,5′-Me2BPY)(DETC)]Cl2
The compound 5 was synthesized according to the procedure as mention above for compound 4. The solution of sodium diethyldithiocarbamate trihydrate 1.0 mM (226 mg) in 10 mL of distilled water was added slowly to the reaction mixture of first step as described above for compound 4 and was stirred for 1 h at room temperature. The final product appeared as deep yellow precipitate in the reaction medium. The product was collected by filtration, washed with fresh distilled water (3 × 10 mL) and dried at room temperature under vacuum for 24 h. The product appeared as yellow crystalline powder. Yield: 88.51% (343.9 g). FT-IR (KBr, υmax, cm−1): 3384 (w), 3035 (w), 2983 (w), 2926 (w), 1572 (s), 1473 (s), 1350 (m), 1220 (s), 1130 (m), 1056 (m), 996 (m), 827 (m), 538 (s), 465 (m). 1H NMR (500 MHz, DMSO-d6): δ = 1.27 (6H, 2 × CH3), 2.37 (6H, 2 × CH3), 3.76 (4H, 2 × CH2), 7.85, 8.31 and 8.53 (2H, 2 × CH, BPY). 13C NMR (125.1 MHz, DMSO-d6): δ = 12.05 (CH3), 17.80 (CH3), 46.55 (CH2), 120.41, 134.23, 138.83 and 148.64 (2,2′-BPY), 193.89 (NC = S). Anal. calc. for C17H22Cl2N3S2Au (600.38): C, 34.01; H, 3.69; N, 7.00; S, 10.68%. Found: C, 33.91; H, 3.64; N, 7.18; S, 10.59%.
[Au(5,5′-Me2BPY)(DBTC)]Cl2
The compound 6 was synthesized according to the procedure as mention above for compound 4. The solution of sodium dibenzyldithiocarbamate hydrate 0.5 mM (148 mg) in 10 mL of distilled water was added slowly to the reaction mixture of first step as described above for compound 4 and was stirred for 1 h at room temperature. The final product appeared as yellowish green precipitates in solution. The product was collected by filtration, washed with fresh distilled water (3 × 10 mL) and dried at room temperature under vacuum for 24 h. Yield: 78.01% (256.6 mg). FT-IR (KBr, υmax, cm−1): 3388 (w), 3031 (w), 2993 (w), 2923 (m), 1553 (s), 1470 (s), 1345 (m), 1224 (s), 1123 (m), 1077 (m), 980 (s), 828 (m), 553 (s), 466 (m). 1H NMR (500 MHz, DMSO-d6): δ = 2.38 (6H, 2× CH3), 5.02 (4H, 2 × CH2), 7.37 (10H, 2 × C6H5), 7.90, 8.33 and 8.57 (2H, 2 × CH, BPY). 13C NMR (125.1 MHz, DMSO-d6): δ = 17.77 (CH3), 55.38 (CH2), 120.78, 134.69, 139.43 and 148.29 (2,2′-BPY), 128.25–132.52 and 150.13 (C6H5), 191.77 (NC = S). Anal. calc. for C27H26Cl2N3S2Au (724.52): C, 44.76; H, 3.62; N, 5.80; S, 8.85%. Found: C, 44.73; H, 3.70; N, 5.77; S, 8.90%.
[Au(6,6′-Me2BPY)(DMTC)]Cl2
The compound 7 was synthesized by following the same procedure as described for compound 1 with minor modifications. The Na[AuCl4]. 2H2O, 0.5 mM (200 mg) and 6,6′-dimethyl-2,2′-dipyridyl 0.5 mM (92 mg) were added simultaneously in 20 mL of ethanol and mixture was stirred for 3 h at room temperature. The sodium dimethyldithiocarbamate dihydrate 0.5 mM (72 mg) in 10 mL distilled water was added slowly in the bright yellow solution of above reaction mixture. The reaction mixture was stirred for 1 h at room temperature. The final product appeared as light yellow precipitates in solution. The precipitates were collected by filtration, washed with fresh distilled water (3 × 10 mL) and dried at room temperature under vacuum. Yield: 90.09% (187.27 mg). FT-IR (KBr, υmax, cm−1): 3378 (m), 3033 (w), 2925 (w), 1575 (s), 1477 (s), 1238 (m), 1162 (m), 1046 (m), 998 (m), 868 (w), 567 (s), 440 (m). 1H NMR (500 MHz, DMSO-d6): δ = 2.49 (6H, 2 × CH3), 3.36 (6H, 2 × CH3), 7.41, 7.93 and 8.22 (2H, 2 × CH, BPY). 13C NMR (125.1 MHz, DMSO-d6): δ = 13.77 (CH3), 45.25 (CH3), 119.15, 125.21, 139.47 and 158.57 (2,2′-BPY), 187.83 (NC = S). Anal. calc. for C15H18Cl2N3S2Au (572.33): C, 31.48; H, 3.17; N, 7.34; S, 11.21%. Found: C, 31.67; H, 3.29; N, 7.38; S, 11.09%.
[Au(6,6′-Me2BPY)(DETC)]Cl2
The compound 8 was synthesized according to the procedure as mention above for compound 7. The solution of sodium diethyldithiocarbamate trihydrate 1.0 mM (226 mg) in 10 mL of distilled water was added slowly to the reaction mixture of first step as described above for compound 4 and was stirred for 1 h at room temperature. The final product appeared as yellow precipitates in the reaction medium. The product was collected by filtration, washed with fresh distilled water (3 × 10 mL) and dried at room temperature under vacuum for 24 h. Yield: 68.51% (343.9 g). FT-IR (KBr, υmax, cm−1): 3418 (w), 3029 (w), 2978 (w), 2925 (w), 1574 (s), 1479 (s), 1351 (m), 1281 (s), 1154 (m), 1081 (m), 988 (m), 847 (m), 585 (s), 415 (m). 1H NMR (500 MHz, DMSO-d6): δ = 1.27 (6H, 2 × CH3), 2.49 (6H, 2 × CH3), 3.76 (4H 2 × CH2), 7.42, 7.92 and 8.23 (2H, 2 × CH, BPY). 13C NMR (125.1 MHz, DMSO-d6): δ = 13.07 (CH3), 24.96 (CH3), 47.71 (CH2), 119.50, 125.28, 139.52 and 158.55 (2,2′-BPY), 188.12 (NC = S). Anal. calc. for C17H22Cl2N3S2Au (600.38): C, 34.01; H, 3.69; N, 7.00; S, 10.68%. Found: C, 34.25; H, 3.51; N, 7.29; S, 10.48%.
[Au(6,6′-Me2BPY)(DBTC)]Cl2
The compound 9 was synthesized according to the procedure as mention above for compound 7. The solution of sodium dibenzyldithiocarbamate hydrate 0.5 mM (148 mg) in 10 mL of distilled water was added slowly to the reaction mixture of first step as described above for compound 7 and was stirred for 1 h at room temperature. The final product appeared as reddish brown precipitates in solution. The product was collected by filtration, washed with fresh distilled water (3 × 10 mL) and dried at room temperature under vacuum for 24 h. Yield: 65.01% (256.6 mg). FT-IR (KBr, υmax, cm−1): 3428 (w), 3035 (w), 2978 (w), 2922 (m), 1566 (s), 1475 (s), 1331 (m), 1251 (s), 1119 (m), 1082 (m), 985 (s), 808 (m), 515 (s), 436 (m). 1H NMR (500 MHz, DMSO-d6): δ = 2.48 (6H, 2 × CH3), 5.01 (4H, 2 × CH2), 7.36 (10H, 2 × C6H5), 7.41, 7.91 and 8.20 (2H, 2 × CH, BPY). 13C NMR (125.1 MHz, DMSO-d6): δ = 24.92 (CH3), 54.88 (CH2), 118.95, 125.18, 138.33 and 157.43 (2,2′-BPY), 128.27–131.52 and 151.11 (C6H5), 199.01 (NC = S). Anal. calc. for C27H26Cl2N3S2Au (724.52): C, 44.76; H, 3.62; N, 5.80; S, 8.85%. Found: C, 44.33; H, 3.490; N, 5.63; S, 8.75%.
Mass spectrometry characterization
Electro spray ionization mass spectrometry (ESI-MS) experiments for the 1–6 compounds were conducted using a 4000QTRAP Linear Ion Trap ( ABsciex) mass spectrometer. Test solutions of 1 to 6 compounds in water /methanol (50%:50%) at concentration of 0.1 μg/mL were infused to Turbo V Ionspray TM ionization source of the mass spectrometer by a syringe pump Harvard apparatus (Holliston MA) and electrosprayed at a flow rate of 10 μL/min. The ESI source parameters use for ionization were: curtain gas CUR: = 20, declastering potential DP = 50, esi voltage: IS = 4000 V, source temperature TEM = 30°C entrance potential EP = :10 V and auxiliary gas G2 = 10 arbitrary unit. Positive ion mass spectra for each compound were acquired with MS-1 scanning from 100–1000 m/z using 2 sec time scan.
Gold compound-protein interaction studies
The interactions of gold compounds with protein have been performed in solution by electrochemical investigations Voltammetry measurements were performed with an electrochemical workstation (CHI1140A, CH Instruments Inc., Austin, TX, USA). The Ag/AgCl reference electrode (in 3M KCl, CHI111, CH Instruments Inc), Glassy carbon working electrode (CHI 112, CH Instruments Inc) and platinum wire counter electrode (CHI115, CH Instruments Inc) inserted into the 5.0 ml glass cell. Due to the poor solubility of the present compounds in water, their solutions were prepared in ethanol. The GCE was polished as a mirror like surface with alumina slurry on the synthetic cloth before every electrochemical analysis. The cyclic voltammetry (CV) and square wave voltammetry (SWV) were scanned from −0.4 to 1.0 V for various analyses for compounds 1–3 in the absence and in the presence of different concentration of lysozyme under physiological environment.
Cell lines and culture conditions
Hodgkin lymphoma (L-540) and human androgen-resistant prostate cancer (PC3) cell lines were obtained from the German Collection of Microorganisms and Cell Cultures (Braunschweig, Germany), human breast adenocarcinoma cell line MCF-7 (HTB-22TM) from the American Type Culture Collection (ATCC, Rockville, MD, USA). Human ovarian epithelial carcinoma-derived cancer cells A2780 and its cisplatin-resistant clone A2780cis were from Sigma Inc (St. Louis, MO,USA) and isogenic cisplatin-resistant 2780CP-16 model developed by sequential exposure of the A2780 cell line to increasing concentrations of cisplatin, as previously described [39]. Cisplatin was obtained from surplus clinical samples from the clinical pharmacy associated with Centro Riferimento Oncologico di Aviano. A panel of non-small cell lung cancer (NSCLC) cell lines H460, A549, H226, H838, H157, H1975, H2122, SKLU1 and H1299 were obtained from Dr. Garth Powis at MD Anderson Cancer Center. The parent cisplatin-resistant subclone (A2780cis) was maintained by weekly treatment with 1 μM cisplatin [38]. The resistance in 2780CP-16 cells was stable and did not require cisplatin maintenance. Cells were cultured at 37°C in 5% CO2 in RPMI (L-540, A2780, A2780cis, 2780CP-16, PC3 and the panel of lung cancer cells) or DMEM (MCF-7) medium supplemented with 10% heat-inactivated fetal calf serum (FCS; Sigma-Aldrich-Italy), 0.2 mg/ml penicillin/streptomycin and 0.1% (w/v) L-glutamine (Biocrom) at 37°C in a 5% CO2 fully humidified atmosphere.
Bone Marrow (BM) derived human Mesenchymal Stromal Cells (MSCs) were from Lonza (Lonza, Verviers, Belgium). MSCs were maintained in MSGM bullet kit (Lonza) and experiments were performed in DMEM medium (Cambrex Bio Science, Milano, Italy) supplemented with 10% FCS. To evaluate effects of compound 1, MSCs (5.0 × 103) were seeded in 96-well flat-bottomed microplates (100 μL) and incubated for 24 h (to allow cell adhesion) before drug testing. The medium was then removed and replaced with fresh medium containing compound 1 to be tested at increasing concentrations (from 0.1 to 1 μM) at 37°C for 72 h. Each treatment was performed in triplicate. Cell proliferation was assayed using the MTT assay.
Cellular uptake of gold compounds
L-540 cells (2.0 × 105/mL) were seeded in 6 well plates and treated for 24 hrs with 0.5 μM of compounds 1, 2 and 3. After treatment cells were washed with ice-cold PBS four times, dry cell pellet digested with 700 μL of HNO3-HCl (1:3 ) solution for 2 h at 100°C and then resuspended in 4 mL of milliQ water. The gold analysis was performed with Agilent 7500 inductively-coupled-plasma mass spectrometer (ICP-MS). Results were expressed as ng/gold/106 cells.
Cell proliferation assay and p53 knockdown
PC3 and MCF-7 cells (2.5 × 103), A2780, A2780cis (4.0 × 103), A2780CP-16 and lung cancer cells (H460, A549, H226, H838, H157, H1975, H2122, SKLU1 and H1299) were seeded in 96-well flat-bottomed microplates (100 μL) and incubated for 24 h (to allow cell adhesion) before drug testing. The medium was then removed and replaced with fresh medium containing the gold(III) complexes (compound 1–9) to be tested at increasing concentrations (from 0.01 to 100 μM) at 37°C for 72 h. Each treatment was performed in triplicate. Cell proliferation was assayed using the MTT assay. Alternatively, L-540 cells (2.0 × 105/mL) were seeded in 96-well plates and treated as previously described [37]. After treatment, cell proliferation was evaluated by the MTS assay (Promega). IC50 (i.e., the half maximal inhibitory concentration representing the concentration of a substance required for 50% in vitro inhibition) IC75 and IC90 values (Supplementary Table S1) were calculated using the CalcuSyn software (Biosoft, Ferguson, MO, USA) [47]. Dependence of activity on p53 was assessed in A2780 cells following p53 knockdown by CRISPR [48].
Evaluation of apoptosis and ROS formation
PC3 (2.5 × 104), MCF-7 (5.0 × 104), A2780 and A2780cis (5.0 × 104), and L-540 cells (2.0 × 105/mL) were incubated in six-well plates with compound 1 (IC75) (Figure 1) for 72 h. Annexin-V binding (Becton-Dickinson [BD] Pharmingen, San Jose, CA) together with propidium iodide (PI) staining was detected in tumor cells by flow cytometry, as described [37, 38]. Caspase 3 and 9 activation was evaluated using the fluorochrome inhibitors of caspases (FLICA) Caspa-TagTM caspase-3/7 (FAM-DEVD-FMK) and caspase 9 (FAM-LETD) (Chemicon International, Milan, Italy). Briefly, cells were treated with compound 1 (IC75), then harvested, washed and resuspended in warmed complete medium supplemented with FLICA for 1 h and then immediately analyzed by flow cytometry. DNA fragmentation was assessed using the Apo-Direct kit (Becton-Dickinson Pharmigen, CA, USA) according to manufacturer’s instructions. For mitochondrial ROS evaluation, cells treated with compound 1 were incubated with 5 μM of MitoSox reagent working solution (Molecular Probes, Invitrogen) for 30 minutes at 37°C. Red fluorescence was immediately analyzed by flow cytometry. Viable cells were identified according to their forward and right-angle scattering, electronically gated and analyzed on a FACScan flow cytometer (BD), using CellQuest software (BD). In another series of experiments, cells were exposed to compound 1 (IC75) in the presence or absence of the antioxidant and ROS scavenger N-acetyl-cysteine (NAC; 5 mM) (Sigma). After 72 h, ROS formation and viable cells number were evaluated by flow cytometry and trypan blue dye exclusion assay, respectively. To evaluate the dissipation of the mitochondrial membrane potential, 200 nM MitoTracker® Red CMXRos (Molecular Probes, Invitrogen, Milan, Italy) was added to the cell culture for 30 min, then cells were washed twice and analyzed by flow cytometry. Cytochrome-c (Cyt-c) release was assessed using the mouse anti-Cytochrome-c antibody (BD), followed by PE-conjugated goat anti-mouse IgG (BD), as explained elsewhere [49].
CONCLUSIONS
Drug resistance to chemotherapeutic agents is a central problem in oncology. Results in this investigation have identified members of a new class of gold(III) compounds as potential candidates for application in cancer chemotherapy. This is based on the demonstration that gold(III) compounds are effective in cisplatin-resistant, as well as in p53-defective cancers cells of different tumor types. Moreover, since our compounds contain a dithiocarbamate group capable of preventing a reaction with other sulphur-containing proteins, we may hypothesize a reduced toxicity in vivo. Thus, a gold(III) drug with an appropriate ligand, such as that presented in this study, has the potential to be an alternative to platinum-based drugs in cancer treatment. Further preclinical testing in vitro and in vivo will be essential to define their mechanism of action, their side effects (toxicity) and to select potential candidates for clinical activity.
CONFLICTS OF INTEREST
The authors reported no potential conflicts of interest.
FUNDING
The project was funded by the National Plan for Science and Technology and Innovation (MAARIFAH)- King Abdul Aziz city for Science and Technology-through the Science and Technology unit at King Fahd University of Petroleum and Minerals (KFUPM)- the Kingdom of Saudi Arabia, Award No. 14-MED64–04. This work was also supported in part by grant from the Italian Association for Cancer Research (AIRC) to D.A. (IG 15844) and by Ministero della Salute, Ricerca Finalizzata FSN, I.R.C.C.S., Rome, Italy. The work was supported in part by Fondazione Umberto Veronesi to G.C. The work was supported in part by the US National Institutes of Health (grant RO1 CA160687 to ZHS).
REFERENCES
1. Galluzzi L, Vitale I, Michels J, Brenner C, Szabadkai G, Harel-Bellan A, Castedo M, Kroemer G. Systems biology of cisplatin resistance: past, present and future. Cell Death Dis. 2014; 5:e1257.
2. Paunescu E, McArthur S, Soudani M, Scopelliti R, Dyson PJ. Nonsteroidal Anti-inflammatory-Organometallic Anticancer Compounds. Inorg Chem. 2016; 55:1788–1808.
3. Muhammad N, Guo Z. Metal-based anticancer chemotherapeutic agents. Curr Opin Chem Biol. 2014; 19:144–153.
4. Dilruba S, Kalayda GV. Platinum-based drugs: past, present and future. Cancer Chemother Pharmacol. 2016; 77:1103–1124.
5. Zou T, Sadler PJ. Speciation of precious metal anti-cancer complexes by NMR spectroscopy. Drug Discov Today Technol. 2015; 16:7–15.
6. Nobili S, Mini E, Landini I, Gabbiani C, Casini A, Messori L. Gold compounds as anticancer agents: chemistry, cellular pharmacology, and preclinical studies. Med Res Rev. 2010; 30:550–580.
7. Nardon C, Boscutti G, Fregona D. Beyond platinums: gold complexes as anticancer agents. Anticancer Res. 2014; 34:487–492.
8. Bergamo A, Gaiddon C, Schellens JH, Beijnen JH, Sava G. Approaching tumour therapy beyond platinum drugs: status of the art and perspectives of ruthenium drug candidates. J Inorg Biochem. 2012; 106:90–99.
9. Wallace HM, Hergenrother PJ, Sadler PJ. Molecular medicine and cancer. Chem Soc Rev. 2015; 44:8771–8772.
10. Massai L, Fernandez-Gallardo J, Guerri A, Arcangeli A, Pillozzi S, Contel M, Messori L. Design, synthesis and characterisation of new chimeric ruthenium(II)-gold(I) complexes as improved cytotoxic agents. Dalton Trans. 2015; 44:11067–11076.
11. Maia PI, Deflon VM, Abram U. Gold(III) complexes in medicinal chemistry. Future Med Chem. 2014; 6: 1515–1536.
12. Serratrice M, Edafe F, Mendes F, Scopelliti R, Zakeeruddin SM, Gratzel M, Santos I, Cinellu MA, Casini A. Cytotoxic gold compounds: synthesis, biological characterization and investigation of their inhibition properties of the zinc finger protein PARP-1. Dalton Trans. 2012; 41:3287–3293.
13. Bertrand B, Spreckelmeyer S, Bodio E, Cocco F, Picquet M, Richard P, Le GP, Orvig C, Cinellu MA, Casini A. Exploring the potential of gold(III) cyclometallated compounds as cytotoxic agents: variations on the C^N theme. Dalton Trans. 2015; 44:11911–11918.
14. Joost M, Amgoune A, Bourissou D. Reactivity of Gold Complexes towards Elementary Organometallic Reactions. Angew chem. 2015; 54:15022–15045.
15. Celegato M, Fregona D, Mongiat M, Ronconi L, Borghese C, Canzonieri V, Casagrande N, Nardon C, Colombatti A, Aldinucci D. Preclinical activity of multiple-target gold(III)-dithiocarbamato peptidomimetics in prostate cancer cells and xenografts. Future Med Chem. 2014; 6:1249–1263.
16. Ronconi L, Aldinucci D, Dou QP, Fregona D. Latest insights into the anticancer activity of gold(III)-dithiocarbamato complexes. Anticancer Agents Med Chem. 2010; 10: 283–292.
17. Milacic V, Chen D, Ronconi L, Landis-Piwowar KR, Fregona D, Dou QP. A novel anticancer gold(III) dithiocarbamate compound inhibits the activity of a purified 20S proteasome and 26S proteasome in human breast cancer cell cultures and xenografts. Cancer Res. 2006; 66:10478–10486.
18. Cattaruzza L, Fregona D, Mongiat M, Ronconi L, Fassina A, Colombatti A, Aldinucci D. Antitumor activity of gold(III)-dithiocarbamato derivatives on prostate cancer cells and xenografts. Int J Cancer. 2011; 128:206–215.
19. Parish RV. Biologically-Active Gold(III) Complexes. Met Based Drugs. 1999; 6:271–276.
20. Giovagnini L, Ronconi L, Aldinucci D, Lorenzon D, Sitran S, Fregona D. Synthesis, characterization, and comparative in vitro cytotoxicity studies of platinum(II), palladium(II), and gold(III) methylsarcosinedithiocarbamate complexes. J Med Chem. 2005; 48:1588–1595.
21. Zou T, Lum CT, Lok CN, Zhang JJ, Che CM. Chemical biology of anticancer gold(iii) and gold(i) complexes. Chem Soc Rev. 2015; 44:8786–8801.
22. Hu D, Liu Y, Lai YT, Tong KC, Fung YM, Lok CN, Che CM. Anticancer Gold(III) Porphyrins Target Mitochondrial Chaperone Hsp60. Angew Chem Int Ed Engl. 2016; 55:1387–1391.
23. Milovanovic M, Djekovic A, Volarevic V, Petrovic B, Arsenijevic N, Bugarcic ZD. Ligand substitution reactions and cytotoxic properties of [Au(L)Cl2](+) and [AuCl2(DMSO)2]+ complexes (L = ethylenediamine and S-methyl-l-cysteine). J Inorg Biochem. 2010; 104:944–949.
24. Ronconi L, Giovagnini L, Marzano C, Bettio F, Graziani R, Pilloni G, Fregona D. Gold dithiocarbamate derivatives as potential antineoplastic agents: design, spectroscopic properties, and in vitro antitumor activity. Inorg Chem. 2005; 44:1867–1881.
25. Saggioro D, Rigobello MP, Paloschi L, Folda A, Moggach SA, Parsons S, Ronconi L, Fregona D, Bindoli A. Gold(III)-dithiocarbamato complexes induce cancer cell death triggered by thioredoxin redox system inhibition and activation of ERK pathway. Chem Biol. 2007; 14: 1128–1139.
26. Nardon C, Schmitt SM, Yang H, Zuo J, Fregona D, Dou QP. Gold(III)-dithiocarbamato peptidomimetics in the forefront of the targeted anticancer therapy: preclinical studies against human breast neoplasia. PLoS One. 2014; 9:e84248.
27. Akhmadullina NS, Churakov AV, Retivov VM, Sandu RA, Shishilov ON. Gold(III) chloride and acetate complexes with bipyridine and phenanthroline. Russian J. Coord. Chem. 2012; 39:589–595.
28. Casini A, Diawara MC, Scopelliti R, Zakeeruddin SM, Gratzel M, Dyson PJ. Synthesis, characterisation and biological properties of gold(III) compounds with modified bipyridine and bipyridylamine ligands. Dalton Trans. 2010; 39:2239–2245.
29. Ogawa T, Sakamoto M, Honda H, Matsumoto T, Kobayashi A, Kato M, Chang HC. Self-association and columnar liquid crystalline phase of cationic alkyl-substituted-bipyridine benzenedithiolato gold(III) complexes. Dalton Trans. 2013; 42:15995–16005.
30. Odola AJ, Woods JA. New Nickel(II) Mixed Ligand Complexes of Dithiocarbamates with Schiff Base. J Chem Pharm Res. 2011; 3:865–871.
31. Altaf M, Isab AA, Vanco J, Dvorak Z, Travnicek Z, Stoeckli-Evans H. Synthesis, characterization and in vitro cytotoxicity of gold(III) dialkyl/diaryldithiocarbamato complexes. RSC Advances. 2015; 5:81599–81607.
32. Jayaraju A, Ahamad MM, Rao RM, Sreeramulu J. Synthesis, characterization and biological evaluation of novel dithiocarbamate metal complexes . J Der Pharma Chemica. 2012; 4:1191–1194.
33. Altaf M, Monom-ul-Mehboob M, Selimam AA, Isab AA, Dhuna V, Bhatia G, Dhuna K. Synthesis, X–ray Structures, Spectroscopic Analysis and Anticancer Activity of Novel Gold(I) Carbene Complexes. J Organometallic Chem. 2014; 765:68–79.
34. Che CM, Sun RW. Therapeutic applications of gold complexes: lipophilic gold(III) cations and gold(I) complexes for anti-cancer treatment. Chem Commun (Camb ). 2011; 47:9554–9560.
35. Altaf M, Monim-ul-Mehboob M, Seliman AA, Sohail M, Wazeer MI, Isab AA, Li L, Dhuna V, Bhatia G, Dhuna K. Synthesis, characterization and anticancer activity of gold(I) complexes that contain tri-tert-butylphosphine and dialkyl dithiocarbamate ligands. Eur J Med Chem. 2015; 95: 464–472.
36. Al-Jaroudi SS, Altaf M, Al-Saadi AA, Kawde AN, Altuwaijri S, Ahmad S, Isab AA. Synthesis, characterization and theoretical calculations of (1,2-diaminocyclohexane)(1,3-diaminopropane)gold(III) chloride complexes. in vitro cytotoxic evaluations against human cancer cell lines. Biometals. 2015; 28:827–844.
37. Celegato M, Borghese C, Casagrande N, Mongiat M, Kahle XU, Paulitti A, Spina M, Colombatti A, Aldinucci D. Preclinical activity of the repurposed drug Auranofin in classical Hodgkin lymphoma. Blood. 2015; 126:1394–1397.
38. Casagrande N, Celegato M, Borghese C, Mongiat M, Colombatti A, Aldinucci D. Preclinical activity of the liposomal Cisplatin lipoplatin in ovarian cancer. Clin Cancer Res. 2014; 20:5496–5506.
39. Ali MS, Whitmire KH, Toyomasu T, Siddik ZH, Khokhar AR. Preparation, characterization, and antitumor activity of new cisplatin analogs with homopiperazines: crystal structure of [PtII(1-methylhomopiperazine)(methylmalonato)].2H2O. J Inorg Biochem. 1999; 77:231–238.
40. Kelland LR, Murrer BA, Abel G, Giandomenico CM, Mistry P, Harrap KR. Ammine/amine platinum(IV) dicarboxylates. a novel class of platinum complex exhibiting selective cytotoxicity to intrinsically cisplatin-resistant human ovarian carcinoma cell lines. Cancer Res. 1992; 52:822–828.
41. Siddik ZH. Cisplatin. mode of cytotoxic action and molecular basis of resistance. Oncogene 2003; 22: 7265–7279.
42. Fritsche M, Haessler C, Brandner G. Induction of nuclear accumulation of the tumor-suppressor protein p53 by DNA-damaging agents. Oncogene. 1993; 8:307–318.
43. Shieh SY, Ikeda M, Taya Y, Prives C. DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell. 1997; 91:325–334.
44. Ahrendt SA, Hu Y, Buta M, McDermott MP, Benoit N, Yang SC, Wu L, Sidransky D. p53 mutations and survival in stage I non-small-cell lung cancer: results of a prospective study. J Natl Cancer Inst. 2003; 95:961–970.
45. Reles A, Wen WH, Schmider A, Gee C, Runnebaum IB, Kilian U, Jones LA, El-Naggar A, Minguillon C, Schonborn I, Reich O, Kreienberg R, Lichtenegger W, et al. Correlation of p53 mutations with resistance to platinum-based chemotherapy and shortened survival in ovarian cancer. Clin Cancer Res. 2001; 7:2984–2997.
46. Liu Y, Li Y, Yu S, Zhao G. Recent advances in the development of thioredoxin reductase inhibitors as anticancer agents. Curr Drug Targets. 2012; 13:1432–1444.
47. Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984; 22:27–55.
48. Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013; 339:819–823.
49. Campos CB, Paim BA, Cosso RG, Castilho RF, Rottenberg H, Vercesi AE. Method for monitoring of mitochondrial cytochrome c release during cell death: Immunodetection of cytochrome c by flow cytometry after selective permeabilization of the plasma membrane. Cytometry A. 2006; 69:515–523.