
INTRODUCTION
Colorectal cancer is the third most common cancer type in men and the second most common in women, and reported as the fourth leading cause of cancer-related mortality worldwide [1, 2]. The issue of whether the poor prognosis and high mortality of CRC are partially attributed to pathogenesis remains unclear. The well-known ‘adenoma-carcinoma sequence’ is known to play a significant role in CRC development [3, 4]. The molecular genetic basis posits that accumulating somatic and germ-line mutations drive epithelial dysplasia and hyperproliferation in the colon, ultimately causing CRC [3, 5]. The most commonly mutated genes include tumor suppressors (APC, P53 and the β-catenin gene) and oncogenes (K-RAS, BRAF and MYC). The triggers for these mutations are multifactorial in origin, associated with infectious agents and high exposure of tissues to microbiota, but remain elusive in many cases [6]. From this perspective, Tjalsmaet et al. [7] proposed a bacterial driver-passenger model explaining microbiota community involvement during CRC development, which may contribute to the genetic paradigm of the ‘adenoma-carcinoma sequence’. The group suggested that the colonic mucosa is colonized by bacterial drivers, defined as intestinal bacteria with procarcinogenic features that initiate CRC development. Bacterial drivers contribute to the initiation of pre-malignant lesions and accumulation of a series of mutations during the adenoma–carcinoma sequence, causing persistent inflammation, increased cell proliferation and production of genotoxic substances. With changes in the tumor microenvironment, pathogenic bacterial drivers are gradually replaced by passenger bacteria with a competitive advantage in the tumor niche. The human colon harbors as many as 36,000 bacterial species and over 100 trillion aerobic and anaerobic bacteria [8, 9], and has been identified as the anatomical location with the highest abundance of microbes [10]. The number of bacterial cells in the gut exceeds all other eukaryotic cells in the human body by a factor of 10 [11, 12]. However, imbalance or microbiota dysbiosis in the gut may induce inflammation or intestinal barrier dysfunction. In addition, existing bacteria may also enhance the susceptibility to disease, such as Helicobacter pylori to gastric cancer and human papilloma virus to cervical cancer.
Previous studies demonstrating an association of one or more microbial species with CRC have implied that gut microbiota may be a driver of CRC tumorigenesis and not connected with colitis [10]. Moreover, the differences in microbial species between CRC tumor and control tissues have been compared. However, no studies to date have successfully identified the exact bacterial strain that causes colorectal cancer. A number of researchers have reported significantly higher abundance of Fusobacterium species colorectal adenomas, compared to controls. For instance, Kostic and colleagues [13–16] revealed enrichment of Fusobacterium species in CRC, relative to adjacent normal tissue. Subsequently, the group reported that Fusobacterium nucleatum enhances intestinal tumorigenesis and modulates the tumor immune microenvironment. A recent study showed a longitudinal shift in the microbial community and molecular networks with colitis-associated CRC, demonstrating that phylotype shifts during this process are complex and highly dynamic [17]. However, the role played by gut microbiota in ‘adenoma-carcinoma sequence’ CRC pathogenesis is yet to be established. In particular, evidence of evolutionary microbiota alterations is scarce. Further studies are therefore necessary to uncover the role of microbiota in the evolutionary process from colorectal atypical hyperplasia to adenoma. The development of next-generation sequencing technologies has allowed the analysis of fecal microbiota at a level of detail that was previously not achievable. The aim of the current study was to investigate the evolutionary biologic changes of the gut microbiota in tumor progression from normal colon epithelium to premalignant adenoma and subsequently invasive adenocarcinoma, with a view to establishing the potential roles of different gut microbes in the specific molecular events characterizing transition to adenocarcinoma in an ‘adenoma–carcinoma sequence’ mouse CRC model.
RESULTS
Progression of the ‘adenoma-carcinoma sequence’ in a mouse model
According to the experimental protocol (Figure 1A), no obvious macroscopic lesions were observed in colon mucosa on week 6 after the first drug injection (Figure 1B), but deeper staining for colon epithelial cell nucleus and atypical hyperplasia upon microscopic examination were observed (Figure 1F). Polyps were first observed on week 12 (Figure 1C, 1G). However, the majority of adenomas were detected on week 18 (Figure 1D, 1H). Unfortunately, 42 mice died due to cachexia. The size and numbers of polyps increased with time. On week 12, only one mouse contained polyps 3 mm diameter in the colon, while on week18, enlarged polyps with diameters of 5 mm were observed (Figure 1E). In the last stage (week 26), the majority of mice (17/20) of the model group had developed polyps, some of which showed integration. Pathological examination disclosed adenocarcinoma (Figure 1I).
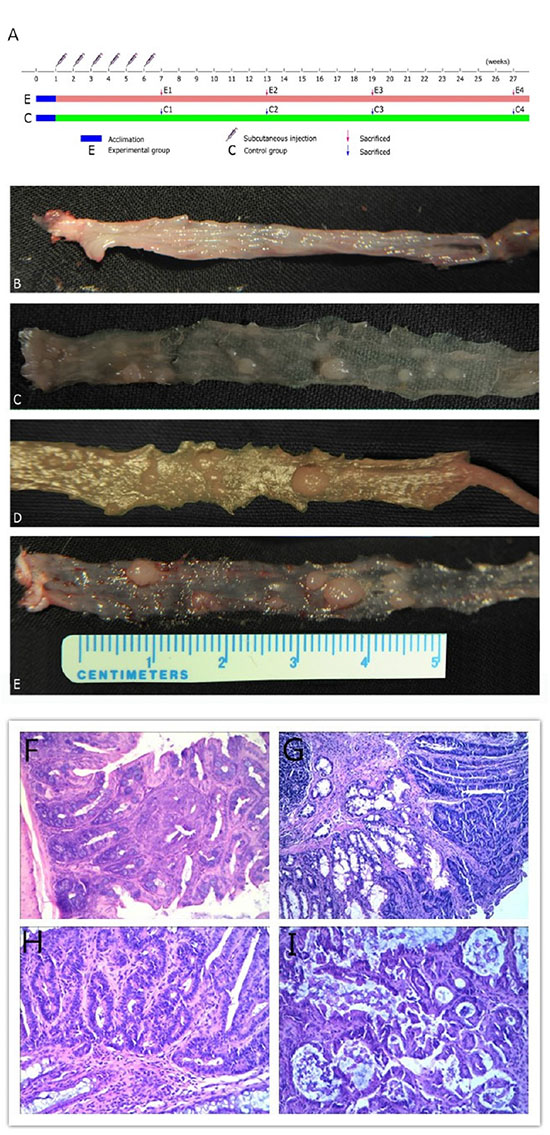
Figure 1: Experimental protocol, representative images of experimental groups and photomicrographs showing pathological characteristics. (A) Experimental procedures. (B) Inflammation group. (C) Hyperproliferation group. (D) Adenoma group. (E) Adenocarcinoma group. Representative photomicrographs showing (F) mucosalinflammation, (G) hyperproliferation tissue, (H) adenoma and (I) adenocarcinoma magnified 20×.
Overview of 454 pyrosequencing
After pyrosequencing on the Roche 454 FLX+ platform, a total of 1,080,304 raw reads were generated for all 151 samples. Following sample date split and read filter, 559,286 effective reads were generated with an average of 3,703 high-quality sequences per sample. The total number of OTUs at 97% identity was 5,689, with an average of 226 OTUs per sample. The observed species index was used to estimate microbial richness, and the Shannon index used to assess the diversity and evenness of gut microbiota in each sample. We have already observed the plateau of the refraction curve and the curve to flatten with current sequencing, suggesting that the Shannon and observed species indices estimated for all samples reached stable values at this sequencing depth. From the observed species indices, statistically significant differences were determined between each group (P = 0.00002).
Structure and diversity variations of fecal microbial communities between control and experimental groups
For beta diversity analysis, Principal Coordinates Analysis (PCoA) based on weighted UniFrac metric was employed to obtain an overview of the microbiota communities of the control and experimental groups of mice. The data revealed significant separation between mice with epithelium dysplasia, those developing cancer and healthy controls (Figure 2).
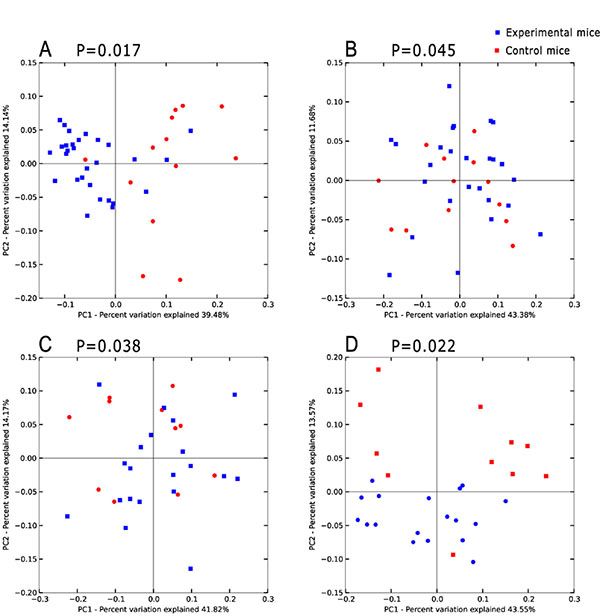
Figure 2: Principal coordinates analysis(PCoA)scores plot basedon relative abundance of OTUs (97% similarity level). (A–D) represent inflammatory, hyperproliferation, adenoma and carcinoma groups and the relevant control group respectively. Each symbol represents a sample. The blue symbol represents experimental mice and the red symbol control mice. Principal coordinates analysis was carried out using QIIME v1.5.0.
At the phylum level, eight phyla were present in all samples, the predominant being Bacteroidetes and Firmicutes. The relative abundance of the two phyla was > 90% (Figure 3A). The relative abundance of Bacteroidetes species was 56.3% (54.5–60.1%) and 64.8% (63.3–74.8%) in the control and experimental groups, while that of Firmicutes was 41.5% (36.4–42.8%) and 33.6% (22.5–40.9%), respectively. The remaining phyla were Verrucomicrobia, Actinobacteria, Proteobacteria, Tenericutes, Deferribacteres, and TM7. Notably, Bacteroidetes was more highly enriched in the gut microbiota of experimental groups than control groups with statistically significant differences (77.65% vs. 56.72%, P = 0.001), whereas Firmicutes was markedly less abundant in the experimental groups (19.44% vs. 40.81%, P = 0.001). Diversity changes were additionally examined. Deferribacteres was absent in the hyperproliferation, adenoma and adenocarcinoma groups, with significant differences from the control group (0.01% vs. 0%, P = 0.010). However, no significant changes were evident with the other phyla.
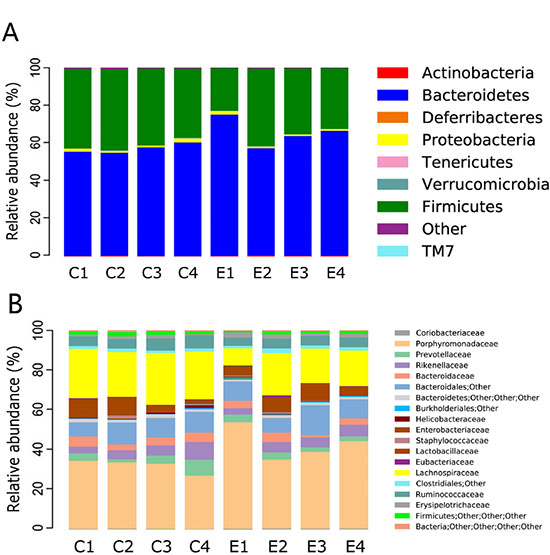
Figure 3: Relative abundance of the dominant phyla and family of each group. ‘C’ representsthe control group, ‘E’ the experimental group. Significant difference analyses were calculated using Metastats (http://metastats.cbcb.umd.edu/), and P values corrected in R (Version 2.15.3).
At the family level, the predominant families in the control group were Porphyromonadaceae and Lachnospiraceae (Figure 3B). Families with relative abundance of > 1% in the control group were Rikenellaceae, Prevotellaceae, Ruminococcaceae, Bacteroidaceae, Lactobacillaceae, Helicobacteraceae, Erysipelotrichaceae, and Staphylococcaceae. The gut microbial community was clearly altered during intestinal tumorigenesis. The predominant families were still Porphyromonadaceae and Lachnospiraceae, but the remaining families with relative abundance > 1% were Lactobacillaceae, Prevotellaceae, Bacteroidaceae, Rikenellaceae, Erysipelotrichaceae and Ruminococcaceae. Porphyromonadaceae (56.01% vs. 35.06%, P= 0.001, FDR = 0.015), Clostridiaceae (0.08% vs. 0%, P = 0.003, FDR = 0.022), Peptostreptococcaceae (0.04% vs. 0%, P = 0.003, FDR = 0.017), Prevotellaceae (3.92% vs. 1.65%, P = 0.018, FDR = 0.066), Coriobacteriaceae (0.37% vs. 0.19%, P = 0.014, FDR = 0.092), and Lactobacillaceae (7.81% vs. 3.47%, P = 0.011, FDR = 0.064) were enriched in the experimental groups whereas Lachnospiraceae (23.31% vs. 6.90%, P = 0.003, FDR = 0.022), Ruminococcaceae (6.95% vs. 4.56%, P = 0.033, FDR = 0.111), and Bacteroidaceae (4.04% vs. 1.16%, P = 0.002, FDR = 0.035) were enriched in the control groups. Interestingly, in our study, Clostridiaceae, Peptostreptococcaceae and Sutterellaceae were only found in the experimental groups. However, Dermabacteraceae, Flavobacteriaceae, Brucellaceae, Caulobacteraceae, Mycoplasmataceae, and Deferribacteraceae were depleted upon CRC development. Although these families exhibited low abundance, variations were statistically significant.
Evolutionary biologic changes and identification of key phylotypes during the process of CRC tumorigenesis
As specified previously, microbiota structure and diversity were obviously changed during carcinogenesis, although the specific phylotypes causing these variations and dynamic patterns of gut microbiota are yet to be identified. Time-dependent analysis of the microbiota community during tumorigenesis with the Kruskal-Wallis rank sum test revealed that at the phylum level, Bacteroidetes was significantly enriched in the inflammatory group (P < 0.01). However, in the hyperprolifarative, adenoma and adenocarcinoma groups, Bacteroidetes displayed decreased abundance. Firmicutes showed lower abundance in the inflammatory group (P < 0.01), but was significantly increased in the hyperprolifarative group (P < 0.01). Proteobacteria was the third most abundant phylum in the model groups, and interestingly, was also enriched in the inflammatory group (P < 0.01) but decreased gradually during the course of tumor development(Figure 4A).
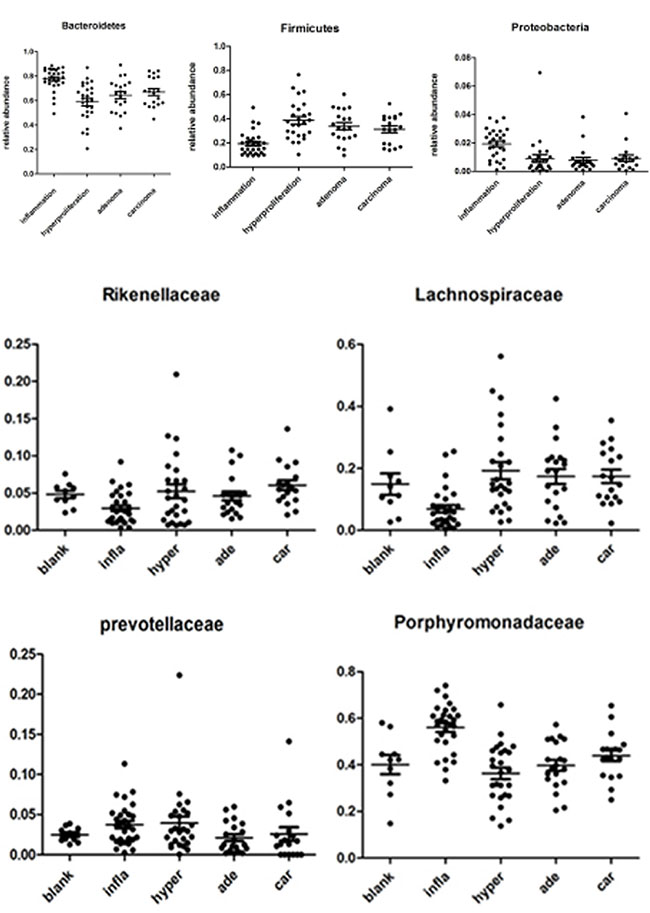
Figure 4: Relative abundance of the dominant phyla and families in samples of experimental groups. The Kruskal-Wallis rank sum test was employed to evaluate the differences between each group.to P0 was color scaled from red (highest) to black (middle) to green (lowest). Grey indicates that no relevant expression was detected.
At the family level, we analyzed the microbial composition of each group. The most abundant family was identified as Porphyromonadaceae, whereas the individual composition was variable, accounting for 13.71–74.03%. Lachnospiraceae was the second most abundant family in the model groups, ranging from 0.37% to 56.06%. In addition, Porphyromonadaceae and Prevotellaceae were enriched in the inflammatory group, but showed a significant decrease in the hyperprolifaration and adenoma groups (P < 0.01). Conversely, Lachnospiraceae was enriched in the hyperproliferation, adenoma and adenocarcinoma groups (P < 0.01). Rikenellaceae and Ruminococcaceae were increased during tumorigenesis. Interestingly, Bacteroidaceae and Enterobacteriaceae were enriched at the early stage, but decreased from the interim of tumor formation (Figure 4B).
To further detect the phylotypes of microbial that contributed to longitudinal microbiota community changes, analysis based on 97% similarity OTU was performed. After filtering OTUs with very low reads, 287 OTUs with97% similarity remained (Figure 5). In an extract test, 19 OTUs were found to be significantly different between the model and control groups (P < 0.05). Among these, 12 were higher and 7 were lower in the model group. Two OTUs were affiliated with the species Alistipes finegoldii (P < 0.01) and three OTUs belonging to the genus Clostridium (P < 0.05) were significantly higher in the model group. An OTU related to the species Lactobacillus animals (P < 0.05) was significantly enriched in the control group and decreased with evolution of the adenoma–carcinoma sequence. In terms of structural changes, six OTUs affiliated with Parasutterella excrementihominis, Akkermansia muciniphila, Odoribacter splanchnicus, Turicibacter sanguinis, Clostridium disporicum and Klebsiellapneumoniae were only shown in the model group and two OTUs related to Odoribacter splanchnicus and Proteusmirabilis were depleted in the model group. We additionally investigated OTU variations during the ‘adenoma-carcinoma’ sequence. The results showed that OTUs belonging to Clostridium lactatifermentans (OTU0201) and Bacteroidesdorei (OTU0051) were significantly different between the hyperproliferation, adenoma and carcinoma groups. However, abundance in the adenoma group was lower. Conversely, three OTUs related to Alistipes finegoldii (OTU0151,0105,0022) showed significant enrichment in the adenoma group, and abundance was consistently increased during tumor development.
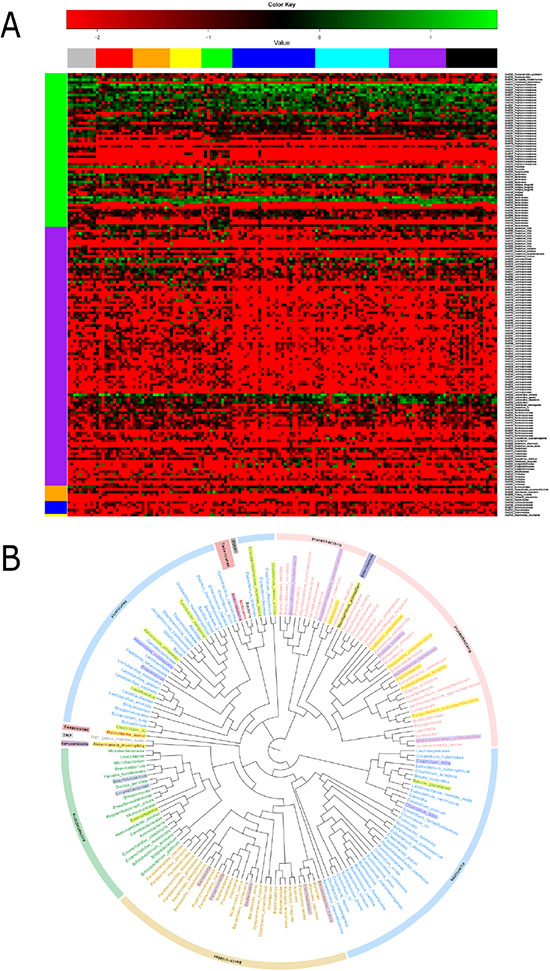
Figure 5: Heatmap and phylogenetic tree of significant differential OTUs. (A) Log-fold-change and heatmap of significant differential OTUs (P < 0.05). (B) Significant differential genera between the experimental and control groups. Generashaded in yellow represent enrichment in the experimental group, and thoseshaded in purple shade represent enrichment in the control group. The analysis was by QIIME (Version 1.50) software, using an iterative algorithm to obtain clustering tree to R (Version 2.15.3) drawing. Clustering method UPGMA (Unweighted Pair Group Method with Arithmetic mean).
Microflora variation at the later period of tumor formation
Although all mice received the same DMH treatment in the experimental group, some mice had no tumor formation by the end of our experiment (26 weeks after DMH treatment). We compared the difference of gut microflora between the mice with tumors and those mice without tumors in experimental group. By beta diversity analysis, we found their microbiota were obviously separated (Figure 6). From the genus level, the abundance of Eubacterium was significantly decreased in the tumor mice (P = 0.013).We also noticed that Streptococcus, Prevotella and Akkermansia were disappeared in the tumor mice.
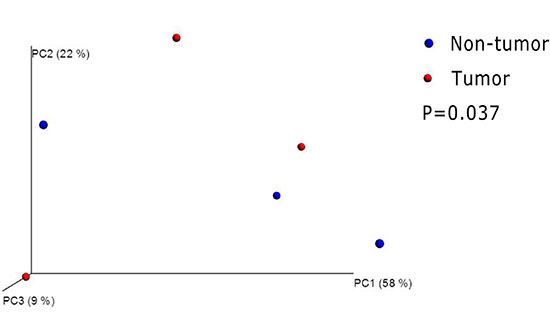
Figure 6: Principal coordinates analysis(PCoA)scores plot basedon relative abundance of OTUs (97% similarity level). Each symbol represents a sample. The blue symbol represents non-tumor mice and the red symbol represents tumor mice. Principal coordinates analysis were carried out using QIIME v1.5.0.
Dynamic detection of the molecular basis of colorectal carcinogenesis
To detect changes in levels of the molecular factors involved in the ‘adenoma-carcinoma’ sequence, we constructed a time-dependent dynamic alteration map of APC, P53, K-RAS and BRAF via immunohistochemical staining. APC and P53 are significant factors in tumor formation [18, 19]. The Wnt signaling pathway is regarded as a key event in initiation of colorectal cancer in which APC plays an important role. In the normal group, P53 staining was almost undetectable. However, expression of P53 was gradually increased in the hyperproliferative, adenoma and carcinoma groups, while APC showed the opposite expression pattern. Increased accumulation of P53 was observed in the villus and cell nucleus (Figure 7A). Additionally, semi-quantification of the expression of these molecules in each group revealed significant variations (P < 0.05) (Figure 7B). K-RAS and BRAF showed a consistent trend of increased expression from the hyperproliferative to adenoma and subsequent carcinoma groups (Figure 2). The observed modifications in expression levels of these molecules indicates dynamic patterns of the ‘adenoma-carcinoma’ sequence of CRC.
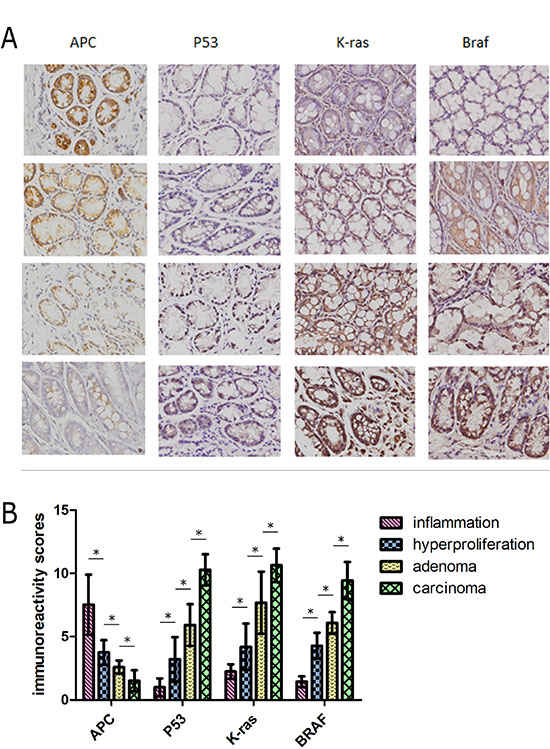
Figure 7: (A) Representative staining for four molecules reflecting the pathological process of CRC. The rows represent the each experimental group (n = 30). Images were magnified 400×. (B) Dynamic changes in expression levels of molecules were semi-quantified with immunoreactivity scores. *: statistically signifcant P < 0.05
DISCUSSION
Populations of gut microbiota in mice and humans are shared to alarge extent, which are representative not only of the same phyla but also a substantial fraction of common genera, and the similarity increases with sequencing depth [20]. Here, we generated a modified ‘adenoma–carcinoma sequence’ mouse colorectal cancer model induced by 1, 2-Dimethylhydrazine to investigate microbiota community variations during tumor formation progression. The initial step in the activation of DMH is series of oxidative steps that occur in the liver. Its metabolism ultimate activated to electrophilic carcinogens by the action of colon tissue enzymes and bacteria hydrolysis. There is need to be claimed that the half-life period of DMH and its metabolisms was over ten hours, however, from the last injection of DMH to the inflammation group was a week. We speculated that DMH has been completely metabolized in the inflammation group and the followed groups, DMH and its metabolites in the host residues have been negligible. Molecular changes were also examined during the ‘adenoma-carcinoma’ sequence. This animal model study may be useful in exploring the steps leading to human disease. In our study, transition from colon epithelium hyperproliferation through adenoma to adenocarcinoma occurred as a series of separate stages, but was a consecutive process complicated with microbe imbalance. The association between microbiota evolution, molecular changes and tumor development at every stage is therefore the key to understanding disease pathogenesis.
The development of next-generation sequencing technology has facilitated determination of the composition of gut microbiota in detail. In our study, 454 pyrosequencing based on the Roche 454 FLX+ platform was employed to detect the bacterial communities in stools. We observed a dynamic and previously undescribed gut microbiota shift in tumorigenesis. At the early stage of tumor formation, significant differences in Bacteroidetes between model and control groups were observed. We detected relative lower abundance of Firmicutes and Proteobacteria in the intestinal lumen of model mice, and variation of the gut microbiota was a dynamic process. Many of the bacterial species displayed increase-decrease trends, in accordance with the ‘driver-passenger’ model of Tjalsma et al. [7]. Clostridium lactatifermentans and Bacteroides dorei, characterized as ‘driver’ bacteria, were enriched in the inflammatory and hyperproliferation groups, and showed an obvious decrease with development of tumor. Conversely, Alistipes finegoldii increased step-by-step during tumorigenesis, supporting a role as a ‘passenger’ bacterium.
The potential roles of infection and inflammation in the pathogenesis of colorectal cancer cannot be overlooked [21]. The family of Enterobacteriaceae also participate in the mechanism of colorectal cancer. Housseau and Mangan [22, 23] showed that the actions of Enterobacteriaceae are similar to the prolonged inflammatory response induced by ETBF. Escherichia coli, a member of this family, contains the polyketide synthase (pks) genotoxic island encoding a genotoxin known as coli bactin that can induce singlestranded DNA breaks [24, 25].Subsequent activation of DNA damageinduced signaling pathways, in turn, increases the mutation rate of infected cells [26, 27]. Our results showed significant enrichment of Enterobacteriaceae in the model group, which was eventually outcompeted by opportunistic passengers during tumorigenesis.
It is likely that the altered microbial communities in our experiments belong to the same family and perform similar roles. These OTU-related species may be considered biological markers that influence longitudinal microbiota community changes during the development of ‘adenoma-carcinoma’ sequence colorectal cancer.
Recent research indicates a complex link between gut microbiome, immunity, and intestinal tumorigenesis. Mima [28] and colleagues confirmed that F. nucleatum is inversely associated with CD3+ T-cell density in colorectal carcinoma tissue. Chronic inflammation and tumorigenesis also probably result in loss of members of the microbial community important for maintaining epithelial health and immune homeostasis, and an increase in some opportunistic pathogens may intensify chronic inflammation and tumorigenesis [16]. Alistipes finegoldii, the first bacterium isolated mainly from appendiceal tissue samples in children with acute appendicitis, is bile-resistant and displays positive indole reaction. Annotation of the complete genome sequence by Konstantinos et al. [29] revealed that the genome is mainly associated with cell cycle control, cell division, replication, recombination and repair, intracellular trafficking and secretion, carbohydrate, amino acid, nucleotide, lipid transport and metabolism. A recent human study showed that Alistipes are enriched in CRC patients, compared with both healthy and advanced adenoma groups [30]. In our study, the three OTUs (OTU0151, 0105, 0022) related to Alistipes finegoldii were consistently increased during tumor development. We speculate that due to dysbiosis of gut microbiota and loss of barrier function of colon epithelium, Alistipes finegoldii may influence the cell cycle of colon epithelium and ultimately affect cell cycle regulation and leading to epithelial and tumor cell proliferation. Additionally, we noted a dramatic decrease in the family Porphyromonadaceae and increase in Rikenellaceae. We hypothesize that Porphyromonadaceae bacteria play a protective role and are important mediators of microbiota balance in the murine gut. One potential mechanism underlying this protective activity may be participation in gut metabolism and providing short-chain fatty acids. SCFA have a critical influence on colonic health, although their contribution to human energy requirement is far lower [31, 32]. Recently, the complete genome sequence of the family Porphyromonadaceae was reported, shown to contain genes catalyzing the production of volatile fatty acid-encoding proteins and enzymes related to the degradation of complex carbohydrates and proteinaceous compounds. Accordingly, Porphyromonadaceae is proposed to be involved in hydrolysis and acidogenesis during anaerobic digestion and biomethanation [33]. It is additionally possible that members of the family Porphyromonadaceae produce anti-inflammatory mediators that modulate the tumor immune microenvironment. Moreover, we speculate that the Rikenellaceae family functions as opportunistic pathogens via intensification of inflammation or production of mutagenic toxins.
CRC development can be effectively described by the genetic ‘adenoma–carcinoma sequence’ model originally developed by Fearon and Markowitz [3, 34]. The hypothesis states that loss of genomic stability, such as accumulating genetic and epigenetic mutations, can drive epithelial dysplasia and hyperplasia in the colon, eventually resulting in CRC. Over the course of progression, some ‘driver’ bacteria, such as enterotoxigenic Bacteroides fragilis or the Enterobacteriaceae family, induce inflammation, dysplasia, hyperplasia and/or genotoxic substances that contribute to the accumulation of mutations during the ‘adenoma–carcinoma sequence’. According to the study of Fearon and Markowitz, mutations start in the APC gene, leading to transition to adenoma, ending with a P53 substitution that triggers carcinoma. With microenvironment alterations during the oncogenic process, driver bacteria are gradually replaced with ‘passengers’ consisting of tumor-foraging opportunistic pathogens, such as Streptococcus spp., commensal or probiotic bacteria (e.g. Coriobacteriaceae) or other bacteria with a competitive advantage in the tumor niche. These previous studies agree with our findings. Bacteroidaceae and Enterobacteriaceae showed higher relative abundance in the early stages of tumor formation, and were gradually replaced with Rikenellaceae, Lachnospiraceae, Ruminococcaceae and Streptococcaceae. In particular, Clostridium lactatifermentans and Bacteroides dorei, characterized as ‘driver’ bacteria, were enriched in the inflammatory and hyperproliferation groups, followed by an obviously decrease with tumor development. Alistipes finegoldii from three related OTUs increased gradually during the process of tumorigenesis, possibly owing to activity as a ‘passenger’ bacterium. CRC has often been associated with activation of the Wnt/β-catenin pathway. Colitis-associated cancer mutations are reported to affect the Wnt/β-catenin pathway following mutations in the TP53 and K-RAS genes [35].
Sears and Goodwin [36, 37] reported that some bacterial pathogens are directly and specifically involved in promoting CRC. For example, Enterotoxigenic Bacteroides fragilis (ETBF) induces spermine oxidase-dependent ROS production and consequent DNA damage. F. nucleatum binds E-cadherin on epithelial cells and activates β-catenin signaling, driving epithelial cell proliferation [38]. These bacterial pathogens are thought to participate in tumorigenesis via several common pathways. In particular, STAT3 is broadly implicated in tumorigenesis, inducing suppression of apoptosis and promotion of cell cycle progression [39]. Apart from inflammatory cytokine signaling, activation of oncogenes and inactivation of suppressor genes are steps involved in the initiation of CRC, such as APC activation of Wnt signaling, due to an inability to decrease β-catenin oncoprotein [18]. Oncogenic mutations of RAS and BRAF activate the mitogen-activated protein kinase (MAPK) signaling pathway in 37% and 13% colorectal cancers, respectively [40–44]. RAS mutations, principally K-RAS, activate GTPase activity that signals directly to RAF. The K-RAS gene encodes a G-protein involved in a constitutively activated signaling pathway that promotes cell survival and apoptotic suppression [45]. Previous studies have reported that BRAF mutations occur, even in small polyps [40].
According to Winter et al. [46], dysbiosis of microbiota produces an abundance of potentially harmful microorganisms that induce inflammatory processes. Inflammation caused by gut bacteria is also considered to have an impact on carcinogenesis, leading to damage of intestinal barrier function, bacterial translocation and secretion of cytokines that maintain an inflammatory environment within the tumor [47]. The interactions between microbiota and molecular pathways underlying CRC require further investigation.
To a considerable extent, this work provides mechanistic insights into the interactions between the host and microbes during the process of tumor-promoting evolutionary changes. However, intestinal microecology is an extremely complex and diverse microbial community, and it is unlikely that one mechanism or hypothesis is applicable to all CRC patients. Clearly many questions remain involving whether the microbial community differences found in the present study are cause or consequence of tumor formation. We still need to provide more detailed information that concerning germ-free animals and transplantation of pathogenic bacteria. Further investigation is warranted to confirm the mechanisms underlying dysbiosis, which would aid in the development of effective methods to pre-diagnose CRC.
In summary, by establishing the network involving microbes and cellular molecules throughout the evolutionary progression of CRC tumorigenesis in the ‘adenoma-carcinoma sequence’ mouse colorectal cancer model, we have identified an imbalance in gut microbiota, characterized by the ‘driver-passenger’ model involve in reduction of butyrate-producing bacteria and increase in DNA-damaging bacteria, which leads to enhanced accumulation of mutations. These steps may be significant features of dysbiosis in animal models of CRC.
MATERIALS AND METHODS
Animals and reagents
Four-week-old male ICR mice (18–22 g) purchased from Qingdao Laboratory Animal Co. Ltd. (Qingdao, China) were used for study. All animals were maintained in plastic cages (five mice/cage) under conditions of humidity (44 ± 5%), light (12 h light/dark cycle) and temperature (22 ± 2°C), and fed certified standard mice chow and tap water ad libitum according to the institutional and National Institutes of Health (NIH) guidelines [48]. 1, 2-Dimethylhydrazine (DMH) was purchased from Beijing J&K Scientific Co. Ltd. DMH solution was prepared freshly before use in 25 mM EDTA/137 mMNaCl carrier (pH 6.4). All animal experiments were approved by the University Committee on the Use and Care of Animals at Qingdao University, and conducted in accordance with the National Institutes of Health guidelines.
Establishment of the ‘adenoma-carcinoma sequence’ animal model and experimental procedures
We modified the ‘adenoma-carcinoma sequence’ mouse CRC model induced by DMH. For generating the model, DMH induction was performed by administering a large dose (~35–40 mg/kg body weight) and increased number of injections (once a week for10–26 weeks) to ensure a high rate of morbidity [49, 50]. The model was modified to decrease the number and dose of injections. Consequently, morbidity was decreased and a percentage of lesion-free mice identified in each group. ‘Adenoma-carcinoma sequence’ CRC is a multistep process, from normal epithelial to neoplasm in a single crypt to the hyperproliferative stage, followed by adenoma, and finally adenocarcinoma [51, 52]. These steps are distinct but continuous in the morphological context.
After one week of acclimatization, mice were randomly divided into two groups. The model group (n = 120) received a subcutaneous (s.c.)injection of DMH solution at a dose of 20 mg/ kg body weight once a week for six consecutive weeks while the control group received a subcutaneous injection with the same volume of EDTA/NaCl carrier. On weeks 6, 12, 18 and 26, all mice in the model and control groups were anaesthetized via peritoneal injection of Ketamine (100 mg/kg), and sacrificed via cervical dislocation. The entire colon and rectum was surgically removed and opened longitudinally along the antimesenteric margin. Fresh feces of animals from both the model and control groups were collected after sacrifice, frozen immediately in liquid nitrogen, and stored at –80°C until DNA extraction. Tissues were washed with normal saline for observation, images obtained, and the total number of lesions counted. For histological examination and immunohistochemistry (IHC), tissues were fixed in 10% neutral phosphate-buffered formalin.
Histological and immunohistochemical examination
IHC was performed using standard protocols [53]. The entire mucosa was examined for macroscopic changes. Portions of the large bowel and tumors in this region were processed for paraffin embedding. Blocks were sectioned at 5 μm intervals, and slides stained with standard hematoxylin and eosin for light microscopic examination. Tissue sections were reviewed independently by two pathologists and re-evaluated by a third examiner in case of ambiguous results. Serial tissue sections 4 mm thick prepared from formalin-fixed and paraffin-embedded tissues were subjected to immunohistochemistry. IHC was completed in accordance with standard protocols using monoclonal antibodies against P53 (sc-6243, Rabbit, Santa Cruz), APC (SAB4200594, Rabbit, Sigma-Aldrich), K-RAS (sc-30, mouse, Santa Cruz) and BRAF (sc-9002, Rabbit, Santa Cruz).
DNA extraction, amplification and pyrosequencing
Genomic DNA was extracted from ~ 0.3 g of each sample in duplicate using the FastDNA SPIN Kit for Soil (Qbiogene-MP Biomedicals, Irvine, CA, USA), according to the manufacturer’s instructions. The V3-V5 hypervariable region of 16S rDNA genes was amplified using the primers 907F (5′-CCGTCAATTCMTTTGAGTTT-3′) and 338R (3′–ACTCCTACGGGAGGCAGCAG-5′). Each forward primer with a unique 10 bp barcode was used to tag each sample. The amplification mixture contained 25 μL Failsafe Premix F (Epicentre Biotechnologies, Madison, WI, U.S.A.), 0.4 μM each primer, 2.5 U of Ex Taq DNA polymerase (Takara, Dalian, China) and 1–2 μL DNA template in a total volume of 50 μL. PCR reactions were performed at 95°C for 5 min, followed by 25 cycles of 95°C for 30 s, 56°C for 30 s and 72°C for 90 s, and final extension at 72°C for 7 min. All samples were amplified in triplicate, pooled, and purified using the QIAquick PCR purification kit (Qiagen, Valencia, CA, USA). Pyrosequencing was performed on a 454 Genome Sequencer FLX platform (Roche) using the GS FLX Titanium XLR70 sequencing kit according to Roche Sequencing Method (Manual GS FLX Titanium Series October 2009 Edition). All DNA extraction, amplification and pyrosequencing procedures were operated by a professional at BGI (Shenzhen, China).
Bioinformatics analysis
Raw pyrosequencing data were processed using Mothur (Version 1.31.2, http://www.mothur.org/) to obtain unique reads. Ribosomal Database Project (RDP) Classifer v.2.2 was used to taxonomically classify OTU representative sequences in the following databases: Greengene V201305; RDP (Release9 201203).
Statistical analysis
Diversity indices, including the nonparametric richness estimator observed_species, Chao, Shannon, and Simpson index, were used to calculated the value of the sample Alpha diversity with Mothur (Version 1.31.2) and dilution curve generated using the Rprogram (Version 2.15.3). The Mann–Whitney test was applied to evaluate differences in the bacterial populations of each group. β-diversity and UniFrac analyses were carried out using QIIME v1.5.0. Significant difference analyses were calculated using Metastats (http://metastats.cbcb.umd.edu/), and P values corrected in R (Version 2.15.3). Data were considered statistically significant at P < 0.05. The Kruskal-Wallis rank sum test was employed to evaluate the differences between each group. Spearman’s rank correlation was used to assess the relationship between variables. Data analyses were performed in SPSS version 19.0.
Abbreviations
CRC, colorectal cancer; DMH, 1,2-Dimethylhydrazine; APC, adenomatous polyposis coli; OTU, operational taxonomic units; ROS, reactive oxygen species; SCFA, short-chain fatty acids
ACKNOWLEDGMENTS
We thank Professor Zhang Dongfeng of the Department of Epidemiology and Health Statistics, The Medical College of Qingdao University, for critical comments and methodological advice. We are grateful to Wang Su, Li Yanmin and Xu Zimin for assistance with performing experiments and sample collection. We additionally acknowledge Chen Jianwei, Wang hui, Jin aixiang, Bian Linling and Zhou Dajie in BGI(Shenzhen) for sequencing support and data analysis.
CONFLICTS OF INTEREST
The Department of General Surgery, Affiliated Hospital of Qingdao University, is the owner of this original research. The other author declares no competing interests.
FUNDING
This study was funded by the National Natural Science Foundation of China (Grant Number:81270449), Shandong Provincial Natural Science Foundation, China (Grant Number: ZR2012HM046), Qingdao Minsheng Science and Technology Foundation, Shandong, China (Grant Number:14-2-3-5-nsh), and the Qingdao Science and Technology Plan Project (Grant Number:13-1-4-220-jch).
REFERENCES
1. Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015; 136: E359–86. doi: 10.1002/ijc.29210.
2. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. Cancer J Clin. 2015; 65:87–108. doi: 10.3322/caac.21262.
3. Fearon ER. Molecular genetics of colorectal cancer. Annu Rev Pathol. 2011; 6:479–507. doi: 10.1146/annurev-pathol-011110-130235.
4. Vogelstein B, Kinzler KW. The multistep nature of cancer. Trends Genet. 1993; 9:138–41.
5. Stratton MR, Campbell PJ, Futreal PA. The cancer genome. Nature. 2009; 458:719–24. doi: 10.1038/nature07943.
6. Dalton-Griffin L, Kellam P. Infectious causes of cancer and their detection. J Biol. 2009; 8:67. doi: 10.1186/jbiol168.
7. Tjalsma H, Boleij A, Marchesi JR, Dutilh BE. A bacterial driver-passenger model for colorectal cancer: beyond the usual suspects. Nat Rev Microbiol. 2012; 10:575–82. doi: 10.1038/nrmicro2819.
8. Frank DN, St Amand AL, Feldman RA, Boedeker EC, Harpaz N, Pace NR. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci USA. 2007; 104: 13780–5. doi: 10.1073/pnas.0706625104.
9. Neish AS. Microbes in gastrointestinal health and disease. Gastroenterology. 2009; 136:65–80. doi: 10.1053/j.gastro.2008.10.080.
10. Sears CL, Garrett WS. Microbes, Microbiota, and Colon Cancer. Cell Host Microbe. 2014; 15:317–28. doi: 10.1016/j.chom.2014.02.007.
11. Chow J, Lee SM, Shen Y, Khosravi A, Mazmanian SK. Host-bacterial symbiosis in health and disease. Adv Immunol. 2010; 107:243–74. doi: 10.1016/B978-0-12-381300-8.00008-3.
12. Savage DC. Microbial ecology of the gastrointestinal tract. Annu Rev Microbiol. 1977; 31:107–33. doi: 10.1146/annurev.mi.31.100177.000543.
13. Kostic AD, Gevers D, Pedamallu CS, Michaud M, Duke F, Earl AM, Ojesina AI, Jung J, Bass AJ, Tabernero J, Baselga J, Liu C, Shivdasani RA, et al. Genomic analysis identifies association of Fusobacterium with colorectal carcinoma. Genome Res. 2012; 22:292–8. doi: 10.1101/gr.126573.111.
14. Castellarin M, Warren RL, Freeman JD, Dreolini L, Krzywinski M, Strauss J, Barnes R, Watson P, Allen-Vercoe E, Moore RA, Holt RA. Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome Res. 2012; 22:299–306. doi: 10.1101/gr.126516.111.
15. Marchesi JR, Dutilh BE, Hall N, Peters WH, Roelofs R, Boleij A, Tjalsma H. Towards the human colorectal cancer microbiome. PLoS One. 2011; 6:e20447. doi: 10.1371/journal.pone.0020447.
16. Kostic AD, Chun E, Robertson L, Glickman JN, Gallini CA, Michaud M, Clancy TE, Chung DC, Lochhead P, Hold GL, El-Omar EM, Brenner D, Fuchs CS, et al. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host Microbe. 2013; 14:207–15. doi: 10.1016/j.chom.2013.07.007.
17. Liang X, Li H, Tian G, Li S. Dynamic microbe and molecule networks in a mouse model of colitis-associated colorectal cancer. Sci Rep. 2014; 4:4985. doi: 10.1038/srep04985.
18. Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, Kinzler KW. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science. 1997; 275:1787–90.
19. Baker SJ, Fearon ER, Nigro JM, Hamilton SR, Preisinger AC, Jessup JM, vanTuinen P, Ledbetter DH, Barker DF, Nakamura Y, White R, Vogelstein B. Chromosome 17 deletions and p53 gene mutations in colorectal carcinomas. Science. 1989; 244:217–21.
20. Krych L, Hansen CH, Hansen AK, van den Berg FW, Nielsen DS. Quantitatively different, yet qualitatively alike: a meta-analysis of the mouse core gut microbiome with a view towards the human gut microbiome. PLoS One. 2013; 8:e62578. doi: 10.1371/journal.pone.0062578.
21. Wu S, Rhee KJ, Albesiano E, Rabizadeh S, Wu X, Yen HR, Huso DL, Brancati FL, Wick E, McAllister F, Housseau F, Pardoll DM, Sears CL. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat Med. 2009; 15:1016–22. doi: 10.1038/nm.2015.
22. Housseau F, Sears CL. Enterotoxigenic Bacteroides fragilis (ETBF)-mediated colitis in Min (Apc+/−) mice: a human commensal-based murine model of colon carcinogenesis. Cell Cycle. 2010; 9:3–5.
23. Mangan PR, Harrington LE, O’Quinn DB, Helms WS, Bullard DC, Elson CO, Hatton RD, Wahl SM, Schoeb TR, Weaver CT. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. 2006; 441: 231–4. doi: 10.1038/nature04754.
24. Arthur JC, Perez-Chanona E, Muhlbauer M, Tomkovich S, Uronis JM, Fan TJ, Campbell BJ, Abujamel T, Dogan B, Rogers AB, Rhodes JM, Stintzi A, Simpson KW, et al. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science. 2012; 338:120–3. doi: 10.1126/science.1224820.
25. de Vos WM, de Vos EA. Role of the intestinal microbiome in health and disease: from correlation to causation. Nutr Rev. 2012; 70:S45–56. doi: 10.1111/j.1753-4887.2012.00505.x.
26. Nougayrede JP, Homburg S, Taieb F, Boury M, Brzuszkiewicz E, Gottschalk G, Buchrieser C, Hacker J, Dobrindt U, Oswald E. Escherichia coli induces DNA double-strand breaks in eukaryotic cells. Science. 2006; 313:848–51. doi: 10.1126/science.1127059.
27. Cuevas-Ramos G, Petit CR, Marcq I, Boury M, Oswald E, Nougayrede JP. Escherichia coli induces DNA damage in vivo and triggers genomic instability in mammalian cells. Proc Natl Acad Sci USA. 2010; 107:11537–42. doi: 10.1073/pnas.1001261107.
28. Mima K, Sukawa Y, Nishihara R, Qian ZR, Yamauchi M, Inamura K, Kim SA, Masuda A, Nowak JA, Nosho K, Kostic AD, Giannakis M, Watanabe H, et al. Fusobacterium nucleatum and T Cells in Colorectal Carcinoma. JAMA Oncol. 2015; 1:653–61. doi: 10.1001/jamaoncol.2015.1377.
29. Mavromatis K, Stackebrandt E, Munk C, Lapidus A, Nolan M, Lucas S, Hammon N, Deshpande S, Cheng JF, Tapia R, Goodwin LA, Pitluck S, Liolios K, et al. Complete genome sequence of the bile-resistant pigment-producing anaerobe Alistipes finegoldii type strain (AHN2437(T)). Stand Genomic Sci. 2013; 8:26–36. doi: 10.4056/sigs.3527032.
30. Feng Q, Liang S, Jia H, Stadlmayr A, Tang L, Lan Z, Zhang D, Xia H, Xu X, Jie Z, Su L, Li X, Li X, et al. Gut microbiome development along the colorectal adenoma-carcinoma sequence. Nat Commun. 2015; 6:6528. doi: 10.1038/ncomms7528.
31. Scheppach W, Luehrs H, Menzel T. Beneficial health effects of low-digestible carbohydrate consumption. Br J Nutr. 2001; 85:S23–30.
32. Mortensen PB, Clausen MR. Short-chain fatty acids in the human colon: relation to gastrointestinal health and disease. Scand J Gastroenterol Suppl. 1996; 216:132–48.
33. Hahnke S, Maus I, Wibberg D, Tomazetto G, Puhler A, Klocke M, Schluter A. Complete genome sequence of the novel Porphyromonadaceae bacterium strain ING2-E5B isolated from a mesophilic lab-scale biogas reactor. J Biotechnol. 2015; 193:34–6. doi: 10.1016/j.jbiotec.2014.11.010.
34. Markowitz SD, Bertagnolli MM. Molecular origins of cancer: Molecular basis of colorectal cancer. N Engl J Med. 2009; 361:2449–60. doi: 10.1056/NEJMra0804588.
35. Lakatos PL, Lakatos L. Risk for colorectal cancer in ulcerative colitis: changes, causes and management strategies. World J Gastroenterol. 2008; 14:3937–47.
36. Sears CL. Enterotoxigenic Bacteroides fragilis: a rogue among symbiotes. Clin Microbiol Rev. 2009; 22:349–69, Table of Contents. doi: 10.1128/CMR.00053-08.
37. Goodwin AC, Destefano Shields CE, Wu S, Huso DL, Wu X, Murray-Stewart TR, Hacker-Prietz A, Rabizadeh S, Woster PM, Sears CL, Casero RA, Jr. Polyamine catabolism contributes to enterotoxigenic Bacteroides fragilis-induced colon tumorigenesis. Proc Natl Acad Sci USA. 2011; 108: 15354–9. doi: 10.1073/pnas.1010203108.
38. Rubinstein MR, Wang X, Liu W, Hao Y, Cai G, Han YW. Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E-cadherin/beta-catenin signaling via its FadA adhesin. Cell Host Microbe. 2013; 14:195–206. doi: 10.1016/j.chom.2013.07.012.
39. Elinav E, Nowarski R, Thaiss CA, Hu B, Jin C, Flavell RA. Inflammation-induced cancer: crosstalk between tumours, immune cells and microorganisms. Nat Rev Cancer. 2013; 13:759–71. doi: 10.1038/nrc3611.
40. Nosho K, Irahara N, Shima K, Kure S, Kirkner GJ, Schernhammer ES, Hazra A, Hunter DJ, Quackenbush J, Spiegelman D, Giovannucci EL, Fuchs CS, Ogino S. Comprehensive biostatistical analysis of CpG island methylator phenotype in colorectal cancer using a large population-based sample. PLoS One. 2008; 3:e3698. doi: 10.1371/journal.pone.0003698.
41. Bos JL, Fearon ER, Hamilton SR, Verlaan-de Vries M, van Boom JH, van der Eb AJ, Vogelstein B. Prevalence of ras gene mutations in human colorectal cancers. Nature. 1987; 327:293–7. doi: 10.1038/327293a0.
42. Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, Davis N, Dicks E, Ewing R, et al. Mutations of the BRAF gene in human cancer. Nature. 2002; 417:949–54. doi: 10.1038/nature00766.
43. Rajagopalan H, Bardelli A, Lengauer C, Kinzler KW, Vogelstein B, Velculescu VE. Tumorigenesis: RAF/RAS oncogenes and mismatch-repair status. Nature. 2002; 418:934. doi: 10.1038/418934a.
44. Siena S, Sartore-Bianchi A, Di Nicolantonio F, Balfour J, Bardelli A. Biomarkers predicting clinical outcome of epidermal growth factor receptor-targeted therapy in metastatic colorectal cancer. J Natl Cancer Inst. 2009; 101: 1308–24. doi: 10.1093/jnci/djp280.
45. Downward J. Ras signalling and apoptosis. Curr Opin Genet Dev. 1998; 8:49–54.
46. Winter SE, Lopez CA, Baumler AJ. The dynamics of gut-associated microbial communities during inflammation. EMBO Rep. 2013; 14:319–27. doi: 10.1038/embor.2013.27.
47. Jobin C. Colorectal cancer: CRC—all about microbial products and barrier function? Nat Rev Gastroenterol Hepatol. 2012; 9:694–6. doi: 10.1038/nrgastro.2012.220.
48. Sandra Newbury, Mary K.blinn, Philip A. Bushby CBC, Julie D Dinnage, Brenda Griffin, Kate F Hurley, Natalie lsaza, Wes Jones, Lila Miller, Jeanette O’Quin, Gary J. Patronek, Martha Smith-blackmore, Miranda Spindel. Guidelines for Standards of Care in Animal Shelters. National Institutes of Health. 2010.
49. Chang WW. Histogenesis of symmetrical 1,2-dimethylhydrazine-induced neoplasms of the colon in the mouse. J Natl Cancer Inst. 1978; 60:1405–18.
50. Zhu Q, Jin Z, Wu W, Gao R, Guo B, Gao Z, Yang Y, Qin H. Analysis of the intestinal lumen microbiota in an animal model of colorectal cancer. PLoS One. 2014; 9:e90849. doi: 10.1371/journal.pone.0090849.
51. Chang WW. Morphological basis of multistep process in experimental colonic carcinogenesis. Virchows Arch B Cell Pathol Incl Mol Pathol. 1982; 41:17–37.
52. Chang WW. The mode of growth and compartmentalization of neoplastic glands during experimental colon carcinogenesis. Am J Pathol. 1986; 124:420–6.
53. Vandeputte DA, Troost D, Leenstra S, Ijlst-Keizers H, Ramkema M, Bosch DA, Baas F, Das NK, Aronica E. Expression and distribution of id helix-loop-helix proteins in human astrocytic tumors. Glia. 2002; 38:329–38. doi: 10.1002/glia.10076.