
INTRODUCTION
Tyrosine kinase inhibitors (TKIs) are the standard front line targeted therapy in CML and an integral part of combination chemotherapy for Philadelphia-positive acute lymphoblastic leukemia (Ph+ ALL). TKIs have led to improvement in rates of complete remission and overall outcomes in both CML as well as Ph+ ALL [1, 2]. The role of allogeneic stem cell transplantation (allo-SCT) in CML is reserved for patients who fail or are intolerant to TKI therapy, or for those with advanced accelerated and blast phase disease [3]. Transplantation remains the standard of care for Ph+ ALL [2].
Relapse rates after allo-SCT are as high as 30–40% and 30–60% for both advanced phase CML and Ph+ ALL, respectively [4, 5]. Therapeutic strategies to prevent or treat relapse after allo-SCT include withdrawal of immunosuppression and donor lymphocyte infusions (DLI) [6, 7]. Given their value in treating relapsed CML or Ph+ ALL [8–15], TKIs were also studied as part of a preemptive or prophylactic approach post allo-SCT. Several groups have evaluated imatinib for the prevention of relapse in high-risk CML and Ph+ALL patients with encouraging results [16–20]. Second generation TKIs, including nilotinib, may be more efficient in preventing and treating relapse of advanced CML and Ph+ ALL [7, 21]. However, data on their use in the post allo-SCT setting are limited [7, 22, 23].
The success of allo-SCT is dependent on both hematopoietic stem cells’ engraftment and immune reconstitution. Immune reconstitution is a complex process requiring functional hematopoietic stem cells in the bone marrow niche and adequate thymic function [24]. Without full reconstitution of the immune system following allo-SCT, patients are more susceptible to life threatening viral and fungal infections. Furthermore, they are also prone to tumor relapse, as graft-versus-leukemia (GVL) depends on a functional immune system [25, 26]. CML is particularly sensitive to allogeneic immune reactivity; there is a dominant GVL effect induced by DLI in treating disease relapse, and improved leukemia-free survival in patients with graft-versus-host disease (GvHD) [27, 28].
The interaction between post-allo-SCT TKI administration and immune reconstitution is of major importance, given the critical role of the latter in the course following transplantation, as detailed above. In several in-vitro studies, inhibition of the innate immune cells activation as well as T-cell proliferation and function were noted [24, 29–32]. However, others have reported that patients treated with TKI have near-normal levels of immunological parameters and ex-vivo response to various cytokine stimuli [27]. Thus, the literature is inconsistent regarding the effects of TKIs on the immune system in the post-allo-SCT setting.
We recently reported the clinical outcomes of a phase 1/2 study in CML and Ph+ ALL patients treated with nilotinb after allogeneic SCT. Nilotininb was safe and partially effective for the prevention of relapse after allo-SCT [23]. In the current study, we further explored nilotinib effect on immune reconstitution post allo-SCT. Our aim was to quantitatively characterize immune subpopulations and evaluate their function including T-cell response to mitogens, NK cytotoxic activity, and T-cell repertoire and thymic activity (TREC) at designated time points up to 1 year after transplantation while on nilotinib therapy.
RESULTS
Total cell numbers
The relation between total white blood cells (WBC) and lymphocytes was analyzed directly from complete blood counts (Figure 1 and Table 1). Mean (± standard error) WBC at day 28 of nilotninb treatment (4014 ± 398 cells/ml) was similar to that measured post allo-SCT and before nilotinib treatment (4137 ± 600 cells/ml), whereas a significant increment of WBC was observed at day 90 of nilotinib treatment (5887 ± 771 cells/ml, p = 0.04). WBC counts continued to increase thereafter, with a mean of 9250 ± 904 cells/ml on day 335 (Figure 2). When compared to their level at day 28 of nilotinib administration, an increase in total lymphocytes was first noted at day 180 (1693.7 ± 166.6 vs. 942.8 ± 120.6 cells/ml, p < 0.001, respectively). Lymphocyte counts were maintained up to day 335 post nilotinib administration (Table 1).
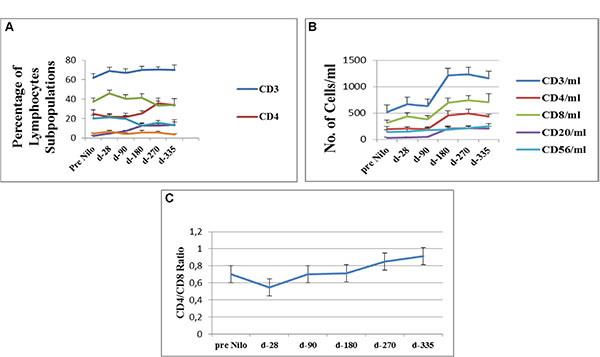
Figure 1: Flow cytometry analysis of lymphocytes subpopulations. (A) Percentage of cells expressing specific lymphocytes surface markers (CD3, CD4,CD8, CD20 and CD56). (B) Average concentration of lymphocytes subpopulations, calculated from their percentage on gated CD45pos cells. (C) CD4/CD8 ratio calculated from their concentration at each study time point. CD - cluster of differentiation.
Table 1: Immune reconstitution after allo-SCT during nilotinib treatment
Pre nilotinib |
Day 28 |
Day 90 |
Day 180 |
Day 270 |
Day 335 |
|
|---|---|---|---|---|---|---|
Total WBC/ml |
4.138 ± 600.5 |
4.014 ± 3.98 |
5.877 ± 771 |
7.081 ± 618.4 |
7.810 ± 608 |
9.250 ± 905 |
Total Lym/ml |
820 ± 148.5 |
943 ± 120.6 |
924.3 ± 102.6 |
1693.7 ± 167 |
1,754 ± 109.4 |
1,647 ± 147 |
CD3/ml |
523.7 ± 78 |
665.3 ± 89.8 |
633.7 ± 87.2 |
1213.3 ± 146 |
1233.3 ± 71.2 |
1160.8 ± 153 |
CD4/ml |
199.6 ± 46.8 |
202.8 ± 37.7 |
197.3 ± 38.7 |
457.1 ± 87.5 |
490.7 ± 77.1 |
434.5 ± 45 |
CD8/ml |
318.1 ± 52 |
437.1 ± 67.8 |
385.3 ± 69.3 |
696.8 ± 88.1 |
738.1 ± 93.0 |
704.5 ± 158.0 |
CD20/ml |
30.0 ± 14 |
39.1 ± 11.9 |
45.7 ± 7.3 |
210.3 ± 44.4 |
218.3 ± 30.1 |
208.0 ± 41.1 |
CD56/ml |
145 ± 35.3 |
146.9 ± 41.5 |
175.2 ± 41.8 |
189.0 ± 48.8 |
215 ± 44.9 |
255.1 ± 49.2 |
Total WBC and total lymphocytes were tested by CBC counts. T cells subpopulation counts were analyzed by Flow Cytometry. Calculations of cells percent and concentrations were performed on gated CD45pos cells.
Abbreviations: WBC - white blood cells, CD - cluster of differentiation.
Lymphocyte subpopulations
CD3pos T-cells
Lymphocyte subpopulations were identified from a gated CD45pos cell population. Starting after allo-SCT pre-nilotinib administration a constant high percentage of CD3pos T lymphocytes was observed (62% ± 4.3) (Figure 1A). CD3pos cells numbers significantly increased at day 180 after nilotinb administration (mean 1213.3 ± 146 × 106/ml; p < 0.001) compared to their numbers at day 28 and at day 90 (665.3 ± 89.8 × 106/ml and 633 ± 87 × 106/ml, respectively). CD3pos T-cell counts were maintained at day 270 and up to the last assessment at day 335 (Figure 1B, Table 1).
CD4pos T-cells
The percentage of CD4pos cells began to increase at day 270 of nilotinib administration (35.8 ± 5.3%; p = 0.06) compared to values measured pre-nilotinb administration. (Figure 1A, Table 1). CD4pos cell counts significantly increased at day 180 (457.1 ± 87.5 × 106/ml; p = 0.01 compared to their values at day 28 (202.8 ± 37.7 × 106/ml). Counts remained stable at day 270 (490.7 ± 77.1 × 106/ml) and at day 335 (434.5 ± 44.9 × 106/ml), respectively (Figure 1B).
CD8pos T-cells
The percentage of CD8pos cells remained stable from post-allo-SCT-pre-nilotinib until the last evaluation at day 335 (Figure 1A). An increase in CD8pos cells was observed after 180 days of nilotinib treatment (696.8 ± 88 × 106/ml), compared to their measurement at the post-allo-SCT-pre-nilotinib time point (318.1 ± 52 × 106/ml; p = 0.001); (Figure 1B, Table 1). These results effect the CD4/CD8 ratio, which was calculated to evaluate immune system activity potential.
CD4/CD8 ratio
The CD4/CD8 ratio consistently increased from day 28 of nilotinib administration (0.55 ± 0.18); 0.7 ± 0.11, 0.85 ± 0.17, and 0.91 ± 0.21 at days 180, 270, and 335, respectively (Figure 1C).
CD20pos B-Cells
The percent of CD20pos cells out of total lymphocytes significantly increased at day 180 (12.9 ± 2.6%; p > 0.001), with no difference at day 270 (12.6 ± 1.8%) and on day 335 (13.7 ± 5.5%), compared to CD20pos percent found post allo-SCT-pre-nilotinib administration. CD20pos counts increased at day 180 (210.3 ± 44.4 × 106/ml; p = 0.002) compared to CD20pos counts found post allo-SCT-pre-nilotinib administration and remained stable up to day 335 (Figure 1A–1B, Table 1).
NK cells (CD56pos, CD3negative)
The percent of CD56pos cells out of the total lymphocyte population was stable from post allo-SCT-pre-nilotinib (20.4 ± 3.14%) up to day 90 post nilotinib administration, following a significant reduction at day 180 (12.5 ± 2.2%; P < 0.04). Counts of Natural Killer cells increased insignificantly from day 90 (P = 0.32) to day 335 (p = 0.18) post nilotinib administration, compared to their values pre-nilotinib and at day 28 (146.9 ± 41.5 × 106/ml) (Figure 1A–1B, Table 1).
Mitogenic responses
Lymphocyte function was assessed by their ability to proliferate in response to stimulation by different mitogens (see material and methods). The average stimulation index (SI) values obtained after anti-CD3 (αCD3) cross-linking decreased insignificantly at day 90 (20.1 ± 5.2) compared to the values obtained pre-nilotinib (30 ± 9.6; p = 0.1) and at day 28 post nilotinib administration (31.6 ± 7.5; p = 0.1), respectively (Figure 2). A decline in the pick stimulation index was observed at day 180 (23 ± 5.4), while a modest insignificant increment was measured at days 270 and 335 post nilotinib administration (42.3 ± 8, p = 0.1; 46.7 ± 11.7, p = 0.1; respectively) (Figure 2).
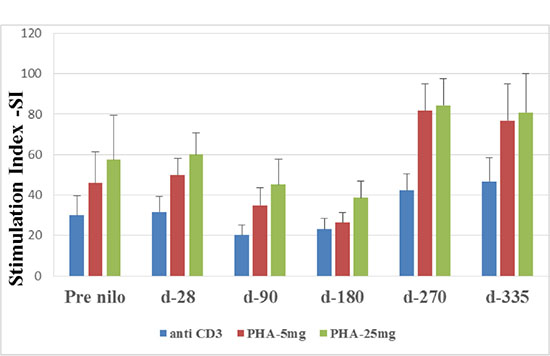
Figure 2: Stimulation Index (SI) of T lymphocytes. T lymphocytes function was tested by their activation abilities to different mytogene at different time points after allo-SCT. SI was calculated as the ratio of (3H) Thymidine uptake of stimulated cells divided by (3H) Thymidine uptake of non-stimulated cells. SI- Stimulation Index, CD- cluster of differentiation, PHA- phytohaemagglutinine.
Similar results were observed in response to PHA at 5 mg and 25 mg. Pick low stimulation index was observed at day 180 (26.3 ± 5.2; and 38.6 ± 8.3; respectively), increased at day 270 (81 ± 13.3; p = 0.02 and 84.5 ± 13.1; p = 0.03, respectively) and remained high at day 335 (76.8 ± 18.3; p = 0.05 and 80.7 ± 19.4; p = 0.09) (Figure 2).
Cytotoxic activity
Natural killer cells function was evaluated by their cytotoxic activity against K562 cells. Figure 3 shows the percentage of dead target cells (K562 cell line) following co-culture with patients NK cells (in different E:T ratios). At all time points, increased E:T ratios, resulted in correlated higher percentage of specific killing, and maximal effect was found at 5:1 E:T ratio. Moreover, a similar percent of target cells death were determined up to day 270 post nilotinib treatment, compared to target cell death prior to nilotinib administration. Specific killing was significantly increased at day 335 post nilotinib administration (49.4 ± 7; p = 0.05), compared to values at day 270 (29.7 ± 3.7) (Figure 3).
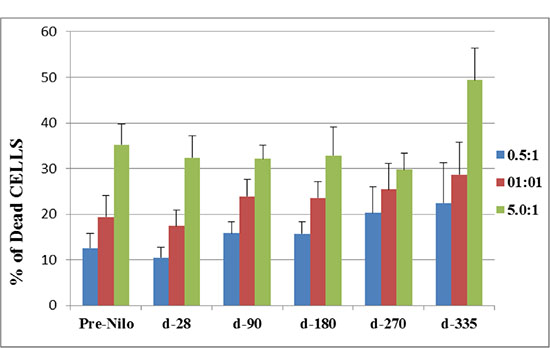
Figure 3: Cytotoxic activity of NK effector cells against K562 cell line as target cells. The cytotoxic activity of NK cells is presented as the percent of dead target cells at d-28, 90, 270 and 335 post nilotinib administration, after co-culture at different E:T ratios. NK cells were obtained from peripheral blood samples of post SCT patients treated with nilotinib. NK–natural killers, E-effector, T- target.
Thymic activity
In order to assess thymic activity, TREC quantification and analysis of the TCR repertoire were performed. Before treatment and despite immunosuppression from previous interventions, the average pre-nilotinb TREC levels were 81.8 ± 108 copies per 0.5 ug DNA. At the end of follow-up, after patients received allo-SCT and nilotinib treatment, TRECs levels increased (142.8 ± 197.4 copies per 0.5 ug DNA). However, this trend did not reach statistical significance. (Table 2) Similarly, the surface membrane expression of 24 different TCR Vβ (TRBV) post-treatment was not different from baseline expression. At the end of follow up, patients had normal expression of 15.1 ± 5.5 receptors (of the 24 that were examined), 2.5 ± 2.2 receptors with clonal expression and 6.4 ± 4.3 receptors with reduced expression. These numbers were similar to the expression of the TCR Vβ before allo-SCT and nilotinib treatment (normal receptors 15.3 ± 5.6, clonal receptors 2.9 ± 3 and reduced receptors 5.8 ± 4.5, respectively).
Table 2: TREC content and TCR repertoire
Pre Nilotinib treatment |
Post Nilotinib treatment |
|
|---|---|---|
TREC content (copies/ 0.5 ug DNA) |
81.8 ± 108 |
142.8 ± 197.4 |
TCR repertoire – normal expression |
15.3 ± 5.6 |
15.1 ± 5.5 |
TCR repertoire – clonal expression |
2.9 ± 3 |
2.5 ± 2.2 |
TCR repertoire – reduced expression |
5.8 ± 4.5 |
6.4 ± 4.3 |
TREC content and surface membrane expression of 24 different T cell receptor Vβ families in patients’ samples collected before and at the last nilotinib treatment last time point. TCR repertoire for each patient was calculated as percentage of normal expression, clonal expression and reduced expression.
Abbreviations: TREC - TCR excision circles, TCR - T cell receptor.
DISCUSSION
We have recently demonstrated the feasibility, safety, and efficacy of nilotinib, a second-generation TKI, as maintenance therapy after allogeneic SCT in patients with advanced CML and Ph+ ALL [23]. In the current study, we show that nilotinb administration up to one year following allogeneic SCT does not jeopardize immune reconstitution and function in the recipients.
TKIs are increasingly used as a complementary therapy after SCT. Several phase 2 studies explored use of imatinib treatment after SCT and have shown promising outcomes [16–20, 33]. Recently, Pfeifer et al., published a randomized trial studying the role of TKIs in the post SCT in patients with Ph+ ALL, comparing prophylactic imatinib and preemptive imatinib therapy, triggered by the detection of minimal residual disease (MRD) [19]. Post-transplant imatinib resulted in low relapse rates and excellent outcomes that were equivalent in both arms [19].
Findings from pre-clinical studies on the effect of imatinib on the immune response are conflicting, ranging from impaired antigen-specific T-cell responses [34–36] to reversal of T-cell tolerance and potentiation of antitumor immune responses [37, 38]. Limited in vitro data that are available with second-generation TKIs, including nilotinib, showed impaired antigen-specific T-cells, NK cell response, and B-cell activation [29, 31, 39–42]. However, recent clinical studies reported that dasatinib enhances NK and NK/T-cell proliferation and activation [43].
The speed of immune recovery after SCT is of central importance for overcoming complications. A rapid immune reconstitution post SCT protects the transplanted patients from severe infections and other transplant related complications as well as relapse [44–47]. The aim of our study was therefore to follow the effect of post-transplantation nilotinib administration on immune reconstitution after allogeneic SCT. Quantitative and functional aspects of the recipients’ immune system were studied during nilotinib administration and throughout the post-transplantation course (from day 28 through day 335). The former was evaluated through characterization of lymphocyte sub-populations, while the latter was investigated by studying T-cell proliferation in response to mitogenic induction and killing assay mediated by NK cells. In addition, thymic function was determined by TREC and TCR repertoire. TREC are by-products of V(D)J recombination at the TCR V/β locus and their presence indicates de-novo lymphopoiesis. This was found to be an excellent predictor for T cell immune reconstitution after SCT [48]. A normal TCR repertoire reflects ability of T-cells to produce a required diversity.
Following SCT there is severe depletion of all hematopoietic cells of the immune system, especially lymphocytes. Previous studies showed that innate immunity recovers within weeks after allogeneic SCT, whereas adaptive immunity (B- and T-cells) recovers within months to years [49, 50]. NK cells (CD16- CD56+), as part of innate immunity, recover both numerically and functionally within the first few weeks following SCT [51]. Our findings confirm the rapid recovery of NK cells, reaching the normal adult level even before nilotinib treatment. Moreover, their level remained stable during nilotinib treatment.
Previous studies indicated that the recovery of lymphocytes and their subpopulation is a major predictor of transplantation success [52]. In this context, our finding that the mean WBC concentration measured on day 28 of niliotinib treatment was similar to that measured post-SCT-pre-nilotinib and remained stable during follow up is therefore of importance. It appears that nilotinib did not impair patients’ leukopoiesis or result in prolonged leukopenia after SCT.
It is well established that balanced recovery of CD4 and CD8 T-cell subsets is needed to control alloimmunity and establish immune tolerance [53, 54]. We demonstrate that CD8 T cells quickly reached normal levels and continued to increase until day 180 after transplant. CD4 T-cells remained below normal until 180–270 days follow-up period. Recovery of CD4 T-cells was somewhat faster in comparison to previous publications, which had demonstrated CD4 T-cells below normal throughout one year after SCT [55, 56]. Similar to T-cells, B-cell counts were shown to be deeply depressed immediately after transplant, reaching normal levels only by nine months after allogeneic SCT [57]. In our cohort, levels of CD20pos B cells were attained after 180 days (approximately 6 months). Again, this indicates the nilotinib does not delay immune recovery and may even facilitate it.
Overall, despite exposure to nilotinib, T-cells responded to mitogenic stimulation and, even more importantly, no functional deficit was observed. There was, however, a decline in T-cell mitogenic response between day 90 to day 180, which might be due the disappearance of graft T-cells that are reported to be present in the recipient blood up to approximately 3 months after transplantation [28, 50, 58, 59]. These cells provide early post-transplant protection against infection, and are reported to be replaced between day 180 to day 335 by the newly reconstituted stem-cell originated T-cells. Our results differ from other studies that showed nilotinib treatment inhibit PHA and IL-2-induced proliferation and activation markers (CD69 and CD25) on CD8+ T lymphocytes in a dose-dependent manner [29, 32]. These differences may be due to our having analyzed T-cell function after relatively prolonged treatment with nilotinib, enabling T-cells to better adapt to and tolerate the drug. Furthermore, measuring T-cells function at several defined time-points during the treatment with nilotinib allowed us to monitor the different tempos of T-cell reconstitution.
Effective reconstitution of T-cells following SCT requires a functional thymus, particularly in the case of CD4+ T cells [60]. With regard the thymic activity, in our study both TREC levels and surface membrane expression of different TCR Vβ families were detected at the end of follow-up in patients who received SCT and nilotinib treatment. Notably, despite being heavily immunosuppressed from previous procedures and leukemia, patients were able to reconstitute their thymic activity. TREC quantification is a suitable and practicable method to monitor thymic output post-SCT in adult patients [61], and has been shown to be an robust assay for determining immune reconstitution and for the prediction of morbidity and mortality after SCT [62]. The process of T-cell neogenesis after SCT becomes evident by 6 months, and normal levels of adult thymic functions are restored at 12 months [63]. In parallel, the TCR repertoire is considerably abnormal during the first year after transplantation, but normalizes by the time of full immune reconstitution, as was seen in our patients.
NK cells sub-population is one of the pillars of innate immunity. Our findings show that NK cells were reconstituted by one month post-transplant. These early reconstituting cells are characterized as donor-derived NK cells that effectively lyse recipient leukemia cells in-vitro [59], and are thus very important in preventing disease relapse. We evaluated NK cell function before and during nilotinib administration (up to 335 days) by their cytotoxic activity against K562 cell line in different E:T ratios. We demonstrated that the percent of target-cell-specific death by patients’ NK cells was stable at post-SCT-pre-nilotinib through day 270 of nilotinib administration. A small but statistically insignificant increment in cell-specific killing was found at day 335. Since it is known that NK cells reconstitute by the first month following allo-HSCT, evaluation of patients’ NK activity pre-nilotinib administration may serve as a valuable control for assessing the effect of nilotinib therapy on NK function. Overall, our results indicate that nilotinib did not attenuate natural killer cytotoxic capabilities. These results align with those presented by Salih et al, which showed no effect of nilotinib administration on cytotoxic activity of NK cells obtained from CML patients, although their cytokine production was significantly reduced [30].
Taken together, our results indicate that use of nilotinib as maintenance therapy following allogeneic SCT in CML and Philadelphia positive ALL does not jeopardize immune reconstitution; WBC counts, lymphocyte subpopulation, and ex-vivo immune functions recovered as expected, concomitant with the nilotinib exposure. Consequently, we suggest that all patients with Ph-positive ALL and advanced stage CML are candidates for the post-transplant use of TKIs to reduce the risk of relapse. The issue of TKI treatment after allo-SCT remains a relatively poorly explored area of investigation with many open questions requiring further research. Rigorous monitoring of MRD allows the identification of the patients who will benefit most from TKI treatment after allo-SCT. A preemptive strategy should be applied only if adequate monitoring of BCR-ABL transcripts is available with the use of real-time quantitative PCR [64]. Patients who cannot be rigorously monitored for the MRD status should be treated prophylactically. Even in cases of strict monitoring, the sensitivity of the methods used for MRD detection varies, with real-time quantitative PCR still insufficiently standardized [65].
Future prospective comparative studies should explore immunological status during treatment with different TKIs after allo-SCT: should TKIs be used as a mean of relapse prophylaxis, or as preemptive therapy triggered by the detection of MRD; what kind of TKI should be used; which TKI is better tolerated in the early period after allo-SCT; and what is the optimal treatment duration with a TKI.
MATERIALS AND METHODS
Patient characteristics
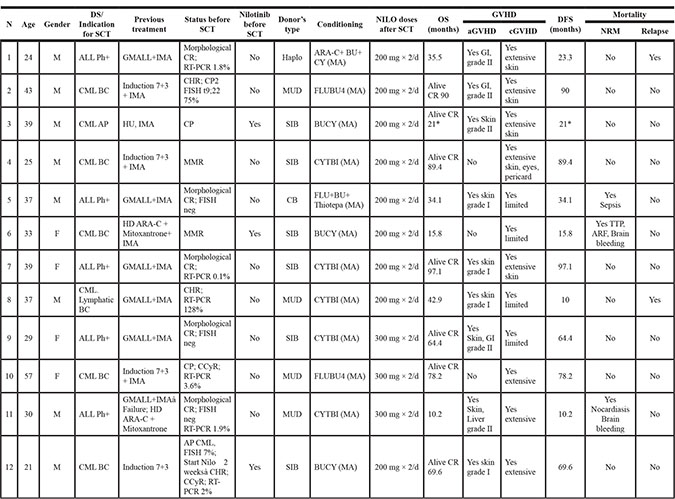
The complete eligibility criteria and the clinical characteristics of the cohorts were previously described by Shimoni et al [23]. Of the original 16 patients who participated in the clinical study, full immunological evaluation was available on 12 patients receiving nilotinib for at least 90 days following allo-SCT (Table 3). Three of the 12 patients also received nilotinib prior to SCT. Patients were included in the study regardless of the number or types of previous therapies. The schedule of post allo-SCT nilotinib administration is described in detail by Shimoni et al. [23]. In the 12 patients included in the current analysis, nilotinib was started at a median of 55 days (range 33–200) after allo-SCT. Nine patients received nilotinib at a dose of 200 mg × 2/day and three patients received nilotinib at a dose of 300 mg × 2/day, respectively. Due to increased toxicity, none of the patients received nilotinib at the highest dose of 400 mg × 2/day [23]. All patients received cyclosporine as part of post-transplant GVHD prophylaxis. At least 50% of the patients received steroids during the study period. The dose and treatment duration were different and the subgroups were too small to stratify patients for immunological assessment.
Table 3: Patients’ characteristics and outcomes
*Dropped from follow up after study was completed
Abbreviations: CML – chronic myeloid leukemia, BC – blast crisis , AP – accelerated phase , ALL – acute lymphoblastic leukemia, Ph+ Philadelphia positive, GMALL – German multicenter ALL protocol, IMA- Imatinib, CR- Complete remission, RT-PCR – Real time polymerase chain reaction , ARA-C – cytosine arabinoside, HD – high dose, BU - Busulfan , CY - Cytoxan, HU - Hydroxyurea, TBI – Total body irradiation, MA - myeloablative, NILO - Nilotinib, OS – Overall survival, DFS – Disease free survival, NRM – Non-relapse mortality, GVHD- Graft versus host disease, CR – Complete remission, F - Female, M - Male, CHR – Complete hematologic response, FISH – Fluorescence in situ hybridization, MMR – Major molecular response, CMR – Complete molecular response, CCyR – Complete cytogenetic response, MUD – Match unrelated donor.
Sampling, cell source and cell culture
All 12 patients received nilotinib for at least 90 days following allo-SCT. Up to 60 mL of whole blood were collected from consenting patients at different time points, as illustrated in Figure 4 (pre-nilotib post-allo-SCT, and within 28, 90, 180, 270, 335 or 365 days of nilotinib administration). The first time-point post allo-SCT pre-nilotinib treatment served as an internal control for each patient.
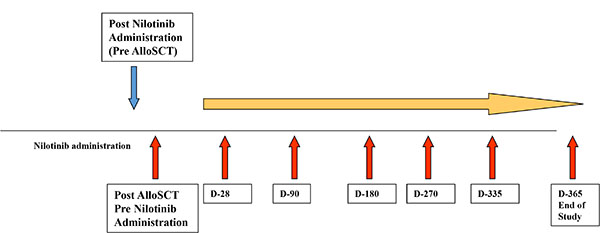
Figure 4: Study design of immunological assessment following SCT and nilotinib administration. Patients’ samples were collected at different time post nilotinib administration (red arrows). Samples obtained from patients post SCT, before nilotinib administration served as a specific control.
Peripheral Blood Mononuclear cells (PBMNCs) were obtained from whole blood samples by density gradient (1.077 g/dl) (LymphoprepTM, cat. #1114547, Fresenius Kabi Norge AS, Norway). Patients’ Natural Killer cells (NK) were isolated immediately using Miltenyi Biotec NK Cell Isolation Kit (Cat # 130-092-657) in accordance with manufacturer instructions. After isolation, cells were resuspended in 1640 RPMI medium supplemented with 10% FCS, 2 mM/ml L-glutamin, 100 U/ml Penicillin-Streptomycin Solution, (all produced by Biological Industries, Beit Haemek, Israel) and 100 IU/ml of IL-2 (Peprotech, Israel). NK cells were incubated overnight in a humidified CO2 incubator at 37°C before being used as effector cells in the killing activity tests performed. A K562 cell-line was used as target cells for killing activity assays. Cells were cultured in 1640 RPMI medium supplemented with 10% FCS, 2 mM/ml L-glutamin, 100 U/ml Penicillin-Streptomycin Solution, (all produced by Biological Industries, Beit Haemek, Israel). Eighteen hours prior to the killing assay, cells were collected and stained with 10 nM CFSE (5–6 Carboxyfluorescein Succinimidyl Ester) (Ebioscience).
Immuno-phenotyping of T cells subpopulations
The immune phenotyping was performed using a multi-color flow cytometry (FACS) with the following antibodies: CD3-FITC (Becman-coulter) for total T-lymphocytes, CD4-PE (BC), CD8-PE (BC), CD16-FITC (BC), CD20-PE (IQP) for total B-lymphocytes and CD56-PE (BC) for natural killer cells, all gated on CD45 bright and SSC low. The following staining panels were performed: CD3-CD4; CD3-CD8 and CD16-CD56-CD3. A total of 1–5 × 105 cells were analyzed with FACS-Canto II (BD Bioscience). The data analysis was performed by FACS-DIVA software (version II, BD bioscience).
T-cell mitogenic response
To evaluate patients’ T-cell mitogenic response, 105 PBMNCs were cultured for 72 hours in the presence of anti-CD3 antibody or phytohaemagglutinine (PHA) at two different doses: 5 μg and 25 μg, respectively. Cell proliferation was assessed by measuring DNA content using a single pulse of 1 μCi [3H] TdR (Rotem Industries, Inc., Israel), eighteen hours before measuring 3H content by β-counter. Stimulation Index (SI) was calculated using the following equation:
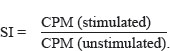
Cytotoxic activity of NK cells
Target and effector cells were collected after overnight incubation, washed once by centrifugation at 1250 rpm for 10 minutes, resuspended in 1 ml complete medium and counted with hemocytometer. A killing assay was performed by co-culturing constant numbers (25,000) of CFSE-labeled K562 and NK cells in 0.5:1, 1:1, 5:1 Effector:Target (E:T) ratios in 96 wells plate (v bottom). Co-cultures were incubated for 4 hours in humidified 5% CO2 incubator at 37°C. Target and effector cells were incubated separately to measure basal cell death. Four hours after co-culture, cells were collected, washed with PBS-1% BSA, and stained with 25 μL/mL 7-amino actinomycin D (7-AAD, E-Biosciences) for 10 minutes at room temperature (R/T). Two-color flow cytometry acquisitions were performed immediately (no longer than 1 hour post staining.) Dead cells were measured by double CFSEpos and 7AADpos cells and cytotoxic activity was expressed as % specific lysis calculated by the following formula:
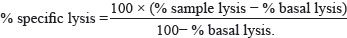
Quantification of thymic activity
Thymic activity was evaluated by analyzing newly-derived T-cells using T-cell receptor excision circles (TREC) and the surface membrane expression of 24 different T-cell receptor (TCR) Vβ families in patient samples collected before and at the last nilotinib treatment time-point. TREC copy numbers were determined using quantitative real-time PCR (qRQ-PCR). PCR reactions were performed as previously described using 0.5 μg genomic DNA (gDNA) extracted from the patients’ PBMCs as template. qRQ-PCR was carried out using an ABI PRISM 7900 Sequence Detector System (Applied Biosystems). A standard curve was constructed by using serial dilutions containing 103 to 106 copies of a known TREC plasmid. Patient and control samples were tested in triplicate, and the number of TRECs in each sample was calculated by comparing the obtained cycle threshold (Ct) value of the sample to the standard curve using an absolute quantification algorithm. Surface expression of individual TCR-Vβ families was analyzed using flow cytometry and a set of Vβ-specific fluorochrome-labeled monoclonal antibodies (Becton-Dickinson, Calibur) as described [48]. Normal control values were obtained from the IOTest Beta Mark-Quick Reference Card (Beckman Coulter).
Statistical analysis
Longitudinal changes in continuous variables were assessed by paired t-test. P-values of 0.05 were considered statistically significant.
Informed consent
Patients gave written informed consent, and the study was approved by the institutional review board. The study was registered at ClinicalTrials.gov as NCT00750659.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
1. Baccarani M, Deininger MW, Rosti G, Hochhaus A, Soverini S, Apperley JF, Cervantes F, Clark RE, Cortes JE, Guilhot F, Hjorth-Hansen H, Hughes TP, Kantarjian HM, et al. European LeukemiaNet recommendations for the management of chronic myeloid leukemia: 2013. Blood. 2013; 122:872–884.
2. Ribera JM. Optimal approach to treatment of patients with Philadelphia chromosome-positive acute lymphoblastic leukemia: how to best use all the available tools. Leuk Lymphoma. 2013; 54:21–27.
3. Goldman JM. How I treat chronic myeloid leukemia in the imatinib era. Blood. 2007; 110:2828–2837.
4. Gratwohl A, Brand R, Apperley J, Crawley C, Ruutu T, Corradini P, Carreras E, Devergie A, Guglielmi C, Kolb HJ, Niederwieser D, Chronic Leukemia Working Party of the European Group for B, Marrow T. Allogeneic hematopoietic stem cell transplantation for chronic myeloid leukemia in Europe 2006: transplant activity, long-term data and current results. An analysis by the Chronic Leukemia Working Party of the European Group for Blood and Marrow Transplantation (EBMT). Haematologica. 2006; 91:513–521.
5. Copelan EA, Crilley PA, Szer J, Dodds AJ, Stevenson D, Phillips G, Elder P, Nivison-Smith I, Avalos BR, Penza S, Topolsky D, Sobecks R, Kalaycio M, et al. Late mortality and relapse following BuCy2 and HLA-identical sibling marrow transplantation for chronic myelogenous leukemia. Biol Blood Marrow Transplant. 2009; 15:851–855.
6. Shimoni A, Kroger N, Zander AR, Rowe JM, Hardan I, Avigdor A, Yeshurun M, Ben-Bassat I, Nagler A. Imatinib mesylate (STI571) in preparation for allogeneic hematopoietic stem cell transplantation and donor lymphocyte infusions in patients with Philadelphia-positive acute leukemias. Leukemia. 2003; 17:290–297.
7. Giebel S, Czyz A, Ottmann O, Baron F, Brissot E, Ciceri F, Cornelissen JJ, Esteve J, Gorin NC, Savani B, Schmid C, Mohty M, Nagler A. Use of tyrosine kinase inhibitors to prevent relapse after allogeneic hematopoietic stem cell transplantation for patients with Philadelphia chromosome-positive acute lymphoblastic leukemia: A position statement of the Acute Leukemia Working Party of the European Society for Blood and Marrow Transplantation. Cancer. 2016.
8. Kantarjian HM, O’Brien S, Cortes JE, Giralt SA, Rios MB, Shan J, Giles FJ, Thomas DA, Faderl S, De Lima M, Garcia-Manero G, Champlin R, Arlinghaus R, et al. Imatinib mesylate therapy for relapse after allogeneic stem cell transplantation for chronic myelogenous leukemia. Blood. 2002; 100:1590–1595.
9. Olavarria E, Ottmann OG, Deininger M, Clark RE, Bandini G, Byrne J, Lipton J, Vitek A, Michallet M, Siegert W, Ullmann A, Wassmann B, Niederwieser D, et al. Chronic Leukaemia Working Party of the European Group of B, Marrow T. Response to imatinib in patients who relapse after allogeneic stem cell transplantation for chronic myeloid leukemia. Leukemia. 2003; 17:1707–1712.
10. DeAngelo DJ, Hochberg EP, Alyea EP, Longtine J, Lee S, Galinsky I, Parekkedon B, Ritz J, Antin JH, Stone RM, Soiffer RJ. Extended follow-up of patients treated with imatinib mesylate (gleevec) for chronic myelogenous leukemia relapse after allogeneic transplantation: durable cytogenetic remission and conversion to complete donor chimerism without graft-versus-host disease. Clin Cancer Res. 2004; 10:5065–5071.
11. Hess G, Bunjes D, Siegert W, Schwerdtfeger R, Ledderose G, Wassmann B, Kobbe G, Bornhauser M, Hochhaus A, Ullmann AJ, Kindler T, Haus U, Gschaidmeier H, et al. Sustained complete molecular remissions after treatment with imatinib-mesylate in patients with failure after allogeneic stem cell transplantation for chronic myelogenous leukemia: results of a prospective phase II open-label multicenter study. J Clin Oncol. 2005; 23:7583–7593.
12. Palandri F, Amabile M, Rosti G, Bandini G, Benedetti F, Usala E, Angelucci E, Tiribelli M, Fanin R, Martinelli G, Baccarani M. Imatinib therapy for chronic myeloid leukemia patients who relapse after allogeneic stem cell transplantation: a molecular analysis. Bone Marrow Transplant. 2007; 39:189–191.
13. Klyuchnikov E, Kroger N, Brummendorf TH, Wiedemann B, Zander AR, Bacher U. Current status and perspectives of tyrosine kinase inhibitor treatment in the posttransplant period in patients with chronic myelogenous leukemia (CML). Biol Blood Marrow Transplant. 2010; 16:301–310.
14. Klyuchnikov E, Schafhausen P, Kroger N, Brummendorf TH, Osanmaz O, Asenova S, Zabelina T, Ocheni S, Ayuk F, Zander AR, Bacher U. Second-generation tyrosine kinase inhibitors in the post-transplant period in patients with chronic myeloid leukemia or Philadelphia-positive acute lymphoblastic leukemia. Acta Haematol. 2009; 122:6–10.
15. Wright MP, Shepherd JD, Barnett MJ, Nantel SH, Sutherland HJ, Toze CL, Hogge DE, Nevill TJ, Song KW, Abou Mourad YR, Narayanan S, Power MM, Smith CA, et al. Response to tyrosine kinase inhibitor therapy in patients with chronic myelogenous leukemia relapsing in chronic and advanced phase following allogeneic hematopoietic stem cell transplantation. Biol Blood Marrow Transplant. 2010; 16:639–646.
16. Olavarria E, Siddique S, Griffiths MJ, Avery S, Byrne JL, Piper KP, Lennard AL, Pallan L, Arrazi JM, Perz JB, O’Shea D, Goldman JM, Apperley JF, et al. Posttransplantation imatinib as a strategy to postpone the requirement for immunotherapy in patients undergoing reduced-intensity allografts for chronic myeloid leukemia. Blood. 2007; 110:4614–4617.
17. Carpenter PA, Snyder DS, Flowers ME, Sanders JE, Gooley TA, Martin PJ, Appelbaum FR, Radich JP. Prophylactic administration of imatinib after hematopoietic cell transplantation for high-risk Philadelphia chromosome-positive leukemia. Blood. 2007; 109:2791–2793.
18. Wassmann B, Pfeifer H, Stadler M, Bornhauser M, Bug G, Scheuring UJ, Bruck P, Stelljes M, Schwerdtfeger R, Basara N, Perz J, Bunjes D, Ledderose G, et al. Early molecular response to posttransplantation imatinib determines outcome in MRD+ Philadelphia-positive acute lymphoblastic leukemia (Ph+ALL). Blood. 2005; 106:458–463.
19. Pfeifer H, Wassmann B, Bethge W, Dengler J, Bornhauser M, Stadler M, Beelen D, Vucinic V, Burmeister T, Stelljes M, Faul C, Dreger P, Kiani A, et al. Randomized comparison of prophylactic and minimal residual disease-triggered imatinib after allogeneic stem cell transplantation for BCR-ABL1-positive acute lymphoblastic leukemia. Leukemia. 2013; 27:1254–1262.
20. Ribera JM, Oriol A, Gonzalez M, Vidriales B, Brunet S, Esteve J, Del Potro E, Rivas C, Moreno MJ, Tormo M, Martin-Reina V, Sarra J, Parody R, et al. Concurrent intensive chemotherapy and imatinib before and after stem cell transplantation in newly diagnosed Philadelphia chromosome-positive acute lymphoblastic leukemia. Final results of the CSTIBES02 trial. Haematologica. 2010; 95:87–95.
21. Kantarjian HM, Giles FJ, Bhalla KN, Pinilla-Ibarz J, Larson RA, Gattermann N, Ottmann OG, Hochhaus A, Radich JP, Saglio G, Hughes TP, Martinelli G, Kim DW, et al. Nilotinib is effective in patients with chronic myeloid leukemia in chronic phase after imatinib resistance or intolerance: 24-month follow-up results. Blood. 2011; 117:1141–1145.
22. DeFilipp Z, Langston AA, Chen Z, Zhang C, Arellano ML, El Rassi F, Flowers CR, Kota VK, Al-Kadhimi Z, Veldman R, Jillella AP, Lonial S, Waller EK, et al. Does Post-Transplant Maintenance Therapy With Tyrosine Kinase Inhibitors Improve Outcomes of Patients With High-Risk Philadelphia Chromosome-Positive Leukemia? Clin Lymphoma Myeloma Leuk. 2016.
23. Shimoni A, Volchek Y, Koren-Michowitz M, Varda-Bloom N, Somech R, Shem-Tov N, Yerushalmi R, Nagler A. Phase 1/2 study of nilotinib prophylaxis after allogeneic stem cell transplantation in patients with advanced chronic myeloid leukemia or Philadelphia chromosome-positive acute lymphoblastic leukemia. Cancer. 2015; 121:863–871.
24. Appel S, Balabanov S, Brummendorf TH, Brossart P. Effects of imatinib on normal hematopoiesis and immune activation. Stem Cells. 2005; 23:1082–1088.
25. Mehta RS, Rezvani K. Immune reconstitution post allogeneic transplant and the impact of immune recovery on the risk of infection. Virulence. 2016:1–16.
26. Fujimaki K, Maruta A, Yoshida M, Kodama F, Matsuzaki M, Fujisawa S, Kanamori H, Ishigatsubo Y. Immune reconstitution assessed during five years after allogeneic bone marrow transplantation. Bone Marrow Transplant. 2001; 27:1275–1281.
27. Jalkanen SE, Vakkila J, Kreutzman A, Nieminen JK, Porkka K, Mustjoki S. Poor cytokine-induced phosphorylation in chronic myeloid leukemia patients at diagnosis is effectively reversed by tyrosine kinase inhibitor therapy. Exp Hematol. 2011; 39:102–113 e101.
28. Mir MA, Battiwalla M. Immune deficits in allogeneic hematopoietic stem cell transplant (HSCT) recipients. Mycopathologia. 2009; 168:271–282.
29. Chen J, Schmitt A, Chen B, Rojewski M, Rubeler V, Fei F, Yu Y, Yu X, Ringhoffer M, von Harsdorf S, Greiner J, Gotzz M, Guillaume P, et al. Nilotinib hampers the proliferation and function of CD8+ T lymphocytes through inhibition of T cell receptor signalling. J Cell Mol Med. 2008; 12:2107–2118.
30. Salih J, Hilpert J, Placke T, Grunebach F, Steinle A, Salih HR, Krusch M. The BCR/ABL-inhibitors imatinib, nilotinib and dasatinib differentially affect NK cell reactivity. Int J Cancer. 2010; 127:2119–2128.
31. de Lavallade H, Khoder A, Hart M, Sarvaria A, Sekine T, Alsuliman A, Mielke S, Bazeos A, Stringaris K, Ali S, Milojkovic D, Foroni L, Chaidos A, et al. Tyrosine kinase inhibitors impair B-cell immune responses in CML through off-target inhibition of kinases important for cell signaling. Blood. 2013; 122:227–238.
32. Chen CI, Koschmieder S, Kerstiens L, Schemionek M, Altvater B, Pscherer S, Gerss J, Maecker HT, Berdel WE, Juergens H, Lee PP, Rossig C. NK cells are dysfunctional in human chronic myelogenous leukemia before and on imatinib treatment and in BCR-ABL-positive mice. Leukemia. 2012; 26:465–474.
33. Kebriaei P, Saliba R, Rondon G, Chiattone A, Luthra R, Anderlini P, Andersson B, Shpall E, Popat U, Jones R, Worth L, Ravandi F, Thomas D, et al. Long-term follow-up of allogeneic hematopoietic stem cell transplantation for patients with Philadelphia chromosome-positive acute lymphoblastic leukemia: impact of tyrosine kinase inhibitors on treatment outcomes. Biol Blood Marrow Transplant. 2012; 18:584–592.
34. Cwynarski K, Laylor R, Macchiarulo E, Goldman J, Lombardi G, Melo JV, Dazzi F. Imatinib inhibits the activation and proliferation of normal T lymphocytes in vitro. Leukemia. 2004; 18:1332–1339.
35. Mumprecht S, Matter M, Pavelic V, Ochsenbein AF. Imatinib mesylate selectively impairs expansion of memory cytotoxic T cells without affecting the control of primary viral infections. Blood. 2006; 108:3406–3413.
36. Seggewiss R, Lore K, Greiner E, Magnusson MK, Price DA, Douek DC, Dunbar CE, Wiestner A. Imatinib inhibits T-cell receptor-mediated T-cell proliferation and activation in a dose-dependent manner. Blood. 2005; 105:2473–2479.
37. Larmonier N, Janikashvili N, LaCasse CJ, Larmonier CB, Cantrell J, Situ E, Lundeen T, Bonnotte B, Katsanis E. Imatinib mesylate inhibits CD4+ CD25+ regulatory T cell activity and enhances active immunotherapy against BCR-ABL- tumors. J Immunol. 2008; 181:6955–6963.
38. Balachandran VP, Cavnar MJ, Zeng S, Bamboat ZM, Ocuin LM, Obaid H, Sorenson EC, Popow R, Ariyan C, Rossi F, Besmer P, Guo T, Antonescu CR, et al. Imatinib potentiates antitumor T cell responses in gastrointestinal stromal tumor through the inhibition of Ido. Nat Med. 2011; 17:1094–1100.
39. Blake SJ, Lyons AB, Hughes TP. Nilotinib inhibits the Src-family kinase LCK and T-cell function in vitro. J Cell Mol Med. 2009; 13:599–601.
40. Fei F, Yu Y, Schmitt A, Rojewski MT, Chen B, Greiner J, Gotz M, Guillaume P, Dohner H, Bunjes D, Schmitt M. Dasatinib exerts an immunosuppressive effect on CD8+ T cells specific for viral and leukemia antigens. Exp Hematol. 2008; 36:1297–1308.
41. Fraser CK, Blake SJ, Diener KR, Lyons AB, Brown MP, Hughes TP, Hayball JD. Dasatinib inhibits recombinant viral antigen-specific murine CD4+ and CD8+ T-cell responses and NK-cell cytolytic activity in vitro and in vivo. Exp Hematol. 2009; 37:256–265.
42. Weichsel R, Dix C, Wooldridge L, Clement M, Fenton-May A, Sewell AK, Zezula J, Greiner E, Gostick E, Price DA, Einsele H, Seggewiss R. Profound inhibition of antigen-specific T-cell effector functions by dasatinib. Clin Cancer Res. 2008; 14:2484–2491.
43. Kreutzman A, Ladell K, Koechel C, Gostick E, Ekblom M, Stenke L, Melo T, Einsele H, Porkka K, Price DA, Mustjoki S, Seggewiss R. Expansion of highly differentiated CD8+ T-cells or NK-cells in patients treated with dasatinib is associated with cytomegalovirus reactivation. Leukemia. 2011; 25:1587–1597.
44. Koehl U, Bochennek K, Zimmermann SY, Lehrnbecher T, Sorensen J, Esser R, Andreas C, Kramm C, Gruttner HP, Falkenberg E, Orth A, Bader P, Schwabe D, et al. Immune recovery in children undergoing allogeneic stem cell transplantation: absolute CD8+ CD3+ count reconstitution is associated with survival. Bone Marrow Transplant. 2007; 39:269–278.
45. Kim DH, Kim JG, Sohn SK, Sung WJ, Suh JS, Lee KS, Lee KB. Clinical impact of early absolute lymphocyte count after allogeneic stem cell transplantation. Br J Haematol. 2004; 125:217–224.
46. Behringer D, Bertz H, Schmoor C, Berger C, Dwenger A, Finke J. Quantitative lymphocyte subset reconstitution after allogeneic hematopoietic transplantation from matched related donors with CD34+ selected PBPC grafts unselected PBPC grafts or BM grafts. Bone Marrow Transplant. 1999; 24:295–302.
47. Kalwak K, Gorczynska E, Toporski J, Turkiewicz D, Slociak M, Ussowicz M, Latos-Grazynska E, Krol M, Boguslawska-Jaworska J, Chybicka A. Immune reconstitution after haematopoietic cell transplantation in children: immunophenotype analysis with regard to factors affecting the speed of recovery. Br J Haematol. 2002; 118:74–89.
48. Lev A, Simon AJ, Bareket M, Bielorai B, Hutt D, Amariglio N, Rechavi G, Somech R. The kinetics of early T, B cell immune recovery after bone marrow transplantation in RAG-2-deficient SCID patients. PLoS One. 2012; 7:e30494.
49. Jaime-Perez JC, Villarreal-Villarreal CD, Mendez-Ramirez N, Vazquez-Garza E, Salazar-Riojas R, Gomez-Almaguer D. Assessment of immune reconstitution status in recipients of a successful hematopoietic stem cell transplant from peripheral blood after reduced intensity conditioning. Blood Cells Mol Dis. 2016; 58:52–56.
50. Storek J, Geddes M, Khan F, Huard B, Helg C, Chalandon Y, Passweg J, Roosnek E. Reconstitution of the immune system after hematopoietic stem cell transplantation in humans. Semin Immunopathol. 2008; 30:425–437.
51. Jacobs R, Stoll M, Stratmann G, Leo R, Link H, Schmidt RE. CD16- CD56+ natural killer cells after bone marrow transplantation. Blood. 1992; 79:3239–3244.
52. Le Blanc K, Barrett AJ, Schaffer M, Hagglund H, Ljungman P, Ringden O, Remberger M. Lymphocyte recovery is a major determinant of outcome after matched unrelated myeloablative transplantation for myelogenous malignancies. Biol Blood Marrow Transplant. 2009; 15:1108–1115.
53. Blazar BR, Murphy WJ, Abedi M. Advances in graft-versus-host disease biology and therapy. Nat Rev Immunol. 2012; 12:443–458.
54. Socie G, Ritz J. Current issues in chronic graft-versus-host disease. Blood. 2014; 124:374–384.
55. Atkinson K, Hansen JA, Storb R, Goehle S, Goldstein G, Thomas ED. T-cell subpopulations identified by monoclonal antibodies after human marrow transplantation. I. Helper-inducer and cytotoxic-suppressor subsets. Blood. 1982; 59:1292–1298.
56. Alho AC, Kim HT, Chammas MJ, Reynolds CG, Matos TR, Forcade E, Whangbo J, Nikiforow S, Cutler CS, Koreth J, Ho VT, Armand P, Antin JH, et al. Unbalanced recovery of regulatory and effector T cells after allogeneic stem cell transplantation contributes to chronic GVHD. Blood. 2016; 127:646–657.
57. Storek J. B-cell immunity after allogeneic hematopoietic cell transplantation. Cytotherapy. 2002; 4:423–424.
58. Toubert A, Glauzy S, Douay C, Clave E. Thymus and immune reconstitution after allogeneic hematopoietic stem cell transplantation in humans: never say never again. Tissue Antigens. 2012; 79:83–89.
59. Ruggeri L, Capanni M, Casucci M, Volpi I, Tosti A, Perruccio K, Urbani E, Negrin RS, Martelli MF, Velardi A. Role of natural killer cell alloreactivity in HLA-mismatched hematopoietic stem cell transplantation. Blood. 1999; 94:333–339.
60. Hakim FT, Memon SA, Cepeda R, Jones EC, Chow CK, Kasten-Sportes C, Odom J, Vance BA, Christensen BL, Mackall CL, Gress RE. Age-dependent incidence, time course, and consequences of thymic renewal in adults. J Clin Invest. 2005; 115:930–939.
61. Mensen A, Ochs C, Stroux A, Wittenbecher F, Szyska M, Imberti L, Fillatreau S, Uharek L, Arnold R, Dorken B, Thiel A, Scheibenbogen C, Na IK. Utilization of TREC and KREC quantification for the monitoring of early T- and B-cell neogenesis in adult patients after allogeneic hematopoietic stem cell transplantation. J Transl Med. 2013; 11:188.
62. Lewin SR, Heller G, Zhang L, Rodrigues E, Skulsky E, van den Brink MR, Small TN, Kernan NA, O’Reilly RJ, Ho DD, Young JW. Direct evidence for new T-cell generation by patients after either T-cell-depleted or unmodified allogeneic hematopoietic stem cell transplantations. Blood. 2002; 100:2235–2242.
63. Hochberg EP, Chillemi AC, Wu CJ, Neuberg D, Canning C, Hartman K, Alyea EP, Soiffer RJ, Kalams SA, Ritz J. Quantitation of T-cell neogenesis in vivo after allogeneic bone marrow transplantation in adults. Blood. 2001; 98:1116–1121.
64. Pfeifer H, Wassmann B, Pavlova A, Wunderle L, Oldenburg J, Binckebanck A, Lange T, Hochhaus A, Wystub S, Bruck P, Hoelzer D, Ottmann OG. Kinase domain mutations of BCR-ABL frequently precede imatinib-based therapy and give rise to relapse in patients with de novo Philadelphia-positive acute lymphoblastic leukemia (Ph+ ALL). Blood. 2007; 110:727–734.
65. Iacobucci I, Lonetti A, Venturi C, Ferrari A, Papayannidis C, Ottaviani E, Abbenante MC, Paolini S, Bresciani P, Potenza L, Parisi S, Cattina F, Soverini S, et al. Use of a high sensitive nanofluidic array for the detection of rare copies of BCR-ABL1 transcript in patients with Philadelphia-positive acute lymphoblastic leukemia in complete response. Leuk Res. 2014; 38:581–585.