
INTRODUCTION
Prostate cancer is the most prevalent cancer in men in Western societies [1]. Although the majority of prostate cancers behave in an indolent manner, a small subset is highly aggressive and requires extensive treatment [2, 3]. Established preoperative prognostic parameters are limited to Gleason grade, tumor extent on biopsies, prostate-specific antigen (PSA), and clinical stage. These data are statistically powerful, but often not sufficient to optimize individual treatment decisions. It is thus hoped that a better understanding of disease biology will eventually lead to the identification of clinically applicable molecular markers that enable a more reliable prediction of prostate cancer aggressiveness.
Deletions of chromosome 8p are of high interest in prostate cancer. After TMPRSS2:ERG fusion, 8p deletions constitute the second most frequent genomic alteration in this tumor type, occurring in about 30% of cancers [4–6]. A specific target gene of the 8p deletion has never been identified. It appears likely, that the tumorigenic effect of 8p deletions occurs through a combined downregulation of multiple relevant genes on 8p. A variety of studies have earlier analyzed 8p deletions in prostate cancer and reported associations with advanced tumor stage and Gleason grade [5–12], metastatic growth [8, 12], early biochemical recurrence [10, 13], and reduced overall survival [14]. Most of these studies were done on patient cohorts ranging from 27 to 195. In an own study on 2,097 cancers we had only found a marginal association with clinical outcome [10].
Because of the still not clarified role of this highly frequent alteration we decided to expand our earlier study in order to better assess the clinical significance of 8p deletion including subgroup analyses and comparisons with other relevant molecular features. For this purpose we took advantage of our current version of a prostate cancer tissue microarray (TMA) now containing more than 12,000 prostate cancers.
RESULTS
Architecture of 8p deletions in prostate cancer
Re-analysis of own [15] and published [4, 16, 17] microarray-based 8p copy number data from 442 prostate cancers using the FISH-Oracle browser [18, 19] shows that 8p deletions typically involve the entire short arm of chromosome 8 (Figure 1).
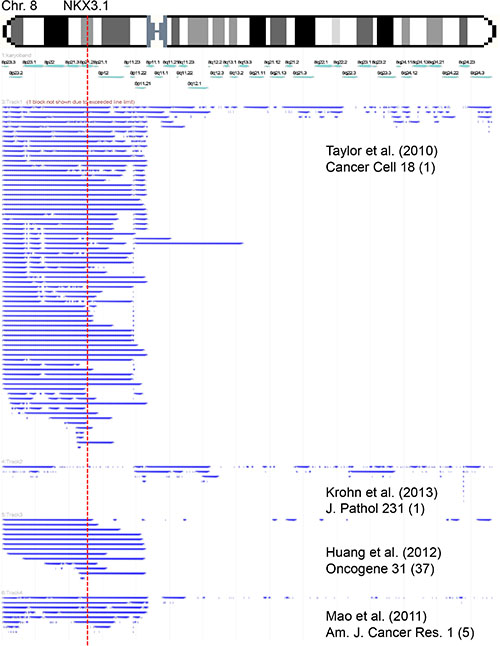
Figure 1: Size and extension of chromosome 8 deletions detected in published microarray-based copy number studies. Each bar represents the deleted area in a single tumor.
Technical aspects
8p (NKX3.1) FISH analysis was successful in 7,017 of 12,427 (56.5%) arrayed cancers. Analysis was not informative in the remaining of 5,410 tumors because lack of tumor cells in the tissue spots, faint or absent FISH signals, or missing tissue spots on the TMA section.
Prevalence and type of 8p deletions and association to prostate cancer phenotype
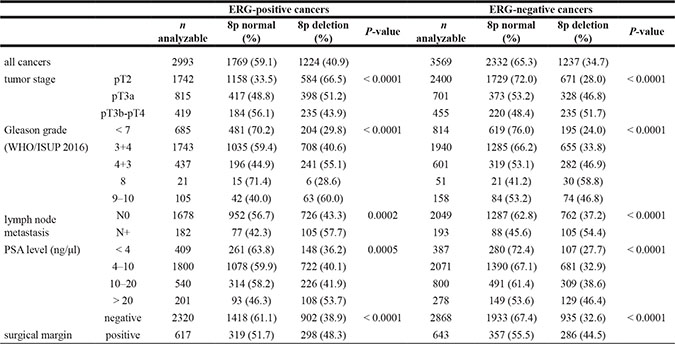
8p deletions were found in 36.8% (2,581/7,017) of all prostate cancers. All deletions were heterozygous. The relationship between 8p deletions and tumor phenotype and clinical parameters is summarized in Table 1. 8p deletions were significantly linked to advanced tumor stage, high Gleason grade, presence of lymph node metastasis, pre-surgical prostate specific antigen (PSA) level and positive surgical margin (P < 0.0001 each). All associations between 8p deletions and clinico-pathological variables held also true in the subsets of 3,569 ERG-negative (P < 0.0001) and 2,993 ERG-positive cancers (P ≤ 0.0005, Table 2).
Table 1: Associations between 8p deletion and prostate cancer phenotype in all cancers
n analyzable |
8p normal (%) |
8p deletion (%) |
P-value |
||
|---|---|---|---|---|---|
all cancers |
7017 |
4436 (63.2) |
2581 (36.8) |
||
tumor stage |
pT2 |
4456 |
3141 (70.5) |
1315 (29.5) |
< 0.0001 |
pT3a |
1598 |
835 (52.3) |
763 (47.8) |
||
pT3b-pT4 |
931 |
438 (47.1) |
493 (53.0) |
||
Gleason grade |
≤ 3 + 3 |
1653 |
1232 (74.5) |
421 (25.5) |
< 0.0001 |
3 + 4 |
3880 |
2462 (63.5) |
1418 (36.6) |
||
4 + 3 |
1090 |
543 (49.8) |
547 (50.2) |
||
≥ 4 + 4 |
354 |
173 (48.9) |
181 (51.1) |
||
lymph node metastasis |
N0 |
3946 |
2386 (60.5) |
1560 (39.5) |
< 0.0001 |
N+ |
398 |
178 (44.7) |
220 (55.3) |
||
PSA level (ng/μl) |
< 4 |
870 |
599 (68.9) |
271 (31.2) |
< 0.0001 |
4–10 |
4138 |
2672 (64.6) |
1466 (35.4) |
||
10–20 |
1417 |
855 (60.3) |
562 (39.7) |
||
> 20 |
510 |
260 (51.0) |
250 (49.0) |
||
surgical margin |
negative |
5559 |
3629 (65.3) |
1930 (34.7) |
< 0.0001 |
positive |
1334 |
726 (54.4) |
608 (45.6) |
Table 2: Associations between 8p deletion and prostate cancer phenotype in the subgroup of ERG-positive and ERG-negative cancers
Association between 8p deletion, ERG fusion and PTEN deletion
8p deletions were marginally more frequent in ERG-positive cancers (40.9% according to IHC and 42.9% to FISH) than in ERG-negative cancers (34.7% according to IHC and 36.4% to FISH%, Figure 2A). To better understand the impact of PTEN deletions on this association we compared the 8p deletion frequency in subsets of cancers defined by their PTEN deletion and ERG fusion status (Figure 2B). This analysis revealed that 8p deletions were massively linked to PTEN deletions independently of the ERG status (P < 0.0001 each in subsets of ERG-positive and ERG-negative cancers), while the ERG status had no relevant further impact on the 8p deletion frequency, neither in cancers with normal PTEN copy numbers (P = 0.021) nor in cancers with PTEN deletion (P = 0.956).
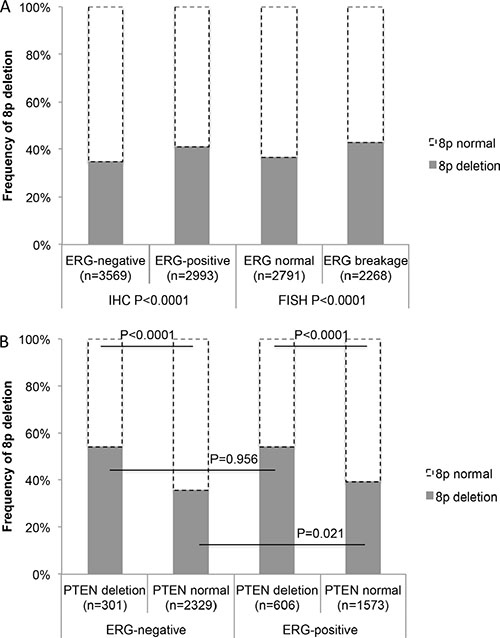
Figure 2: (A) Associations between 8p deletion and ERG-fusion by IHC and FISH analysis. (B) Association between deletion of 8p and PTEN in ERG-negative and ERG-positive prostate cancers.
Associations with prognosis
The prognostic impact of pT and pN category as well as “classical” Gleason grade groups and quantitative Gleason grade is given for the 6,375 patients with interpretable 8p FISH for comparison (P < 0.0001 each, Supplementary Figure S1A–S1D). 8p deletion was significantly linked to early biochemical (PSA) recurrence in 6,375 cancers with interpretable 8p FISH results and follow-up data (P < 0.0001, Figure 3A). Multivariate analysis revealed that this was independent from established prognostic parameters including pathological tumor stage, Gleason grade, presence of lymph node metastases, status of the resection margin and pre-operative PSA level (P = 0.0100, Table 3). The relationship with patient outcome was similar in the subsets of 3,231 ERG-negative (P < 0.0001, Figure 3B) and 2,738 ERG-positive cancers (P < 0.0001, Figure 3C).
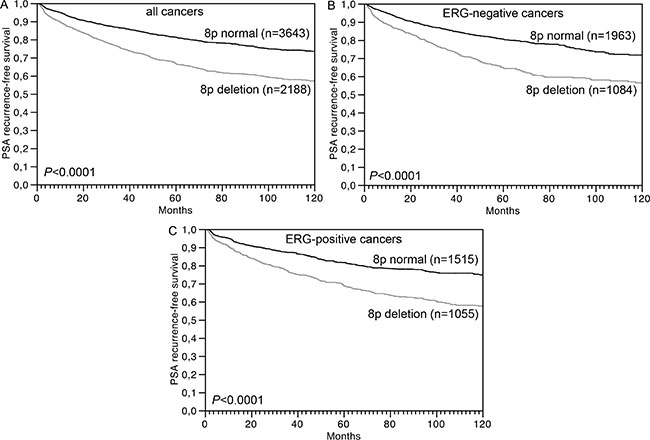
Figure 3: Association between 8p deletion and biochemical (PSA) recurrence in (A) all cancers (n = 6,375), (B) ERG-negative cancers (n = 3,231), and (C) ERG-positive cancers (n = 2,738).
Table 3: Multivariate analysis (Cox regression) including clinical and pathological parameters in addition to the 8p deletion status in all prostate cancers
Parameter |
RR |
95% CI |
P-value |
|---|---|---|---|
Tumor stage |
|||
pT3a vs pT2 |
2.0 |
1.7–2.3 |
< 0.0001 |
pT3b vs pT3a |
1.4 |
1.2–1.7 |
|
pT4 vs pT3b |
2.1 |
1.4–2.8 |
|
Gleason grade |
|||
3 + 4 vs ≤ 3+ 3 |
2.3 |
1.8–3.0 |
< 0.0001 |
4 + 3 vs 3 + 4 |
2.1 |
1.8–2.4 |
|
≥ 4 + 4 vs 4 + 3 |
1.2 |
1.0–1.5 |
|
Nodal stage |
|||
pN1 vs pN0 |
1.5 |
1.3–1.8 |
< 0.0001 |
Resection margin status |
|||
R1 vs R0 |
1.2 |
1.0–1.3 |
0.0254 |
Pre-operative PSA (ng/ml) |
|||
4–10 vs < 4 |
1.1 |
0.9–1.4 |
< 0.0001 |
10–20 vs 4–10 |
1.2 |
1.0–1.4 |
|
> 20 vs 10–20 |
1.4 |
1.2–1.7 |
|
8p status |
|||
8p deletion vs 8p normal |
1.2 |
1.0–1.3 |
0.0100 |
A strong prognostic impact was also seen for the subgroups R0 and R1 cancers (Figure 4A). However, a variable impact on prognosis was seen for tumors with different pN stage (Figure 4B), pT stage (Figure 4C) or “classical” Gleason grade groups (Figure 4D). Here, 8p deletions could not distinguish prognostic subgroups in categories with a particular good (Gleason 3 + 3) or bad prognosis (Gleason ≥ 8, pN1, pT3b). A further analysis of tumors characterized by a comparable quantitative Gleason grade revealed, that the prognostic impact of 8p deletions was only retained in a few subgroups including cancers with 6–10%, 21–30% and ≥ 61% Gleason 4 (Figure 5A–5G).
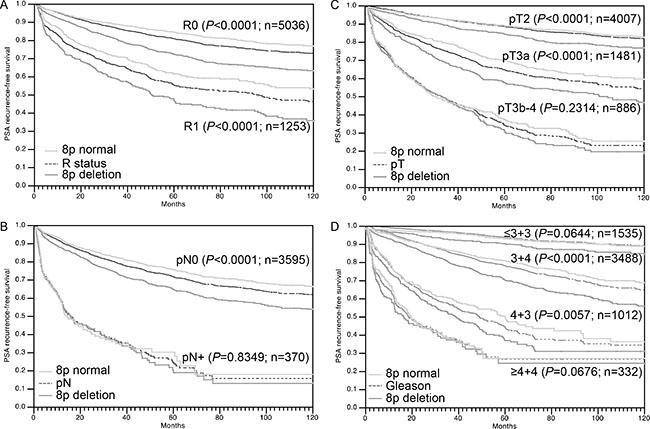
Figure 4: Association between 8p deletion and biochemical (PSA) recurrence in dependence on (A) resection margin status (R), (B) pathological lymph node status (pN), (C) pathological tumor stage(pT) and (D) classical Gleason grade.
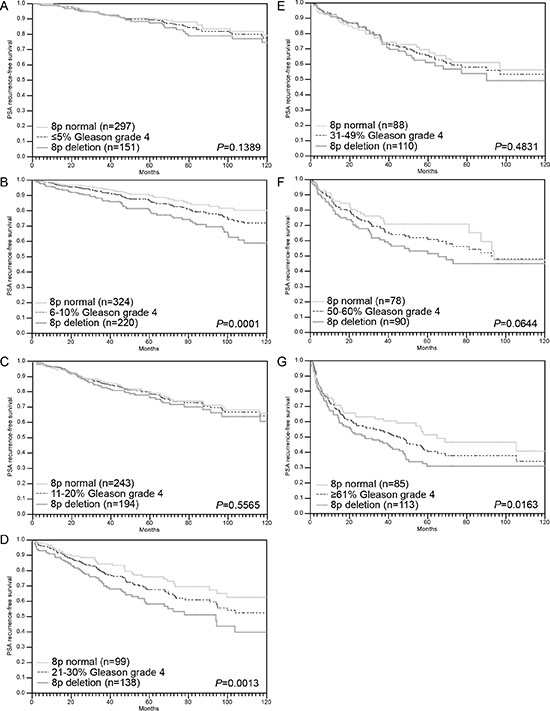
Figure 5: Association between 8p deletion and biochemical recurrence in dependence on quantitative Gleason grading subgroups (A–G).
Because of the strong association between 8p deletion and PTEN deletion, the prognostic impact of both deletions was also studied in combination (Figure 6). In this analysis, prognosis deteriorated continuously from 2,285 cancers without deletions to 1,324 cancers with 8p deletion only, to 400 cancers with PTEN deletion only, to 467 cancers with 8p/PTEN co-deletions (P = 0.0003 for PTEN deletion vs. 8p/PTEN co-deletion).
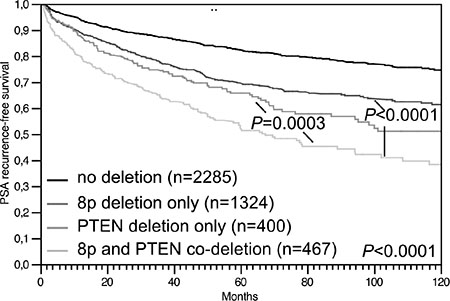
Figure 6: Association between 8p/PTEN co-deletion and biochemical (PSA) recurrence in all prostate cancers.
DISCUSSION
Chromosome 8p deletion is one of the most frequent alterations in many different cancer types (reviewed in [20]). 8p deletions are typically large and often involve the entire short arm of the chromosome. This made the search for 8p deletion target genes difficult. Extensive studies searching for the underlying tumor suppressor gene have failed to identify a universal 8p tumor suppressor but have revealed multiple 8p genes with documented tumor suppressive properties, such as NKX3.1 [21, 22], CSMD1 [23], DLC1 [24], PPP2CB and PPP3CC [25], MSR1 [26], TNFRSF10C and TNFRSF10D [25]. Studies employing whole genome or exome sequencing have meanwhile clarified that 8p deletion is - in the vast majority of cases - not accompanied by a mutation of the coding area of any 8p gene [27–30]. It is still possible that one or several 8p-deleted genes are completely silenced by epigenetic or other mechanisms. It is an appealing alternative, however, that reduced expression of a combination of 8p genes could exert biologically relevant consequences in case of heterozygous 8p deletion. Studies in murine models of hepatocellular carcinomas, another cancer type frequently affected by large 8p deletions, have shown that partial inactivation of several 8p genes can cooperatively drive cancer development [31], thus supporting a model of compound haplo-insufficiency which might also apply for 8p-deleted prostate cancers. A significant biologic impact of haplo-insufficiency has recently also been demonstrated for NKX3.1 [32].
Our meta-analysis of 8p copy number data demonstrates that deletions are typically large and often involve loss of the entire 8p chromosome arm. The successful analysis of more than 7,000 prostate cancers using a FISH probe directed against the NKX3.1 locus, thus, indicates 8p deletions in 37% of tumors. This frequency is comparable to the 32% 8p deletions found in an own earlier study on a subset of 2,097 of our cancers [10] but somewhat lower than in most other FISH studies, which reported deletion frequencies between 40 and 69% [11–13, 33–35]. Substantially lower deletion rates (1.9%-15.6%) have been reported from studies on 56-333 prostate cancers analyzed within the frameworks of The Cancer Genome Atlas (TCGA) and the International Cancer Genome Consortium (ICGC) [36, 37]. These discrepant findings are most likely attributable to the comparatively small sample sets and the different thresholds for 8p deletion calling. For example, reported deletion rates in the TCGC/ICGC data portal are limited to “deep deletions” extending a certain threshold (-2) in Genomic Identification of Significant Targets in Cancer (GISTIC) analysis [36, 37], while “shallow deletions” (threshold -1) are not reported. Although “deep” and “shallow” deletions are defined as “possibly homozygous” and “possibly heterozygous” deletions (http://www.cbioportal.org [36, 37]) such data are difficult to compare with FISH findings that allow for precise copy number counting. The higher deletion rates in earlier FISH studies are likely due to the use of thresholds for defining 8p deletions that were based on FISH results in normal epithelial cells [11, 13, 33]. This is a potential source of error, because cancer cell nuclei are markedly larger than nuclei from normal epithelial cells and consequently, the rate of FISH signal loss through nuclear truncation will generally be higher in cancer cells than in normal epithelial cells on tissue sections. The cut-off levels selected for our FISH deletion analyses are based on a 100% concordance with CGH array data found for PTEN deletions in an earlier study [38]. High rates of 8p deletion were also described in studies using conventional or array-based comparative genomic hybridization (CGH). For example, two meta-analysis of 872 and 622 prostate cancers analyzed by classical CGH [5] or array CGH (aCGH) [6] revealed 34%-62% 8p deletions, and a large array CGH study reported 53–78% 8p deletions in 181 prostate cancers [4]. In a comparison with studies from our own laboratory using identical criteria for FISH deletion scoring, 8p deletion (36%) is the second most frequent genomic alteration right after TMPRSS2:ERG fusions (52%). The next frequent deletions are 16q (21%) [39], PTEN (20%) [38] and 6q (18%) [40].
Our data identify 8p deletions as a strong prognostic parameter, which is independent of established clinical and pathological prognosticators. This is generally in line with earlier studies on 27-2,097 patients suggesting associations of 8p deletions with unfavorable tumor phenotype [5–12], metastasis [8, 12], hormone refractory disease [10], and PSA recurrence [10, 13]. The high number of cancers analyzed in this study enabled a selective analysis of clinically relevant subgroups. Here, the complete absence of a prognostic impact of 8p deletions in certain high-risk subpopulations such as pT3b, N1 or Gleason ≥ 8 cancers demonstrates, that the dismal prognosis of these cancers is already quite reliably recognized by established histological parameters. That 8p deletion does not distinguish a particularly aggressive subgroup among these tumors further suggests, that 8p deletion does not lead to the activation of a “key prognostic pathway”, which – even in advanced cancers - would result in a massive tumor growth or devastating metastatic spread.
The variable prognostic impact of 8p deletions in cancers with different Gleason grades is of particular interest. That 8p deletions were prognostically most relevant in the subgroup of 3 + 4 carcinomas in univariate as well as in multivariate analysis (when combined with Gleason score) is encouraging because Gleason 3 + 4 represents the clinically most difficult group with treatment options ranging from active surveillance to prostatectomy. These data also illustrate, that the subgroup Gleason 3 + 4 is more heterogeneous then others in clinical outcome. Based on the morphologic analysis of more than 10.000 prostate cancers, we had recently shown, that prognostic Gleason Grade information can be expanded in Gleason 3 + 4 and 4 + 3 cancers by using the percentage of Gleason 4 patterns as a continuous variable. Both in biopsies and in prostatectomy samples, prostate cancer prognosis continuously deteriorated with increasing percentage of Gleason 4 pattern (quantitative Gleason Grade) [41]. That a statistically significant prognostic impact of 8p deletion only remained in few subgroups defined by a comparable quantitative Gleason grade illustrates how difficult it is for a molecular parameter to outperform morphological parameters of malignancy in prostate cancer.
That 8p deletions were not pinpointing towards an unfavorable clinical course in Gleason 3 + 3 carcinomas is of particular interest. It demonstrates that a morphologically benign appearing prostate cancer is not necessarily aggressive, if molecular features with documented prognostic relevance are detected. The high frequency of 8p deletion in 3 + 3 carcinomas (25%) further argues against a clinical importance of this alteration in such early cancers. The fraction of 3 + 3 carcinomas that are thought to develop an aggressive disease course is far lower than 25% [42]. The strong (and independent) overall prognostic impact of 8p deletions in prostate cancer in combination with the high frequency but lack of clinical relevance in 3 + 3 cancers leads us to hypothesize, that 8p deletion represents an early alteration in prostate cancer needing additional factors to drive a cancer towards high aggressiveness.
Our data suggest PTEN inactivation as a possible candidate for a molecular aberration that could particularly efficiently interplay with 8p deletion. PTEN deletion is the strongest single molecular prognostic feature in prostate cancer known as to yet. The strong statistical association between PTEN and 8p deletions and the particularly poor clinical course in case of a co-deletion of PTEN and 8p argues for synergistic effects between these lesions. The 8p genes possibly involved in such an interaction remain unclear. However, mouse models suggest that PTEN loss could drive invasion and metastasis in cooperation with loss of NKX3.1 [43, 44]. Earlier studies had demonstrated that the panel of chromosomal deletions occurring in prostate cancer is largely dependent on the ERG status. Deletions of PTEN, 16q, and 3p13 are tightly linked to ERG expression while deletions of 6q15 and 5q21 is largely restricted to ERG-negative cancers [4, 7, 15, 27, 38, 40, 45, 46]. The data of the present study delineate 8p deletion as the exceptional case of an “ERG-independent” deletion. This is also supported by CGH array data from a cohort of 181 prostate cancers, where no significant link between 8p deletion and ERG fusion was reported [4]. Our data demonstrate, that the slightly higher rate of 8p deletions in ERG-positive (40.9%) than in ERG-negative cancers (34.7%) is entirely driven by its strong association with the ERG-linked PTEN deletion.
In summary, 8p deletion is the second most common genomic alteration in prostate cancer. Several lines of evidence suggest an interplay with PTEN deletions. 8p deletion is a strong and independent prognostic factor in prostate cancer. That 8p deletion lacks prognostic significance in several morphologically defined subgroups with very poor or very good prognosis demonstrates the power of established criteria – such as the Gleason grade - for assessing prostate cancer aggressiveness.
MATERIALS AND METHODS
Patients
A set of prostate cancer tissue microarrays (TMA) was used in this study containing one tissue core each from 12,427 consecutive radical prostatectomy specimens from patients undergoing surgery at the Department of Urology, and the Martini Clinic, Prostate Cancer Center, University Medical Center Hamburg-Eppendorf. This TMA is based on our previous 3,261 samples prostate prognosis TMA [10], with additional 9,166 tumors and updated clinical data from 12,344 patients with a median follow-up of 36.4 months (range: 1 to 241 months; Table 4). In all patients, prostate specific antigen (PSA) values were measured quarterly in the first year, followed by biannual measurements in the second and annual measurements after the third year following surgery. Recurrence was defined as a postoperative PSA of 0.2 ng/ml and rising thereafter. The first PSA value above or equal to 0.2 ng/ml was used to define the time of recurrence. Patients without evidence of tumor recurrence were censored at the time of the last follow-up. All prostate specimens were diagnosed according to a standard procedure, including complete embedding of the entire prostate for histological analysis [47]. The TMA manufacturing process was described earlier in detail [48, 49]. In short, one 0.6 mm core was taken from a representative tissue block from each patient. The tissues were distributed among 27 TMA blocks, each containing 144 to 522 tumor samples. Presence or absence of cancer tissue was validated by immunohistochemical AMACR and 34BE12 analysis on adjacent TMA sections. For internal controls, each TMA block also contained various control tissues, including normal prostate tissue. The molecular database attached to this TMA contained results on ERG expression in 10,678, ERG break apart fluorescence in-situ hybridization (FISH) analysis in 7,099 (expanded from [27, 50]), and deletion status of PTEN in 6,704 (expanded from [38]) tumors.
Table 4: Clinico-pathological features of 12,427 arrayed prostate cancers
No. of patients (%) |
||
|---|---|---|
Study cohort on TMA (n = 12427) |
Biochemical relapse among categories |
|
Follow-up (mo) |
||
n |
11665 (93.9%) |
2769 (23.7%) |
Mean |
48.9 |
- |
Median |
36.4 |
- |
Age (y) |
||
≤ 50 |
334 (2.7%) |
81 (24.3%) |
51–59 |
3061 (24.8%) |
705 (23.0%) |
60–69 |
7188 (58.2%) |
1610 (22.4%) |
≥ 70 |
1761 (14.3%) |
370 (21.0%) |
Pre-operative PSA (ng/ml) |
||
< 4 |
1585 (12.9%) |
242 (15.3%) |
4–10 |
7480 (60.9%) |
1355 (18.1%) |
10–20 |
2412 (19.6%) |
737 (30.6%) |
> 20 |
812 (6.6%) |
397 (48.9%) |
pT category (AJCC 2002) |
||
pT2 |
8187 (66.2%) |
1095 (13.4%) |
pT3a |
2660 (21.5%) |
817 (30.7%) |
pT3b |
1465 (11.8%) |
796 (54.3%) |
pT4 |
63 (0.5%) |
51 (81.0%) |
Gleason grade (WHO/ISUP 2016) |
||
< 7 |
2997 (24.2%) |
368 (12.3%) |
3 + 4 |
6964 (56.1%) |
1288 (18.5%) |
4 + 3 |
1849 (14.9%) |
789 (42.7%) |
8 |
127 (1.0%) |
50 (39.4%) |
9–10 |
469 (3.8%) |
262 (55.7%) |
pN category |
||
pN0 |
6970 (91.0%) |
1636 (23.5%) |
pN+ |
693 (9.0%) |
393 (56.7%) |
Resection margin status |
||
Negative |
9990 (81.9%) |
1848 (18.5%) |
Positive |
2211 (18.1%) |
853 (38.6%) |
NOTE: Numbers do not always add up to 12427 in the different categories because of cases with missing data. Abbreviation: AJCC, American Joint Committee on Cancer.
The usage of archived diagnostic left-over tissues for manufacturing of tissue microarrays and their analysis for research purposes as well as patient data analysis has been approved by local laws (HmbKHG, §12,1) and by the local ethics committee (Ethics commission Hamburg, WF-049/09 and PV3652). All work has been carried out in compliance with the Helsinki Declaration.
Fluorescence in-situ hybridization
Four micrometer TMA sections were used for fluorescence in-situ hybridization (FISH). For proteolytic slide pretreatment, a commercial kit was used (paraffin pretreatment reagent kit; Abbott, Chicago, USA) TMA sections were deparaffinized, air-dried, and dehydrated in 70%, 85%, and 100% ethanol, followed by denaturation for 5 min at 74°C in 70% formamid 2× SSC solution. The FISH probe set consisted of a spectrum-orange labeled NKX3.1 probe (made from a mixture of BAC RP11-625E02 and BAC RP11-116M17), and a commercial spectrum-green labeled centromere 8 probe (#6J37-08; Abbott, Chicago, USA) as a reference. Hybridization was overnight at 37°C in a humidified chamber. Slides were subsequently washed and counterstained with 0.2μmol/L 4′-6-diamidino-2-phenylindole in antifade solution. Stained slides were manually interpreted with an epifluorescence microscope, and the predominant FISH signal numbers were recorded in each tissue spot. Homozygous deletion of 8p was defined as complete absence of NKX3.1 FISH probe signals in ≥ 60% of tumor nuclei, with the presence of one or two NKX3.1 FISH signals in adjacent normal cells. Tissue spots with a lack of NKX3.1 signals in all (tumor and normal cells) or lack of any normal cells as an internal control for successful hybridization of the NKX3.1 probe were excluded from analysis. Heterozygous deletion of NKX3.1 was defined as the presence of fewer NKX3.1 signals than centromere 8 probe signals of ≥ 60% tumor nuclei (Figure 7). These thresholds were based on a previous study comparing PTEN deletion data obtained by FISH and SNP in a subset of cancers included into this TMA set [38].
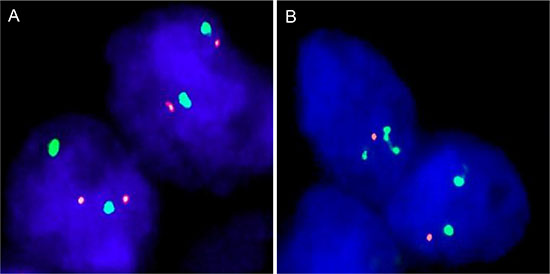
Figure 7: Examples of FISH findings using the 8p deletion probe. (A) Normal 8p copy numbers as indicated by two orange 8p signals and two green centromere 8 signals. (B) Heterozygous deletion as indicated by the lack of one orange 8p signal.
8p copy number data sources and analysis
Raw data were obtained from 4 large studies employing array CGH or SNP array analysis in a total of 442 prostate cancers [4, 15–17]. Data were imported into the FISH Oracle browser [18, 19] and visualized in different tracks corresponding to each study. A global threshold of -0.3 was applied to all 4 datasets to display deletions.
Statistics
For statistical analysis, the JMP 9.0 software (SAS Institute Inc., NC, USA) was used. Contingency tables and Chi-square (Likelihood) tests were utilized to study the relationship between 8p deletion and categorical clinico-pathological variables. Kaplan Meier curves were generated for PSA recurrence free survival. The log-Rank test was applied to test the significance of differences between stratified survival functions. Cox proportional hazards regression analysis was performed to test the statistical independence and significance between pathological, molecular, and clinical variables. Because of the high number of samples included in our study, we considered differences as being statistically relevant at an alpha niveau of 0.01.
ACKNOWLEDGMENTS
We thank Janett Lütgens, Sünje Seekamp, Inge Brandt, Bianca Kelp, Anne Meyer and Lisa Paustian for excellent technical assistance.
CONFLICTS OF INTEREST
We certify that there is no actual or potential conflicts of interest in relation to this article.
FUNDING
This work was supported by the Federal Ministry of Education and Research, grant 01ZX1302C.
REFERENCES
1. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA-Cancer J Clin. 2011; 61:69–90.
2. Sakr WA, Grignon DJ, Crissman JD, Heilbrun LK, Cassin BJ, Pontes JJ, Haas GP. High grade prostatic intraepithelial neoplasia (HGPIN) and prostatic adenocarcinoma between the ages of 20–69: an autopsy study of 249 cases. In Vivo. 1994; 8:439–443.
3. Yin M, Bastacky S, Chandran U, Becich MJ, Dhir R. Prevalence of incidental prostate cancer in the general population: a study of healthy organ donors. J Urol. 2008; 179:892–895; discussion 895.
4. Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, Arora VK, Kaushik P, Cerami E, Reva B, Antipin Y, Mitsiades N, Landers T, et al. Integrative genomic profiling of human prostate cancer. Cancer cell. 2010; 18:11–22.
5. Sun J, Liu W, Adams TS, Li X, Turner AR, Chang B, Kim JW, Zheng SL, Isaacs WB, Xu J. DNA copy number alterations in prostate cancers: a combined analysis of published CGH studies. Prostate. 2007; 67:692–700.
6. Williams JL, Greer PA, Squire JA. Recurrent copy number alterations in prostate cancer: an in silico meta-analysis of publicly available genomic data. Cancer Genet. 2014; 207:474–488.
7. Lapointe J, Li C, Giacomini CP, Salari K, Huang S, Wang P, Ferrari M, Hernandez-Boussard T, Brooks JD, Pollack JR. Genomic profiling reveals alternative genetic pathways of prostate tumorigenesis. Cancer Res. 2007; 67:8504–8510.
8. Ribeiro FR, Diep CB, Jeronimo C, Henrique R, Lopes C, Eknaes M, Lingjaerde OC, Lothe RA, Teixeira MR. Statistical dissection of genetic pathways involved in prostate carcinogenesis. Gene Chromosomes Cancer. 2006; 45:154–163.
9. Matsuda K, Matsuyama H, Hara T, Yoshihiro S, Oga A, Kawauchi S, Furuya T, Izumi H, Naito K, Sasaki K. DNA sequence copy number aberrations in prostate cancers: a comparison of comparative genomic hybridization data between Japan and European countries. Cancer Genet. Cytogen. 2004; 152:119–123.
10. El Gammal AT, Bruchmann M, Zustin J, Isbarn H, Hellwinkel OJ, Kollermann J, Sauter G, Simon R, Wilczak W, Schwarz J, Bokemeyer C, Brummendorf TH, Izbicki JR, et al. Chromosome 8p deletions and 8q gains are associated with tumor progression and poor prognosis in prostate cancer. Clin Cancer Res. 2010; 16:56–64.
11. Matsuyama H, Pan Y, Yoshihiro S, Kudren D, Naito K, Bergerheim US, Ekman P. Clinical significance of chromosome 8p, 10q, and 16q deletions in prostate cancer. Prostate. 2003; 54:103–111.
12. Oba K, Matsuyama H, Yoshihiro S, Kishi F, Takahashi M, Tsukamoto M, Kinjo M, Sagiyama K, Naito K. Two putative tumor suppressor genes on chromosome arm 8p may play different roles in prostate cancer. Cancer Genet Cytogen. 2001; 124:20–26.
13. Tsuchiya N, Slezak JM, Lieber MM, Bergstralh EJ, Jenkins RB. Clinical significance of alterations of chromosome 8 detected by fluorescence in situ hybridization analysis in pathologic organ-confined prostate cancer. Gene Chromosomes Cancer. 2002; 34:363–371.
14. Sato K, Qian J, Slezak JM, Lieber MM, Bostwick DG, Bergstralh EJ, Jenkins RB. Clinical significance of alterations of chromosome 8 in high-grade, advanced, nonmetastatic prostate carcinoma. J Nat Cancer Inst. 1999; 91:1574–1580.
15. Krohn A, Seidel A, Burkhardt L, Bachmann F, Mader M, Grupp K, Eichenauer T, Becker A, Adam M, Graefen M, Huland H, Kurtz S, Steurer S, et al. Recurrent deletion of 3p13 targets multiple tumour suppressor genes and defines a distinct subgroup of aggressive ERG fusion-positive prostate cancers. J Pathol. 2013; 231:130–141.
16. Mao X, Boyd LK, Yanez-Munoz RJ, Chaplin T, Xue L, Lin D, Shan L, Berney DM, Young BD, Lu YJ. Chromosome rearrangement associated inactivation of tumour suppressor genes in prostate cancer. Am J Cancer Res. 2011; 1:604–617.
17. Huang S, Gulzar ZG, Salari K, Lapointe J, Brooks JD, Pollack JR. Recurrent deletion of CHD1 in prostate cancer with relevance to cell invasiveness. Oncogene. 2012; 31:4164–4170.
18. Mader M, Simon R, Kurtz S. FISH Oracle 2: a web server for integrative visualization of genomic data in cancer research. J Clin Bioinforma. 2014; 4:5.
19. Mader M, Simon R, Steinbiss S, Kurtz S. FISH Oracle: a web server for flexible visualization of DNA copy number data in a genomic context. J Clin Bioinforma. 2011; 1:20.
20. Birnbaum D, Adelaide J, Popovici C, Charafe-Jauffret E, Mozziconacci MJ, Chaffanet M. Chromosome arm 8p and cancer: a fragile hypothesis. Lancet Oncol. 2003; 4:639–642.
21. Bowen C, Bubendorf L, Voeller HJ, Slack R, Willi N, Sauter G, Gasser TC, Koivisto P, Lack EE, Kononen J, Kallioniemi OP, Gelmann EP. Loss of NKX3.1 expression in human prostate cancers correlates with tumor progression. Cancer Res. 2000; 60:6111–6115.
22. He WW, Sciavolino PJ, Wing J, Augustus M, Hudson P, Meissner PS, Curtis RT, Shell BK, Bostwick DG, Tindall DJ, Gelmann EP, Abate-Shen C, Carter KC. A novel human prostate-specific, androgen-regulated homeobox gene (NKX3.1) that maps to 8p21, a region frequently deleted in prostate cancer. Genomics. 1997; 43:69–77.
23. Zhang R, Song C. Loss of CSMD1 or 2 may contribute to the poor prognosis of colorectal cancer patients. Tumour Biol. 2014; 35:4419–4423.
24. Xue W, Krasnitz A, Lucito R, Sordella R, Vanaelst L, Cordon-Cardo C, Singer S, Kuehnel F, Wigler M, Powers S, Zender L, Lowe SW. DLC1 is a chromosome 8p tumor suppressor whose loss promotes hepatocellular carcinoma. Genes Dev. 2008; 22:1439–1444.
25. Hornstein M, Hoffmann MJ, Alexa A, Yamanaka M, Muller M, Jung V, Rahnenfuhrer J, Schulz WA. Protein phosphatase and TRAIL receptor genes as new candidate tumor genes on chromosome 8p in prostate cancer. Cancer Genomics Proteomics. 2008; 5:123–136.
26. Beuten J, Gelfond JA, Franke JL, Shook S, Johnson-Pais TL, Thompson IM, Leach RJ. Single and multivariate associations of MSR1, ELAC2, and RNASEL with prostate cancer in an ethnic diverse cohort of men. Cancer Epidemiol Biomarkers Prev. 2010; 19:588–599.
27. Weischenfeldt J, Simon R, Feuerbach L, Schlangen K, Weichenhan D, Minner S, Wuttig D, Warnatz HJ, Stehr H, Rausch T, Jäger N, Gu L, Bogatyrova O, et al. Integrative genomic analyses reveal androgen-driven somatic alteration landscape in early-onset prostate cancer. Cancer Cell. 2013; 23:159–170.
28. Berger MF, Lawrence MS, Demichelis F, Drier Y, Cibulskis K, Sivachenko AY, Sboner A, Esgueva R, Pflueger D, Sougnez C, Onofrio R, Carter SL, Park K, et al. The genomic complexity of primary human prostate cancer. Nature. 2010; 470:214–220.
29. Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, Quist MJ, Jing X, Lonigro RJ, Brenner JC, Asangani IA, Ateeq B, Chun SY, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012; 487:239–243.
30. Barbieri CE, Baca SC, Lawrence MS, Demichelis F, Blattner M, Theurillat JP, White TA, Stojanov P, Van Allen E, Stransky N, Nickerson E, Chae SS, Boysen G, et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat Genet. 2012; 44:685–689.
31. Xue W, Kitzing T, Roessler S, Zuber J, Krasnitz A, Schultz N, Revill K, Weissmueller S, Rappaport AR, Simon J, Zhang J, Luo W, Hicks J, et al. A cluster of cooperating tumor-suppressor gene candidates in chromosomal deletions. Proc Natl Acad Sci. 2012; 109:8212–8217.
32. Locke JA, Zafarana G, Ishkanian AS, Milosevic M, Thoms J, Have CL, Malloff CA, Lam WL, Squire JA, Pintilie M, Sykes J, Ramnarine VR, Meng A, et al. NKX3.1 haploinsufficiency is prognostic for prostate cancer relapse following surgery or image-guided radiotherapy. Clin Cancer Res. 2012; 18:308–316.
33. Macoska JA, Trybus TM, Wojno KJ. 8p22 loss concurrent with 8c gain is associated with poor outcome in prostate cancer. Urol. 2000; 55:776–782.
34. Bethel CR, Faith D, Li X, Guan B, Hicks JL, Lan F, Jenkins RB, Bieberich CJ, De Marzo AM. Decreased NKX3.1 protein expression in focal prostatic atrophy, prostatic intraepithelial neoplasia, and adenocarcinoma: association with gleason score and chromosome 8p deletion. Cancer Res. 2006; 66:10683–10690.
35. Das K, Lau W, Sivaswaren C, Ph T, Fook-Chong S, Sl T, Cheng C. Chromosomal changes in prostate cancer: a fluorescence in situ hybridization study. Clin. Genet. 2005; 68:40–47.
36. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012; 2:401–404.
37. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, Cerami E, Sander C, Schultz N. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013; 6:pl1.
38. Krohn A, Diedler T, Burkhardt L, Mayer PS, De Silva C, Meyer-Kornblum M, Kotschau D, Tennstedt P, Huang J, Gerhauser C, Mader M, Kurtz S, Sirma H, et al. Genomic deletion of PTEN is associated with tumor progression and early PSA recurrence in ERG fusion-positive and fusion-negative prostate cancer. Am J Pathol. 2012; 181:401–412.
39. Kluth M, Runte F, Barow P, Omari J, Abdelaziz ZM, Paustian L, Steurer S, Christina Tsourlakis M, Fisch M, Graefen M, Tennstedt P, Huland H, Michl U, et al. Concurrent deletion of 16q23 and PTEN is an independent prognostic feature in prostate cancer. Int J Cancer. 2015; 137:2354–2363.
40. Kluth M, Hesse J, Heinl A, Krohn A, Steurer S, Sirma H, Simon R, Mayer PS, Schumacher U, Grupp K, Izbicki JR, Pantel K, Dikomey E, et al. Genomic deletion of MAP3K7 at 6q12–22 is associated with early PSA recurrence in prostate cancer and absence of TMPRSS2:ERG fusions. Mod Pathol. 2013; 26:975–983.
41. Sauter G, Steurer S, Clauditz TS, Krech T, Wittmer C, Lutz F, Lennartz M, Janssen T, Hakimi N, Simon R, von Petersdorff-Campen M, Jacobsen F, von Loga K, et al. Clinical Utility of Quantitative Gleason Grading in Prostate Biopsies and Prostatectomy Specimens. Eur Urol. 2016; 69:592–598.
42. Klotz L. Active surveillance for low-risk prostate cancer. Curr Urol Rep. 2015; 16:24.
43. Kim MJ, Cardiff RD, Desai N, Banach-Petrosky WA, Parsons R, Shen MM, Abate-Shen C. Cooperativity of Nkx3.1 and Pten loss of function in a mouse model of prostate carcinogenesis. Proc Natl Acad Sci. 2002; 99:2884–2889.
44. Abate-Shen C, Banach-Petrosky WA, Sun X, Economides KD, Desai N, Gregg JP, Borowsky AD, Cardiff RD, Shen MM. Nkx3.1; Pten mutant mice develop invasive prostate adenocarcinoma and lymph node metastases. Cancer Res. 2003; 63:3886–3890.
45. Kluth M, Harasimowicz S, Burkhardt L, Grupp K, Krohn A, Prien K, Gjoni J, Hass T, Galal R, Graefen M, Haese A, Simon R, Huhne-Simon J, et al. Clinical significance of different types of p53 gene alteration in surgically treated prostate cancer. Int J Cancer. 2014.
46. Burkhardt L, Fuchs S, Krohn A, Masser S, Mader M, Kluth M, Bachmann F, Huland H, Steuber T, Graefen M, Schlomm T, Minner S, Sauter G, et al. CHD1 Is a 5q21 Tumor Suppressor Required for ERG Rearrangement in Prostate Cancer. Cancer research. 2013; 73:2795–2805.
47. Erbersdobler A, Fritz H, Schnoger S, Graefen M, Hammerer P, Huland H, Henke RP. Tumour grade, proliferation, apoptosis, microvessel density, p53, and bcl-2 in prostate cancers: differences between tumours located in the transition zone and in the peripheral zone. Eur Urol. 2002; 41:40–46.
48. Mirlacher M, Simon R. Recipient block TMA technique. Methods Mol Biol. 2010; 664:37–44.
49. Bubendorf L, Kononen J, Koivisto P, Schraml P, Moch H, Gasser TC, Willi N, Mihatsch MJ, Sauter G, Kallioniemi OP. Survey of gene amplifications during prostate cancer progression by high-throughout fluorescence in situ hybridization on tissue microarrays. Cancer Res. 1999; 59:803–806.
50. Minner S, Enodien M, Sirma H, Luebke AM, Krohn A, Mayer PS, Simon R, Tennstedt P, Muller J, Scholz L, Brase JC, Liu AY, Schluter H, et al. ERG Status Is Unrelated to PSA Recurrence in Radically Operated Prostate Cancer in the Absence of Antihormonal Therapy. Clin. Cancer Res. 2011; 17:5878–5888.