
Introduction
Enhanced activity of the class IA phosphoinositide 3-kinase (PI3K) pathway is frequent in cancer [1-6]. PI3K are lipid kinases encoded by PIK3 genes, of which PIK3CA and CB are ubiquitous [1-6]. After activation by growth factors, PI3K generate phosphoinositide (PI)(3,4,5)P3 at the plasma membrane; this stimulates downstream targets such as PKB and mTOR, which trigger cell survival, division, and migration. The phosphatase PTEN reduces PI(3,4,5)P3 levels [6]. PIK3CA mutations appear at early stages in cancer progression, whereas PTEN inactivation is a later event [3-10]. The role of PI3Kβ in cancer is less understood; PIK3CB mutations in cancer appear at lower frequency than those of PIK3CA [5, 11], but enhanced PI3Kβ expression is sufficient to induce transformation [12]. PI3Kβ levels are increased in colon, ovarian, endometrial, breast and bladder carcinoma [3, 5, 9, 10].
In the mouse, expression of active PI3Kβ leads to intraepithelial prostate neoplasia and Pik3cb deletion inhibits prostate cancer in Pten+/- mice [13, 14]. These observations led to studies in xenograft models, which showed that PI3Kβ inhibitors decelerate tumor growth in PTEN-mutant tumors [15-17]. The correlation between PTEN loss of function and PI3Kβ inhibitor-mediated cytostatic effect is not universal, and the mechanism underlying PTEN and PI3Kβ cooperation is unclear [5, 18-20]. PI3K is considered a key target for cancer therapy, but only enzymatic inhibitors have been tested clinically [21-23]. Some of these studies are promising, but others have failed due to blockade of feedback mechanisms or acquisition of resistance [5, 21-23]. Since PI3Kβ generates PI(3,4,5)P3 but also has kinase-independent functions [24-26], we tested the effect of depleting PI3Kβ using interfering RNA (siRNA) on a collection of diverse tumor types and in a set of urothelial bladder carcinoma (UBC) lines.
UBC is the fifth most frequent tumor in developed countries; although the majority of these tumors (75%) are non muscle-invasive (NMI) and treatable by transurethral resection, many recur (70%) and up to 10-15% progress to muscle-invasive (MI) metastatic carcinoma, with poor prognosis [1, 2, 5. The molecular mechanisms that underlie UBC invasiveness must thus be identified, as well as new therapeutic strategies for MI-UBC.
We tested the effect of depleting PI3Kβ in these tumors, since UBC tumors show enhanced PI3Kβ expression, loss of PTEN function at advanced stages, and accessibility for local administration of siRNA [4, 7, 10]. We show that PI3Kβ depletion is cytotoxic for most tumors with mutant PTEN. Treatment of PTEN-mutant tumor xenografts by intratumor (i.t.) injection of PIK3CB siRNA (siPIK3CB) or by expression of doxycycline (doxy)-inducible PIK3CB shRNA was markedly more potent than PI3Kβ inhibition. Some PTEN-mutant tumor cells were resistant to siPIK3CB; we took advantage of this phenotype to study what determines resistance. We show that PTEN inactivation and E-cadherin (E-cad) loss are necessary for sensitivity. These observations support development of PIK3CB-interfering approaches for treatment of advanced UBC with E-cad loss and PTEN mutations.
Results
PTEN mutation is not sufficient for tumor PI3Kβ dependence
PI3Kβ enzymatic inhibition retards the growth of most PTEN-mutant tumor xenografts, but does not trigger cell death [16, 18-20]. Since PI3Kβ has enzymatic and non-enzymatic activities [24-26], we evaluated the effect of depleting PIK3CB in xenografts. We optimized PI3Kβ depletion using siRNA in PTEN-mutant prostate cancer PC3 cells. Transfection of 0.5x106 cells with 6 to 12 pmol of siRNA1 or siRNA2 was sufficient for efficient silencing and for reduction of cell viability (Supplementary Figure S1a). We also grew PC3 cell-derived xenografts in immunodeficient mice; when tumors reached ~75 mm3, mice were treated with siRNA. As systemic siRNA administration by intraperitoneal injection (i.p.) did not deplete PI3Kβ efficiently in tumors (not shown), we optimized administration by direct injection into the tumor center of PIK3CB siRNA1 (siPIK3CB1) (12.5, 50, 120 pmol/mm3 tumor) on days 1, 3 and 5, and effects were tested on day 7. The 50pmol/mm3 dose of siPIK3CB1optimally reduced PI3Kβ levels in PC3 xenografts and the PI3Kβ -dependent [25] PCNA binding to chromatin (Supplementary Figure S1b).
To study the effect of PI3K depletion on PC3 tumor growth, we divided a set of mice bearing ~75 mm3 tumors into random groups, which we treated with PBS, liposomes, or liposomes plus control or siPIK3CB1 (50pmol/mm3). Treatment was administered three times a week for two weeks. Repeated injection with liposomes decelerated tumor growth; nonetheless, siPIK3CB delivery triggered tumor regression (Supplementary Figure S1c). siRNA delivery at the end of treatment might have been suboptimal due to the small tumor size; optimal PIK3CB depletion might have yielded additional effects. Using representative PC3 tumor extracts, we showed that PI3Kβ knockdown reduced pPKB levels and PCNA binding to chromatin (Supplementary Figure S1d). Histopathology analysis of representative samples showed that whereas necrotic areas in vehicle and control siRNA-treated tumors did not exceed 10% of the tumor, the cell death index in siPIK3CB1-treated tumors reached 90-100% of cells in 60% of samples; all liposome-treated samples showed an inflammatory neutrophil infiltration, which might explain the tumor growth deceleration in the presence of vehicle (Supplementary Figure S1e). TUNEL staining of tumor sections showed that those treated with control siRNA had few positive cells, and that siPIK3CB1 treatment significantly increased TUNEL+ cells (Supplementary Figure S1f).
We extended the analysis to a collection of cell lines representative of distinct tumor types that expressed wild-type (WT) PTEN (HT-29, SW480, NCI-H226, HT1080), mutant PTEN (U87MG, MDA-MB468, BLM, H520), or showed PTEN promoter methylation (CaLu-1) [27]. We performed two transfections (days 1 and 3) using siPIK3CB1 (0.2 nmol/0.5x106 cells) and tested the consequences on day 5. PIK3CB silencing did not affect WT PTEN lines but reduced viability of the cells with PTEN loss of function, with the exception of MDA-MB468 and H520 cells, which were resistant (Figure 1a-1c). This shows that PTEN inactivation is necessary but insufficient for PI3Kβ dependence.
To define which pathways could synergize with PTEN inactivation to render the cells PI3Kβ-dependent, we examined features frequently altered in cancer, including E-cad levels, TP53 mutation and ERK activation [28, 29]. All cells were pre-checked for TP53 mutations (cansar.icr.uk/cansar/cell lines); we used Western blot (WB) to evaluate pERK and E-cad levels (Figure 1d). The siPIK3CB-responsive cells shared PTEN mutation and low E-cad levels but presented variable pERK and TP53 status; accordingly, the two PTEN mutant resistant cells (MDA-MB468 and H520) expressed high E-cad levels (Figure 1d, 1e).
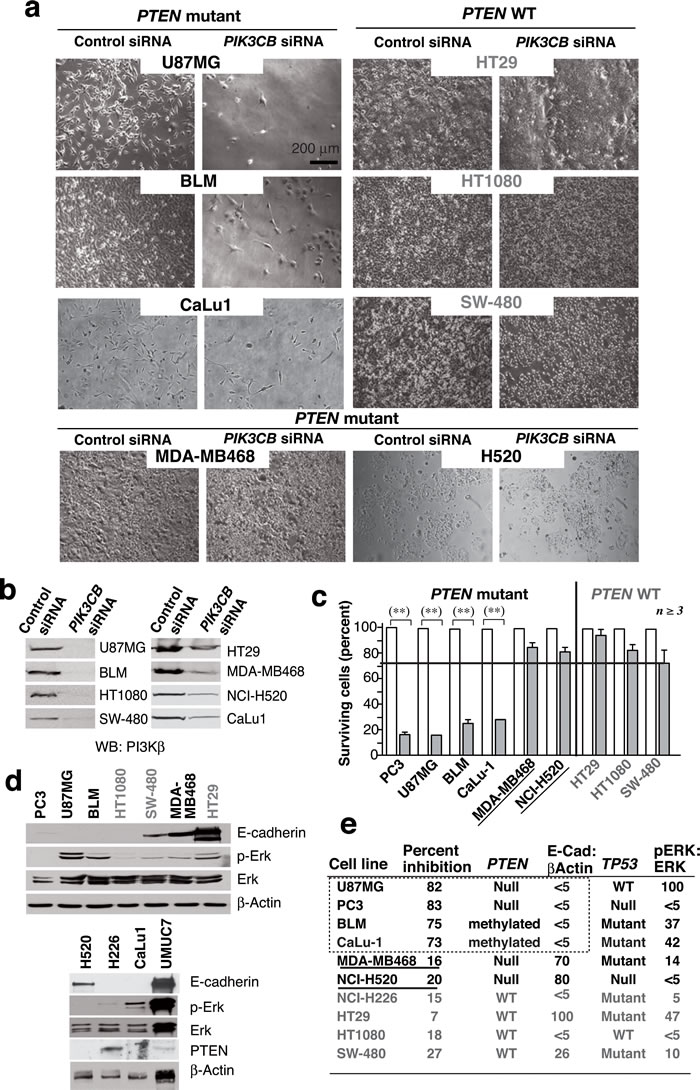
Figure 1: PTEN mutation is necessary but not sufficient for PI3Kβ dependence of cancer cell lines. a. We transfected various WT or PTEN mutant cancer cell lines (0.5x106 cells) with 12 or 20 pmol siPIK3CB (depending on cell line) on days 1 and 3 and examined them at day 5. Representative images for 8 cell lines tested. b. Western blot (WB) analysis of extracts from tumor cells as in a.. c. Viability of cells treated with siPIK3CB as in a. and compared to controls (100%). ** P < 0.01, Student’s t-test (n ≥ 3). d. Extracts of various tumor cells were tested in WB (indicated). e. Summary of tumor cell responses to PI3Kβ silencing (as a percent of cell survival inhibition), the TP53 and PTEN mutation status, and the ratio of pErk:Erk or E-cad:β-actin signals in d. expressed as a percentage of maximum levels (100%). Cell lines with mutant PTEN and high E-cad levels are underlined.
PTEN mutation is not sufficient for PI3Kβ dependence in bladder cancer
Previous studies using a few cell lines suggest that PI3Kβ expression increases in UBC [9]; we examined a large UBC collection from reported DNA microarrays [30-32]. Samples were grouped as “normal tissue”, “low-grade NMI” (G1-2, Ta-T1), and “high-grade tumors” including NMI (G3, Ta-T2) and MI tumors (T3-T4). “Low-grade tumors” had higher PIK3CB mRNA levels than normal bladder tissue, and “high-grade tumors” had the highest levels (Figure 2a). Data were confirmed in two independent datasets; PIK3CB gene expression levels were higher in “high-grade” than in “low-grade” tumors [31, 32] (Figure 2a). These results show that increased PIK3CB expression is frequent at advanced stages UBC.
We next studied 11 well-characterized UBC lines [33]. We optimized siPIK3CB treatment in 639V UBC cells (Supplementary Figure S2a-S2f); transfection (0.5x106 cells) on day 1 and 3 with 0.2 nmol siPIK3CB1 efficiently depleted PI3Kβ by day 5 and reduced cell viability (Figure 2b-2d), expression of PIK3CB shRNA had a similar effect (Supplementary Figure S2a-S2f). WT PTEN lines were resistant to PIK3CB silencing; of the six PTEN-mutant or -low-expression lines, four were sensitive, but UMUC-9 and -7 cells were resistant to PI3Kβ depletion (Figure 2b-2d) confirming that PTEN loss is insufficient for PI3Kβ dependency.
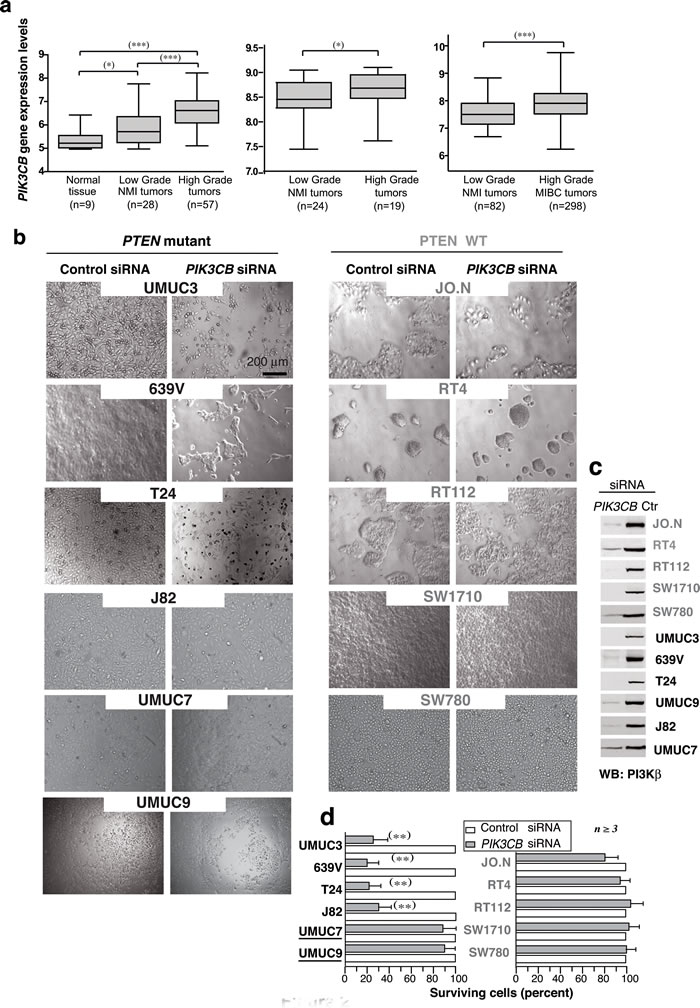
Figure 2: Advanced clinical UBC express high PIK3CB levels; PIK3CB depletion reduces cell survival in UBC lines. a. Box plots show PIK3CB gene expression (mean ± SEM) in clinical UBC. Gene expression data are expressed in log2 Robust Multichip Average (RMA) intensity units. Corresponding GEO accession numbers are GSE5287 (left panel) and GSM1838824 (center). The third dataset (right) is available from ArrayExpress (see Methods). MI, muscle-invasive; NMI, non-muscle invasive. For statistical analysis, we used the Kruskal-Wallis test with two-tailed Mann-Whitney post hoc test for paired group comparison; the one-tailed Mann-Whitney test was applied for group comparison. *** P ≤0.0001, ** P < 0.01, * P < 0.05. (b, c) Bladder carcinoma cell lines (0.5 x 106 cells) were transfected on days 1 and 3 with 0.2 nmol control or siPIK3CB and were analyzed on day 5. Images show representative fields b. and WB analysis of tumor extracts c.. d. Percentage of surviving cells at experiment termination in siPIK3CB- vs. control siRNA-transfected cells (100%). ** P < 0.01 Student’s t-test.
We also analyzed the sensitivity of the UBC lines to a PI3Kβ inhibitor, AZD8186 [20]. 639V cells responded to AZD8186 in a dose-dependent manner (72 h), but cell death was lower than that detected upon siPIK3CB transfection, examined in parallel; both siPIK3CB and AZD8186 moderately reduced pPKB levels (Figure 3a-3c). Analysis of the remaining lines (Supplementary Figure S3a-S3c) showed that AZD8186 did not impair the growth of WT PTEN cells; in mutant-PTEN lines (UMUC-3, T24, J82), AZD8186 was less potent than siPIK3CB in reducing cell numbers; the siPIK3CB-resistant UMUC-9 and -7 lines were also resistant to PI3Kβ inhibitors; responsive cells showed moderate reduction of pPKB levels.
To confirm that a PI3Kβ scaffold function is more important than its kinase activity for UBC cell survival, we prepared an inactive siPIK3CB-resistant mutant KR-PI3Kβ (KR-PI3Kβ-si-res) that was not depleted by siPIK3CB (Figure 3d-3f). Although KR-PI3Kβ expression reduced pPKB more than siPIK3CB; KR- and KR-PI3Kβ-si-res reduced viability moderately while PI3Kβ depletion had a greater effect that was corrected by KR-PI3K-si-res expression (Figure 3d-3f). Thus, a PI3Kβ kinase-independent action contributes to cell survival in PTEN-mutant UBC.
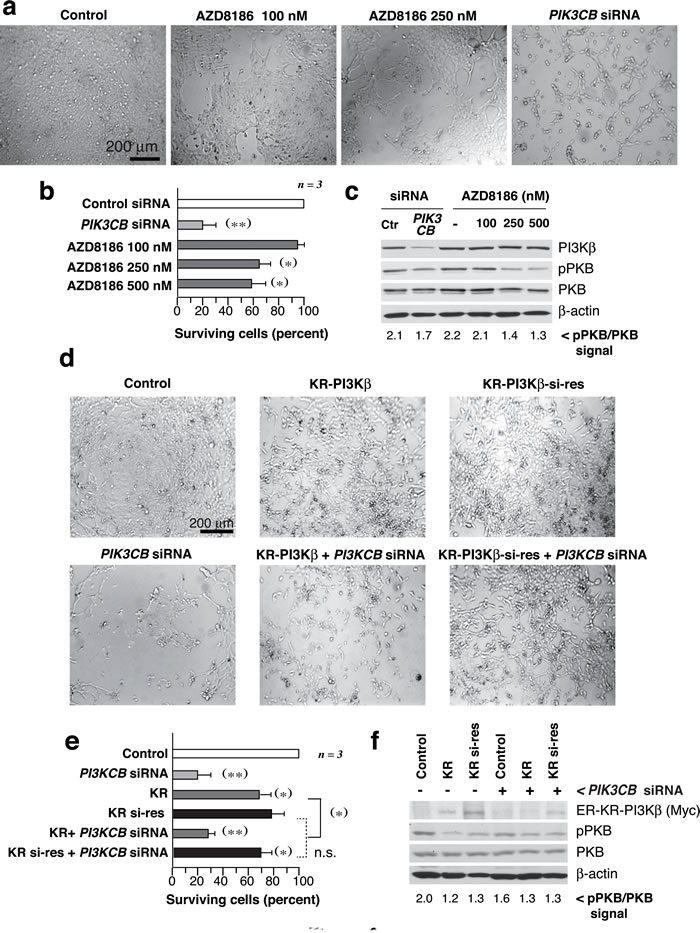
Figure 3: PI3Kβ regulates UBC cell survival in a kinase-independent manner. a.-c. 639V cells were cultured with AZD8186 (dose indicated) or were transfected with siPIK3CB at days 1 and 3, and examined at day 5. Images show representative fields a., percentage of surviving cells at experiment termination compared to control cells (100%) b., and WB examination of cell extracts c.; pPKB signal normalized to PKB is indicated at the bottom c.. d., e. 639V cells were transfected with control or siPIK3CB alone or in combination with ER-myc-K805R-p110β (KR-PI3Kβ) or the siPIK3CB-resistant mutant (KR-PI3Kβ-si-res) (72 h). All KR-p110β-transfected cells were treated with tamoxifen (1 µM) for 12 h before analysis. Representative images d. and percentage of surviving cells e. analyzed as in b.. f. WB showing recombinant KR-PI3Kβ (examined with anti-myc Ab), pPKB, and controls; pPKB signal was quantitated as in c.. ** P < 0.01, *P < 0.05; Student’s t-test.
PI3Kβ depletion induces tumor regression in a PTEN-mutant UBC
To determine whether UBC also require PI3Kβ for tumor growth in vivo, we treated 639V xenografts with siPIK3CB. Tumors grew exponentially and when they reached ~75 mm3, they were treated by i.t. injection of PBS, liposomes, or liposomes plus control or siPIK3CB (50pmol/mm3). Although liposomes decelerated tumor growth, siPIK3CB was able to induce a partial tumor regression (Figure 4a). Considering that liposomes decelerated tumor growth, to confirm the effect of depleting PIK3CB in an alternative method, we generated tumors using 639V cells that expressed doxy-inducible shPIK3CB. When tumors were ~75 mm3, mice were treated by addition of doxy in the drinking water (4 mg/ml) for two weeks. PI3Kβ depletion was partial (see below); nonetheless, tumors from untreated mice grew exponentially, while tumors from doxy-treated mice failed to grow or reduced its size (~50%) (Figure 4b). We also treated the tumors by i.p. injection with the PI3Kβ inhibitor AZD8186 [20] (40 mg/kg, every 12 h, 16 days). Pharmacological inhibition only moderately decelerated tumor growth (Figure 4c) and was associated with greater toxicity as 20% of treated mice had to be sacrificed prematurely due to weight loss.
PI3Kβ depletion in the tumors induced caspase 3 cleavage, reduced PI3Kβ expression and decreased PCNA levels in chromatin; it also slightly lowered pPKB levels; AZD8186 treatment reduced pPKB and PCNA chromatin binding and had a low effect on caspase 3 (Figure 4d). PIK3CB shRNA only partially depleted PI3Kβ; optimal PIK3CB depletion might have yielded additional effects. Histopathology analysis showed a moderate inflammatory infiltration in liposome-treated samples, few necrotic cells in controls and a large percentage of dead cells in siPIK3CB-treated tumors (50-100%) (Figure 4e). This analysis shows that PI3Kβ depletion reduced UBC tumor growth more markedly than PI3Kβ inhibition.
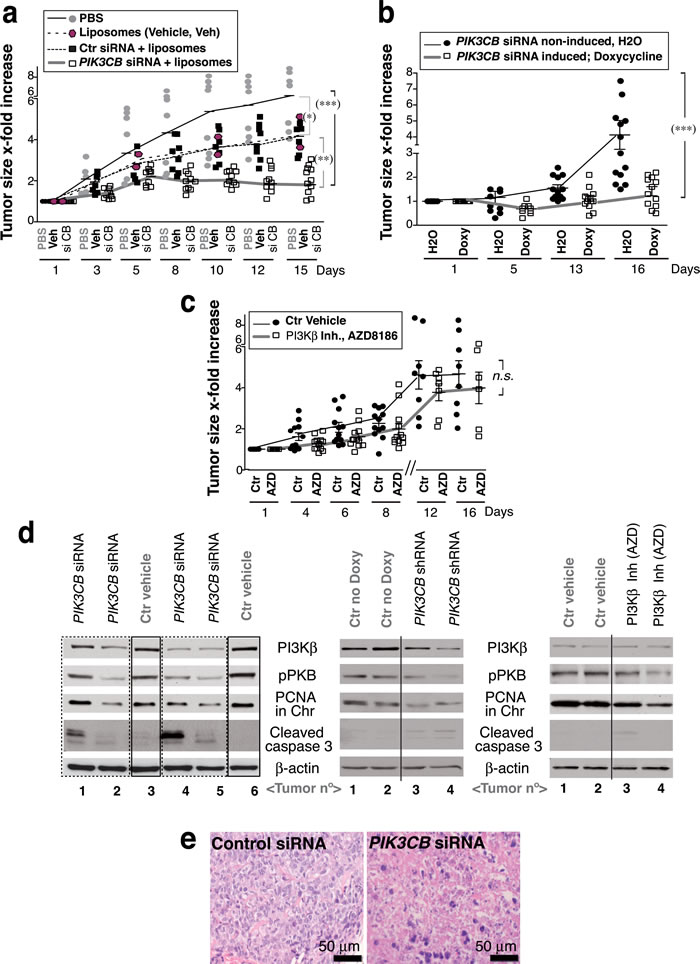
Figure 4: PTEN-negative, E-cadherin-low UBC cells are sensitive to PIK3CB depletion. a. To generate xenografts, 639V cells were injected subcutaneously into immunodeficient mice. When tumors reached 75 mm3, mice were treated by i.t. injection of PBS (control), vehicle alone (Invivofectamine, liposomes), or vehicle plus control or siPIK3CB (50pmol/mm3), every other day for two weeks. b. Xenografts were generated as in a. using 639V cells expressing doxycycline (Doxy)-inducible PIK3CB shRNA. When tumors reached ~75 mm3, ~half of the mice were treated by addition of Doxy (4 mg/ml) in the drinking water. c. Another set of tumors was generated similarly and was treated by i.p. injection of AZD8186 (40 mg/kg) in vehicle (methylcellulose) or with vehicle alone (control) every 12 h (days 1 to 5 each week) and once a day (days 6 and 7) for two weeks. (a-c) Graphs show the x-fold increase in tumor size (mean ± SD) at various times relative to volume at treatment initiation (day 1). *** P < 0.001; ** P < 0.01; * P < 0.05; n.s. not significant; one-way ANOVA, Tukey’s comparison test. d. WB analysis of extracts from representative tumors (a-c). e. Hematoxylin/eosin staining of representative tumors from a..
E-cad/N-cad expression determines PTEN-mutant UBC dependence on PI3Kβ
PI3Kβ depletion in UBC showed that PTEN loss of function is necessary but not sufficient for PI3Kβ dependence. We compared the TP53 mutation status [33] and E-cad levels in the UBC lines. Of the six lines exhibiting PTEN loss of function, the four PI3Kβ-dependent cells had little or no E-cad, and expressed 90 or 130 kDa N-cad forms; in contrast, the siPIK3CB-resistant cells (UMUC-9 and -7) expressed high E-cad levels (Figure 5a), suggesting that also in UBC low E-cad expression is needed for PI3Kβ dependence.
Pten loss of function cooperates with Lkb1 or Tp53 mutations to initiate EMT, which is associated with E-cad loss and N-cad expression [29, 34]. Using the Cancer Genome Atlas UBC tumor collection, we evaluated whether PTEN loss of function was linked to E-cad (CDH1) downregulation or N-cad (CDH2) expression. PTEN alterations in UBC were associated with CDH2 upregulation (P = 0.01, www.cbioportal.org) (Supplementary Figure S4). Of the samples with altered PTEN and CDH1, 75% showed parallel gain or loss of function in both genes; in contrast, 75% of samples with alterations in PTEN and CDH2 showed an inverse pattern of alterations in both genes (Figure 5b). This suggests that PTEN loss of function is associated to E-cad loss and N-cad upregulation in UBC.
The association of PTEN loss of function with CDH2 upregulation made difficult to find mutant PTEN UBC lines that expressed E-cad (two in the UBC collection; two in the diverse tumor set). To explore the hypothesis that low E-cad levels and PTEN mutation are both necessary for UBC dependence on PI3Kβ, we used a genetic approach and examined the effect of E-cad reconstitution in a responsive cell line lacking E-cad expression (639V) or its depletion in an E-cad-expressing resistant cell line (UMUC-9). E-cad reconstitution rendered 639V cells resistant to PIK3CB shRNA, whereas E-cad reduction yielded UMUC-9 cells sensitive to PIK3CB depletion (Figure 5c-5e). pPKB levels were moderately reduced by PI3Kβ depletion in E-cad-low 639V cells, but pPKB was unaffected by PI3Kβ depletion in WT or E-cad-depleted UMUC-9 cells (Figure 5c). We confirmed that PIK3CB depletion induced apoptosis in E-cad-depleted but not in control UMUC-9 cells (Supplementary Figure S5).
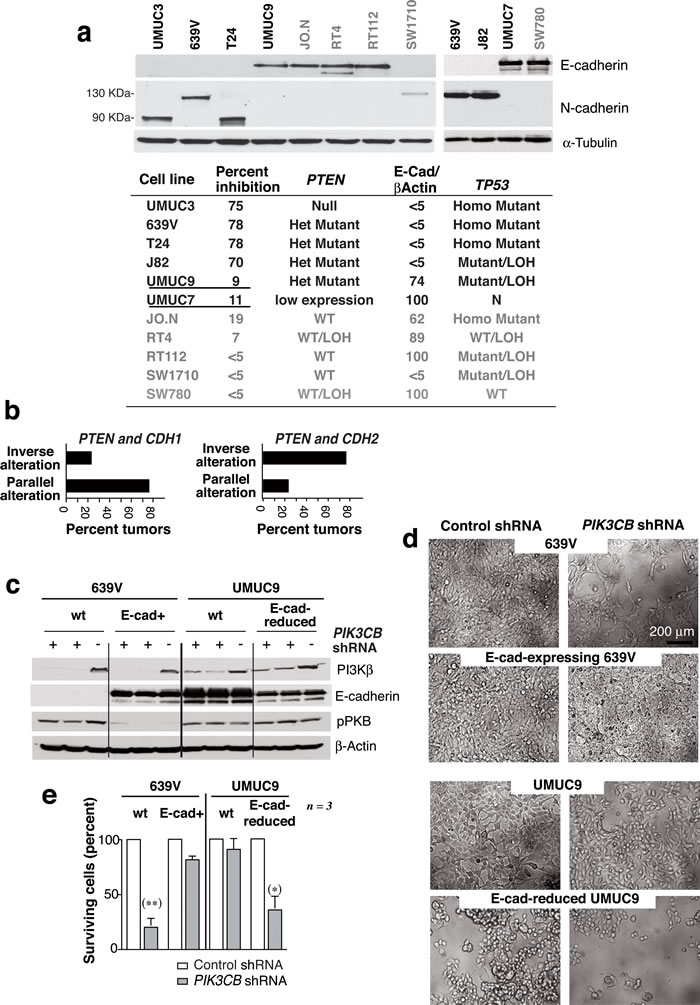
Figure 5: E-cadherin expression determines UBC line sensitivity to siPIK3CB. a. E-cad and N-cad levels in UBC cell line extracts were examined in WB. The table summarizes the E-cad/β-actin signal ratio relative to maximal levels (100%), the TP53 and PTEN mutational status, and the UBC cell responses to PI3Kβ silencing expressed as a percentage of cell survival inhibition (shown in Figure 2). Mutant PTEN, E-cad-high expressing cells are underlined. Het, heterozygous; Homo, homozygous; LOH, loss of heterozygosity; N, normal copy number. b. Graphs show UBC (from TCGA) with PTEN and CDH1 (E-cad) alterations or with PTEN and CDH2 (N-cad) alterations classified as tumors with parallel or inverse alterations in these genes (see Suppl. Figure S4). (c-e) 639V cells and 639V clones expressing E-cad were transfected with cDNA encoding doxy-inducible-control or -PIK3CB shRNA. Control and E-cad-depleted UMUC-9 cells were also transfected with control or inducible-PIK3CB shRNA. Extracts were tested in WB after doxy induction (5 µg/ml, 96 h) c.; images show representative fields d.. e. Percentage of cells after PIK3CB depletion (96 h) relative to controls. ** Student’s t-test P < 0.01, *P < 0.05.
E-cadherin depletion sensitizes PTEN-mutant UBC tumors to PIK3CB silencing in vivo
To confirm the sensitivity of control and E-cad-low UMUC-9 cells to PIK3CB silencing in vivo, we used UMUC-9 cells stably expressing CDH1 shRNA and doxy-inducible PIK3CB shRNA. When xenografts reached ~75 mm3, we administered doxycycline. PIK3CB-depletion moderately affected UMUC-9 xenografts growth; moreover, PIK3CB-depletion induced regression of E-cad-low UMUC-9 tumors (Figure 6a). Histopathology analysis of representative tumors at assay termination showed that, at difference from controls, tumor vestiges from E-cad-low PIK3CB shRNA-treated UMUC-9 cells were composed essentially of dying or dead cells; TUNEL analysis of these samples confirmed apoptosis in PIK3CB shRNA-treated tumors from E-cad-low UMUC-9 cells (Figure 6b, 6c).
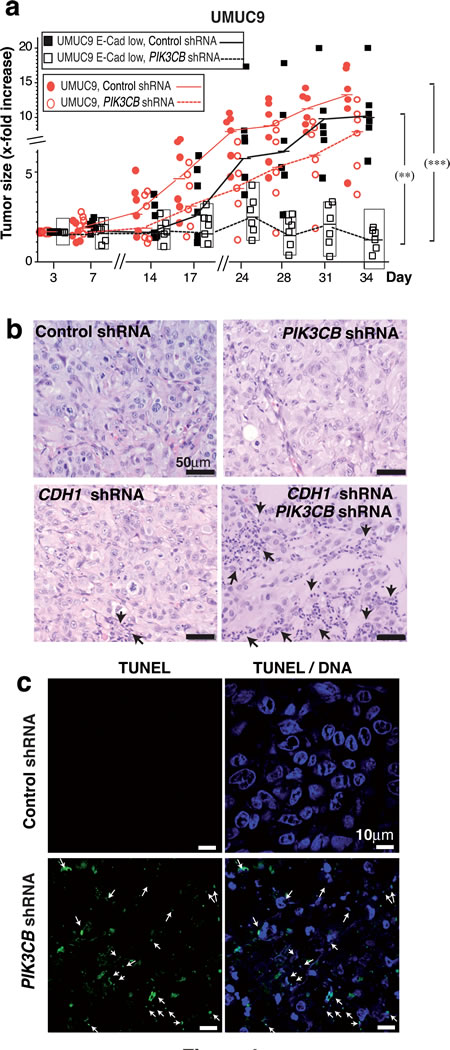
Figure 6: E-cadherin depletion sensitizes PTEN mutant UBC cells for PIK3CB silencing. a. Xenografts were generated by injecting E-cad-depleted UMUC-9 cells (5 x 106) in PBS:Matrigel (1:1) into both flanks of immunodeficient mice. When most tumors reached ~75 mm3, doxycycline was added to drinking water and tumor size measured every 3 days. The graph shows the x-fold increase in tumor size (mean ± SD) at different times relative to the volume at treatment initiation (day 1). *** P < 0.001; ** P < 0.01; one-way ANOVA Tukey’s multiple comparison test. (b, c) Representative histochemistry b. and TUNEL analysis c. of indicated tumor sections.
PI3Kβ controls N-cadherin adhesions
E-cad-negative UBC form N-cad adhesions [35, 36] that are essential regulators of survival [37; we showed that PTEN loss of function was frequently linked with N-cad increase in UBC. Since E-cad loss and the accompanying increase in N-cad levels are needed for PI3Kβ dependence in UCB, we postulated that PI3Kβ might regulate N-cad adhesions. We treated PI3Kβ-dependent (639V or J82) or PI3Kβ-independent UBC cells (SW780 or RT112) (0.5x106) with AZD8186 for three days, or transfected them with siPIK3CB1 (as above) and we examined cell-cell adhesions by immunofluorescence (IF).
E-cad adhesions in SW780 cells were virtually unaffected by PI3Kβ depletion or inhibition. E-cad and β-catenin (β-cat) colocalized at cell-cell interaction regions similarly in controls than in cells treated with AZD8186 or with siPIK3CB1, as examined by E-cad/β-cat signal quantitation at cell membranes (Figure 7a). We also detected N-cad/β-cat colocalization at plasma membranes of 639V and J82 cells treated with control siRNA or AZD8186; in contrast, PI3Kβ depletion severely disrupted N-cad localization at cell-cell adhesions (Figure 7a). Examination of the percentage of cells exhibiting N-cad (or E-cad) colocalization with β-cat at cell-cell adhesions confirmed that PI3Kβ did not affect E-cad adhesions but impaired N-cad localization in ~75% of 639V and J82 cells (Figure 7b).
To test whether interference of N-cad adhesions could promote cell death, we used interfering peptides for N-cad and E-cad in 639V and J82 cells; incubation with specific peptides for N-cad reduced cell viability in 639V and J82 cells (Supplementary Figure S6). PTEN alterations in UBC associate with N-cad upregulation; our findings suggest that PI3Kβ controls cell survival in these UBC by regulating N-cad adhesion integrity.
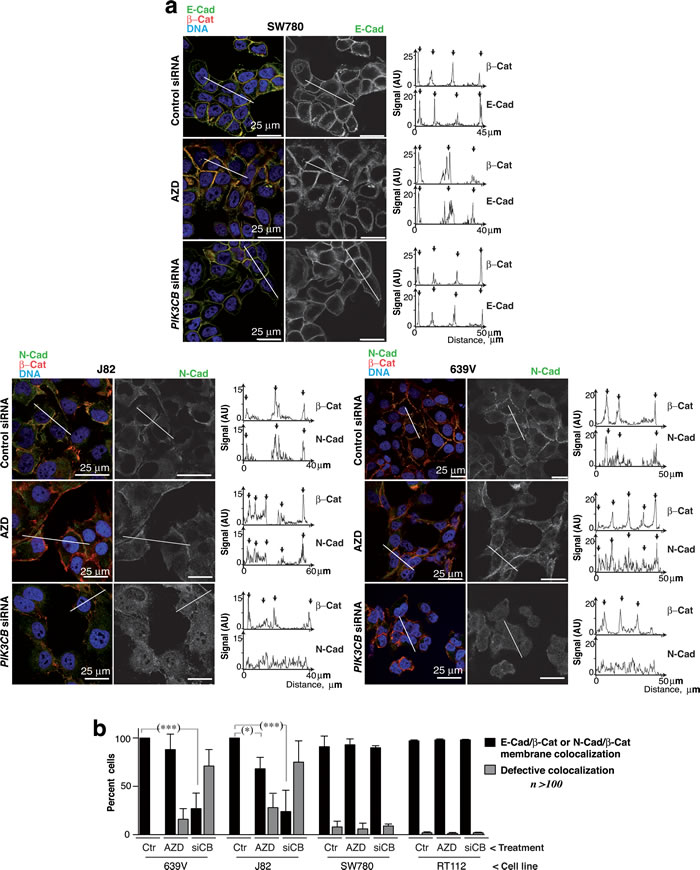
Figure 7: PI3Kβ depletion selectively impairs N-cadherin-mediated cell adhesions. a. 639V, J82 and SW780 cells were transfected with control or siPIK3CB1 shRNA or cultured with 250 nM AZD8186 (72 h). Cells were stained with the indicated antibodies and examined by confocal microscopy (indicated). Representative z-sections are shown. Quantitation of N-cad or β-cat signal in arbitrary units (AU) in the linear plots (indicated). b. Percentage of 639V, J82, RT112 and SW780 cells with normal cell adhesions defined as those showing N-cad (or E-cad) and β-cat colocalization in the membrane region between adjacent cells in at least in one side of the cell. *** P < 0.001, * P < 0.05; Student’s t-test.
PI3Kβ associates with N-cadherin
PI3Kβ expression is needed for N-cad adhesions integrity; we tested whether PI3Kβ could bind to N-cad and thereby exert a scaffold function for N-cad membrane localization. We first examined PI3Kα and PI3Kβ levels in E-cad+ SW780 cells and N-cad+ 639V cells. To determine the PI3Kα / PI3Kβ ratios in the cells, we immunoprecipitated PI3Kα and PI3Kβ from extracts of SW780 and 639V cells (900 µg). As PI3K associates 1:1 with p85, we compared the amount of p85/PI3Kα and p85/PI3Kβ complexes by examining in WB the associated p85 subunit. To check that the immunoprecipitation (IP) was efficient we confirmed that the supernatants of the IP were depleted of the PI3Kα or β; both cell lines showed moderately higher levels of p85/PI3Kβ than p85/PI3Kα (Figure 8a). E-cad IP from SW780 cell extracts followed by WB with PI3Kα or PI3Kβ Ab showed that E-cad preferentially associates PI3Kα (Figure 8b). A similar analysis after N-cad IP from 639V extracts showed that N-cad preferentially associates PI3Kβ (Figure 8b). For quantitation, we referred the amount of PI3Kα or β associated to N-cad or E-cad to the total PI3Kα or β cell content (in cell extracts). We confirmed that the PI3Kβ signal in complex with N-cad was specific, since PIK3CB depletion abolished this signal (Figure 8c).
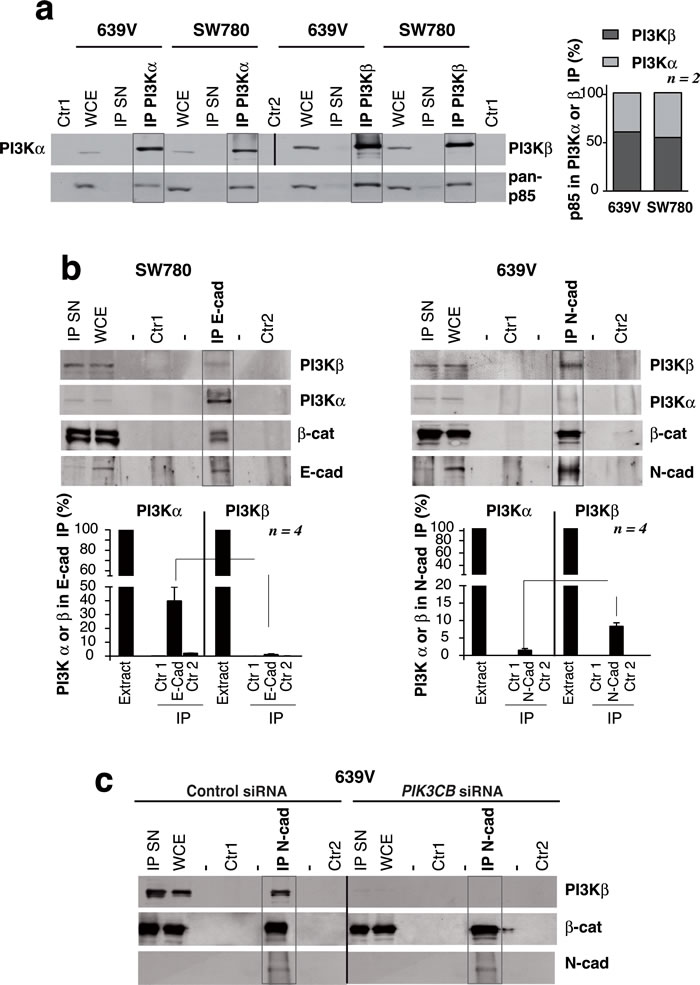
Figure 8: PI3Kβ associates with N-cadherin in PTEN mutant, E-cadherin-low UBC cells. a. Total extracts from 639V and SW780 cells (50 µg) or immunoprecipitates (IP) from these extracts (1 mg) were prepared with anti-PI3Kα or anti-PI3Kβ Ab and were tested in WB. PI3Kα IP and its controls were tested using PI3Kα Ab; PI3Kβ IP and its controls were blotted with PI3K Ab; all the samples were simultaneously examined with a pan-p85 Ab. Control 1 (Ctr1), non-specific signal in immunoprecipitates without antibody; Control 2 (Ctr2), lysis buffer incubated with antibody; IP SN, supernatant of IP (~30 µg). WB using pan-p85 Ab showed that 639V and SW780 cells had similar PI3Kα /PI3Kβ proportion as form both we isolated moderately higher amounts of p85/PI3Kβ than p85/PI3Kα complexes. The graph represents the percent of p85 signal (mean SEM) associated to PI3Kα or PI3Kβ referred to total p85 signal in both IP (100%) in each cell line. b. Extracts (900 µg) from 639V or SW780 cells were immunoprecipitated using N-cad or E-cad Ab, respectively; complexes were analyzed in WB. Total cell extracts (30 µg) were resolved in parallel. We quantitated the PI3Kα signal per µg in WCE (considered 100%) as well as PI3Kα signal per µg in the IP that was referred to maximal. We performed a similar quantitation of PI3Kβ signal. Graphs show the percent of PI3Kα (or of PI3Kβ signal) associated to cadherins in 639V or SW780 cells (mean SEM, n = 4). Controls as in a.. c. Specificity of the PI3Kβ band in complex with N-cad was tested as in b. using control or PI3Kβ-depleted 639V cell extracts. Controls as in a.. **P < 0.001, *P < 0.05; Student’s t-test.
Discussion
There is increasing evidence that inhibition of oncogenic kinases activities does not equate to the reduction of their expression. This is the case of PI3Kβ, whose expression, rather than its activity, is necessary from the onset of embryonic development [24-26]. We explored the therapeutic potential of local PIK3CB siRNA delivery on PTEN-mutant tumors, since PI3Kβ is the main isoform activated in these tumors [15]. After examining a set of 10 unrelated cancer cell lines, we focused on a panel of 11 bladder carcinoma cells (UBC), as these tumors have PTEN mutations and increased PI3Kβ expression. In most PTEN-mutant UBC cells, PIK3CB knockdown induced apoptosis and effectively led to tumor regression; PI3Kβ depletion was markedly more efficient than PI3Kβ inhibition. The apoptotic response was absent in a few samples, which provided an opportunity to determine the molecular requirements for PI3Kβ dependence. Our results suggest that PTEN mutation and concomitant E-cad loss (found in invasive UBC) are needed for PI3Kβ dependence. In parallel to E-cad loss, UBC express N-cad [35], which controls cell survival [38]. We show that PI3Kβ associates with and regulates N-cad adhesion.
Approximately half of human cancers have mutations in PI3K or in its negative regulator PTEN; the latter are more frequent at advanced tumor phases [3, 4, 39]. PI3K inhibitors have been tested clinically; some of these studies hold promise, but others have failed due to blockade of PI3K feedback pathway regulation or generation of resistance [21-23, 40-42], making necessary the development of additional therapeutic approaches. As proof of concept for siRNA-based local treatment of UBC, we optimized i.t. injection of siRNA in UBC xenografts. Local siRNA delivery is anticipated to reduce dose requirements and minimize toxicity [43, 44]. i.t. siRNA delivery reduced xenograft size, triggered apoptosis, and was more effective than PI3Kβ inhibition, which supports siRNA injection as a feasible approach. The vehicle used, however, showed partial toxicity, been necessary development of optimal siRNA vehicles. Since all vehicle- (liposome) treated samples showed an inflammatory neutrophil infiltration, it is possible that an action of PIK3CB siRNA in the tumor environment contributes to tumor growth deceleration.
Local delivery appears to be suitable for tumors with increased PIK3CB expression that are accessible to local administration, such as bladder cancer. Indeed, high-risk non-MI UBC is currently treated by intravesical therapy with Bacille-Calmette Guerin [7], and siPIK3CB treatment can be reduced to two injections a week for two weeks; intravesical infusion with a defective virus encoding PIK3CB shRNA or the use of novel non-toxic vehicles, currently under development [45], might be suitable reagents for PIK3CB depletion. Several siRNA targets are currently in clinical trials, and the large number of successful phase II and III trials for human disease predicts the incorporation of these therapies for medical applications [46, 28]. Development of antisense oligonucleotides [47] or application of the successful CRISPR-Cas approach [48] for PIK3CB deletion might also be useful for local treatment of advanced UBC (T2b and beyond) bearing PTEN mutations.
PI3Kβ depletion was more effective than its inhibition in reducing UBC tumor growth; whereas the inhibitor AZD8186 decelerated tumor expansion, PI3Kβ depletion induced caspase 3 cleavage and tumor regression. The observation that PI3Kβ has both enzymatic and non-enzymatic functions [24-26] might explain the effectiveness of siPIK3CB in the PTEN-mutant tumors. We tested the contribution of the PI3Kβ scaffold function in several ways. We compared the effect of depleting or inhibiting PI3Kβ in the growth of UBC lines in culture. PTEN loss of function was necessary for sensitivity to PI3Kβ inhibition or depletion, however, cell numbers were moderately reduced by inhibitor and markedly reduced by PI3Kβ depletion. pPKB levels were unchanged or only modestly diminished by PI3Kβ depletion. Moreover, whereas expression of an inactive KR-PI3Kβ reduced 639V cell viability moderately, PI3Kβ depletion had a greater effect on cell death induction, which was reduced by co-expression of a siPIK3CB-resistant inactive PI3Kβ mutant. We also found that PI3Kβ depletion has a more potent effect than its inhibition on disruption of N-cad adhesions. These observations support a PI3Kβ scaffold function in UBC survival.
A small proportion of PTEN-defective cells were resistant to PIK3CB silencing; we evaluated the molecular characteristics of these cell lines. The all four siPIK3CB-resistant PTEN-mutant cells (two UBC and two from the diverse tumor set) exhibited had high E-cad levels. Invasive UBC show PTEN defects (58% of tumors) [9, 10, 39], PIK3CB upregulation, and mesenchymal phenotypes [35, 49-51]. In the mouse, Pten loss of function cooperates with Lkb1 or Tp53 mutation to initiate EMT, which induces E-cad downregulation and N-cad expression [29, 34-36]. We examined the Cancer Genome Atlas bladder cancer collection (www.cancergenome.nih.gov) to evaluate the possible linkage of PTEN mutation and E-cad or N-cad expression in UBC. Simultaneous TP53 and PTEN mutation did not correlate with changes in E-cad or N-cad expression, nonetheless, PTEN alterations in UBC were associated with CDH2 upregulation (P = 0.01, www.cbioportal.org) supporting a link between PTEN and E-cad/N-cad expression changes. Moreover, using a genetic approach, we found that E-cad-expression in 639V-PI3Kβ-dependent cells made them resistant to PI3Kβ depletion while E-cad silencing sensitized UMUC-9 cells to PIK3CB depletion. This suggests that siPIK3CB resistance in PTEN mutant UBC is due to E-cad expression.
siPIK3CB-sensitive and insensitive lines can be distinguished by E-cad/N-cad levels; we studied the consequences of depleting or inhibiting PI3Kβ on cell-cell adhesions. We used two E-cad-high (PI3Kβ-insensitive) and two E-cad-low/N-cad-positive (PI3Kβ-sensitive) UBC lines. In insensitive lines, cell adhesions were unaffected by PI3Kβ depletion or inhibition; in sensitive lines, PI3Kβ inhibition moderately reduced, but PI3Kβ depletion abrogated, N-cad-adhesions in more than 75% of the cells. PI3Kβ expression thus regulates N-cad adhesions in UBC. We show that E-cad bound preferentially PI3Kα in PTEN-WT SW780 cells; in PTEN-mutant 639V cells, N-cad bound to preferentially PI3Kβ. As cell adhesions are needed for UBC cell survival (Figure S6), these results suggest that PI3Kβ exhibits a scaffold function for N-cad adhesions stabilization, which regulate UBC survival.
Acquisition of resistance in 639V cells after E-cad expression can be explained by considering that PI3Kβ depletion does not affect E-cad adhesions. We also show that E-cad reduction in UMUC-9 cells renders them sensitive to PI3Kβ depletion although these cells do not express N-cad. The reduction in E-cad levels and an additional survival effect mediated by PI3Kβ (such as Bim regulation, data not shown) might explain their sensitivity to PIK3CB depletion.
PTEN alterations in UBC are associated with CDH2 upregulation. In these cells, PI3Kβ associates with and is needed to maintain N-cad cell adhesions. As these adhesions regulate cell survival in UBC, disruption of N-cad adhesions is at least part of the mechanism for the greater cytotoxic effect of PI3Kβ depletion compared to PI3Kβ inhibition in PTEN mutant UBC. There is increasing evidence that high-risk non-MI bladder tumors are precursors of MI cancers, with whom they share genomic features [7]. High-risk tumors confined to the bladder are generally treated by intravesical therapy, but up to 30% progress to muscle-invasive UBC, which have poor prognosis and often develop metastases. Our results support the use of strategies that decrease PIK3CB expression as a treatment for high-risk non-muscle-invasive bladder cancer with PTEN mutation and low E-cad levels, a frequent phenotype in advanced urothelial carcinoma.
Materials and Methods
Antibodies, reagents, siRNA, shRNA and cDNA
Antibodies used were anti-p110β, -p110α and -pPKB (Ser 473) (Cell Signaling), pan-p85 (Millipore), -E-cad, PCNA (BD Biosciences), -PKB (Upstate Biotechnology), -β-actin, -α-tubulin (Sigma), -N-cad (Abcam) and --catenin (BD). For in vivo assays, we used Stealth PIK3CB siRNA that targeted the sequences AACCACTGGAATTTGATATTAATAT (siRNA1) or GATTCACAGATAGCATCTGAT (siRNA2), or control (scrambled-siRNA1) and vehicle (Invivofectamine), (Invitrogen). We prepared doxycycline (doxy)-inducible-pLKO-tet-PIK3CB shRNA vector by subcloning in the EcoRI site the oligonucleotide CCGGGATTCACAGATAGCATC TGATCTCGAGATCAGATGCTATCTGTGAATCTTTTT and its reverse complementary oligonucleotide. We used RNAiMAX (Invitrogen) and OptiMem (Life Technologies) for siRNA transfection. pLKO-Puro-CDH1 shRNA and the dexamethasone (Dex)-inducible pLK-pac vector encoding CDH1 have been described [52]. The PI3Kβ inhibitor AZD8186 was from Astra Zeneca [20] . PSG5-ER-myc-K805R-p110β was prepared as described [25]. To generate a siPIK3CB resistant-K805R-p110β, we used the oligonucleotide GGAATGA ACCACTCGAATTTCATATTAATATTTGTGACTT ACCAAGAATGG and its reverse complementary in a PCR reaction using Quickchange (Agilent). ER-myc-KR-p110β was cotransfected with siRNA using Lipofectamine 2000 (Invitrogen); these cells were treated with tamoxifen (1 µM, Sigma) 12 h before analysis.
Cell lines
We used the cell lines PC3 (ATCC-CRL-1435), U87MG (ATCC-HTB-14), BLM (lung metastasis of BRO melanoma cells, confirmed by typing the short tandem repeat DNA profiles at Genomics Service, Instituto de Investigaciones Biomédicas Alberto Sols, Madrid), HT1080, HT-29, SW680, MDA-MB468 (ATCC-CCL121, -HTB38, -228 and HTB-132, respectively), and NCI-H226, -H520 and CaLu-1 (ATCC-CRL-5826, -HTB182 and HTB-54, respectively). We also used 639V, UMUC-3, UMUC-7, UMUC-9, J82, T24, RT4, RT112, JON, SW1710 and SW780 urethral bladder carcinomas [33]. Cells were maintained in DMEM or RPMI (Gibco-BRL) with 10%fetal bovine serum, 2mM glutamine, 10mM HEPES, 100 U/ml penicillin, and 100µg/ml streptomycin (37ºC, 5%CO2). We prepared UMUC-9 cell clones expressing shRNA for CDH1 using lentiviral particles containing the pLKO-Puro-CDH1 shRNA. Clones were selected in medium with puromycin (1-2µg/mL). We similarly generated 693V lines overexpressing E-cad using lentiviral particles containing the Dex-inducible pLK-pac-CDH1 cDNA. Stable clones were selected with puromycin (5µg/mL); E-cad expression was induced in medium with 10 nM Dex (48 h). Clones expressing pLKO-tet-PIK3CB-shRNA vector (doxy-inducible) were prepared similarly and were selected in medium containing G418 (0.8 mg/ml, 10 days).
Mice wearing xenografts treatment
All procedures using mice were approved by the Ethics Committee of the CNB/CSIC in accordance with EU/Spanish legislation (RD53/2013). To generate xenografts, cells (2.5-to-5x106) were suspended in 100 µl PBS containing 25% (639V and PC3 cells) or 50% Matrigel (UMUC-9 cells) and were injected subcutaneously (s.c.) into both flanks of 2-to-3-month-old immunodeficient CB17/lcrHsd-Prkdcscid/Lys bg-j mice (Harlan). When tumors reached a mean size of 75 mm3, mice were treated by direct central intratumor injection (Hamilton syringe 702N, 25 µl, 22-g bevel tip needle) with vehicle, PBS alone, or either control or siPIK3CB in Invivofectamine. siPIK3CB was dissolved in PBS, followed by Invivofectamine (0.8:1 Invivofectamine:siRNA in PBS). siRNA doses were 12.5, 50 or 125 pmol/mm3 of tumor volume. For optimal PIK3CB depletion, 50 pmol per mm3 of tumor volume and dose siPIK3CB1 was sufficient and was used for subsequent experiments. Mice were treated three times weekly for two weeks, although two injections in two weeks were similarly efficient.
When tumors of cells that expressed inducible shPIK3CB reached ~75 mm3, silencing was induced by doxy (4 mg/ml in H2O with 5% glucose) to drinking water. Tumor-bearing mice also were treated by i.p. injection with the PI3Kβ inhibitor AZD8186 (40 mg/kg) in 0.5% methylcellulose, 0.2% Tween-20. Tumor size was measured with a caliper and volume was calculated as (V) = (smaller side length2 x larger side length)/2.
TUNEL, annexin V, histology, western blot, flow cytometry, immunofluorescence, immunoprecipitation and peptide assays
TUNEL assays were performed using the DeadEnd Fluorometric TUNEL System Kit (Promega); the Annexin V-FITC assay was from Southern Biotech. Histology was as described [18]. Cell extracts were prepared in RIPA buffer as described [25]; WB and cell fractionation were as described [25]. Sub-G1 DNA-containing cells were tested by flow cytometry [25]. Immunofluorescence procedures were described [26]; for immunoprecipitation, cells were lysed in buffer containing 10 mM Tris-HCl pH7.4, 150 mM NaCl, 10 mM KCl, 1% NP-40 with phosphatase and protease inhibitors, and precipitated as reported [25]. We used three synthetic peptides bearing E-cad (LFSHAVSSNG) or N-cad (LRAHAVDING) sequences or a “scrambled” sequence with no known matches (ARLHDVAING). Peptides were N-acetylated and C-amidated (Biomatik); UBC cells were cultured with peptides (400 µg/ml) for different periods prior to analysis.
Analysis of PIK3CB expression levels and of PTEN, CDH1 and CDH2 changes in UBC
We evaluated PIK3CB mRNA levels based on previously reported DNA microarray data. The first dataset (94 samples), includes 9 normal bladder biopsies, 56 NMI tumors and 29 MI tumors [30, 31] (GEO:GSE5287). The second dataset (44 tumor samples, includes 34 NMI and 8 MI tumors [32] (GEO:GSE71576). These analyses were performed using Affymetrix U133A:212688 and Affymetrix 1.0 ST 8091009 probe hybridization data. The third dataset was from ArrayExpress (www.ebi.ac.uk/arrayexpress/) with accession numbers E-MTAB-1803 for MI-UBC and E-MTAB-1940 for non-MI-UBC. RMA software (v1.20) from Bioconductor was used to normalize gene expression with express function and parameters by default. We used the Cancer Genome Atlas [42] (TCGA) bladder cancer collection (www.cancergenome.nih.gov) and the software at http://www.cbioportal.org/index.do to examine the relationship between PTEN, CDH1 and CDH2.
Statistical analyses
Gel band intensity was quantitated with ImageJ software (NIH). Significance was calculated (GraphPad Prism 5.0 Software) using one-way ANOVA followed by Tukey’s multiple comparison test, Kruskal-Wallis, one- or two-tailed Mann-Whitney tests, and Student’s t-tests.
Acknowledgments
We thank C Mark for editorial assistance.
Conflicts of interest
The authors declare no conflict of interest.
FUNDING
This work was financed by grants from the Spanish Ministry of Science and Innovation (SAF2011-29530 to FXR, SAF2013-48657 to ACC; Consolider ONCOBIO to FXR, Network of Cooperative Research in Cancer cofinanced by the European Regional Development Fund (RTICC RD12/0036/0059 to ACC and RD12/0036/0034 to FXR), the Madrid regional government (BMD2502 to ACC), and an AECC (Spanish Association against Cancer) grant to FXR.
Editorial note
This paper has been accepted based in part on peer-review conducted by another journal and the authors’ response and revisions as well as expedited peer-review in Oncotarget.
References
1. Dinney CP, McConkey DJ, Millikan RE, Wu X, Bar-Eli M, Adam L, Kamat AM, Siefker-Radtke AO, Tuziak T, Sabichi AL, Grossman HB, Benedict WF, Czerniak B. Focus on bladder cancer. Cancer Cell 2004; 6: 111-116.
2. Shah JB, McConkey DJ, Dinney CP. New strategies in muscle-invasive bladder cancer: on the road to personalized medicine. Clin Cancer Res 2011; 17: 2608-12.
3. Wymann MP, Marone R. Phosphoinositide 3-kinase in disease: timing, location, and scaffolding. Curr Opin Cell Biol. 2005; 17:141-9.
4. Liu P, Cheng H, Roberts TM, Zhao JJ. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov. 2009; 8:627-44.
5. Thorpe LM, Yuzugullu H, Zhao JJ. PI3K in cancer: divergent roles of isoforms, modes of activation and therapeutic targeting. Nat Rev Cancer. 2015; 15:7-24.
6. Worby CA, Dixon JE. PTEN. Annu Rev Biochem. 2014; 83:641-69.
7. Knowles MA, Hurst CD. Molecular biology of bladder cancer: new insights into pathogenesis and clinical diversity. Nat Rev Cancer. 2015; 15:25-41.
8. López-Knowles E, Hernández S, Malats N, Kogevinas M, Lloreta J, Carrato A, Tardón A, Serra C, Real FX. PIK3CA mutations are an early genetic alteration associated with FGFR3 mutations in superficial papillary bladder tumors. Cancer Res. 2006; 66: 7401-4.
9. Benistant C, Chapuis H, Roche S. A specific function for phosphatidylinositol 3-kinase α (p85α-p110α) in cell survival and for phosphatidylinositol 3-kinase β (p85α-p110b) in de novo DNA synthesis of human colon carcinoma cells. Oncogene 2000; 19: 5083-90.
10. Castillo-Martin M, Domingo-Domenech J, Karni-Schmidt O, Matos T, Cordon-Cardo C. Molecular pathways of urothelial development and bladder tumorigenesis. Urol Oncol. 2010; 28: 401-8.
11. Kan Z, Jaiswal BS, Stinson J, Janakiraman V, Bhatt D, Stern HM, Yue P, Haverty PM, Bourgon R, Zheng J, Moorhead M, Chaudhuri S, Tomsho LP. Diverse somatic mutation patterns and pathway alterations in human cancers. Nature. 2010; 466:869-73.
12. Kang S, Denley A, Vanhaesebroeck B, Vogt PK. Oncogenic transformation induced by the p110beta, -gamma, and -delta isoforms of class I phosphoinositide 3-kinase. Proc Natl Acad Sci USA 2006; 103:1289-94.
13. Lee SH, Poulogiannis G, Pyne S, Jia S, Zou L, Signoretti S, Loda M, Cantley LC, Roberts TM. A constitutively activated form of the p110beta isoform of PI3-kinase induces prostatic intraepithelial neoplasia in mice. Proc Natl Acad Sci U S A 2010; 107:11002-7.
14. Jia S, Liu Z, Zhang S, Liu P, Zhang L, Lee SH, Zhang J, Signoretti S, Loda M, Roberts TM, Zhao JJ. Essential roles of PI(3)K-p110beta in cell growth, metabolism and tumorigenesis. Nature 2008; 454:776-9.
15. Wee S, Wiederschain D, Maira SM, Loo A, Miller C, deBeaumont R, Stegmeier F, Yao YM, Lengauer C. PTEN-deficient cancers depend on PIK3CB. Proc Natl Acad Sci U S A 2008; 105: 13057-62.
16. Ni J, Liu Q, Xie S, Carlson C, Von T, Vogel K, Riddle S, Benes C, Eck M, Roberts T, Gray N, Zhao J. Functional characterization of an isoform-selective inhibitor of PI3K-p110β as a potential anticancer agent. Cancer Discov. 2012; 2: 425-33.
17. Czauderna F, Santel A, Hinz M, Fechtner M, Durieux B, Fisch G, Leenders F, Arnold W, Giese K, Klippel A, Kaufmann J. Inducible shRNA expression for application in a prostate cancer mouse model. Nucleic Acids Res 2003; 31: e127.
18. Schmit F, Utermark T, Zhang S, Wang Q, Von T, Roberts TM, Zhao JJ. PI3K isoform dependence of PTEN-deficient tumors can be altered by the genetic context. Proc Natl Acad Sci U S A. 2014; 111: 6395-400.
19. Weigelt B, Warne PH, Lambros MB, Reis-Filho JS, Downward J. PI3K pathway dependencies in endometrioid endometrial cancer cell lines. Clin Cancer Res. 2013; 19: 3533-44.
20. Hancox U, Cosulich S, Hanson L, Trigwell C, Lenaghan C, Ellston R, Dry H, Crafter C, Barlaam B, Fitzek M, Smith PD, Ogilvie D, D’Cruz C et al. Inhibition of PI3Kβ signaling with AZD8186 inhibits growth of PTEN-deficient breast and prostate tumors alone and in combination with docetaxel. Mol Cancer Ther. 2015; 14 (1): 48-58.
21. Klempner SJ, Myers AP, Cantley LC. What a tangled web we weave: emerging resistance mechanisms to inhibition of the phosphoinositide 3-kinase pathway. Cancer Discov. 2013; 3: 1345-1354.
22. Fruman DA, Rommel C. PI3K and cancer: lessons, challenges and opportunities. Nat Rev Drug Discov. 2014; 13:140-156.
23. Shapiro GI, Rodon J, Bedell C, Kwak EL, Baselga J, Braña I, Pandya SS, Scheffold C, Laird AD, Nguyen LT, Xu Y, Egile C, Edelman G. Phase I safety, pharmacokinetic, and pharmacodynamic study of SAR245408 (XL147), an oral pan-class I PI3K inhibitor, in patients with advanced solid tumors. Clin Cancer Res. 2014; 20:233-245.
24. Ciraolo E, Iezzi M, Marone R, Marengo S, Curcio C, Costa C, Azzolino O, Gonella C, Rubinetto C, Wu H, Dastrù W, Martin EL, Silengo L, et al. Phosphoinositide 3-kinase p110beta activity: key role in metabolism and mammary gland cancer but not development. Sci Signal 2008; 1 (36): ra3.
25. Marqués M, Kumar A, Poveda AM, Zuluaga S, Hernández C, Jackson S, Pasero P, Carrera AC. Specific function of phosphoinositide 3-kinase beta in the control of DNA replication. Proc Natl Acad Sci U S A. 2009; 106: 7525-30.
26. Kumar A, Fernandez-Capetillo O, Carrera AC. Nuclear phosphoinositide 3-kinase beta controls double-strand break DNA repair. Proc Natl Acad Sci U S A. 2010; 107: 7491-6.
27. Soria JC, Lee HY, Lee JI, Wang L, Issa JP, Kemp BL, Liu DD, Kurie JM, Mao L, Khuri FR. Lack of PTEN expression in non-small cell lung cancer could be related to promoter methylation. Clin Cancer Res. 2002; 8: 1178-84.
28. Roberts PJ, Der CJ. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene. 2007; 26: 3291-310.
29. Shorning BY, Griffiths D, Clarke AR. Lkb1 and Pten synergise to suppress mTOR-mediated tumorigenesis and epithelial-mesenchymal transition in the mouse bladder. PLoS One. 2011;6(1):e16209.
30. Aaboe M, Marcussen N, Jensen KM, Thykjaer T, Dyrskjot L, Orntoft TF. Gene expression profiling of noninvasive primary urothelial tumours using microarrays. Br J Cancer 2005; 93: 1182-90.
31. Als AB, Dyrskjot L, von der Maase H. Emmprin and survivin predict response and survival following cisplatin-containing chemotherapy in patients with advanced bladder cancer. Clin Cancer Res 2007; 13: 4407-14.
32. Pineda S, Gomez-Rubio P, Picornell A, Bessonov K, Márquez M, Kogevinas M, Real FX, Van Steen K, Malats N. Framework for the Integration of Genomics, Epigenomics and Transcriptomics in Complex Diseases. Hum Hered. 2015; 79: 124-36.
33. Earl J, Rico D, Carrillo-de-Santa-Pau E, Rodríguez-Santiago B, Méndez-Pertuz M, Auer H, Gómez G, Grossman HB, Pisano DG, Schulz WA, Pérez-Jurado LA, Carrato A, Theodorescu D, et al. The UBC-40 Urothelial Bladder Cancer Cell Line Index: a genomic resource for functional studies. BMC Genomics. 20; 16: 403.
34. Puzio-Kuter AM, Castillo-Martin M, Kinkade CW, Wang X, Shen TH, Matos T, Shen MM, Cordon-Cardo C, Abate-Shen C. Inactivation of p53 and Pten promotes invasive bladder cancer. Genes Dev. 2009; 23: 675-80.
35. Derycke LD, Bracke ME. N-cadherin in the spotlight of cell-cell adhesion, differentiation, embryogenesis, invasion and signalling. Int J Dev Biol. 2004; 48:463-76.
36. Ye X, Weinberg RA. Epithelial-Mesenchymal Plasticity: A Central Regulator of Cancer Progression. Trends Cell Biol. 2015; 25: 675-86.
37. Aceto N, Toner M, Maheswaran S, Haber DA. En Route to Metastasis: Circulating Tumor Cell Clusters and Epithelial-to-Mesenchymal Transition. Trends in Cancer. 2015; 1:44-52.
38. Lelièvre EC, Plestant C, Boscher C, Wolff E, Mège RM, Birbes H. N-cadherin mediates neuronal cell survival through Bim down-regulation. PLoS One. 2012; 7(3): e33206. doi: 10.1371/journal.pone.0033206.
39. Cancer Genome Atlas Research Network. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature. 2014; 507:315-322.
40. Costa C, Ebi H, Martini M, Beausoleil SA, Faber AC, Jakubik CT, Huang A, Wang Y, Nishtala M, Hall B, Rikova K, Zhao J, Hirsch E, et al. Measurement of PIP3 levels reveals an unexpected role for p110β in early adaptive responses to p110α-specific inhibitors in luminal breast cancer. Cancer Cell. 2015; 27:97-108.
41. Schwartz S, Wongvipat J, Trigwell CB, Hancox U, Carver BS, Rodrik-Outmezguine V, Will M, Yellen P, de Stanchina E, Baselga J, Scher HI, Barry ST, Sawyers CL, et al. Feedback suppression of PI3Kα signaling in PTEN-mutated tumors is relieved by selective inhibition of PI3Kβ. Cancer Cell. 2015; 27: 109-122.
42. Cirone P, Andresen CJ, Eswaraka JR, Lappin PB, Bagi CM. Patient-derived xenografts reveal limits to PI3K/mTOR- and MEK-mediated inhibition of bladder cancer. Cancer Chemother Pharmacol. 2014; 73:525-538.
43. Castanotto D, Rossi JJ. The promises and pitfalls of RNA-interference-based therapeutics. Nature. 2009; 457:426-33.
44. Fujita Y, Kuwano K, Ochiya T. Development of small RNA delivery systems for lung cancer therapy. Int J Mol Sci. 2015; 16: 5254-70.
45. Lee SJ, Kim MJ, Kwon IC, Roberts TM. Delivery strategies and potential targets for siRNA in major cancer types. Adv Drug Deliv Rev. 2016; 104: 2-15.
46. Tefferi A, Lasho TL, Begna KH, Patnaik MM, Zblewski DL, Finke CM, Laborde RR, Wassie E, Schimek L, Hanson CA, Gangat N, Wang X, Pardanani A. A Pilot Study of the Telomerase Inhibitor Imetelstat for Myelofibrosis. N Engl J Med. 2015; 373: 908-19.
47. Hong D, Kurzrock R, Kim Y, Woessner R, Younes A, Nemunaitis J, Fowler N, Zhou T, Schmidt J, Jo M, Lee SJ, Yamashita M, Hughes SG, et al. AZD9150, a next-generation antisense oligonucleotide inhibitor of STAT3 with early evidence of clinical activity in lymphoma and lung cancer. SciTransl Med. 2015; 7(314): 314ra185.
48. Tabebordbar M, Zhu K, Cheng JK, Chew WL, Widrick JJ, Yan WX, Maesner C, Wu EY, Xiao R, Ran FA, Cong L, Zhang F, Vandenberghe LH, et al. In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science 2016; 351:407-411.
49. Kastritis E, Murray S, Kyriakou F, Horti M, Tamvakis N, Kavantzas N, Patsouris ES, Noni A, Legaki S, Dimopoulos MA, Bamias A. Somatic mutations of adenomatous polyposis coli gene and nuclear β-catenin accumulation have prognostic significance in invasive urothelial carcinomas: evidence for Wnt pathway implication. Int. J. Cancer 2009; 124: 103-8.
50. Keck B, Wach S, Goebell PJ, Kunath F, Bertz S, Lehmann J, Stöckle M, Taubert H, Wullich B, Hartmann A. SNAI1 protein expression is an independent negative prognosticator in muscle-invasive bladder cancer. Ann Surg Oncol. 2013; 20: 3669-3674.
51. Ahmad I, Morton JP, Singh LB, Radulescu SM, Ridgway RA, Patel S, Woodgett J, Winton DJ, Taketo MM, Wu XR, Leung HY, Sansom OJ. β-catenin activation synergizes with PTEN loss to cause bladder cancer formation. Oncogene 2011; 30: 178-89.
52. Sarrió D, Palacios J, Hergueta-Redondo M, Gómez-López G, Cano A, Moreno-Bueno G. Functional characterization of E- and P-cadherin in invasive breast cancer cells. BMC Cancer. 2009; 9: 74.