
INTRODUCTION
Lung cancer (LC) is one of the leading causes of cancer-associated deaths worldwide [1]. LC is broadly divided into two main groups: small cell lung cancer (SCLC) and non-small cell lung cancer (NSCLC). NSCLC accounts for 85% of all lung cancer cases, for which improvement of 15.9% has been reported in 5-year survival rate during the past few decades [2]. NSCLC is currently subdivided into two predominant histologic phenotypes: adenocarcinoma (ADC; 50%) and squamous cell carcinoma (SQC; 40%) [3, 4].
The current American Joint Committee on Cancer (AJCC) staging system serves as the best predictor of prognosis and a standard to guide treatment decisions in NSCLC [5]. Complete surgical resection is the most effective for patients in the early stage [6], even though 30-60% of patients diagnosed with stage IB to IIIA relapse and die within 5-year of survival [7]. For patients in stage II-III, adjuvant chemotherapy (ACT) is the standard treatment with survival rate from 4% to 15% [8, 9]. However, due to the heterogeneous nature of NSCLC, the current AJCC staging cannot accurately classify patients who would benefit from chemotherapy [10]. Prognostic biomarkers with transcriptomic data and the mutation status of genes which are important in cancer development need to be investigated [11]. Previous studies identified three major genes (EGFR, KRAS, and ALK) for the development of lung cancer [12-15]. Mutations in the EGFR have been associated with enhanced overall survival, whereas KRAS mutations may predict shorter survival for lung adenocarcinoma patients [16]. Molecular tests for these prognostic biomarkers have been started for preclinical and clinical applications to advance the treatment of NSCLC [17-20].
Recent advances in microarray gene expression profiling have demonstrated possibility of screening gene expression signatures to predict the prognosis of patients. Previously, this approach successfully identified prognostic and predictive gene signatures in the breast cancer [21]. To date, several studies based on gene expression signatures have been shown to classify various cancer patients into different prognostic groups with distinct clinical features by supervised or unsupervised methods [22-28]. However, the identified survival-related signatures lack consistency among studies, likely due to genetic alteration among patients, technical factors such as differences in microarray platforms, and limited number of patients. Therefore, it is important to establish a prognostic gene signature that could predict patient’s survival and guide decisions of adjuvant therapy for individual patient.
In this study, we identified an 8-gene signature to distinguish two prognostic groups (low- vs high- risk), using an unbiased gene expression profiling and bioinformatics analysis. The 8-gene signature was further validated in five microarray retrospective and independent data sets and one RNA-seq data set. Furthermore, we assessed the associations of the identified prognostic gene signature with clinicopathological factors and molecular alterations. Finally, we investigated whether our 8-gene signature could predict patients who might have benefits from ACT in the patients diagnosed as stage II-III NSCLC. Our findings suggest that the 8-gene signature can be rapidly implemented in a clinical setting and demonstrated excellent predictive power for NSCLC.
RESULTS
Development of a prognostic gene signature and a risk predictor
In order to identify a prognostic gene signature that distinguished low- and high-risk NSCLC patients, gene expression profiling was analyzed in relation to survival data. GSE50081 was used as the training data set. As shown in the flow chart of the procedure (Figure 1A), after filtering for probe set intensity, 3,294 probe sets were analyzed in univariate Cox regression analysis with overall survival (OS) as the prognostic survival end point. A gene signature with 21-probe set was developed. However, the microarray chip type for the GSE50081 [29], GSE31210 [25, 30], GSE30219 [31], GSE29013 [32] and E-MTAB-923 data sets was Affymetrix GeneChip Human Genome U133 Plus 2.0 (HG-U133_Plus_2), and the other chip types for the GSE68465 and GSE42127 were Affymetrix GeneChip Human Genome (HG-U133A) and Illumina HumanWG-6 v3.0 Expression BeadChip (IlluminaHuman-WG6 V3), respectively, as described in ‘Methods’ section (Supplementary Table S1). Among the 21-probe set, 8 probes, which were corresponded to 8 annotated genes, were commonly found both in the training and all validation data sets. Thus, this model was termed the 8-gene signature, including STAT1, CLU, GTSE1, NUSAP1, ABCA8, TNNT1, ENTPD3 and CPA3 (Supplementary Table S2). Prognostic index for each patient was calculated based on the 8-gene signature (Figure 1B). Patients were dichotomized according to the risk score into low (n=89) and high (n=92) risk groups on their prognostic index in the training data set. The heatmap showed different expression patterns of the 8-gene signature for the low- and high-risk patient groups into two clusters (Figure 1C). The Kaplan-Meier analysis confirmed that overall survival rate was different between the predicted low- and high-risk groups based on the 8-gene signature (p=4.49e-05, Figure 1D).
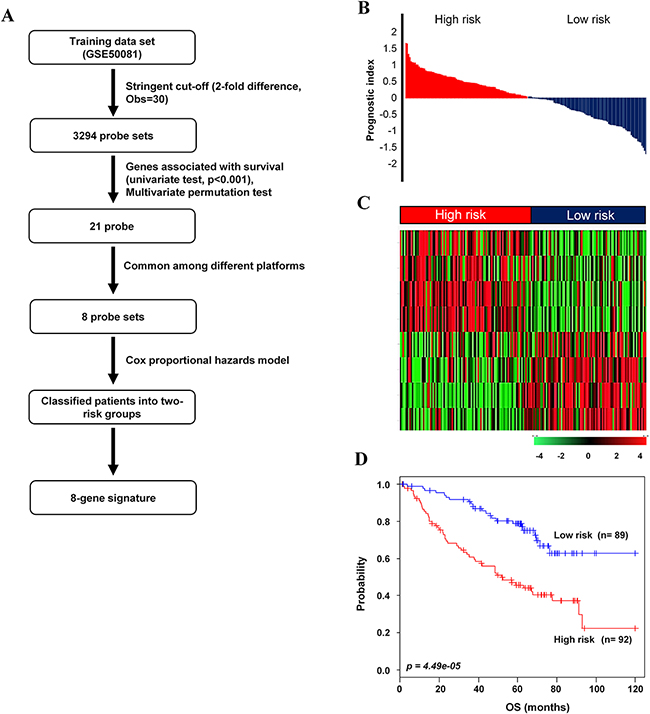
Figure 1: Survival analysis of the training data set. A. Schematic overview of the procedure used to construct the 8-gene signature based on gene expression data. B. The relative prognostic index based on the 8-gene signature expression of each patient. C. The heatmap of the median centered 8 genes’ expression profiles (red, relative high expression; green, relative low expression) between low- and high-risk groups. D. Kaplan-Meier plots for OS of two risk groups in the training data set. The p values were computed by log-rank test.
The 8-gene signature can be used as an independent clinical parameters
We next tested whether the prognostic gene signature was associated with clinical parameters, including age, gender, smoking, stage and survival. Chi-square (χ2) test revealed that patient survival time (p=4.02e-5), stage (p=0.006) and smoking (p=0.003) were significantly correlated with our signature, while other parameters were not associated (Supplementary Table S4). To evaluate the prognostic accuracy of the 8-gene signature on overall survival (OS), univariate and multivariate Cox proportional regression analyses were performed in the training data set. In univariate and multivariate analyses, the stage was significantly associated with OS (HR: 1.68, 95% CI 1.04-2.71, p=0.031 and HR: 1.81, 95% CI 1.08-3.01, p=0.023, respectively). Univariate and multivariate analyses also showed that the 8-gene signature had the stronger prognostic ability than stage (HR: 2.84, 95% CI 1.74-4.65, p=3.06e-05 and HR: 2.62, 95% CI 1.51-4.53, p=0.001, respectively) (Table 1). No significant difference was obtained in other parameters.
Table 1: Univariate and multivariate Cox proportional hazard regression analyses in the training set
Variable |
Univariate |
Multivariate |
||||
|---|---|---|---|---|---|---|
HR |
95% CL |
p-value |
HR |
95% CL |
p-value |
|
Age |
1.25 |
0.60-2.61 |
0.547 |
1.02 |
0.48-2.18 |
0.941 |
Gender |
0.51 |
0.31-0.84 |
0.008 |
0.58 |
0.33-0.99 |
0.047 |
Smoking (N vs E)a |
1.38 |
0.65-2.91 |
0.389 |
0.79 |
0.36-1.71 |
0..552 |
Stage (I, II) |
1.68 |
1.04-2.71 |
0.031 |
1.81 |
1.08-3.01 |
0.023 |
8-gene signature |
2.84 |
1.74-4.65 |
3.06e-05 |
2.62 |
1.51-4.53 |
0.001 |
aN; Never smoking, E; Ever smoking
The 8-gene signature was validated in five independent data sets
To evaluate the robustness of the newly identified 8-gene classifier, validation was done on five independent microarray and one RNA-seq data sets of NSCLC. A flow chart of the procedure used to validate the external data sets is summarized in Figure 2A. The leave-one-out cross-validation (LOOCV) in five validation data sets resulted in the specificity and the sensitivity of 0.972 and 0.932, respectively. To identify whether the gene signature could be a more accurate prediction marker, we validated in the combined five validation data sets. As expected, the 8-gene signature significantly stratified patients into low- and high-risk groups (p=1.15e-07, Figure 2B). The three validation data sets (GSE31210, GSE30219 and GSE29013/E-MTAB-923) were derived from the same platform as the training data set. The 8-gene signature significantly classified patients into low- and high-risk groups for these data sets (p=0.006, p=5.13e-04 and p=0.009, Figure 2C-2E), respectively. Furthermore, cross-platform validation of the gene signature was demonstrated in two data sets. The Kaplan-Meier plots also predicted significant differences in prognosis among independent validation data sets: GSE68465 (p=0.01, Figure 2F) and GSE42127 (p=0.04, Figure 2G). Low- and high-risk groups were distinguished, based on the prognostic index of each patient (Supplementary Figure S1A-S1E). We also validated RNA-seq data from TCGA based on the 8-gene signature (p=0.005, Supplementary Figure S2). Moreover, univariate and multivariate analyses demonstrated that the 8-gene signature was a prognostic factor in combined validation sets (HR: 1.77, 95% Cl 1.43-2.20, p=1.71e-7 and HR: 1.34, 95% Cl 1.02-1.77, p=0.034, respectively) (Supplementary Table S5).
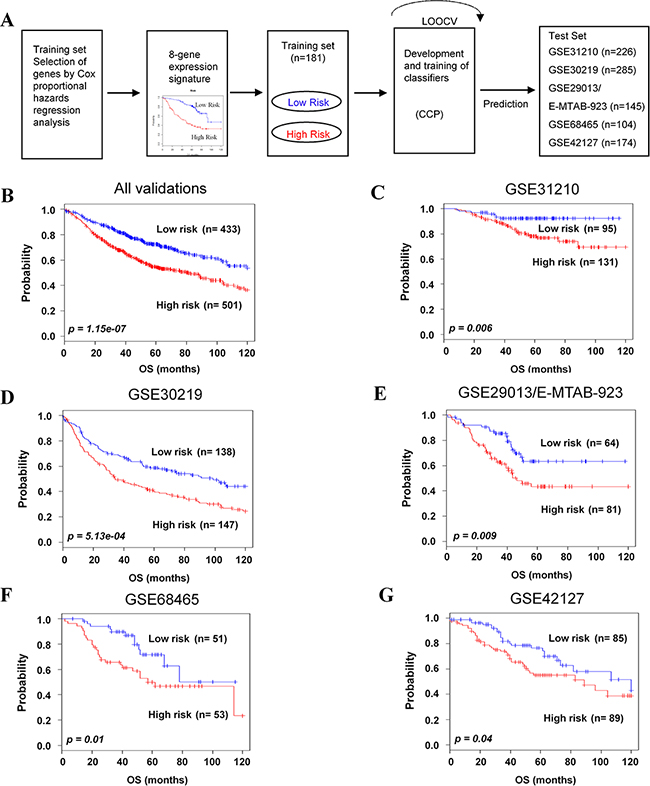
Figure 2: Validation of the 8-gene signature classifying independent data sets. A. Schematic overview of the strategy used for the construction of the prediction model and evaluation of predicted outcomes in five independent data sets by the 8-gene signature. B. All combined validation data sets. C-G. GSE31210, GSE30219, GSE29013/E-MTAB-923, GSE68465, and GSE42127 were classified by the 8-gene signature into low- and high-risk groups, and evaluated by Kaplan-Meier analyses. The p values were computed by log-rank test.
Prognostic value of the 8-gene signature in association with stages
To evaluate whether the 8-gene signature could classify patients in each stages into two risk groups in the training and validation data sets, patients in each stage were combined as followed: stage I (n=733), II (n=227), and III (n=149). As expected, the 8-gene signature clearly stratified patients into low- and high-risk groups in combined patients in stage I-III (p=1.02e-10, Figure 3A; p=1.43e-11 in 5-year OS, Supplementary Figure S4A). Moreover, the 8-gene signature significantly separated stage I NSCLC patients into low- (n=406, 55.3%) and high-risk groups (n=327, 44.6%) (p=1.44e-04, Figure 3B; p=1.65e-05 in 5-year OS, Supplementary Figure S4B). In addition, our gene signature classified patients in stage II and III into low- and high-risk groups (p=0.01 and p=0.04, Figure 3C and 3D; p=0.0371 and p=0.0268 in 5-year OS, Supplementary Figure S4A and S4D, respectively).
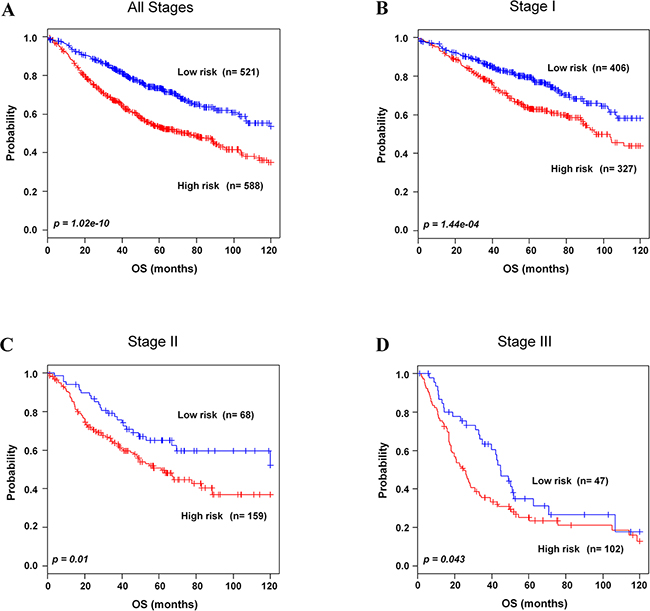
Figure 3: Kaplan-Meier survival analysis of the 8-gene signature with stages. A. Patients in all stages in the combined training and validation data sets. B. Patients in stage I in the combined training and validation data sets. C. Patients in stage II in the combined training and validation data sets. D. Patients in stage III in the validation data sets were classified by the 8-gene signature into low- and high-risk groups. The p values were computed by log-rank test.
The 8-gene signature predicts clinical outcomes for adjuvant chemotherapy
For NSCLC patients of stage II-III, ACT has improved survival rate and has become standard therapy [8, 9]. To find association of the 8-gene signature with response to chemotherapy, subset analysis was performed in stage II-III patients. By incorporating the 8-gene signature into chemotherapy information, the combined patients in stage II-III with high-risk group showed better survival with chemotherapy compared to without chemotherapy. In high-risk group, seventy six (42.4 %) patients improved survival from chemotherapy (p=0.04, Figure 4A; p=0.0382 in 5-year OS, Supplementary Figure S5A). On the contrary, low-risk group patients in stage II-III did not get any significant benefit from chemotherapy (p=0.42, Figure 4B. Similarly, among high-risk group of stage III, fifty (50 %) patients had benefit from chemotherapy (p=0.01, Figure 4C; p=0.0218 in 5-year OS, Supplementary Figure 5SB), which was not observed among low-risk group patients of stage III (p=0.93, Figure 4D). Our gene signature was also applied to patients with stage I or stage II. We found that all patients in these stages did not get benefit from chemotherapy (Supplementary Figure S3A-S3D).
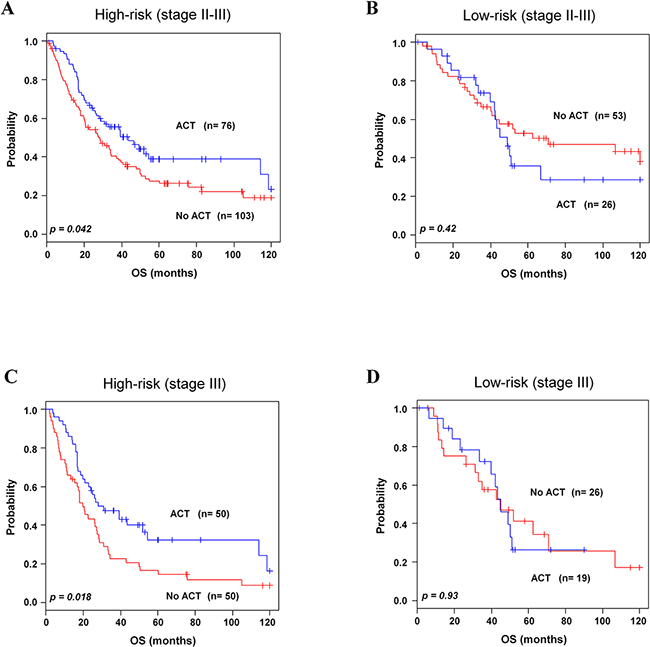
Figure 4: Kaplan-Meier survival analysis of the 8-gene signature with adjuvant chemotherapy. Patients from combined validation data sets with available ACT data were included for analysis. A-B. Patients in high-and low-risk groups with chemotherapy in stage II-III. C-D. Patients in high- and low-risk groups with chemotherapy in stage III. Patients were plotted according to presence and absence of ACT. The p values were computed by log-rank test.
Association of the 8-gene signature with EGFR and KRAS mutated/wild-type groups
Accumulation of EGFR and KRAS genetic alterations leads to the pathogenesis of lung cancer [12-15]. Based on the information of these genetic alterations available in validation data sets GSE31210 and GSE29013/E-MTAB-923, we investigated whether the 8-gene signature could further stratify lung cancer patients. In association analysis using χ2 tests, the 8-gene signature was significantly interrelated with EGFR status (p=0.007, Figure 5A) but barely with KRAS status (p=0.07, Figure 5B). These results strongly supported that the 8-gene signature would be helpful for prediction of prognosis particularly with EGFR alteration in NSCLC patients.
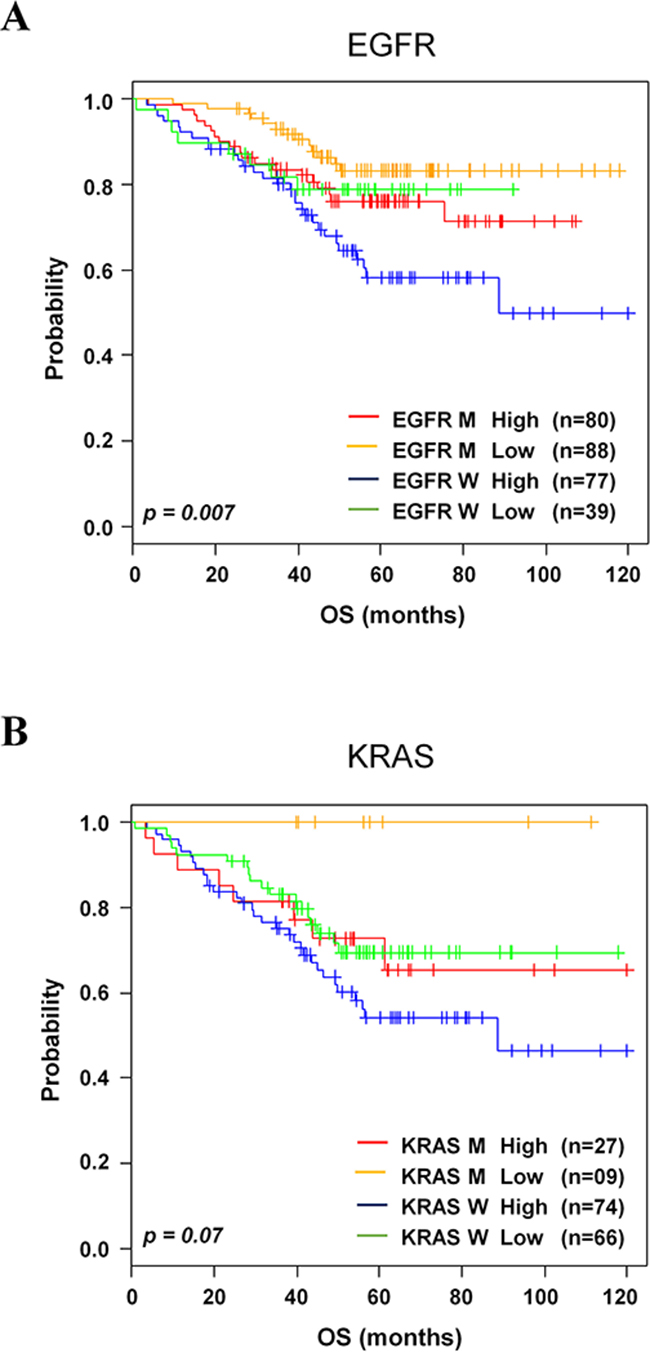
Figure 5: Kaplan-Meier survival analysis of the 8-gene signature with gene mutations. A. Kaplan-Meier curves of patients in EGFR. B. KRAS in the validation data sets. Each group was classified by the 8-gene signature into low- and high-risk groups. The p values were computed by log-rank test.
Association of the 8-gene signature with histological subtypes
To further determine whether lung cancer histology was associated with our 8-gene signature, we incorporated the gene signature into histological information in GSE30219, GSE29013, E-MTAB-923 and GSE42127. The 8-gene signature significantly classified the adenocarcinoma patients into low- and high-risk groups (p=8.76e-03, Figure 6A). However, it could not stratify the squamous cell carcinoma patients (Figure 6B).
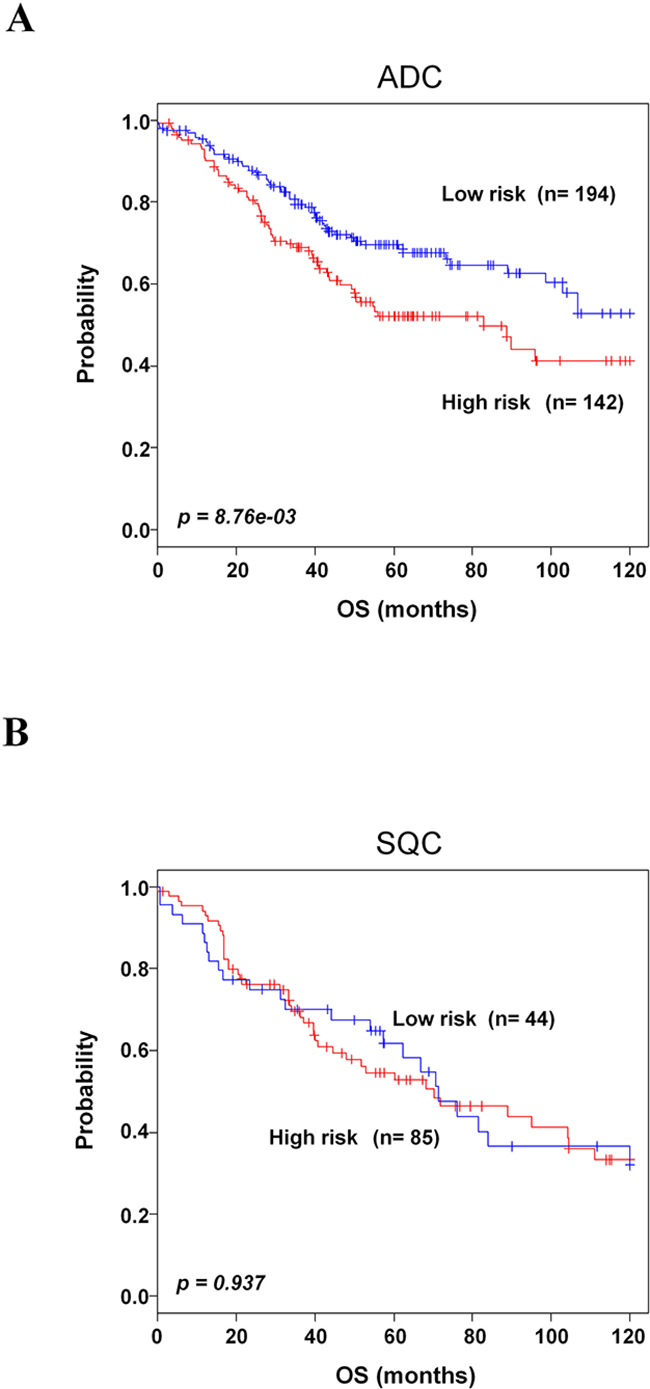
Figure 6: Kaplan-Meier survival analysis of the 8-gene signature with histological subtype. A. Kaplan-Meier curves of patients with adenocarcinoma (ADC). B. Kaplan-Meier curves of patients with squamous cell carcinoma (SQC). Each group was classified by the 8-gene signature into low- and high-risk groups. The p values were computed by log-rank test.
DISCUSSION
In this study, we have developed a novel 8-gene signature for NSCLC using computational approaches in tissues derived from patients. A supervised approach was integrated to construct the signature refined by LOOCV. Furthermore, the prognostic value of the 8-gene signature was determined in six microarray independent data sets (n=934) and one RNA-seq data set (n=543) patients. The robustness of the gene signature was supported by the high sensitivity (>0.90) and specificity (>0.90) values, and a significant association of predicted outcomes was found with patient prognosis in those data sets. Using univariate Cox analysis, the 8-gene signature was found to be one of the most reliable predictive factors for survival. Univariate and multivariate analyses performed after adjusting the clinical parameters showed a significant association of this prognostic gene signature with survival rate. Additionally, the 8-gene signature had the ability to identify stage II-III patients benefiting from ACT. Our gene signature strongly supported that 8 genes are also highly informative for prediction of patients with EGFR-mutated and wild type. These results suggest that our signature might be helpful in clinical management.
In clinical oncology, identification of individual patients who need ACT in NSCLC still represents a major concern. To date, only AJCC stage has been validated as the predictive factor to identify which patients should be treated with, or spared from chemotherapy. The benefit of ACT was previously demonstrated in patients at stage II and III [8, 9, 33]. In the context of survival benefit from ACT, a 15 gene-signature was first reported in resected NSCLC [24] in the JBR.10 trial [9]. Malignancy-risk gene signature was also developed as a predictive signature for ACT in lung cancer [34]. Recently, a 12-gene signature predicted ACT benefits with stage I-III NSCLC in two different data sets [35]. However, these gene signatures were studied in a small number of patients who received ACT and only tested on the JBR.10 trial data. None of the previously published findings showed a survival advantage in stage II-III patients. In our study, from the predictive point of view, the 8-gene signature has confirmed the potential to identify patients who would be likely to receive benefits from ACT. In subset analysis, the 8-gene signature clearly showed the benefit in stage II-III NSCLC patients. Patients in the high-risk group benefited significantly from ACT (HR, 1.47; 95% Cl, 1.01 to 2.14; p=0.044). In contrast, benefit from ACT was not statistically significant in low-risk group patients (HR, 0. 77; 95% Cl, 0.41 to 1.45; p=0.42). Our findings also confirmed the benefits of the ACT for patients with stage III. Therefore, we think that our 8-gene signature has the capability to facilitate clinical decisions for stage II-III NSCLC patients who might benefit from ACT.
Molecular alterations in EGFR, KRAS, and ALK genes are involved in lung cancer pathogenesis [12-15], but clinical use of these biomarkers is still a debatable issue. Because several prognostic gene signatures could not separate EGFR-mutated, KRAS-mutated, and wild-type patients into distinct subgroups, prognostic performance of these subgroups showed conflicting results [25, 36]. Consistent with previously published findings [17, 37], the 8-gene signature was interrelated with EGFR alteration. In contrast, the 8-gene signature was not able to be associated with KRAS alteration. At this time, we do not know why this happens, but we guess this discrepancy may be due to a small number of patients in this category [38]. We could not analyze our gene signature to classify patients with ALK-mutation due to small number of available data. Further studies are required to evaluate the 8-gene signature in response to chemotherapy in these mutation patients in independent and larger data sets. Therefore, our study demonstrated that the supervised analysis approach can identify patients with both mutation-specific and wild types into patients at higher risk with worse prognosis.
In the analyses by incorporating the 8-gene signature into histological information, our gene signature further stratified the patients with adenocarcinoma into high- and low-risk groups. However, unfortunately, the 8-gene signature could not significantly predict the prognosis for patients with squamous cell carcinoma. The 8-gene signature might imply the potential benefit of individual treatment in patients with adenocarcinoma, although we agree that it would not be enough to make a strong conclusion on the predictive power for squamous cell carcinoma due to the small number of patients.
Notably, some genes in the 8-gene signature (STAT1, CLU, GTSE1 and NUSAP1) are involved in angiogenesis, invasion, migration, and proliferation. Overexpression of STAT1 was observed in lung cancer progression [39]. STAT1 promotes tumor growth by diverse processes that range from suppression of tumor immune surveillance and an increase in invasiveness/metastasis to acquisition of resistance against irradiation and chemotherapy [40]. It is also related to purinergic signaling which has immunologic consequences in patients with neoplastic disease [41]. CLU is upregulated after exposure to chemo- and radiotherapy in studies for lung cancer cell lines and animal models. In NSCLC prognostic research, CLU-positive patients with lung cancer had a better overall survival and disease-free survival than those with CLU-negative tumors [42]. STAT1 and CLU are also involved in hypoxia and inflammation which are two inseparable hallmarks in tumorigenesis [43], indicating that they really play important roles in NSCLC pathogenesis. GTSE1, a negative regulator of p53, facilitates the proteasomal degradation of p53 during cellular recovery from DNA damage [44]. NUSAP1 expression is positively correlated with tumor progression and recurrence [45, 46]. Thus, we read that these genes have significant roles in the NSCLC tumorigenesis. In our current study, their expression patterns in NSCLC patients of our current study were corresponded to results from the previous studies [39, 42, 47, 48]. In addition, our gene signature identified new promising biomarkers such as ATP binding cassette subfamily A member 8 (ABCA8), troponin T1 (TNNT1), ectonucleoside triphosphate diphosphohydrolase 3 (ENTPD3) and carboxypeptidase A3 (CPA3).
Here, we report the identification of the 8-gene signature by system biology approaches using highly reliable NSCLC data sets. The 8-gene signature predicted patients at high-risk of mortality in all validation data sets. Moreover, our gene signature predicted which patients would respond to ACT. In clinical context, the gene signature stratified patients into two distinct prognostic risk groups, and thus overcomes limitations in conventional classification. Therefore, the 8-gene signature can preferentially be valuable as an independent and accurate prognostic predictor and provides an opportunity for future clinical trial to test the benefit of chemotherapy in NSCLC patients.
MATERIALS AND METHODS
Patient and gene expression data
All data sets were downloaded from the National Center for Biotechnology Information Gene Expression Omnibus database (http://www.ncbi.nlm.nih.gov/geo) and Array express (https://www.ebi.ac.uk/arrayexpress/). Data were selected based on the chip type [Affymetrix U133 Plus 2.0 (GPL570), HG-133A (GPL96) and Illumina HumanWG-6 v3.0 expression beadchip (GPL6884)] (Supplementary Table S1). Raw data were preprocessed using robust multiarray averaging (RMA) method for normalization. GSE50081 (n=181, University Health Network) [29] was used as the training data set. GSE31210 (n=226, National Cancer Center Hospital) [25], GSE30219 (n=285, INSERM-UJF) [31], GSE29013 (n=55, UT Southwestern Medical Center) [32], E-MTAB-923 (n=90, French National League against Cancer) [36], GSE68465 (n=104, Memorial Sloan-Kettering Cancer Center) [23] and GSE42127 (n=174, UT Southwestern Medical Center) [35] were used as validation data sets (Supplementary Table S3). To test the prognostic significance of gene signature, only gene expression data with available survival data were used. ACT information was available for 170 patients from the validation data sets.
Development of the prognostic gene expression signature
A gene expression signature to predict prognostic risk was developed from the training data set (GSE50081). Gene expression and overall survival (OS) data were combined to build a gene expression profiling-based survival classifier. The 54,675 probe sets were filtered by gene filtration using at least 2 absolute value of log2 scale, which represented the same gene expression level. The univariate Cox proportional hazard regression (p < 0.001) was then used to identify OS-associated gene expression signature from the training data set. Regarding prediction of prognosis, genes from the survival signature were applied to the survival risk prediction analysis [49]. This method used the principal component from the training data set and produced a prognostic index (PI) for each patient. The PI was computed by the formula ∑iwi xi - 0.00895 where wi and xi were the weight and logged gene expression for the i-th gene, respectively. Patients were classified into two groups based on a median prognostic index of 0.047018. Patients were assigned to the high-risk group if their prognostic indices were greater than the median value, whereas the low-risk group was composed of patients with prognostic indices that were equivalent to or less than the median value.
Validation of the prognostic signature
The validation of the gene signature was accomplished on independent data sets. Gene expression data from different data sets were adjusted individually by subtracting the median expression value across the samples. To further refine this model and to sub-stratify the predicted outcomes, Compound Covariate Predictor (CCP) was utilized as a class prediction algorithm [50]. The robustness was estimated by the misclassification rate that was determined during the leave-one-out cross-validation (LOOCV) in the training data set.
Kaplan-Meier survival analyses were performed after patient classification into two risk groups, and Chi-square (χ2) and log-rank tests were used to evaluate the survival risk between two predicted subgroups of patients. The univariate and multivariate Cox proportional hazard regression analyses were used to evaluate independent prognostic factors associated with survival. Gene signature, stage, smoking, gender, and age were employed as covariates.
Statistical methods of microarray data
Microarray data and heatmap were analyzed using BRB-Array Tools Version 3.0 (http://linus.nci.nih.gov/BRB-ArrayTools.html) [51]. All other statistical analyses were accomplished in the R language environment (http://www.r-project.org) and Statistical Package for Social Sciences (SPSS) software (version 20, SPSS Inc, Chicago, IL, USA). In all statistical analyses, p value of less than 0.05 was considered significant.
ACKNOWLEDGMENTS
This study was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MEST) (No. 2011-0030072).
CONFLICTS OF INTEREST
Conflict of interest relevant to this article was not reported.
REFERENCES
1. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J and Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015; 65:87-108.
2. Ettinger DS, Akerley W, Borghaei H, Chang AC, Cheney RT, Chirieac LR, D’Amico TA, Demmy TL, Govindan R, Grannis FW, Jr., Grant SC, Horn L, Jahan TM, et al. Non-small cell lung cancer, version 2.2013. J Natl Compr Canc Netw. 2013; 11:645-653; quiz 653.
3. Davidson MR, Gazdar AF and Clarke BE. The pivotal role of pathology in the management of lung cancer. J Thorac Dis. 2013; 5 Suppl 5:S463-478.
4. Langer CJ, Besse B, Gualberto A, Brambilla E and Soria JC. The evolving role of histology in the management of advanced non-small-cell lung cancer. J Clin Oncol. 2010; 28:5311-5320.
5. Chansky K, Sculier JP, Crowley JJ, Giroux D, Van Meerbeeck J, Goldstraw P, International Staging C and Participating I. The International Association for the Study of Lung Cancer Staging Project: prognostic factors and pathologic TNM stage in surgically managed non-small cell lung cancer. J Thorac Oncol. 2009; 4:792-801.
6. Kelsey CR, Marks LB, Hollis D, Hubbs JL, Ready NE, D’Amico TA and Boyd JA. Local recurrence after surgery for early stage lung cancer: an 11-year experience with 975 patients. Cancer. 2009; 115:5218-5227.
7. Rami-Porta R, Crowley JJ and Goldstraw P. The revised TNM staging system for lung cancer. Ann Thorac Cardiovasc Surg. 2009; 15:4-9.
8. Group NM-aC, Arriagada R, Auperin A, Burdett S, Higgins JP, Johnson DH, Le Chevalier T, Le Pechoux C, Parmar MK, Pignon JP, Souhami RL, Stephens RJ, Stewart LA, Tierney JF, Tribodet H and van Meerbeeck J. Adjuvant chemotherapy, with or without postoperative radiotherapy, in operable non-small-cell lung cancer: two meta-analyses of individual patient data. Lancet. 2010; 375:1267-1277.
9. Winton T, Livingston R, Johnson D, Rigas J, Johnston M, Butts C, Cormier Y, Goss G, Inculet R, Vallieres E, Fry W, Bethune D, Ayoub J, et al. Vinorelbine plus cisplatin vs. observation in resected non-small-cell lung cancer. N Engl J Med. 2005; 352:2589-2597.
10. Herbst RS, Heymach JV and Lippman SM. Lung cancer. N Engl J Med. 2008; 359:1367-1380.
11. Hanahan D and Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011; 144:646-674.
12. Pao W and Girard N. New driver mutations in non-small-cell lung cancer. Lancet Oncol. 2011; 12:175-180.
13. Janku F, Stewart DJ and Kurzrock R. Targeted therapy in non-small-cell lung cancer--is it becoming a reality? Nat Rev Clin Oncol. 2010; 7:401-414.
14. Gerber DE and Minna JD. ALK inhibition for non-small cell lung cancer: from discovery to therapy in record time. Cancer Cell. 2010; 18:548-551.
15. Oxnard GR, Lo PC, Nishino M, Dahlberg SE, Lindeman NI, Butaney M, Jackman DM, Johnson BE and Janne PA. Natural history and molecular characteristics of lung cancers harboring EGFR exon 20 insertions. J Thorac Oncol. 2013; 8:179-184.
16. Johnson ML, Sima CS, Chaft J, Paik PK, Pao W, Kris MG, Ladanyi M and Riely GJ. Association of KRAS and EGFR mutations with survival in patients with advanced lung adenocarcinomas. Cancer. 2013; 119:356-362.
17. Lopez-Chavez A, Thomas A, Rajan A, Raffeld M, Morrow B, Kelly R, Carter CA, Guha U, Killian K, Lau CC, Abdullaev Z, Xi L, Pack S, et al. Molecular profiling and targeted therapy for advanced thoracic malignancies: a biomarker-derived, multiarm, multihistology phase II basket trial. J Clin Oncol. 2015; 33:1000-1007.
18. Martin P, Leighl NB, Tsao MS and Shepherd FA. KRAS mutations as prognostic and predictive markers in non-small cell lung cancer. J Thorac Oncol. 2013; 8:530-542.
19. Yang P, Kulig K, Boland JM, Erickson-Johnson MR, Oliveira AM, Wampfler J, Jatoi A, Deschamps C, Marks R, Fortner C, Stoddard S, Nichols F, Molina J, Aubry MC, Tang H and Yi ES. Worse disease-free survival in never-smokers with ALK+ lung adenocarcinoma. J Thorac Oncol. 2012; 7:90-97.
20. Lindeman NI, Cagle PT, Beasley MB, Chitale DA, Dacic S, Giaccone G, Jenkins RB, Kwiatkowski DJ, Saldivar JS, Squire J, Thunnissen E and Ladanyi M. Molecular testing guideline for selection of lung cancer patients for EGFR and ALK tyrosine kinase inhibitors: guideline from the College of American Pathologists, International Association for the Study of Lung Cancer, and Association for Molecular Pathology. J Thorac Oncol. 2013; 8:823-859.
21. Paik S, Shak S, Tang G, Kim C, Baker J, Cronin M, Baehner FL, Walker MG, Watson D, Park T, Hiller W, Fisher ER, Wickerham DL, Bryant J and Wolmark N. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N Engl J Med. 2004; 351:2817-2826.
22. Lu Y, Lemon W, Liu PY, Yi Y, Morrison C, Yang P, Sun Z, Szoke J, Gerald WL, Watson M, Govindan R and You M. A gene expression signature predicts survival of patients with stage I non-small cell lung cancer. PLoS Med. 2006; 3:e467.
23. Director’s Challenge Consortium for the Molecular Classification of Lung A, Shedden K, Taylor JM, Enkemann SA, Tsao MS, Yeatman TJ, Gerald WL, Eschrich S, Jurisica I, Giordano TJ, Misek DE, Chang AC, Zhu CQ, et al. Gene expression-based survival prediction in lung adenocarcinoma: a multi-site, blinded validation study. Nat Med. 2008; 14:822-827.
24. Zhu CQ, Ding K, Strumpf D, Weir BA, Meyerson M, Pennell N, Thomas RK, Naoki K, Ladd-Acosta C, Liu N, Pintilie M, Der S, Seymour L, Jurisica I, Shepherd FA and Tsao MS. Prognostic and predictive gene signature for adjuvant chemotherapy in resected non-small-cell lung cancer. J Clin Oncol. 2010; 28:4417-4424.
25. Okayama H, Kohno T, Ishii Y, Shimada Y, Shiraishi K, Iwakawa R, Furuta K, Tsuta K, Shibata T, Yamamoto S, Watanabe S, Sakamoto H, Kumamoto K, et al. Identification of genes upregulated in ALK-positive and EGFR/KRAS/ALK-negative lung adenocarcinomas. Cancer Res. 2012; 72:100-111.
26. Nguyen MN, Choi TG, Nguyen DT, Kim JH, Jo YH, Shahid M, Akter S, Aryal SN, Yoo JY, Ahn YJ, Cho KM, Lee JS, Choe W, Kang I, Ha J and Kim SS. CRC-113 gene expression signature for predicting prognosis in patients with colorectal cancer. Oncotarget. 2015; 6:31674-31692. doi: 10.18632/oncotarget.5183.
27. Akter S, Choi TG, Nguyen MN, Matondo A, Kim JH, Jo YH, Jo A, Shahid M, Jun DY, Yoo JY, Nguyen NN, Seo SW, Ali L, et al. Prognostic value of a 92-probe signature in breast cancer. Oncotarget. 2015; 6:15662-15680. doi: 10.18632/oncotarget.3525.
28. Shahid M, Cho KM, Nguyen MN, Choi TG, Jo YH, Aryal SN, Yoo JY, Yun HR, Lee JW, Eun YG, Lee JS, Kang I, Ha J, et al. Prognostic value and their clinical implication of 89-gene signature in glioma. Oncotarget. 2016; 7:51237-51250. doi: 10.18632/oncotarget.9983.
29. Der SD, Sykes J, Pintilie M, Zhu CQ, Strumpf D, Liu N, Jurisica I, Shepherd FA and Tsao MS. Validation of a histology-independent prognostic gene signature for early-stage, non-small-cell lung cancer including stage IA patients. J Thorac Oncol. 2014; 9:59-64.
30. Yamauchi M, Yamaguchi R, Nakata A, Kohno T, Nagasaki M, Shimamura T, Imoto S, Saito A, Ueno K, Hatanaka Y, Yoshida R, Higuchi T, Nomura M, et al. Epidermal growth factor receptor tyrosine kinase defines critical prognostic genes of stage I lung adenocarcinoma. PLoS One. 2012; 7:e43923.
31. Rousseaux S, Debernardi A, Jacquiau B, Vitte AL, Vesin A, Nagy-Mignotte H, Moro-Sibilot D, Brichon PY, Lantuejoul S, Hainaut P, Laffaire J, de Reynies A, Beer DG, et al. Ectopic activation of germline and placental genes identifies aggressive metastasis-prone lung cancers. Sci Transl Med. 2013; 5:186ra166.
32. Xie Y, Xiao G, Coombes KR, Behrens C, Solis LM, Raso G, Girard L, Erickson HS, Roth J, Heymach JV, Moran C, Danenberg K, Minna JD and Wistuba, II. Robust gene expression signature from formalin-fixed paraffin-embedded samples predicts prognosis of non-small-cell lung cancer patients. Clin Cancer Res. 2011; 17:5705-5714.
33. Scagliotti GV, Parikh P, von Pawel J, Biesma B, Vansteenkiste J, Manegold C, Serwatowski P, Gatzemeier U, Digumarti R, Zukin M, Lee JS, Mellemgaard A, Park K, et al. Phase III study comparing cisplatin plus gemcitabine with cisplatin plus pemetrexed in chemotherapy-naive patients with advanced-stage non-small-cell lung cancer. J Clin Oncol. 2008; 26:3543-3551.
34. Chen DT, Hsu YL, Fulp WJ, Coppola D, Haura EB, Yeatman TJ and Cress WD. Prognostic and predictive value of a malignancy-risk gene signature in early-stage non-small cell lung cancer. J Natl Cancer Inst. 2011; 103:1859-1870.
35. Tang H, Xiao G, Behrens C, Schiller J, Allen J, Chow CW, Suraokar M, Corvalan A, Mao J, White MA, Wistuba, II, Minna JD and Xie Y. A 12-gene set predicts survival benefits from adjuvant chemotherapy in non-small cell lung cancer patients. Clin Cancer Res. 2013; 19:1577-1586.
36. Fouret R, Laffaire J, Hofman P, Beau-Faller M, Mazieres J, Validire P, Girard P, Camilleri-Broet S, Vaylet F, Leroy-Ladurie F, Soria JC and Fouret P. A comparative and integrative approach identifies ATPase family, AAA domain containing 2 as a likely driver of cell proliferation in lung adenocarcinoma. Clin Cancer Res. 2012; 18:5606-5616.
37. Planck M, Isaksson S, Veerla S and Staaf J. Identification of transcriptional subgroups in EGFR-mutated and EGFR/KRAS wild-type lung adenocarcinoma reveals gene signatures associated with patient outcome. Clin Cancer Res. 2013; 19:5116-5126.
38. Starmans MH, Pintilie M, Chan-Seng-Yue M, Moon NC, Haider S, Nguyen F, Lau SK, Liu N, Kasprzyk A, Wouters BG, Der SD, Shepherd FA, Jurisica I, et al. Integrating RAS status into prognostic signatures for adenocarcinomas of the lung. Clin Cancer Res. 2015; 21:1477-1486.
39. Yao R, Wang Y, Lubet RA and You M. Differentially expressed genes associated with mouse lung tumor progression. Oncogene. 2002; 21:5814-5821.
40. Meissl K, Macho-Maschler S, Muller M and Strobl B. The good and the bad faces of STAT1 in solid tumours. Cytokine. 2015.
41. Eltzschig HK, Sitkovsky MV and Robson SC. Purinergic signaling during inflammation. N Engl J Med. 2013; 368:1260.
42. Panico F, Rizzi F, Fabbri LM, Bettuzzi S and Luppi F. Clusterin (CLU) and lung cancer. Adv Cancer Res. 2009; 105:63-76.
43. Eltzschig HK and Carmeliet P. Hypoxia and inflammation. N Engl J Med. 2011; 364:656-665.
44. Liu XS, Li H, Song B and Liu X. Polo-like kinase 1 phosphorylation of G2 and S-phase-expressed 1 protein is essential for p53 inactivation during G2 checkpoint recovery. EMBO Rep. 2010; 11:626-632.
45. Okamoto A, Higo M, Shiiba M, Nakashima D, Koyama T, Miyamoto I, Kasama H, Kasamatsu A, Ogawara K, Yokoe H, Tanzawa H and Uzawa K. Down-Regulation of Nucleolar and Spindle-Associated Protein 1 (NUSAP1) Expression Suppresses Tumor and Cell Proliferation and Enhances Anti-Tumor Effect of Paclitaxel in Oral Squamous Cell Carcinoma. PLoS One. 2015; 10:e0142252.
46. Gulzar ZG, McKenney JK and Brooks JD. Increased expression of NuSAP in recurrent prostate cancer is mediated by E2F1. Oncogene. 2013; 32:70-77.
47. Yan Y, Luo K, Zhang H and Chai W. RNA interference-mediated secretory clusterin gene silencing inhibits proliferation and promotes apoptosis of human non-small cell lung cancer cells. Hepatogastroenterology. 2013; 60:70-75.
48. Tian T, Zhang E, Fei F, Li X, Guo X, Liu B, Li J, Chen Z and Xing J. Up-regulation of GTSE1 lacks a relationship with clinical data in lung cancer. Asian Pac J Cancer Prev. 2011; 12:2039-2043.
49. Bair E and Tibshirani R. Semi-supervised methods to predict patient survival from gene expression data. PLoS Biol. 2004; 2:E108.
50. Radmacher MD, McShane LM and Simon R. A paradigm for class prediction using gene expression profiles. J Comput Biol. 2002; 9:505-511.
51. Simon R, Lam A, Li MC, Ngan M, Menenzes S and Zhao Y. Analysis of gene expression data using BRB-ArrayTools. Cancer Inform. 2007; 3:11-17.