
INTRODUCTION
Although the incidence and mortality of gastric cancer have steadily declined in recent decades, it remains the fourth most common type of cancer and the second leading cause of cancer mortality worldwide [1]. Surgery is an effective treatment for gastric cancer. Recent research has also shown that chemotherapy following radical surgery is an effective adjuvant therapy for East Asian patients [2]. Cisplatin is one of the most widely used drugs for chemotherapy and improves the overall survival for cancer patients [3-7]. However, cancer treatment is limited by the different efficacies of chemotherapeutic regimens, diverse disease states of patients and response rate to drugs, drug-related side-effects, acquisition of drug resistance, and cancer recurrence with metastasis [8-11]. To this end, the targeted approaches that focus on drug resistance-associated molecules are required to improve the efficacy of chemotherapy against advanced gastric cancers.
RhoGDI2 belongs to a family of Rho GTPase dissociate inhibitors (RhoGDIs). They are pivotal regulators of the function of Rho GTPase and typically exert their effects by forming a complex with Rho GTPase, and thereby modulating their nucleotide exchange and membrane association. Therefore, RhoGDIs play significant roles in regulating the actin cytoskeleton, cell polarity, microtubule dynamics, membrane transport pathways, and transcription factor activities [12, 13]. Unlike other members of the family (such as RhoGDI1 and RhoGDI3), RhoGDI2 is preferentially expressed in hematopoietic cells, and appears to have a narrow selectivity and lower binding affinity for Rho GTPases [14]. RhoGDI2 associates with and negatively regulates Rac1 and Rac3 in breast cancer cells, but not RhoA, Cdc42, and RhoC [15]. In contrast, it positively regulates Rac1 in human bladder cancer cells [16]. The significant role of RhoGDI2 in cancer has been previously noted in several lines of study. RhoGDI2 expression is inversely correlated with invasive capacity in bladder cancer cell lines [17]. Furthermore, reduced RhoGDI2 protein expression has been associated with poor prognosis for patients with advanced bladder cancer [18]. In contrast, RhoGDI2 mRNA expression is significantly higher in ovarian adenocarcinomas than in benign adenomas [19]. Consistent with this finding, RhoGDI2 is overexpressed in human breast cancer cell lines, and it increases cancer cell invasiveness and motility in vitro [20]. We also demonstrated that RhoGDI2 expression is positively correlated with tumor progression and metastatic potential in gastric cancer [21]. In addition, our recent work demonstrated that RhoGDI2 is associated with the acquisition of resistance to chemotherapeutic agents (such as cisplatin), which is a hallmark of aggressive cancers, in gastric cancer cells [22].
To delineate the mechanism by which RhoGDI2 promotes gastric cancer cell invasion and chemoresistance, we performed two-dimensional gel electrophoresis (2-DE) on proteins that were derived from a RhoGDI2-overexpressing SNU-484 human gastric cancer cell line and control cells, and noted that levels of 14-3-3σ, which is a member of the multifunctional 14-3-3 protein family, were significantly reduced [23]. In this study, we demonstrated that the downregulation of 14-3-3σ is largely correlated with the cisplatin-resistant phenotype of RhoGDI2-overexpessing gastric cancer cells. Of note, the restoration of 14-3-3σ is associated with impaired RhoGDI2-induced chemoresistance of gastric cancer cells through the activation of p38 and Erk.
RESULTS
RhoGDI2 downregulates 14-3-3σ expression
Previously, we identified 14 downregulated proteins in RhoGDI2-overexpressing SNU-484(GDI2-4) gastric cancer cells compared with control SNU-484(Mock) cells by using comparative 2-DE [23]. For further analysis, we selected 14-3-3σ that was previously implicated in cancer cell proliferation, metastasis, and apoptosis. To validate our mass spectrometry results, we performed reverse transcription-polymerase chain reaction and western blot analyses to determine the mRNA and protein expression levels of 14-3-3σ in RhoGDI2-overexpressing SNU-484 cells and RhoGDI2-depleted MKN-28 cells. Consistent with the results of 2-DE and imaging analysis (Fig. 1A), the mRNA and protein expression of 14-3-3σ were significantly downregulated in RhoGDI2-overexpressing SNU-484 cells and upregulated in RhoGDI2-depleted MKN-28 cells, compared to its expression in control cells (Fig. 1B). We also examined the mRNA expression levels of the other 14-3-3 isoforms (β, γ, ε, ζ, η, and τ) in RhoGDI2-overexpressing SNU-484 cells, but could not find any significant differences between these cells and the control cells (Fig. 1C). To further elucidate whether the decreased expression of 14-3-3σ is associated with RhoGDI2 expression, we observed 14-3-3σ expression levels in HeLa cells and MCF-7 cells after transient transfection with a Flag-tagged RhoGDI2 expression vector. As shown in Fig. 1D, transient expression of RhoGDI2 caused the markedly reduced expression of 14-3-3σ compared with the expression level in the vector-transfected control cells, which suggests that 14-3-3σ is a direct target of RhoGDI2.
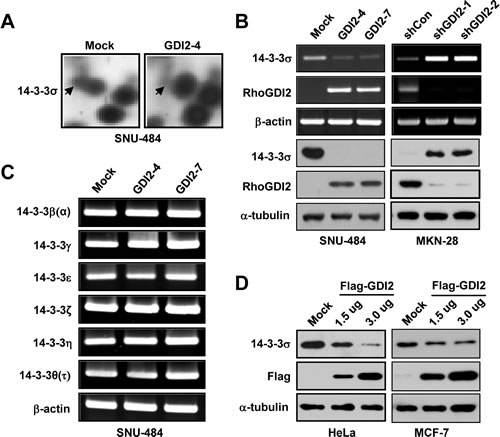
Figure 1: RhoGDI2 downregulates 14-3-3σ expression. (A) Detailed 2-DE images of 14-3-3σ protein spots in RhoGDI2-overexpressing SNU-484(GDI2-4) cells compared with SNU-484(Mock) cells. (B) mRNA (upper panel) and protein (lower panel) expression of 14-3-3σ in RhoGDI2-overexpressing SNU-484(GDI2-4 and GDI2-7) cells. (C) mRNA expression of 14-3-3 isoforms in RhoGDI2-overexpressing SNU-484(GDI2-4 and GDI2-7) cells. (D) HeLa and MCF-7 cells were transiently transfected with RhoGDI2-expressing plasmid (Flag-RhoGDI2) and immunoblotted with indicated antibodies.
Depletion of 14-3-3σ expression enhances cisplatin resistance of gastric cancer cells
Since 14-3-3σ is known to enhance the chemosensitivity of some types of cancer cells [24-27], we first examined whether depletion of endogenous 14-3-3σ expression could enhance chemoresistance of gastric cancer cells. For this purpose, we used SNU-484(Mock) cells in which 14-3-3σ, which is highly expressed (Fig. 1B), was depleted by employing siRNA transfection. As shown in Fig. 2A, the expression of 14-3-3σ was markedly reduced in 14-3-3σ-specific siRNA transfected cells, but not in control siRNA transfected cells. To determine whether the depletion of 14-3-3σ expression affects cisplatin-induced apoptosis, we analyzed the cells by TUNEL staining. Control cells were highly sensitive to cisplatin-induced apoptosis (Fig. 2B and C), as previously reported.22 However, depletion of 14-3-3σ significantly attenuated cisplatin-induced apoptosis (Fig. 2B and C) and cleavage of poly(ADP-ribose) polymerase (PARP) (Fig. 2D) in SNU-484(Mock) cells. These results led us to hypothesize that the downregulation of 14-3-3σ contributes to the RhoGDI2-induced chemoresistance of gastric cancer cells.
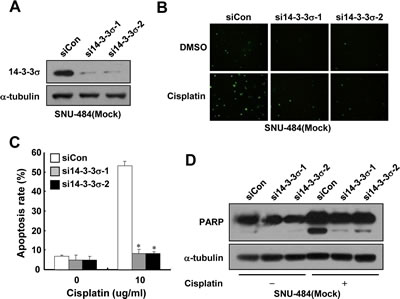
Figure 2: Depletion of 14-3-3σ expression enhances cisplatin resistance in gastric cancer cells. (A) Representative immunoblot for 14-3-3σ in 14-3-3σ-depleted SNU-484(Mock) cells. (B) Representative images for TUNEL staining of 14-3-3σ-depleted SNU-484(Mock) cells after cisplatin treatment (10 µg/ml) for 24 h. (C) Histogram shows the ratio of TUNEL-positive 14-3-3σ-depleted SNU-484(Mock) cells after cisplatin treatment (10 µg/ml) for 24 h. Data are mean ± SD of three individual experiments, each in triplicate. *, P 0.01 as determined by paired Student t test. (D) Representative immunoblot for PARP cleavage after cisplatin treatment in 14-3-3σ-depleted SNU-484(Mock) cells.
Ectopic expression of 14-3-3σ restores chemosensitivity to cisplatin in RhoGDI2-overexpressing gastric cancer cells
To determine whether 14-3-3σ expression can alter cellular sensitivity to cisplatin in RhoGDI2-overexpressing gastric cancer cells, we established 14-3-3σ-overexpressing cells in RhoGDI2-overexpressing SNU-484(GDI2-7) cells. To this end, a myc-tagged 14-3-3σ expression vector (pcDNA4-myc-His/14-3-3σ) or empty vector (pcDNA4-myc-His) was stably transfected into RhoGDI2-overexpressing SNU-484(GDI2-7) cells. The expression levels of 14-3-3σ in the respective cells were verified by performing western blot analysis using anti-myc antibody (Fig. 3A). RhoGDI2-overexpressing SNU-484(GDI2-7/Mock) control cells were highly resistant to cisplatin-induced apoptosis (Fig. 3B and C), as previously reported [22]. However, the overexpression of 14-3-3σ significantly increased cisplatin-induced apoptosis (Fig. 3B and C) and PARP cleavage (Fig. 3D) in RhoGDI2-overexpressing SNU-484(GDI2-7) cells. These results suggest that the downregulation of 14-3-3σ expression confers resistance to RhoGDI2-overexpressing gastric cancer cells against cisplatin-induced apoptosis.
To determine if 14-3-3σ expression affects the chemosensitivity of RhoGDI2-overexpressing gastric cancer cells in vivo, 14-3-3σ-overexpressing SNU-484(GDI2-7/14-3-3σ-2) cells and control cells (GDI2-7/Mock) were injected subcutaneously into the flanks of nude mice. Growth of the 14-3-3σ-overexpressing SNU-484(GDI2-7/14-3-3σ-2) cell-injected tumor did not differ from that of control cells (Fig. 3E and F). Cisplatin significantly inhibited tumor growth in mice that had been injected with 14-3-3σ-overexpressing SNU-484(GDI2-7/14-3-3σ-2) cells, but not in mice that had been injected with control cells (GDI2-7/Mock) (Fig. 3E and F). These findings indicate that 14-3-3σ contributes to the inhibition of resistance to cisplatin of RhoGDI2-expressing gastric cancer cells in xenograft tumor models.
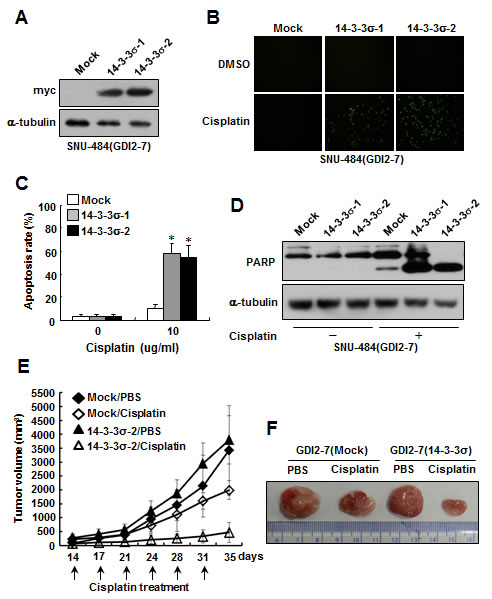
Figure 3: Ectopic expression of 14-3-3σ restores chemosensitivity to cisplatin in RhoGDI2-overexpressing gastric cancer cells. (A) Representative immunoblot for myc in 14-3-3σ-overexpressing SNU-484(GDI2-7) cells. (B) Representative images for TUNEL staining of 14-3-3σ-overexpressing SNU-484(GDI2-7) cells after cisplatin treatment (10 µg/ml) for 24 h. (C) Histogram shows the ratio of TUNEL-positive 14-3-3σ-overexpressing SNU-484(GDI2-7) cells after cisplatin treatment for 24 h. Data are mean ± SD of three individual experiments, each in triplicate. *, P < 0.01 as determined by paired Student t test. (D) Representative immunoblot for PARP cleavage after cisplatin treatment in 14-3-3σ-overexpressing SNU-484(GDI2-7) cells. (E) In vivo chemoresistance assay of 14-3-3σ-overexpressing SNU-484(GDI2-7) cells. Xenograft tumor volumes in mice treated with cisplatin or PBS are shown. Tumor volume is presented as the mean ± SD (n = 12 mice in each group). (F) Representative images of tumor derived from 14-3-3σ-overexpressing SNU-484(GDI2-7) and control cells in mice treated with cisplatin or PBS for 35 days.
Suppression of Erk and p38 activities enhances cisplatin resistance of gastric cancer cells
Since mitogen-activated protein kinase (MAPK) pathways are implicated in the execution of apoptosis by different cytotoxic agents and p38/JNK kinase activities have been known to be increased in RhoGDI2-depleted breast cancer cells [15], we first assessed whether the activities of these apoptosis-related kinases are suppressed in RhoGDI2-overexpressing gastric cancer cells. To this end, we examined MAPK activation in RhoGDI2-overexpressing SNU-484 cells by assessing their phosphorylation states by using antibodies specific to the phosphorylated species of each enzyme. As shown in Fig 4A, the phosphorylation levels of Erk and p38, but not JNK, were significantly decreased in RhoGDI2-overexpressing SNU-484(GDI2-4 and GDI2-7) cells compared with control cells (Mock) under normal culture condition. We next examined whether suppression of Erk or p38 activity affects cisplatin-induced apoptosis in gastric cancer cells. Suppression of Erk and p38 activity by U0126 and SB203580, respectively, significantly inhibited cisplatin-induced apoptosis (Fig. 4B and C) and PARP cleavage (Fig. 4D) in SNU-484(Mock) cells. These results suggest that suppression of Erk and p38 activity is critical for the cisplatin resistance of gastric cancer cells.
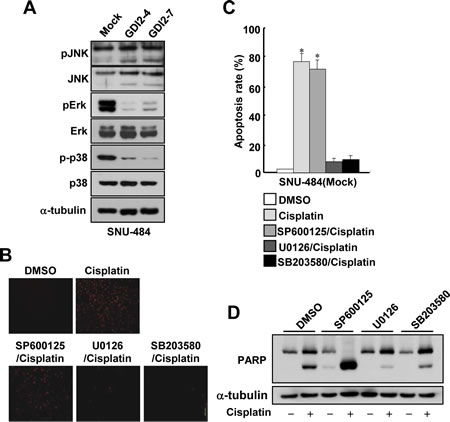
Figure 4: Suppression of Erk and p38 activation attenuates cisplatin-induced apoptosis in gastric cancer cells. (A) Representative immunoblot for phosphorylated and total Erk, JNK, and p38MAPK in RhoGDI2-overexpressing SNU-484(GDI2-4 and GDI2-7) cells. (B) Representative images for TUNEL staining of SNU-484(Mock) cells in the presence or absence of respective inhibitor after cisplatin treatment (10 µg/ml) for 24 h. (C) Histogram shows the ratio of TUNEL-positive SNU-484(Mock) cells in the presence or absence of respective inhibitor after cisplatin treatment (10 µg/ml) for 24 h. Data are mean ± SD of three individual experiments, each in triplicate. *, P < 0.01 as determined by paired Student t test. (D) Representative immunoblot for PARP cleavage of SNU-484(Mock) cells in the presence or absence of respective inhibitor after cisplatin treatment.
Ectopic expression of 14-3-3σ attenuates RhoGDI2-induced cisplatin resistance of gastric cancer cells through Erk and p38 activation
Since 14-3-3σ regulates MAPK activity and the activities of Erk and p38 were repressed in RhoGDI2-overexpressing SNU-484(GDI2-4 and GDI2-7) cells (Fig. 4A), we examined whether the downregulation of 14-3-3σ expression is critical for the repression of Erk and p38 activities in RhoGDI2-overexpressing SNU-484(GDI2-7) cells. As we expected, the levels of phospho-Erk and phospho-p38 markedly increased in 14-3-3σ-overexpressing SNU-484(GDI2-7) cells (14-3-3σ-1 and 14-3-3σ-2) compared with control (Mock) cells (Fig. 5A). As shown in Figure 3, the overexpression of 14-3-3σ significantly increased cisplatin-induced apoptosis in RhoGDI2-overexpressing SNU-484(GDI2-7) cells. We next examined whether 14-3-3σ-mediated activation of Erk and p38 activities is critical for the recovery of cisplatin sensitivity in RhoGDI2-overexpressing SNU-484(GDI2-7) cells. Suppression of Erk and p38 activities by their specific inhibitors, respectively, markedly inhibited cisplatin-induced apoptosis (Fig. 5B and C) and PARP cleavage (Fig. 5D) in 14-3-3σ-overexpressing SNU-484(GDI2-7) cells (14-3-3σ-1 and 14-3-3σ-2). All of these results suggest that the suppression of 14-3-3σ-mediated Erk and p38 activation is critical for the cisplatin resistance of RhoGDI2-expressing gastric cancer cells.
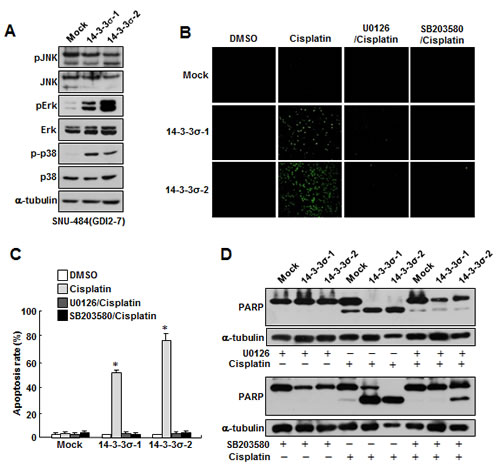
Figure 5: Ectopic expression of 14-3-3σ attenuates RhoGDI2-induced cisplatin resistance in gastric cancer cells through Erk and p38 activation. (A) Representative immunoblot for phosphorylated and total MAPK in 14-3-3σ-restored RhoGDI2-overexpressing SNU-484(GDI2-7) cells. (B) Representative images for TUNEL staining of 14-3-3σ-restored RhoGDI2-overexpressing SNU-484(GDI2-7) cells in the presence or absence of respective inhibitor after cisplatin treatment (10 µg/ml) for 24 h. (C) Histogram shows the ratio of TUNEL-positive 14-3-3σ-restored RhoGDI2-overexpressing SNU-484(GDI2-7) cells in the presence or absence of respective inhibitor after cisplatin treatment (10 µg/ml) for 24 h. Data are mean ± SD of three individual experiments, each in triplicate. *, P < 0.01 as determined by paired Student t test. (D) Representative immunoblot for PARP cleavage of 14-3-3σ-restored RhoGDI2-overexpressing SNU-484(GDI2-7) cells in the presence or absence of respective inhibitor after cisplatin treatment.
14-3-3σ suppresses RhoGDI2-induced migration and invasion abilities of gastric cancer cells
Since RhoGDI2 promotes gastric cancer cell invasion21 as well as enhances cisplatin resistance, we next examined whether the restoration of 14-3-3σ could alter the migration and invasive properties in RhoGDI2-overexpressing gastric cancer cells. Ectopic expression of 14-3-3σ considerably decreased the invasiveness of RhoGDI2-overexpressing SNU-484(GDI2-7) cells (14-3-3σ-1 and 14-3-3σ-2) compared with control (Mock) cells (Figure 6A). Next, we checked the migrating ability of 14-3-3σ-overexpressing SNU-484(GDI2-7) cells by using a wound healing assay. For this analysis, we used a culture-insert, where a non-bias cell-free gap is produced as the “wound” when the culture-insert is removed. Immediately after removal of the culture-insert, cell images were obtained at various time points (0-30 h) under a light microscope. After 30 h, complete wound closure (100%) was achieved in the RhoGDI2-overexpressing SNU-484 control (Mock) cells, whereas only 14.9% (14-3-3σ-1) and 41% (14-3-3σ-2) wound closure was achieved in 14-3-3σ-overexpressing SNU-484(GDI2-7) cells compared with the control cells (Figure 6B and C). Taken together, these results suggest that the downregulation of 14-3-3σ expression plays a key role in RhoGDI2-induced gastric cancer cell migration and invasion. In an effort to exclude the possibility that the effect of 14-3-3σ on the migration and invasion of RhoGDI2-overexpressing gastric cancer cells was attributable to different proliferation rates, we compared the growth rates of 14-3-3σ-overexpressing SNU-484(GDI2-7) cells (14-3-3σ-1 and 14-3-3σ-2) with those of control (Mock) cells. Under the same growth conditions, all of the cells exhibited similar growth rates (Figure 6D), thereby indicating that decreased tumor cell migration and invasion via the expression of 14-3-3σ in the respective cells was not associated with their proliferation rates.
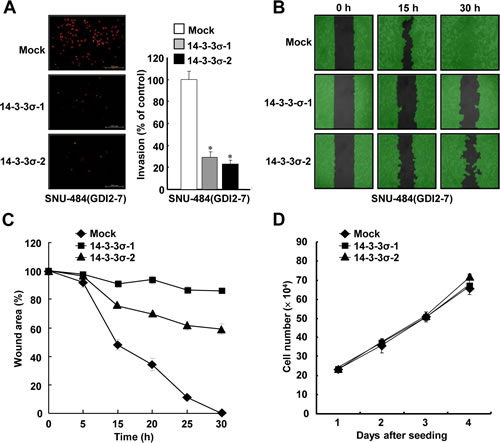
Figure 6: Ectopic expression of 14-3-3σ suppresses RhoGDI2-induced gastric cancer cell migration and invasion. (A) Representative images of invading cells through the matrigel-coated membrane stained by propidium iodide. Quantitative data of invasion assay are expressed relative to the invasion ability of SNU-484(GDI2-4/Mock) cells. Data are mean ± SD of three individual experiments, each in triplicate. *, P < 0.01 as determined by paired Student t test. (B) Representative images of migrating cells obtained at indicated time points after wound formation by phase contrast microscopy and using Wimasis Image Analysis software. (C) Quantitative analysis of wound healing assay by using the WimScratch software (Wimasis). Data represent the percentage of wound area at indicated time points in control and 14-3-3σ-overexpressing SNU-484(GDI2-4) cells. (D) Effect of 14-3-3σ overexpression on the proliferation of RhoGDI2-overexpressing SNU-484(GDI2-7) cells.
DISCUSSION
Several studies over the last decade have linked RhoGDI1 expression to apoptosis and chemoresistance of various human cancer cells. For example, the reduction of RhoGDI1 expression is associated with astrocytoma cell protection and tamoxifen resistance of breast cancer cells [28, 29]. In addition, Ronneburg et al. demonstrated that RhoGDI1 may sensitize invasive breast cancer to treatment with CMF (cyclophosphamide, methotrexate, and 5-fluorouracil), and higher RhoGDI1 expression tends to be correlated with a better clinical outcome [30]. However, RhoGDI1 was also described as an antiapoptotic protein in breast, lymphoma, fibrosarcoma, and lung cancer cells [31-33]. In contrast to RhoGDI1, several groups have focused their research on elucidating the explicit mechanisms by which RhoGDI2 regulates aggressive features of cancer cells, particularly motility, invasiveness, and metastasis. However, we recently suggested that RhoGDI2 enhances the chemoresistance of gastric cancer through the upregulation of Bcl-2 expression as well as promotes tumor growth and malignant progression.22 Consistent with our results, Zheng et al. suggested that the knockdown of RhoGDI2 expression significantly increases the sensitivity of colon cancer cells to 5-FU [34], and that the ectopic expression of RhoGDI2 in gastric cancer cells induces resistance to 5-FU and reverses 5-FU-induced G2/M phase arrest [35].
RhoGDI1 protects breast cancer cells from drug-induced apoptosis through the inhibition of Rac1 cleavage that is mediated by capase-3 [31]. Although RhoGDI1 is completely resistant to degradation during apoptosis, RhoGDI2 is well characterized as being a substrate for caspases and is cleaved in various cells during apoptosis [36]. Therefore, RhoGDI2 may act as an antiapoptotic molecule via a mechanism that is distinct from RhoGDI1 and may do so prior to caspase activation during drug-induced apoptosis. To delineate the mechanism by which RhoGDI2 contributes to chemoresistance and tumor metastasis, we performed 2-DE on proteins that were derived from a RhoGDI2-overexpressing SNU-484 human gastric cancer cell line and control cells. We found that the expression levels of 14-3-3σ were significantly downregulated in the RhoGDI2-overexpressing gastric cancer cells [23]. The results of this study indicate that 14-3-3σ is a direct target of RhoGDI2 and that the downregulation of 14-3-3σ is important for enhancing the chemoresistance of RhoGDI2-expressing gastric cancer cells. 14-3-3σ was first identified as being a human mammary epithelium-specific marker 1 (HME1) and participates in the regulation of subcellular localization, protein stability, cell apoptosis, proliferation, and the cell cycle [37-39]. However, 14-3-3σ is directly associated with human cancers, and the downregulation of 14-3-3σ expression has been observed in various human cancers, including those of the lung, prostate, breast, and liver [40, 41]. Consistent with our findings, several previous findings have shown that 14-3-3σ expression is elevated in response to different cellular stresses, and that it enhances the chemosensitivity of human colorectal, breast, and nasopharyngeal cancer [24-27].
We also suggested that the ectopic expression of 14-3-3σ could activate Erk and p38 MAPK, and that suppression of 14-3-3σ-mediated Erk and p38 activation is critical for the cisplatin resistance of RhoGDI2-overexpressing gastric cancer cells. We showed that the phosphorylation levels of Erk and p38 kinase are markedly downregulated in RhoGDI2-overexpressing (14-3-3σ downregulated) gastric cancer cells. However, the restoration of 14-3-3σ expression reverses this phenomenon. It is believed that MAPK activation is a major component that decides the fate of a cell in response to cisplatin. The pro-death or pro-survival roles of MAPKs in response to cisplatin could depend on the type of activated MAPK. While the activation of p38 plays only a pro-death role, the induction of Erk can take on both roles (survival or cell death) as a consequence [42]. Consistent with our results, the knockdown of RhoGDI2 expression significantly increases the phosphorylation levels of p38 kinase, and pretreatment with p38 kinase inhibitor significantly inhibits the apoptosis of RhoGDI2-depleted breast cancer cells [15]. Benzinger et al. also showed that the ectopic expression of 14-3-3σ enhances Erk1/2 activity in colon cancer cells [43]. The increase in MAPK activity that was observed after 14-3-3σ expression may be due to interactions with multiple ligands. Among others, A-RAF, B-RAF, and c-RAF seem to be good candidates because previous studies on the interaction between RAF proteins and 14-3-3 isoforms have shown that 14-3-3 proteins are critical modulators of RAF activity [44].
We also examined the possible pathways that lead to RhoGDI2-induced 14-3-3σ downregulation. Recently, we demonstrated that phospholipase C-gamma (PLCγ) is activated in RhoGDI2-overexpressing SNU-484 cells, and that it is required for RhoGDI2-mediated cisplatin resistance and cancer cell invasion in gastric cancer [45]. To determine whether PLC is required for RhoGDI2-induced 14-3-3σ downregulation, we examined the expression levels of 14-3-3σ in PLCγ-depleted SNU-484(GDI2) and control cells, but did not observe any difference (data not shown). Alternatively, ongoing studies in our laboratory have revealed that Rac1, but not RhoA or Cdc42, is activated in RhoGDI2-overexpressing gastric cancer cells. Therefore, we are now examining whether the activation of Rac1 is involved in RhoGDI2-induced 14-3-3σ downregulation.
MATERIALS AND METHODS
Cell cultures and reagents
Human gastric cancer cell lines SNU-484 derived RhoGDI2-overexpressing cells (GDI2-4 and GDI2-7) were maintained in RPMI-1640 medium (SIGMA). Human cervical cancer cell line HeLa and breast cancer cell line MCF-7 were cultivated in DMEM (SIGMA). All cell lines were maintained as mono-layer cultures in each optimal medium supplemented with 10% heat-inactivated FBS (Gibco, Invitrogen) and 2% of a penicillin-streptomycin (antibiotic-antimycotic) mixture (Gibco, Invitrogen) The SNU-484 cells stably transfected with RhoGDI2 and MKN-28 cells stably transfected with shRNA-expressing lentiviral vector for targeting RhoGDI2 were described in our previous report [21, 22]. Cisplatin was purchased from SIGMA. SP600125 (JNK inhibitor), U0126 (MEK1/2 inhibitor), and SB203580 (p38 inhibitor) were purchased from Cell Signaling Technology.
Reverse Transcription-PCR analysis
Total RNA was isolated using an RNeasy mini kit (Qiagen, Valencia, CA, USA) according to the manufacturer’s instructions. RT-PCR was performed using a Maxime RT-PCR PreMix kit (Intron, Taejon, Korea). 200 ng of total RNA and specific primer were added into the Maxime RT-PCR PreMix tubes and RNase-free water was added to a total volume of 20 µl. RT-PCR was performed using a Thermo Electron PCR thermal cycler. Amplified products were separated on 1~1.5% agarose gels. The RT-PCR conditions were as follows: 45oc for 30 min (reverse transcription), 94oc for 5 min (inactivation of RTase), 94oc for 1 min, 55oc for 1 min, 72oc for 1 min for 23~38 cycles, followed by 10 min of incubation at 72oc. The used primers were described in Supplementary Table 1.
Construction of the 14-3-3σ expression plasmid and transfection
Human 14-3-3σ cDNA was amplified by PCR using the following primers: 5’-GATCGGAATCCAGAGCGAAACCTGCTCTCAG-3’ and 5’-GATCGGGATCCTGATGAGGGTGCTGTCTTTG-3’. PCR products were cloned into the pcDNA4/myc-His© (Invitrogen). RhoGDI2-overexpressing SNU-484(GDI2-7) cells were transfected with 14-3-3σ expressing plasmid by using the FuGENE® 6 reagent (Promega) following the manufacturer’s instructions. After 48h of incubation, cells were treated with ZeocinTM (100 ug/ml) for selection. For transient transfection, 3 × 105/ml HeLa and MCF-7 cells were seeded in 6-well plate for 24 h and transfected with the indicated plasmids by using FuGENE® HD reagent (Promega). After 48 h, the cells were harvested and analyzed by western blot.
Antibodies and western blot analysis
Rabbit anti-PARP, anti-SAPK/JNK, anti-Phospho-SAPK/JNK (Thr183/Tyr185), anti- p44/42 Map Kinase, anti-Phospho-p44/42 Map Kinase (Thr202/Tyr204), anti-p38 MAP Kinase, and anti-Phospho-p38 MAP Kinase (Thr180/Tyr182) antibodies were purchased from Cell Signaling Technology. Anti-14-3-3 sigma antibody was purchased from Thermo Scientific. Mouse anti-α-tubulin and anti-Flag antibodies were purchased from Sigma. Mouse anti-myc antibody was purchased from IGTHERAPY. For western blot analysis, cells were harvested after defined time and lysed in lysis buffer (20 Mm Tris (pH 7.4), 2 mM EDTA, 150 mM sodium chloride, 1 mM sodium deoxycholate, 1% Triton X-100, 10% glycerol, 2 pills protease inhibitor cocktail (Roche)) on ice for 1 h and centrifuged at 13,000 rpm for 15 min. Cell lysates were separated by 8–12% SDS-PAGE and transferred to a polyvinylidene difluoride membrane (Amersham Bioscience). Subsequently, the membrane was incubated in TBST supplemented with 5% non-fat dry milk and probed with the appropriate primary antibodies. The bound antibodies were visualized with a suitable secondary antibody conjugated with horseradish peroxidase using enhanced chemiluminescence (ECL) reagent WESTSAVE up (AbFRONTIER, Korea).
RNA interference experiments
Two different siRNA oligo duplexes for targeting 14-3-3σ were purchased from Bioneer (Daejeon, Korea). The sequence was as follows; si14-3-3σ-1: 5’-GGAUCCCACUCUUCUUGCA-3’, si14-3-3σ-2: 5’-GACCAUGUUUCCUCUCAAU-3’. Transient transfection of siRNA oligo duplex was accomplished using siLentFectTM Reagent (BIO-RAD) followed by instructions of manufacturers. After incubation for 48hrs, the cells were harvested and efficiency of each siRNA oligo duplex was confirmed by western blotting using anti-14-3-3σ antibody.
Apoptosis detection
Apoptosis was measured by the terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick-end labeling (TUNEL) assay using the In Situ Cell Death Detection Kit, Fluorescein or TMR red (Roche Applied Science, Germany) following the manufacturer’s instruction. Cisplatin-treated or non-treated cells were washed with cold PBS and fixed with 4% paraformaldehyde. Fixed cells were permeabilized and stained using the TUNEL reaction mixture in the dark. The cells were then stained with 1 µg/ml DAPI solution for 5 min at room temperature in the dark and observed under a fluorescence microscope. The apoptosis rate was quantified by the TUNEL-positive rate.
Tumorigenicity in nude mice
For tumorigenicity experiments, six-week-old female BALB/cSlc-nude mice were injected subcutaneously with 5.5×106 SNU-484(GDI2-7/14-3-3σ-2) or SNU-484(GDI2-7/Mock) cells. When tumors measured an average volume of 50 mm3, the mice (12 per group) were treated with cisplatin (5mg/kg, 2 times a week) or physiological saline for three weeks. Tumors were measured with calipers to estimate volume(0.5×width2×length). All animal experiments were approved by the Institutional Animal Care and Use Committees (IACUC) of Gyeongsang National University and followed National Research Council Guidelines.
Invasion and migration assay
The invasion ability of cancer cells was assessed using a matrigel-based transwell system. Briefly, 24-well cell culture plate inserts with 8-μm pore size polycarbonate membrane (Corning, NY, USA) were precoated with 100 μl matrigel/DMEM solution (2.2 mg/ml, BD Bioscience, Bedford, MA) and incubated at 37oc for 2 h or overnight at 4oc. All the cells were preincubated in serum-free media with or without inhibitors for 24 h. 2.5 × 105 cells in 250 µl of medium (no serum) were placed in the insert and allowed to invade for 48 h. The lower chamber was filled with 750 µl of appropriated media containing 20% FBS. After incubation, medium remaining on top of the insert were removed by pipetting and non-invading cells on the upper surface of the insert membrane were removed with cotton swab. After washing twice with PBS, the insert membranes were fixed for 10 min with MeOH/Acetic acid (3:1) at -20oc and stained with 50 µg/ml propidium iodide (SIGMA) for 20 min at 37oc. The upper surface of the insert membrane was gently scrubbed with cotton swab again and washed with distilled water. Membranes were cut and mount on slide glass and the number of invaded cells was counted microscopically at 100-200 × magnification. For wound healing assays, 4.9 × 104 cells in 70 µl of medium were seeded into Culture-Insert (Ibidi, Munich, Germany). After the cells were confluent, to inhibit the effect of cell proliferation, the cells were pretreated with 10 µg/ml mitomycin C (SIGMA) for 2 h, and washed with culture medium. After removal of Culture-Insert, cells were incubated with fresh media and photographs of the migration assay were taken at 0, 5, 15, 20, 25 and 30 h using a phase-contrast microscope with digital camera. The cell migration was quantified by calculating the cell-covered area using WimScratch software (Wimasis, Munich, Germany).
Proliferation assay
The cells were placed in a 6-well plate at a concentration of 3 × 104 cells per well. After incubation for 1 to 4 days, cells were trypsinized and resuspended in 3 ml of appropriate medium. Cell suspensions were centrifuged at 1000 rpm for 5 min. Cell pellets were resuspended in 1 ml of appropriate medium. The viable cells were counted with a hemocytometer after trypan blue staining.
Statistical analysis
We performed statistical analysis using the unipolar, paired Student t-test. The significance of the data was accepted when the P value was less than 0.05.
Funding
This work was supported by grant from Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (NRF-2011-0010805), and National R&D Program for Cancer Control, Ministry of Health, Welfare and Family affairs, Republic of Korea (0820050).
CONFLICT OF INTEREST
Authors have no conflict of interests
References
1 Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011; 61: 69–90.
2 Sakuramoto S, Sasako M, Yamaguchi T, Kinoshita T, Fujii M, Nashimoto A, Furukawa H, Nakajima T, Ohashi Y, Imamura H, Higashino M, Yamamura Y, Kurita A, Arai K; ACTS-GC Group. Adjuvant chemotherapy for gastric cancer with S-1, an oral fluoropyrimidine. N Engl J Med. 2007; 357: 1810-1820.
3 Barocas DA, Clark PE. Bladder cancer. Curr Opin Oncol. 2008; 20: 307-314.
4 Ferraldeschi R, Baka S, Jyoti B, Faivre-Finn C, Thatcher N, Lorigan P. Modern management of small-cell lung cancer. Drugs. 2007; 67: 2135-2152.
5 Kelland L. The resurgence of platinum-based cancer chemotherapy. Nat Rev Cancer. 2007; 7: 573-584.
6 Moehler M, Galle PR, Gockel I, Junginger T, Schmidberger H. The multidisciplinary management of gastrointestinal cancer. Multimodal treatment of gastric cancer. Best Pract Res Clin Gastroenterol. 2007; 21: 965-981.
7 Yuan J N, Chao Y, Lee WP, Li CP, Lee RC, Chang FY, Yen SH, Lee SD, Whang-Peng J. Chemotherapy with etoposide, doxorubicin, cisplatin, 5-fluorouracil, and leucovorin for patients with advanced hepatocellular carcinoma. Med Oncol. 2008; 25: 201-206.
8 Wagner AD, Grothe W, Haerting J, Kleber G, Grothey A, Fleig WE. Chemotherapy in advanced gastric cancer: a systematic review and meta-analysis based on aggregate data. J Clin Oncol. 2006; 24: 2903-2909.
9 Price TJ, Shapiro JD, Segelov E, Karapetis CS, Pavlakis N, Van Cutsem E, Shah MA, Kang YK, Tebbutt NC. Management of advanced gastric cancer. Expert Rev Gastroenterol Hepatol 2012; 6: 199-208.
10 Andrews PA, Jones JA, Varki NM, Howell SB. Rapid emergence of acquired cis-diamminedichloroplatinum(II) resistance in an in vivo model of human ovarian carcinoma. Cancer Commun. 1990; 2: 93-100.
11 Saka M, Katai H, Fukagawa T, Nijjar R, Sano T. Recurrence in early gastric cancer with lymph node metastasis. Gastric Cancer. 2008; 11: 214-218.
12 Heasman SJ, Ridley AJ. Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat Rev Mol Cell Biol. 2008; 9: 690–701.
13 Jaffe AB, Hall A. Rho GTPases: biochemistry and biology. Annu Rev Cell Dev Biol. 2005; 21: 247–269.
14 Cho HJ, Baek KE, Yoo J. RhoGDI2 as a therapeutic target in cancer. Expert Opin Ther Targets. 2010; 14: 67-75.
15 Zhang Y, Rivera Rosado LA, Moon SY, Zhang B. Silencing of D4-GDI inhibits growth and invasive behavior in MDA-MB-231 cells by activation of Rac-dependent p38 and JNK signaling. J Biol Chem. 2009; 284: 12956–12965.
16 Moissoglu K, McRoberts KS, Meier JA, Theodorescu D, Schwartz MA. Rho GDP dissociation inhibitor 2 suppresses metastasis via unconventional regulation of Rho GTPases. Cancer Res. 2009; 69: 2838–2844.
17 Gildea JJ, Seraj MJ, Oxford G, Harding MA, Hampton GM, Moskaluk CA, Frierson HF, Conaway MR, Theodorescu D. RhoGDI2 is an invasion and metastasis suppressor gene in human cancer. Cancer Res. 2002; 62: 6418-6423.
18 Theodorescu D, Sapinoso LM, Conaway MR, Oxford G, Hampton GM, Frierson HF Jr. Reduced expression of metastasis suppressor RhoGDI2 is associated with decreased survival for patients with bladder cancer. Clin Cancer Res. 2004; 10: 3800-3806.
19 Tapper J, Kettunen E, El-Rifai W, Seppälä M, Andersson LC, Knuutila S. Changes in gene expression during progression of ovarian carcinoma. Cancer Genet Cytogenet. 2001; 128: 1-6.
20 Zhang Y, Zhang B. D4-GDI, a Rho GTPase regulator, promotes breast cancer cell invasiveness. Cancer Res. 2006; 66: 5592-5598.
21 Cho HJ, Baek KY, Park SM, Kim IK, Choi YL, Cho HJ, Nam IK, Hwang EM, Park JY, Han JY, Kang SS, Kim DC, Lee WS, Lee MN, Oh GT, Kim JW et al. RhoGDI2 expression is associated with tumor growth and malignant progression of gastric cancer. Clin Cancer Res. 2009; 15: 2612-2619.
22 Cho HJ, Baek KE, Park SM, Kim IK, Nam IK, Choi YL, Park SH, Im MJ, Choi J, Ryu J, Kim JW, Lee CW, Kang SS, Yoo J. RhoGDI2 confers gastric cancer cells resistance against cisplatin-induced apoptosis by upregulation of Bcl-2 expression. Cancer Lett. 2011; 311: 48-56.
23 Cho HJ, Baek KE, Kim IK, Park SM, Choi YL, Nam IK, Park SH, Im MJ, Yoo JM, Ryu KJ, Oh YT, Hong SC, Kwon OH, Kim JW, Lee CW, Yoo J. Proteomics-based strategy to delineate the molecular mechanisms of RhoGDI2-induced metastasis and drug resistance in gastric cancer. J Proteome Res. 2012; 11: 2355-2364.
24 Sang M, Li Y, Ozaki T, Ono S, Ando K, Yamamoto H, Koda T, Geng C, Nakagawara A. p73-dependent induction of 14-3-3sigma increases the chemo-sensitivity of drug-resistant human breast cancers. Biochem Biophys Res Commun. 2006; 347: 327-333.
25 Zheng G, Xiong Y, Yi S, Zhang W, Peng B, Zhang Q, He Z. 14-3-3σ regulation by p53 mediates a chemotherapy response to 5-fluorouracil in MCF-7 breast cancer cells via Akt inactivation. FEBS Lett. 2012; 586: 163-168.
26 Satoh J, Tabunoki H, Nanri Y, Arima K, Yamamura T. Human astrocytes express 14-3-3 sigma in response to oxidative and DNA-damaging stresses. Neurosci Res. 2006; 56: 61-72.
27 Feng XP, Yi H, Li MY, Li XH, Yi B, Zhang PF, Li C, Peng F, Tang CE, Li JL, Chen ZC, Xiao ZQ. Identification of biomarkers for predicting nasopharyngeal carcinoma response to radiotherapy by proteomics. Cancer Res. 2010; 70: 3450-3462.
28 Abbracchio MP, Camurri A, Ceruti S, Cattabeni F, Falzano L, Giammarioli AM, Jacobson KA, Trincavelli L, Martini C, Malorni W, Fiorentini C. The A3 adenosine receptor induces cytoskeleton rearrangement in human astrocytoma cells via a specific action on Rho proteins. Ann N Y Acad Sci. 2001; 939: 63-73.
29 Barone I, Brusco L, Gu G, Selever J, Beyer A, Covington KR et al. Loss of Rho GDIα and resistance to tamoxifen via effects on estrogen receptor α. J Natl Cancer Inst. 2011; 103: 538-552.
30 Ronneburg H, Span PN, Kantelhardt E, Dittmer A, Schunke D, Holzhausen HJ, Sweep FC, Dittmer J. Rho GDP dissociation inhibitor alpha expression correlates with the outcome of CMF treatment in invasive ductal breast cancer. Int J Oncol. 2010; 36: 379-386.
31 Zhang B, Zhang Y, Dagher MC, Shacter E. Rho GDP dissociation inhibitor protects cancer cells against drug-induced apoptosis. Cancer Res. 2005; 65: 6054-6062.
32 Sinha P, Hütter G, Köttgen E, Dietel M, Schadendorf D, Lage H. Search for novel proteins involved in the development of chemoresistance in colorectal cancer and fibrosarcoma cells in vitro using two-dimensional electrophoresis, mass spectrometry and microsequencing. Electrophoresis. 1999; 20: 2961-2969.
33 Rong F, Li W, Chen K, Li DM, Duan WM, Feng YZ, Li F, Zhou XW, Fan SJ, Liu Y, Tao M. Knockdown of RhoGDIα induces apoptosis and increases lung cancer cell chemosensitivity to paclitaxel. Neoplasma. 2012; 59: 541-550.
34 Zheng Z, Li J, He X, Chen X, Yu B, Ji J, Zhang J, Wang T, Gu Q, Zhu Z, Liu B. Involvement of RhoGDI2 in the resistance of colon cancer cells to 5-fluorouracil. Hepatogastroenterology. 2010; 57: 1106-1112.
35 Zheng Z, He XY, Li JF, Yu BQ, Chen XH, Ji J, Zhang JN, Gu QL, Zhu ZG, Liu BY. RhoGDI2 confers resistance to 5-fluorouracil in human gastric cancer cells. Oncol Lett. 2013; 5: 255-260.
36 Essmann F, Wieder T, Otto A, Muller EC, Dorken B, Daniel PT. GDP dissociation inhibitor D4-GDI, but not the homologous RhoGDI1, is cleaved by caspase-3 during drug-induced apoptosis. Biochem J. 2000; 346: 777-783.
37 Prasad GL, Valverius EM, McDuffie E, Cooper HL. Complementary DNA cloning of a novel epithelial cell marker protein, HME1, that may be down-regulated in neoplastic mammary cells. Cell Growth Differ. 1992; 3: 507-513.
38 Yaffe MB. How do 14-3-3 proteins work? Gatekeeper phosphorylation and the molecular anvil hypothesis. FEBS Lett. 2002; 513: 53-57.
39 Bridges D, Moorhead GB. 14-3-3 proteins: a number of functions for a numbered protein. Sci STKE. 2005; 2005: re10.
40 Li Z, Liu JY, Zhang JT. 14-3-3sigma, the double-edged sword of human cancers. Am J Transl Res. 2009; 1: 326-340.
41 Lodygin D, Hermeking H. Epigenetic silencing of 14-3-3sigma in cancer. Semin Cancer Biol. 2006; 16: 214-224.
42 Brozovic A, Osmak M. Activation of mitogen-activated protein kinases by cisplatin and their role in cisplatin-resistance. Cancer Lett. 2007; 251: 1-16.
43 Benzinger A, Muster N, Koch HB, Yates JR 3rd, Hermeking H. Targeted proteomic analysis of 14-3-3 sigma, a p53 effector commonly silenced in cancer. Mol Cell Proteomics. 2005; 4: 785-795.
44 Tzivion G, Avruch J. 14-3-3 proteins: active cofactors in cellular regulation by serine/threonine phosphorylation. J Biol Chem. 2002; 277: 3061– 3064.
45 Cho HJ, Baek KE, Nam IK, Park SM, Kim IK, Park SH, Im MJ, Ryu KJ, Yoo JM, Hong SC, Kim JW, Lee CW, Yoo J. PLCγ is required for RhoGDI2-mediated cisplatin resistance in gastric cancer. Biochem Biophys Res Commun. 2011; 414: 575-580.