
Introduction
Nowadays, atrial fibrillation (AF) is the most common arrhythmic condition encountered in clinical practice, producing significant morbidity and mortality via the development of various concurrent diseases and consequences, such as strokes and myocardial infarction [1]. Unfortunately, its prevalence and incidence continue to accelerate, placing a significant financial burden on the healthcare systems worldwide. Current available clinical treatment strategies, such as pharmacological therapy or invasive procedures such as catheter ablation have limited efficacy and are not without adverse effects, especially for patients with persistent AF. As one independent risk factor of AF, diabetes mellitus (DM) is a clinical condition that is becoming increasingly prevalent, in part due to a rising level of obesity, and has been shown to increase atrial arrhythmogenicity through structural and electrophysiological remodeling [2-4]. The exact underlying pathophysiological mechanisms of such remodeling processes are not fully understood [5]. In an alloxan-induced diabetic rabbit model, our group had previously demonstrated that hyperglycemic conditions lead to increased interstitial fibrosis and higher likelihood of AF development[7], as well as reduction of the Na+ current (INa) and increasement of the Ca2+ current (ICaL) in the atria [8]. In the past, it had been demonstrated that an increase in oxidative stress leads to shortened action potential durations (APDs) in the atria due to the elevated transient outward K+ current [10] and promotes Ca2+ release from the intracellular store [11]. Furthermore, reduction of INa current due to reduced mRNA and protein levels was also observed [12]. Together, these changes can lead to both triggered and re-entrant arrhythmogenesis.
Probucol is an anti-hyperlipidemic medication with antioxidant effects by reducing lipid peroxidation and increasing the activities of antioxidant enzymes. Our previous study has demonstrated that probucol exerted anti-inflammatory effects in the atria, through downregulation of a number of key genes involved in the pro-inflammatory pathways, such as NF-κB, TGF-β, HSP70, TNF-α in DM rabbit models [9]. However, electrical remodeling under DM and the impact of probucol have not been studied and reported. In this present study, by using an integrative approach of molecular biology and electrophysiology techniques on well established animal models, we tested the hypothesis that probucol can prevent ion channel remodeling in the atria by its anti-inflammatory and anti-oxidative impact.
Results
Serum oxidative stress parameters
The levels of biochemical and oxidative stress markers in the four experimental groups are illustrated in Table 1. We discovered that DM led to increased levels of cholesterol, triglyceride and LDL-c, as well as higher MPO, and lower SOD and CAT concentrations compared to control (P < 0.05), but not significance in renal function (BUN and creatinine values) (P > 0.05). The probucol treatment reversed the rise in the lipid peroxidation product, malondialdehyde (MDA) in the DM group (P < 0.05).
Table 1: Biochemical and oxidative stress parameters
Control group (n= 10) |
DM group (n= 10) |
DM-P group (n= 10) |
control-P group (n= 10) |
|
Weight, Kg |
2.19±0.17 |
2.29±0.47 |
2.29±0.20 |
2.34±0.08 |
Glu level at 8W,mmol/L |
5.58± 0.75 |
20.86±4.37* |
19.28±8.82* |
6.54±0.52# |
BUN, mmol/L |
5.65±2.00 |
6.87±2.23 |
5.08±1.01 |
4.49±1.40 |
Cr, umol/L |
98.5±16.31 |
103.7±29.22 |
95.13±27.18 |
89.66±9.48 |
TG, mmol/L |
1.19±0.31 |
1.52±1.23 |
1.30±0.29 |
1.25±0.38 |
TC, mmol/L |
1.14±0.57 |
1.25±0.39 |
0.87±0.35 |
0.92±0.36 |
HDL-c, mmol/L |
0.56±0.16 |
0.49±0.20 |
0.43±0.14 |
0.47±0.17 |
LDL-c, mmol/L |
0.36±0.16 |
0.38±0.16 |
0.31±0.15 |
0.24±0.10 |
INS, uIU/ml |
16.24±7.87 |
5.71±3.01* |
7.33±1.56* |
13.89±5.11# |
Serum CAT, U/ml |
6.40±2.46 |
4.79±2.06 |
4.80±0.96 |
5.35±1.74 |
Serum SOD, U/ml |
588.08±82.09 |
556.29±47.7 |
589.18±63.44 |
640.12±82.02 |
Serum MPO, U/L |
63.66±29.44 |
76.41±27.02 |
56.66±20.58 |
64.30±14.35 |
Serum MDA, nmol/ml |
17.69±3.59 |
22.17±4.15* |
18.32±1.63# |
18.14±2.62# |
CAT in atrium, U/mg prot |
4.30±1.29 |
3.82±1.87 |
4.00±1.13 |
4.78±1.33 |
SOD in atrium, U/mg prot |
98.41±12.07 |
72.11± 11.08* |
88.48±8.81# |
93.03±20.04# |
MPO in atrium, U/mg prot |
0.51±0.13 |
1.00±0.38* |
0.78±0.08* |
0.55±0.91# |
MDA in atrium, mol/mg prot |
0.78±0.12 |
1.10±0.24* |
0.84±0.32# |
0.64±0.17# |
Values are mean ± SD; Abbreviations: LA-left atrial; LV-left ventricular; *:Compared with Control group p < 0.05; #: Compared with DM group p < 0.05
INa densities
The average value of cell capacitance is 68.51±10.52 pF (n = 11) in the Control group, 68.91±14.23 pF (n = 10) in the DM group, 67.19±11.17 pF (n = 8) in the DM-P group, and 66.20±9.88 pF (n = 9) in the Control-P group. Figure 1(A) shows INa properties in response to the applied protocol, whereas Figure 1 (B) depicts INa densities and peak INa density as a function of test potential. Compared to the control group, the DM group did not exhibit differing morphology of the I-V curve, but demonstrated lower INa densities at all voltages (–60 mV to +35 mV) (P < 0.05), with a 72.29% reduction in INa density at –40 mV compared to the Control group. Peak INa density in the DM-P group increased by 157.92% compared with the DM group.
Activation and inactivation properties of INa channels
The steady-state INa activation curve is shown in Figure 1(C) and the different activation and inactivation parameters are summarized in Table 2. k is the slope factor of the inactivation curve. The experimental data of the voltage dependent activation were fitted by Boltzmann equation. The membrane potential at which half-activation occurs, V1/2act, was not significantly different between the Control and DM groups (P > 0.05). It was not altered by probucol in either group (P > 0.05). The steady-state INa inactivation curve is shown in Figure 1(D). Together, the above data indicate that neither DM nor probucol modified the voltage-dependent activation or inactivation properties of INa.
Table 2: The shifts in activation and inactivation variables of INa (x±s)
Group |
Control |
DM |
DM-P |
Control-P |
k |
1.49±0.37 |
2.03±0.69 |
1.98±0.28 |
2.63±0.34 |
V1/2 (mV) |
-56.97±0.78 |
-57.01±1.12 |
-54.48±0.74 |
-52.93±0.48 |
V1/2inact(mV) |
5.53±0.52 |
5.20±0.59 |
4.38±0.42 |
4.33±0.31 |
k inact |
-84.00±0.57 |
-77.91±0.70 |
-77.32±0.49 |
-86.86±0.35 |
V1/2act (membrane voltage [Vm] at which half-activation occurs;V1/2inact: Vm at which half-inactivation occurs; k: the activation slope factor; k inact: inactivation slope factor
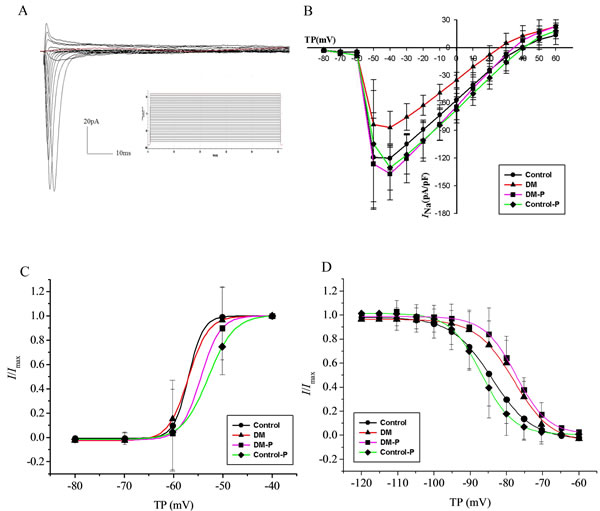
Figure 1: A. representative INa evoked by the applied protocol; B. INa densities and peak INa density as a function of test potential; C. The steady-state activation curve of INa; D. The steady-state inactivation curve of INa, V1/2inact, TP: Test potential.
ICaL changes
The mean cell capacitance was 55.22±10.37pF (n = 12) in the Control group, 57.63±14.49 pF (n = 10) in the DM group, 57.79±14.38 pF (n = 10) in the DM-P group, and 59.57±9.24 pF (n = 13) in the Control-P group. Figure 2(A) shows IcaL properties in response to the applied protocol. Figure 2(B) illustrates IcaL densities and peak IcaL as a function of the test potential. Compared to the Control group, the DM group did not show differing morphology in the I-V curve, but had larger IcaL at all voltages steps tested between –60 mV to +35 mV (P < 0.05). Peak IcaL currents in the DM-P group were reduced by 62.68% compared to the DM group. The maximal IcaL amplitude was between 0 mV and 10 mV in all four experimental groups.
Activation characteristics of ICaL in isolated atrial myocytes
The steady-state IcaL activation curve is shown in Figure 2 (C), in which the data were fitted by Boltzmann equation. V1/2act was not significantly different between the Control and DM groups (P > 0.05). Probucol increased V1/2act compared to the Control or DM groups (Control-11.21±0.51mv, DM-11.63±0.49mv, DM-P-5.91±0.71mv, Control-P-10.69±0.56mv, P < 0.05). A right shift in the activation curve was observed in the DM-P group, i.e. a given value of Vm the degree of current activation was smaller in the DM-P group. k in four groups are Control 4.67±0.49, DM 4.50±0.46, DM-P 5.37±0.57, Control-P 5.00±0.54, respectively, P > 0.05.
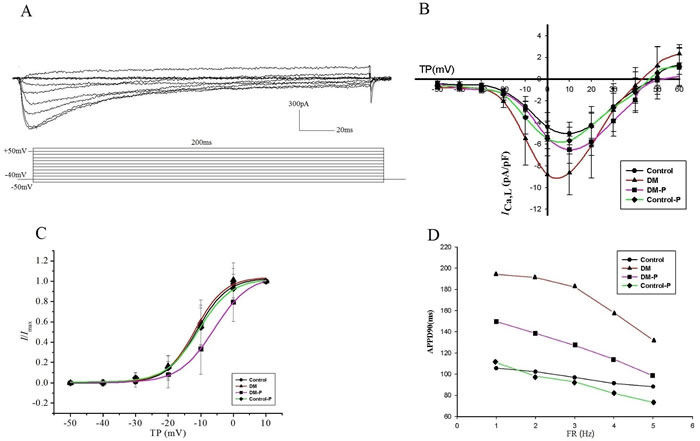
Figure 2: A. representative IcaL evoked by the applied protocol; B. IcaL densities and peak IcaL density as a function of test potential, TP: Test potential; C. The steady-state activation curve of IcaL. TP: Test potential; D. Rate-dependent APD90 changes.
Properties of the cellular action potential
The resting membrane potential was not altered in DM group with an average of −68.41±6.04 mV (n = 12 cells) in control rabbits compared with −67.70±6.20 mV (n = 12), −64.26±6.94 mV (n = 13), and −64.14±5.28 mV (n = 10) in DM, DM-P, and Control-P rabbits, respectively (P > 0.05). APD90 was shortened with increasing stimulation frequencies in all four experimental groups (Figure (2D)). Prolonged APD90 and APD50 values in the DM group were observed compared to the Control group; these changes were abolished by probucol (Figure 3, Table 3). Mean APD, AP amplitude and maximum upstroke velocity are displayed in DM did not significantly affect rate-dependent APD properties.
Table 3: Mean values of APD90 and APD50(x±s)
Control (n= 12) |
DM(n= 12) |
DM-P (n= 13) |
Control-P(n= 10) |
|
RP(mv) |
-78.41±6.04 |
-77.70±6.20 |
-74.26±6.94 |
-74.14±5.28 |
APD90-1Hz |
105.69±29.77 |
194.34±45.47* |
149.75±52.24# |
110.93±23.37# |
APD90-2Hz |
102.34±26.39 |
191.19±42.04* |
138.62±48.76# |
98.08±30.55# |
APD90-3Hz |
96.92±25.01 |
182.40±40.93* |
127.25±43.13# |
92.95±30.41# |
APD90-4Hz |
91.38±23.33 |
158.28±16.09* |
114.11±35.16# |
81.95±26.03# |
APD90-5Hz |
88.27±22.2 |
132.21±11.62* |
98.51±25.32# |
73.62±25.11# |
APD50-1Hz |
35.26±20.34 |
69.65±24.02* |
70.22±35.90* |
44.50±11.53 |
APD50-2Hz |
29.80±13.57 |
64.10±20.15* |
57.71±25.89* |
35.21±13.17 |
APD50-3Hz |
27.97±8.14 |
182.40±40.93* |
46.32±12.04# |
30.26±10.76# |
APD50-4Hz |
24.50±6.23 |
63.05±28.62* |
44.63±12.39 |
24.27±11.23 |
APD50-5Hz |
20.51±5.15 |
55.83±28.40* |
37.62±9.59# |
22.06±10.68 |
AP-amplitude(mv) |
204.96±20.14 |
134.54±24.58* |
134.69±14.01* |
188.85±14.79# |
APA (mV) |
204.96±20.14 |
134.54±24.58* |
134.69±14.01* |
188.85±14.79# |
Vmax (V/s) |
81.44±7.42 |
42.37±10.64* |
52.23±9.39*# |
82.31±7.74# |
Abbreviations: RP: resting potential ;APA: action potential amplitude; Vmax: maximum rate of rise of the action potential; APD50, APD90: action potential duration at 50 and 90% repolarization; * Compared with Control group P < 0.05; # Compared with DM group P < 0.05
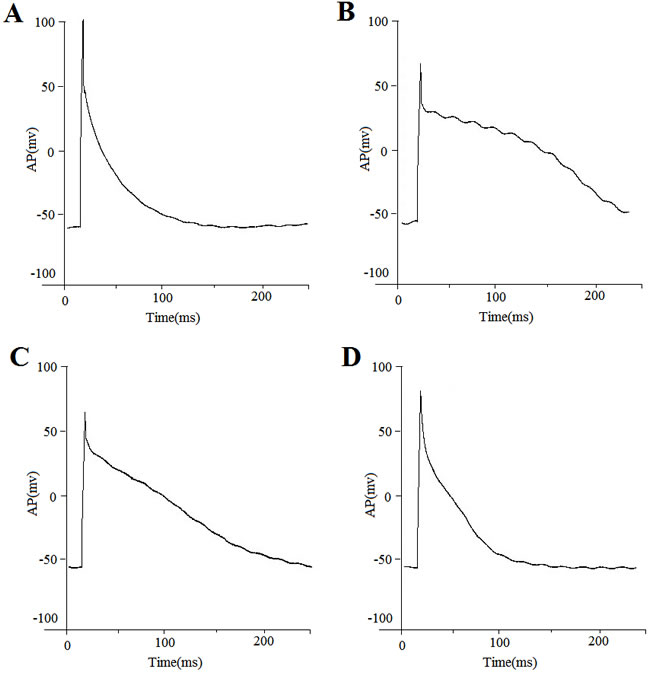
Figure 3: Action potential recordings from representative cells obtained from a A. control group; B. DM group; C. DM-P group and D. Control-P group. Action potentials were recorded in current-clamp mode at the frequencies indicated.
Discussion
In this study, we demonstrated: 1) diabetes reduced INa densities in atrial tissue, while leaving V1/2act or kact of INa intact; 2) Whilst diabetes did not alter the morphology of the IcaL I-V curve, it increased IcaL densities. Probucol prevented the reduction of INa densities and the increase in IcaL densities, increased V1/2act and produced a right shift in the activation curve, and IcaL was smaller at any given value of Vm compare with the DM group; 3) DM prolonged APD90 and APD50, and these changes were abolished by probucol.
Effects of diabetes mellitus and probucol on INa
Many isoforms of the voltage-gated Na+ channels have been described in many current literatures and the predominant isoform was found in cardiac tissue. It is known that Nav1.5 is encoded by SCN5A and INa activation contributes to the upstroke of cardiac AP [15]. Our previous experiments have shown that increased atrial interstitial fibrosis and higher vulnerability to AF was evident under hyperglycemic conditions in a DM rabbit model induced by alloxan [8]. These pathological changes were associated with higher expression levels of several proteins involved in inflammation, including transforming growth factor-β (TGF-β), TNF-α and NF-κB [7-9]. Moreover, prolonged APD90 and APD50 values in isolated left atrial cardiomyocytes from diabetic rabbit hearts were demonstrated, and these changes were associated with INa downregulation and ICaL upregulation [8]. We argue the ion channel remodeling can be explained by altered cellular signaling, such as protein kinase C (PKC) activation [16-17]. The activity of the cardiac Na+ channel can be regulated by post-translational modification such as phosphorylation events, through increased protein kinase A (PKA) and PKC [18]. This reduction in INa can be prevented by the antioxidant enzyme SOD or a PKC inhibitor. PKCα-mediated phosphorylation of the Na+ channel has been reported to result in immediate reduction of Nav1.5 channels [19]. Also, previous studies suggest that PKC decreases INa, but this was not due to altered channel gating mechanisms [20]. Instead, this may involve alterations in transcriptional activity of the channel. In supporting of this notion, the promoter region of the SCN5a gene has a consensus NF-κB binding site [21]. Indeed, angiotensin II, which is elevated in cardiometabolic disorders, can lower Na+ channel expression mediated by higher levels of oxidative stress from H2O2. The latter can promote NF-κB binding to the Na+ channel promoter in turn leading to a decrease in Na+ channel expression in the plasma membrane [22]. Therefore, multiple interacting molecular mechanisms can alter the activity and expression of Na+ channels to mediate adverse electrophysiological remodeling.
Probucol is a potent antioxidant compound that has promising therapeutic effects in patients with DM in randomized clinical trials. It is an anti-hyperlipidemic agent that can reduce redox imbalances by reducing myocardial lipid peroxidation and increasing the antioxidant system [23, 24] and by downregulating a number of key pro-inflammatory mediators such as NF-κB, TGF-β, HSP70 and TNF-α in a diabetes rabbit model produced by alloxan [9].Therefore, it is likely that probucol exerts multiple effects on Na+ channel during pathophysiological conditions through reduction in NF-κB- and PKC-mediated oxidative stress.
Effects of diabetes mellitus and probucol on Ica and action potential
Voltage-gated Ca2+ channels (VGCCs) modulated by cytosolic Ca2+ determine myocardial contractility and APDs in cardiac tissue. Normally, VGCCs are closed during electrical diastole and activated by membrane depolarization, and it contributes a significant amount of inward current during the plateau of the APs. This inward Ca2+ influx is responsible for inducing further release from the intracellular store, the sarcoplasmic reticulum, leading to contractile activation [25].
Previous reports found reduced expression of VGCCs and SERCA2 in AF patients [26-30]. However, the role of VGCCs in postoperative AF is less clear. Van Wagoner et al [31] observed that higher ICa,L in cardiomyocytes isolated from the right atria in postoperative AF patients compared to those without AF, but a different study found no significant difference in ICa,L or APD in these two groups [32]. We believe that these conflicting results are likely due to different baseline characteristics of the subjects involved and different methods of detecting AF [33]. In our study, prolongations in both APD90 and APD50 values were observed in left atrial cardiomyocytes isolated from diabetic rabbit hearts. These changes were linked to higher ICa,L densities. These findings are consistent with those by Van Wagoner et al [31]. Prophylactic treatment using calcium channel antagonists can reduce or prevent alterations in ion channel expression in the diabetic atria [34, 35]. Since calcium overload is an important mechanism in mediating electrical instability [36], we hypothesized that higher ICa in DM is responsible for this observation. AF can be caused by triggered activity or by re-entry [37-39]. EADs develop in the context of APD prolongation, when inward currents (Ica.L or late Na+ current (INaL)) are increased, or when outward currents (IK+) decreased [46, 47]. The causal relationship between abnormal Ca2+ handling and AF development is bidirectional [32]. Thus, hydrogen peroxide (H2O2), a reactive oxygen species, can alter molecular signaling mechanisms that ultimately lead to altered cellular calcium homeostasis [47]. The data generated from our group demonstrate that through a reduction in oxidative stress, as shown using the anti-oxidant compound probucol, can prevent or reverse electrophysiological remodeling in the atria [9].
materials and Methods
This study was approved by the Experimental Animal Administration Committee of Tianjin Medical University and Tianjin Municipal Commission for Experimental Animal Control, which follow the guidelines established by the U.S. National Institutes of Health.
Experimental animals
Forty Japanese rabbits, weighted ~1.5–2.0 kg at the beginning of the study (Beijing Medical Animals Research Institute, Beijing, China), were randomly assigned to control group (Control, n = 10), alloxan-induced diabetic group (DM, n = 10), probucol-treated group (Control-P, n = 10), and probucol-treated diabetic group (DM-P, n = 10). In the DM and DM-P groups, alloxan monohydrate (Sigma ldrich Chemical, Saint Louis, MO, USA) was dissolved in sterile sodium chloride to achieve a concentration of 5% (W/V) and 120 mg/kg, and then was immediately administered into the marginal ear vein. The diabetic state was confirmed 48 hours later by measuring blood glucose levels of ≥14mmol/L. Rabbits in the DM-P and Control-P groups received probucol (Lunan Pharmaceutical Company, Shandong, China) orally (1,000 mg/day) for 8 weeks. The animals in the four groups were housed in cages on a standard laboratory pellet diet during the 8 week experimental period.
Serum biochemical and oxidative stress parameters
Serum cholesterol, triglyceride, low density lipoprotein cholesterin (LDL-c), high density lipoprotein cholesterol (HDL-c), fasting insulin blood urea nitrogen (BUN), and creatinine (Cr) levels were measured using automated techniques. Antioxidant enzyme activities levels in serum and left atrial tissue, including superoxide dismutase (SOD), catalase (CAT), myeloperoxidase (MPO) and malondialdehyde (MDA), were detected using antioxidant enzyme activities kits (Nanjing Jianchen Bioengineering Institute, Jiangsu province, China).
Single-cell electrophysiology
Cell isolation
The left atrium (LA) was used for isolation of single cardiomyocytes. The procedure for isolating cells was followed from a previously developed method [13]. A rabbit was anesthetized with 3% Pelltobarbitalum Natricum (30 mg/kg), and then the rabbit’s heart was quickly removed from the torso and was placed in cold perfusion fluid (4 °C). The aorta was cannulated and connected to a Langendorff perfusion system filled with warmed (37 ±0.5 °C) Ca2+-free Tyrode’s solution. Next, the heart was perfused at 30ml/min for 20 minutes, followed by a 20-minute perfusion containing collagenase (0.075%, CLS II, Worthington Biochemical, Lakewood, CO, USA) and 0.2% bovine serum albumin (BSA; Sigma Chemical Co., St. Louis, MO, USA). Cardiac tissue became opaque and mottled in appearance. Tissue from a well-perfused region of the LA free wall was removed using forceps, gently triturated, and maintained at room temperature in a high-K+ storage solution (KB solution). A small aliquot of the solution containing isolated cells was placed in a 2-mL chamber mounted on the stage of an inverted microscope (IX-50, Olympus Co., Tokyo, Japan). Five minutes following cell adhesion to the bottom of the chamber, cells were superfused with Tyrode’s solution (3 mL/min) for 5 minutes, followed by extracellular solution for the recording of INa or IcaL at the same velocity and time of flow. Recordings of INa and IcaL were obtained 5 minutes following membrane rupture.
Data acquisition
Membrane currents were measured at room temperature (22±1oC) using the whole-cell configuration of the patch-clamp technique [14]. Briefly, whole-cell configuration was made in Tyrode’s solution. Pipette resistances were 2-4 mV. After achieving a gigaseal, the test-pulse current was nulled by adjusting the pipette capacitance compensator with both fast and slow components. After break-in, the whole-cell charging transient was nulled by adjusting whole cell capacitance and series resistance. Voltage control protocols were generated with Axopatch 200B amplifier/Digidata 1200B acquisition system using pCLAMP-10 software (Molecular Devices/Axon, Sunnyvale,CA). Whole-cell recording was analyzed using Clampfit 10.2. INa, ICaL and action potentials (APs) were recorded and filtered at 0.5 kHz using a low pass filter. The sampling frequency was set at 0.5 Hz for recording INa and 0.2 Hz for ICaL. The current-voltage relation was determined in the extracellular solution over a voltage range of -80 to +60 mV increased in 10-mV steps from a hold potential (HP) of -90mV for INa and -40 to +60 mV increased in 10-mV steps from a HP of -50mV for ICaL. The APs were recorded at 60, 120, 180, 240 and 300 bpm. APD was stabilized within 5 action potentials at each cycle length, and was measured at 50% (APD50) and 90% (APD90) of full repolarization.
Statistical analysis
All results are presented as mean ± standard deviation. Comparisons between the groups were analyzed for statistical significance using the one-way analysis of variance (ANOVA) and Fisher’s exact test, respectively. Differences with P < 0.05 were considered statistically significant. In order to ensure data validity from voltage clamp studies, similar numbers of cells from different regions of each heart were studied utilizing the same protocol (i.e., cells were distributed evenly across rabbits).
Acknowledgments and funding
This work was supported by grants (30900618, 81270245, 81570298 to T.L.) from the National Natural Science Foundation of China, Tianjin Natural Science Foundation (16JCYBJC25000 to H.F., 16JCZDJC34900 to T.L.), the Croucher Foundation of Hong Kong (Diabetes Project Grant and Clinical Assistant Professorship to G.T.) and the Health Research Council of New Zealand (J.Z.).
CONFLICTS OF INTEREST
All authors declare no potential conflicts of interest.
References
1. Chugh SS, Havmoeller R, Narayanan K, Singh D, Rienstra M, Benjamin EJ, Gillum RF, Kim YH, McAnulty JH Jr, Zheng ZJ, Forouzanfar MH, Naghavi M, Mensah GA, Ezzati M, and Murray CJ. Worldwide epidemiology of atrial fibrillation: a Global Burden of Disease 2010 Study. Circulation. 2014; 129:837-847.
2. Movahed MR, Hashemzadeh M, and Jamal MM. Diabetes mellitus is a strong, independent risk for atrial fibrillation and flutter in addition to other cardiovascular disease. Int J Cardiol. 2005; 105:315-318
3. Watanabe H, Tanabe N, Watanabe T, Darbar D, Roden DM, Sasaki S, and Aizawa Y. Metabolic syndrome and risk of development of atrial fibrillation: the Niigata preventive medicine study. Circulation. 2008;117:1255-1260.
4. Nichols GA, Reinier K, and Chugh SS. Independent contribution of diabetes to increased prevalence and incidence of atrial fibrillation. Diabetes Care. 2009; 32:1851-1856.
5. Zhang Q, Liu T, Ng CY, and Li G. Diabetes mellitus and atrial remodeling: mechanisms and potential upstream therapies. Cardiovasc Ther. 2014; 32:233-241.
6. Goette A, Honeycutt C, and Langberg JJ. Electrical remodeling in atrial fibrillation. Time course and mechanisms. Circulation.1996; 94:2968-2974.
7. Liu T, Zhao H, Li J, Korantzopoulos P, and Li G. Rosiglitazone attenuates atrial structural remodeling and atrial fibrillation promotion in alloxan-induced diabetic rabbits. Cardiovasc Ther. 2014; 32:178-183.
8. Liu C, Fu H, Li J, Yang W, Cheng L, Liu T, and Li G. Hyperglycemia aggravates atrial interstitial fibrosis, ionic remodeling and vulnerability to atrial fibrillation in diabetic rabbits. Anadolu Kardiyol Derg .2012; 12:543-550.
9. Fu H, Li G, Liu C, Li J, Wang X, Cheng L, and Liu T. Probucol Prevents Atrial Remodeling by Inhibiting Oxidative Stress and TNF-α/NF-κB/TGF-β Signal Transduction Pathway in Alloxan-induced Diabetic Rabbits. J Cardiovasc Electrophysiol. 2015, 26:211-222.
10. Sridhar A, Nishijima Y, Terentyev D, Khan M, Terentyeva R, Hamlin RL, and Nakayama T. Chronic heart failure and the substrate for atrial fibrillation. Cardiovasc Res.2009; 84:227-236.
11. Vest JA1, Wehrens XH, Reiken SR, Lehnart SE, Dobrev D, Chandra P, Danilo P, Ravens U, Rosen MR, and Marks AR. Defective cardiac ryanodine receptor regulation during atrial fibrillation. Circulation. 2005; 111:2025-2032.
12. Shang LL, Sanyal S, Pfahnl AE, Jiao Z, Allen J, Liu H, and Dudley SC Jr. NF-kappaB-dependent transcriptional regulation of the cardiac scn5a sodium channel by angiotensin II. Am J Physiol Cell Physiol. 2008; 294:C372-379.
13. Li D, Melnyk P, Feng J, Wang Z, Petrecca K, Shrier A, and Nattel S. Effects of experimental heart failure on atrial cellular and ionic electrophysiology. Circulation. 2000; 101:2631-2638
14. Karmazínová M, and Lacinová L. Measurement of cellular excitability by whole cell patch clamp technique. Physiol Res. 2010; 59 Suppl 1: S1-7.
15. Zimmer T, Haufe V, and Blechschmidt S. Voltage-gated sodium channels in the mammalian heart. Glob Cardiol Sci Pract. 2014; 2014:449-463.
16. Xin X, Khan ZA, Chen S, and Chakrabarti S. Extracellular signal-regulated kinase (ERK) in glucose-induced and endothelin-mediated fibronectin synthesis. Lab Invest. 2004; 84:1451-1459.
17. Khan ZA, Barbin YP, Farhangkhoee H, Beier N, Scholz W, and Chakrabarti S.Glucose-induced serum- and glucocorticoid-regulated kinase activation in oncofetal fibronectin expression. Biochem Biophys Res Commun. 2005; 329:275-280.
18. Zhou J, Yi J, Hu N, George AL Jr, and Murray KT. Activation of protein kinase A modulates trafficking of the human cardiac sodium channel in Xenopus oocytes. Circ Res. 2000; 87:33-38.
19. Shin HG, and Murray KT. Conventional protein kinase C isoforms and cross-activation of protein kinase A regulate cardiac Na+ current. FEBS Lett .2001; 495:154-158.
20. Yanagita T, Kobayashi H, Yamamoto R, Kataoka H, Yokoo H, Shiraishi S, Minami S, Koono M, and Wada A. Protein kinase C-alpha and -epsilon down-regulate cell surface sodium channels via differential mechanisms in adrenal chromaffin cells. J Neurochem. 2000; 74:1674-1684.
21. Shang LL, and Dudley SC Jr. Tandem promoters and developmentally regulated 5’- and 3’-mRNA untranslated regions of the mouse Scn5a cardiac sodium channel. J Biol Chem. 2005, 280: 933-940.
22. Shang LL, Sanyal S, Pfahnl AE, Jiao Z, Allen J, Liu H, and Dudley SC Jr. NF-kappaB-dependent transcriptional regulation of the cardiac scn5a sodium channel by angiotensin II.Am J Physiol Cell Physiol. 2008;294:C372-937.
23. Li T, Chen W, An F, Tian H, Zhang J, Peng J, Zhang Y,and Guo Y. Probucol attenuates inflammation and increases stability of vulnerable atherosclerotic plaques in rabbits. Tohoku J Exp Med. 2011; 225:23-34.
24. Colle D, Santos DB, Moreira EL, Hartwig JM, dos Santos AA, Zimmermann LT, Hort MA, and Farina M. Probucol increases striatal glutathione peroxidase activity and protects against 3-nitropropionic acid-induced pro-oxidative damage in rats.PLoS One. 2013; 8:e67658.
25. Richard S, Leclercq F, Lemaire S, Piot C,and Nargeot J. Ca2+ currents in compensated hypertrophy and heart failure.Cardiovasc Res.1998;37:300-311.
26. Lai LP, Su MJ, Lin JL, Lin FY, Tsai CH, Chen YS, Huang SK, Tseng YZ, and Lien WP.Down-regulation of L-type calcium channel and sarcoplasmic reticular Ca(2+)-ATPase mRNA in human atrial fibrillation without significant change in the mRNA of ryanodine receptor, calsequestrin and phospholamban: an insight into the mechanism of atrial electrical remodeling. J Am Coll Cardiol.1999;33:1231-1237.
27. Ohkusa T, Ueyama T, Yamada J, Yano M, Fujumura Y, Esato K, Matsuzaki M. Alterations in cardiac sarcoplasmic reticulum Ca2+ regulatory proteins in the atrial tissue of patients with chronic atrial fibrillation. Am Coll Cardiol.1999; 34:255-63.
28. Grammer JB, Zeng X, Bosch RF, and Kühlkamp V. Atrial L-type Ca2+-channel, beta-adrenorecptor, and 5-hydroxytryptamine type 4 receptor mRNAs in human atrial fibrillation.Basic Res Cardiol. 2001; 96:82-90.
29. van der Velden HMW, van der Zee L, Wijffels MC, van Leuven C, Dorland R, Vos MA, Jongsma HJ,and Allessie MA. Atrial fibrillation in the goat induces changes in monophasic action potential and mRNA expression of ion channels involved in repolarization.J Cardiovasc Electrophysiol. 2000;11:1262-1269.
30. Grammer JB, Bosch RF, Kühlkamp V,and Seipel L. Molecular and electrophysiological evidence for “remodeling” of the L-type Ca2+ channel in persistent atrial fibrillation in humans.Z Kardiol. 2000; 89 Suppl 4:IV23-9.
31. Van Wagoner DR, Pond AL, Lamorgese M, Rossie SS, McCarthy PM,and Nerbonne JM. Atrial L-type Ca2+ currents and human atrial fibrillation.Circ Res.1999; 85:428-436.
32. Workman AJ, Pau D, Redpath CJ, Marshall GE, Russell JA, Kane KA, Norrie J, and Rankin AC. Post-operative atrial fibrillation is influenced by beta-blocker therapy but not by pre-operative atrial cellular electrophysiology. J Cardiovasc Electrophysiol.2006; 17:1230-1238.
33. Greiser M, Lederer WJ, and Schotten U. Alterations of atrial Ca(2+) handling as cause and consequence of atrial fibrillation.Cardiovasc Res. 2011;89:722-733.
34. Tieleman RG, De Langen C, Van Gelder IC, de Kam PJ, Grandjean J, Bel KJ, Wijffels MC, Allessie MA, and Crijns HJ. Verapamil reduces tachycardia-induced electrical remodeling of the atria.Circulation. 1997; 95:1945-1953.
35. Daoud EG, Knight BP, Weiss R, Bahu M, Paladino W, Goyal R, Man KC, Strickberger SA, and Morady F. Effect of verapamil and procainamide on atrial fibrillation-induced electrical remodeling in humans. Circulation. 1997; 96:1542-1550.
36. Ravens U, and Himmel HM. Drugs preventing Na+ and Ca2+ overload. Pharmacol Res.1999; 39:167-174.
37. Nattel S. New ideas about atrial fibrillation 50 years on. Nature. 2002; 415:219-226.
38. Wakili R, Voigt N, Kääb S, Dobrev D, and Nattel S. Recent advances in the molecular pathophysiology of atrial fibrillation. J Clin Invest. 2011; 121:2955-2968.
39. Tse G. Mechanisms of Cardiac Arrhythmias. J Arrhythm. 2015; 32(2):75-81.
40. Tse G, Tse V and Yeo JM. Ventricular anti-arrhythmic effects of heptanol in hypokalaemic, Langendorff-perfused mouse hearts. Biomed Rep. 2016; 4:313-324.
41. Tse G, Tse V, Yeo JM, and Sun B. Atrial anti-arrhythmic effects of heptanol in Langendorff-perfused mouse hearts. PLoS One. 2016; 11(2):e0148858.
42. Tse G, Wong ST, Tse V, and Yeo JM. Restitution analysis of alternans using dynamic pacing and its comparison with S1S2 restitution in heptanol-treated, hypokalaemic Langendorff-perfused mouse hearts. Biomed Rep. 2016; 4:673-680.
43. Tse G, Sun B, Wong ST, Tse V, and Yeo JM. Ventricular anti-arrhythmic effects of hypercalcaemia treatment in hyperkalaemic, Langendorff-perfused mouse hearts. Biomed Rep. 2016; 5:301-310.
44. Tse G, Yeo JM, Tse V, Kwan SK, and Sun B. Gap junction inhibition by heptanol increases ventricular arrhythmogenicity by decreasing conduction velocity without affecting repolarization properties or myocardial refractoriness in Langendorff-perfused mouse hearts. Mol Med Rep. 2016; 14:4069-4074.
45. Tse G, Hothi SS, Grace AA, and Huang CL. Ventricular arrhythmogenesis following slowed conduction in heptanol-treated, Langendorff-perfused mouse hearts. J Physiol Sci. 2012; 62:79-92.
46. Song Y, Shryock JC, and Belardinelli L. An increase of late sodium current induces delayed afterdepolarizations and sustained triggered activity in atrial myocytes. Am J Physiol Heart Circ Physiol. 2008; 294:H2031-2039.
47. Barrington PL. Interactions of H2O2, EGTA and patch pipette recording methods in feline ventricular myocytes. J Mol Cell Cardiol. 1994; 26:557-568.
48. Qu Y, Fang M, Gao B, Amagasu S, Crumb WJ, and Vargas HM. Oxytocin does not directly alter cardiac repolarization in rabbit or human cardiac myocytes. Pharmacol Res Perspect.2015; 3:e00102.