
INTRODUCTION
Avian leukosis virus (ALV) is a highly oncogenic alpha-retrovirus of Retroviridae family, causing avian leukosis (AL) in chickens. ALVs can be classified as endogenous (ALV-E) and exogenous (A, B, C, D, and J) based on their mode of transmission, host range, viral envelope interference, and cross-neutralization patterns [1, 2]. ALV-J was first isolated from meat-type chickens in 1988 [2], and has primarily been associated with myeloid leukosis (ML) in broiler breeders [3, 4]. In recent years, however, various tumors including hemangiomas induced by ALV-J have emerged among parent and commercial layer flocks [5, 6], leading to enormous economic losses, and indicating an evolution of ALV-J oncogenicity.
Tumorigenesis is a complex process caused by a variety of factors. Some viruses contain oncogenes in their genomes; therefore induce tumors via the multiple functions of these oncogenes. However, ALV-J does not carry a viral oncogene. Most studies regarding the ALV-J oncogenicity have focused on the insertional mechanisms of ALV-J, which activates or inactivates the tumor-associated genes of the host [7-11]. However, as ALV integrates in a largely random fashion with only a slight preference for active transcriptional units [12, 13], there must be some other mechanisms for ALV tumorigenicity.
It has been reported that VEGF-A and its receptor, VEGFR-2, are involved in ALV-J tumorigenesis [14]. VEGF is the most important proangiogenic agent that activates receptors on vascular endothelial cells (VECs) and promotes blood vessel regeneration. VEGF and VEGFR have been associated with the pathogenesis of leukemia. The VEGF/VEGFR-dependent pathways regulate angiogenesis, vasculogenesis, and recruitment of endothelial progenitor cells, and have been associated with progression and metastasis of solid tumors [15-17]. Furthermore, VEGF/VEGFR interactions may stimulate proliferation, migration, and survival of leukemia and lymphoma cells via autocrine and paracrine loops [18]. Notably, a previous study has indicated that acute leukemia cells secret large amounts of VEGF into the serum and that malignant hematopoietic cells express VEGF and VEGFRs [19]. We have previously shown that ALV-J infection induces expression of VEGF-A and VEGFR-2. A newly isolated ALV-J strain, with a stronger replication and oncogenesis capability, induced higher expression of VEGF/VEGFR in vascular cells and tissues than other ALV-J strains [14].
The expression of VEGF/VEGFR is associated with interleukin 6 (IL-6) signaling pathways in many cancers, such as breast and intestinal cancers [20, 21]. IL-6 is a multifunctional cytokine with central roles in immune and inflammatory reactions, as well as in cancer development [20-24]. IL-6 plays an important role in host immune system, wherein it has been considered to facilitate elimination of pathogens during virus-host interactions. However, through evolution, viruses have developed a number of strategies to avoid such an outcome and successfully establish chronic infections through hijacking the host immune system [25-27].
Our previous study has demonstrated that ALV-J infection promotes IL-6 expression in vivo in chickens [28]. Here, we tested the role of IL-6 in ALV-J-induced VEGF/VEGFR expression, and examined the underlying mechanisms.
RESULTS
ALV-J promotes IL-6 production in splenocytes, lymphocytes, and VECs
We have previously shown that ALV-J promotes IL-6 expression in vivo [28]; in this study, we have investigated whether ALV-J induces IL-6 production in vitro. Chicken splenocytes, peripheral blood lymphocytes (PBLs), and VECs were infected with ALV-J strain HLJ09SH02. Enzyme linked immunosorbent assay (ELISA) was performed to confirm the successful ALV-J infection in these three cell types (data not shown). Infection with ALV-J strain HLJ09SH02 significantly induced IL-6 mRNA and protein levels in all three cell types (Figure 1). For splenocytes, the expression of IL-6 peaked at 6 h post-infection, with almost a 40-fold higher level compared to controls (p < 0.01). However, at 3, 12, and 24 h post-infection, the infected group showed no significant difference in IL-6 expression compared to control group (Figure 1A and 1B). For PBLs at 3 and 6 h post-infection, the IL-6 levels were similar between infected and control groups. At 12 h post-infection, IL-6 mRNA expression in the infected group was approximately 4-fold higher than in the control group (p < 0.01), with a similar trend exhibited for protein expression (p < 0.01) (Figure 1C and 1D). In VECs, the IL-6 expression differences appeared from 3 h post-infection and were maintained over the following 22 h. The expression of IL-6 mRNA in infected VECs peaked at 12 h post-infection, at a level of almost 3.5-fold higher than in the control cells (p < 0.01). ELISA results showed that IL-6 protein expression exhibited a similar trend (Figure 1E and 1F).
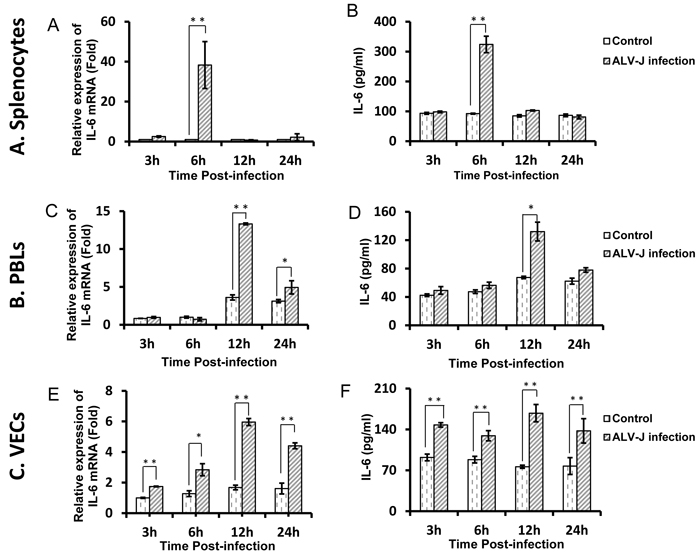
Figure 1: ALV-J promotes IL-6 expression in vitro. A. Splenocytes, B. PBLs, or C. VECs were infected with ALV-J for 3, 6, 12, and 24 h and IL-6 mRNA and protein levels were determined using real-time RT-PCR (1) or ELISA (2).
ALV-J gp85 and p27 proteins promote IL-6 expression
To examine whether the ALV-J-induced IL-6 expression was caused by ALV-J proteins, we analyzed the effect of p27, gp85, integrase, reverse transcriptase, and gp37 on IL-6 levels in splenocytes. Expression vectors of p27, gp85, integrase, reverse transcriptase, and gp37 were constructed, and the protein expression was confirmed by western blotting (Figure 2A). Splenocytes were then transfected with each respective vector. Cells and supernatants were harvested 48 h post-transfection for IL-6 mRNA and protein measurement by real-time reverse transcription-PCR (RT-PCR) and ELISA, respectively. The results showed that only gp85 and p27 increased the production of IL-6, with gp85 playing a more significant role. As shown in Figures 2B and 2C, ALV-J gp85 and p27 proteins increased IL-6 mRNA levels by approximately 8-fold and 4-fold, respectively, (p < 0.01). At protein levels, the increase was smaller, but still significant (p < 0.01). None of the other tested ALV-J proteins were able to increase IL-6 gene expression. These results indicate that ALV-J gp85 and p27 proteins promote the IL-6 expression.
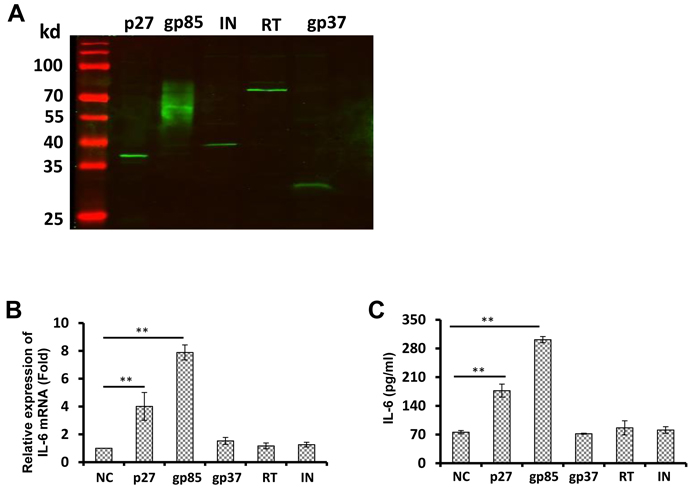
Figure 2: The ALV-J capsid protein p27 promotes IL-6 expression in a dose-dependent manner in splenocytes. A. The expression of p27, gp85, integrase, reverse transcriptase, and gp37 were confirmed by western blot. Splenocytes were transfected with the pCAGGS vector or ALV-J p27, gp85, gp37, reverse transcriptase, or integrase expression vectors for 48 h. Then, IL6 mRNA levels were determined using real-time RT-PCR B. and IL-6 protein levels were determined using ELISA C.
NF-κB and PI3K mediate ALV-J-induced chicken IL-6 expression
To dissect the signaling pathways involved in ALV-J-induced IL-6 production, splenocytes were pretreated for 1 h with inhibitors of key signaling pathways, including NF-κB, MEK, p38 MAPK, PI3K, or PKC prior to ALV-J infection. At 48 h post-infection, IL-6 expression was analyzed by real-time RT-PCR and ELISA. As shown in Figure 3A and 3B, NF-κB inhibitor BAY11-7082, PI3K inhibitor LY294002, and p38 MAPK inhibitor SB203580 significantly inhibited the IL-6 expression after ALV-J infection at both mRNA and protein levels, while inhibition of MEK (PD98059) and PKC (GF-109203X) exhibited no significant effect on the IL-6 production. However, a decrease in IL-6 production was also observed in cells treated with p38 MAPK inhibitor SB203580 without the ALV-J infection. These results suggested that p38 pathway was involved in the constitutive IL-6 expression in splenocytes, but NF-κB and PI3K pathways were specifically involved in the ALV-J-induced IL-6 expression.
To further verify these results and explore the relationship between NF-κB and PI3K pathways, a Dual-glo Luciferase Assay was utilized to test the activity of NF-κB with and without the activation of PI3K pathway. As illustrated in Figure 3C, although ALV-J was able to activate NF-κB on its own, NF-κB activity was decreased when activation of PI3K pathway was inhibited. This result suggested that activation of the PI3K signaling pathway was important for the ALV-J-induced NF-κB activation.
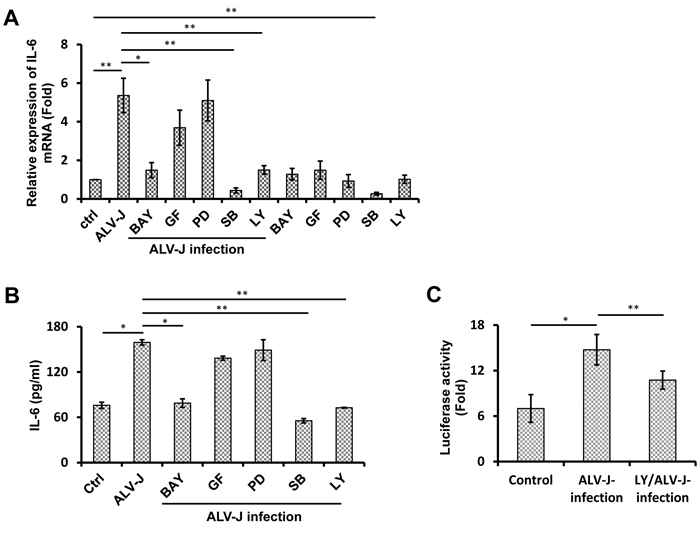
Figure 3: Effect of signal transduction inhibitors on IL-6 expression induced by ALV-J infection. A., B. Splenocytes were pretreated with inhibitors of NF-κB (BAY11-7082 [BAY]), PKC (GF-109203 [GF]), MEK (PD98059 [PD]), MAPK (SB202190 [SB]), PI3K (LY294002 [LY]), or DMSO control for 1 h and then inoculated with or without ALV-J (MOI = 0.1). After 6 h, total RNA was extracted for detecting IL6 mRNA by real-time RT-PCR (A) and the cell culture supernatants were harvested to test IL-6 protein by ELISA B. C. Splenocytes were pretreated with LY or DMSO control for 1 h and then the pretreated splenocytes were transfected with plasmids to test NF-κB activity and infected with ALV-J (MOI = 0.1) or DMEM. After another 6 h, cells were harvested for luciferase analysis.
IL-6 increases VEGF-A and VEGFR-2 expression via signal transducer and activator of transcription 3 (STAT3) signaling pathway
Previous studies have demonstrated that IL-6 serves as a link between inflammation and tumorigenesis, since it induces the expression of VEGF-A/VEGFR-2 [29]. As ALV-J is an important oncogenic virus, it was of interest to determine whether the ALV-J-induced IL-6 induces VEGF-A/VEGFR-2 expression in chickens. IL-6 gene expression vector was transfected into VECs to determine the effect of IL-6 on VEGF-A and VEGFR-2 expression. After 48 h post-transfection, VEGF-A expression was over 2-fold elevated compared to control cells transfected with empty pCAGGS vector (p < 0.01), and VEGFR-2 expression was nearly 1-fold elevated compared to control cells (p < 0.05) (Figure 4A and 4B). These results thus indicate that IL-6 induces VEGF-A and VEGFR-2 expression in VECs in vitro.
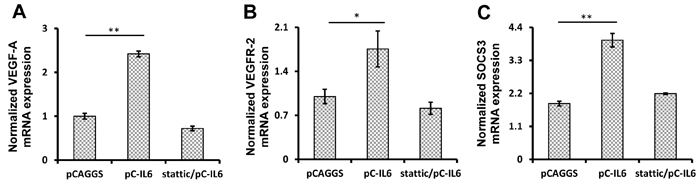
Figure 4: IL-6 promotes VEGF-A and VEGFR-2 expression via STAT3 signaling pathway in VECs in vitro. VECs were pretreated with an inhibitor of STAT3 (stattic) or DMSO control for 1 h. Then, the pretreated VECs were transfected with the pCAGGS vector or IL-6 expression vector for 48 h. Total RNAs were extracted and VEGF-A A., VEGFR-2 B., or SOCS3 C. mRNA expression was detected by real-time RT-PCR.
To validate these results in vivo, embryos were used to test the effect of IL-6 on VEGF-A/VEGFR-2 production. Recombinant IL-6 protein was inoculated into 7-day-old embryos intravenously, and vascular tissues were collected 24, 48, and 72 h post-inoculation. As shown in Figure 5A, VEGF-A expression in the inoculated embryos was significantly increased compared to control embryos 24 h post-inoculation (p < 0.01), and this difference became even more obvious over the following 48 and 72 h post-inoculation (p < 0.01). For VEGFR-2, at 24 h post-inoculation, VEGFR-2 expression showed no difference between inoculated and control embryos. At 48 h post-inoculation, VEGFR-2 expression in the inoculated embryos increased nearly 2-fold compared to control embryos (p < 0.05), with the difference reaching almost 5-fold at 72 h post-inoculation (Figure 5B). These results thus demonstrate that IL-6 increases VEGF-A and VEGFR-2 expression in embryos in vivo.
Previous studies have indicated that STAT3 represents a downstream signaling pathway of IL-6 [29-31]. In order to investigate the signaling pathways involved in the VEGF-A and VEGFR-2 regulation by IL-6, STAT3 inhibitor stattic was used to pretreat VECs prior to IL-6 transfection. The results showed that upon stattic pretreatment, VEGF-A and VEGFR-2 expression in IL-6-transfected VECs was reduced compared to cells transfected with control vectors (Figure 4A and 4B), indicating that STAT3 was involved in the IL-6-induced VEGF-A and VEGFR-2 expression. Suppressor of cytokine signaling 3 (SOCS3) is considered as an indicator of STAT3 activation as it is often over-expressed during STAT3 activation [32-34]. Therefore, to further verify the activation of STAT3, we analyzed the expression of SOCS3 in both VECs and vascular tissues of embryos. Real-time RT-PCR results indicated that upon IL-6 treatment, STAT3 signaling was activated both in vitro and in vivo (Figure 4C and 5C). These results suggest that STAT3 signaling is involved in the IL-6-induced VEGF-A and VEGFR-2 expression.
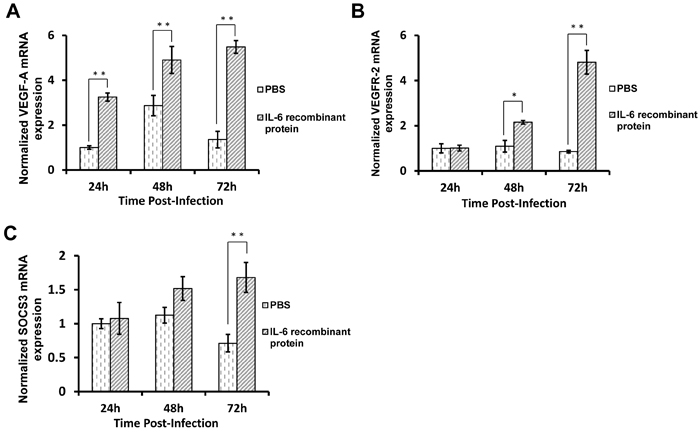
Figure 5: IL-6 promotes VEGF-A and VEGFR-2 expression by activating STAT3 in embryos in vivo. A. Embryos were intravenously inoculated with IL-6 recombinant protein or PBS control. After 24, 48, or 72 h post-inoculation, the vascular tissues of the inoculated embryos were collected and used to extract total RNAs. Then, the levels of VEGF-A A., VEGFR-2 B., and SOCS3 C. mRNA were determined by real-time RT-PCR.
ALV-J induces VEGF-A and VEGFR-2 expression via IL-6
The above results have indicated that ALV-J induces IL-6 expression, which stimulates VEGF-A and VEGFR-2 expression. Since we have previously shown that ALV-J increases VEGF-A and VEGFR-2 expression [14], we next explored whether IL-6 represented the key factor inducing the VEGF-A/VEGFR-2 expression in response to ALV-J infection. VECs transfected with IL-6 siRNA or negative control were infected with ALV-J. IL-6 siRNA-transfected VECs had significantly decreased IL-6 mRNA compared with cells transfected with control siRNA (Figure 6A). Infection with ALV-J increased the VEGF-A expression by approximately 2-fold (p < 0.01) in cells transfected with control siRNA. However, in cells transfected with IL-6 siRNA, ALV-J failed to induce VEGF-A expression (Figure 6B). Quantitative VEGFR-2 mRNA determination showed a similar result (Figure 6C). Together, these results demonstrate that ALV-J induces VEGF-A and VEGFR-2 expression via IL-6 in VECs.
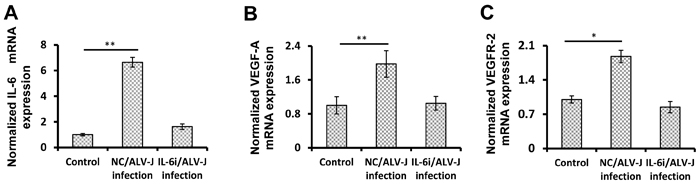
Figure 6: ALV-J increases VEGF-A and VEGFR-2 expression by inducing IL-6 expression. A. VECs were transfected with IL6 siRNA or negative control siRNA for 24 h, and then the transfected VECs were infected with or without ALV-J (MOI = 0.1) for another 48 h. Total RNAs were extracted from the VECs and the mRNA levels of IL6 (to test the effect of the IL6 siRNA) A., VEGF-A B., and VEGFR-2 were determined by real-time RT-PCR.
DISCUSSION
Previous studies have demonstrated that IL-6 induces in vivo tumor growth in prostate, breast, and lung cancers [35], and that elevated IL-6 levels stimulate development of inflammation-associated cancers [36, 37]. As we have found that ALV-J infection promotes IL-6 expression in vivo [28], we hypothesized that IL-6 might be associated with ALV-J pathogenesis. To test this hypothesis, in the present study, we have examined the ability of ALV-J to induce the IL-6 expression in vitro, and we have analyzed the biological impact of the induced IL-6. Our results demonstrate that ALV-J induces VEGF-A and VEGFR-2 expression through activating the IL-6 expression. As VEGF-A and VEGFR-2 are important pro-tumorigenic factors, our results suggest that IL-6 induces ALV-J tumorigenesis through its induction of VEGF-A and VEGFR-2 expression.
We initially demonstrated that ALV-J increased IL-6 expression in vitro. In both splenocytes and PBLs, the induction of IL-6 expression in ALV-J infected cells was brief and transient. However, in VECs, the ALV-J-induced IL-6 expression was persistent and remained increased over time. As splenocytes and PBLs are immune cells, we suspect that there might exist additional immune responses in these cells that regulate the IL-6 expression. In VECs, which do not exhibit pleiotropic immune functions, however, the ALV-J-induced IL-6 increase was continuous. Although the effects of ALV-J appeared to be effectively countered by host immune responses in splenocytes and PBLs, over the course of long-term ALV-J infection in vivo, ALV-J may overcome host immune responses to develop a chronic infection. Such a process might be conditional; e.g., affected by the host immune conditions; which is evinced by the low oncogenicity of ALV-J. However, this assumption requires further investigation.
Envelope and capsid proteins are important proteins for ALV-J infection. Gp85 protein is responsible for recognizing specific virus receptors on host cell membranes, and determines the specificity of different subgroups and host range [38-40]. Notably, it has previously been reported that the HIV envelope protein gp120 increases the production of IL-6 in host immune cells, thereby suppressing the immunity of the host [41]. Similarly, in the current study, we found that the ALV-J envelope protein gp85 also induced the production of IL-6 in chicken splenocytes. In addition, we demonstrated that ALV-J capsid protein p27 could be recognized by splenocytes and stimulate IL-6 expression as a host immune response to ALV-J infection. Capsid protein also plays a major role in virus infections; for example, it has been shown to trigger host innate immune responses to HIV via interacting with tripartite motif protein isoform 5 alpha [42, 43]. In comparison, for ALV, p27 acts as a major group-specific antigen [44] and an indicator of ALV infection [45-48]. A prior study has suggested that ALV p27 is involved in the immunosuppression induced by ALV in chickens [49].
This study also demonstrated that PI3K and NF-κB signaling pathways were involved in the ALV-J-induced IL-6 expression. NF-κB plays a central role in the activation of numerous pro-inflammatory cytokines in multiple cell types, including macrophages, T cells, and epithelial cells [50]. Similarly to our study, Ankit et al. reported that the HIV-1 envelope protein gp120 induced IL-6 expression through an NF-κB-dependent mechanism [41]. Other studies have also indicated that IL-6 expression is regulated via NF-κB [51-53]. Consistent with our results, chicken IL-6 promoter sequence (GenBank: AJ250838.2) contains a putative NF-κB binding site (using TRANSFAC). Thus, we speculate that IL-6 is a target gene for NF-κB. In addition, the PI3K signaling pathway is closely related to tumorigenesis via inhibiting apoptosis [54] and promoting angiogenesis [55]. In this study, inhibition of PI3K or NF-κB signaling suppressed the ALV-J-induced IL-6 production, suggesting that both pathways are essential for IL-6 expression, and that a cross talk might exist between these two pathways.
When investigating the significance of ALV-J-induced IL-6 expression, we first demonstrated that IL-6 transfection was able to promote VEGF-A and VEGFR-2 expression in VECs. IL-6 promotes VEGF-A and VEGFR-2 production in humans via trans-signaling, where soluble and extracellular IL-6 initially binds the surrounding soluble IL-6 receptors and then activates gp130 protein on the surface of target cells to trigger a series of responses [56]. Based on this theory, the embryo assays performed in this study further indicated that the IL-6 present in blood was able to increase VEGF-A and VEGFR-2 expression in vivo. Furthermore, our results indicated that STAT3 was involved in this process. Many studies have indicated that VEGF-A and VEGFR-2 are important for tumor growth in different cancers [57-61]. Previous studies in our laboratory have shown that some ALV-J strains have stronger oncogenicity in hemangiomas than other ALV-J strains, with their higher VEGF-A and VEGFR-2 expression in vascular tissues [62]. However, further studies are required to validate the correlation between VEGF and ALV-J oncogenesis.
In summary, the present study indicates that ALV-J increases IL-6 expression, which then induces VEGF-A and VEGFR-2 production. These results suggest that IL-6 plays a central role in ALV-J-induced VEGF-A and VEGFR-2 expression. IL-6 is a proinflammatory cytokine with important roles in inflammation as well as in innate immunity. Innate immune response provides the first line of defense against invading pathogens, leading to the halt of initial spread of infection and activating adaptive immunity and other secondary defense mechanisms [63, 64]. However, many viruses have evolved to hijack the host immune system to facilitate their own proliferation and invasion, such as human herpesviruses, hepatitis C virus, Epstein Barr virus, and human papillomavirus [26, 27]. Inflammation and immunity have been suggested to affect different stages of cancer development, with inflammation and innate immunity exerting protumorigenic effects [65]. Based on our results, we speculate that ALV-J hijacks IL-6 as a means of its tumorigenicity. These findings provide new insight into the tumorigenic mechanisms of ALV-J and suggest potential targets for the development of prophylactic or treatment strategies to mitigate ALV-J infection and pathogenicity.
MATERIALS AND METHODS
Cells, viruses, and virus preparation
Splenocytes were isolated from the spleens of 21-day-old specific-pathogen-free (SPF) chickens as described previously [66], and maintained in RPMI 1640 supplemented with 10% FBS, penicillin-streptomycin, and pyruvic acid sodium salt. PBLs were isolated from the anticoagulate of 21-day-old SPF chickens and maintained in RPMI 1640 supplemented with 10% FBS, penicillin-streptomycin, and pyruvic acid sodium salt. VECs were isolated from the 14-day-old SPF embryos as described previously [62], and maintained in Dulbecco’s minimum essential medium (DMEM) supplemented with 20% FBS and penicillin-streptomycin.
ALV-J strain HLJ09SH02 was isolated and stored at −70°C in our laboratory and propagated in DF-1 cells as described [67].
Construction and expression of ALV-J structural protein gene expression vectors and IL-6 gene expression vector
The ALV-J genes for structural proteins including p27, gp85, gp37, reverse transcriptase, and integrase of ALV-J strain HLJ13SH01 were amplified by PCR using specific primers and the PCR products were subcloned into the pcAGGS vector (Addgene) with a Flag tag at the C-terminus. All construction vectors were confirmed by sequencing as compared with ALV-J strain HLJ13SH01 sequence, GenBank accession number KM376510.1), and the protein expression of each was confirmed by western blot (Figure 2A). The IL-6 gene was amplified from chicken spleen using specific primers and the PCR product was then subcloned into the pcAGGS vector. All primers used in this study are listed in Table 1. IL-6 expression was confirmed by ELISA (data not shown).
Table 1: Sequences of the primers and probes used in the study.
Name |
Sequences |
p27 forward |
5’-GCCGGTACCATGCCTGTAGTGATTAAGACAGAGG-3’ |
p27 reverse |
5’-CAACTCGAGTCAGGCCGCGGCTATGCCTCG-3’ |
gp85 forward |
5’-GCCATCGATATGGGAGTTCATCTGTTGCA-3’ |
gp85 reverse |
5’-CAACTCGAGTCAGCGCCTGCTACGGCGGT-3’ |
gp37 forward |
5’-GAAGGTACCATGTCGCTGAGTCGTCTCTCGCC-3’ |
gp37 reverse |
5’-GCCCTCGAGTCACAGTTGCTCCCTAATTCTAT-3’ |
IN forward |
5’-GCCATCGATATGCCCTTGAGAGAGGCTAAA-3’ |
IN reverse |
5’-GAACTCGAGTCATGCAAAGAGAGGACTCGC-3’ |
RT forward |
5’-GCCGGTACCATGACTGCTGCGCTACATCTG-3’ |
RT reverse |
5’-GGCCTCGAGTCAATACGCTTGAAAGGTGGC-3’ |
IL-6 forward |
5’-GCCGGTACCGTGTGCAGCGGTTGAACC-3’ |
IL-6 reverse |
5’-CGGCTCGAGAGTTCATGGTGCGGCTTC-3’ |
Abbreviations: IN: integrase; RT: reverse transcriptase
IL-6 mRNA quantification
Splenocytes, PBLs, and VECs were infected with ALV-J strain HLJ09SH02 at a multiplicity of infection (MOI) of 0.1. To confirm successful ALV-J infection, cells infected with ALV-J were collected at 3, 6, 12 and 24h post-infection. Cell lysates were prepared after three freeze-thaw cycles and tested for ALV group specific antigen by an antigen capture enzyme-linked immunosorbent assay with anti-p27 antibody-coated plates (IDEXX Inc., MA).
Total RNA was extracted from the infected cells using TRIzol reagent (Invitrogen Life Technologies, Carlsbad, CA, USA) and then purified using the RNeasy Mini kit (Qiagen, Venlo, The Netherlands). Real-time RT-PCR was performed on the Roche LightCycler real-time PCR system (Madison, WI, USA) using specific primers and probe for chicken IL-6 (forward 5′-GAT GGG ACG GCG GCC GGG GAG-3′, reverse 5′- TAA CGG CGG CGG GCA GCG GGA-3′, probe 5′-(FAM)-GGA CGG GGC GCT CTC CGG CGG-(TAMRA)-3′) and the One Step PrimeScript RT-PCR Kit (Perfect Real Time) (TaKaRa, Dalian, China) following the manufacturer’s instructions. IL-6 gene expression was normalized to that of the 28S rRNA gene (forward 5′-ATC CTG CCA GTA GCA TAT G-3′, reverse 5′-GCC GTG CGT ACT TAC ACG T-3′, probe 5′-(FAM)-GCA TGG CTT AAT CTT TGA GAC AA-(TAMRA)-3′) and presented as fold induction relative to medium control.
Splenocytes seeded in 12-well plates were transfected with p27, gp85, gp37, reverse transcriptase, and integrase gene expression vectors (2 μg/5 × 106 cells) using Lonza Nucleofector Kits. IL-6 mRNA was measured at 48 h post transfection as described above.
To determine whether ALV-J induced VEGF-A and VEGFR-2 expression via IL-6, VECs were transfected with IL-6 siRNA or negative control siRNA and 24 h later were infected with ALV-J over another 24 h. The expression of IL-6 was then tested as described above to confirm siRNA efficacy.
ELISA for IL-6
The levels of secreted IL-6 were measured in cell culture supernatants using commercially available chicken IL-6 ELISA kits (Uscn Life Science Inc., Aachen, Germany) according to the manufacturer’s instructions.
Inhibition of signal transduction pathways
Splenocytes were pretreated with control DMSO, PI3K inhibitor LY294002 (Enzo Life Sciences; 5 μM), MEK inhibitor PD98059 (Enzo Life Sciences; 10 μM), p38 mitogen-activated protein kinase (MAPK) inhibitor SB203580 (Enzo Life Sciences; 5 μM), PKC inhibitor GF-109203X (Enzo Life Sciences; 1 μM), or NF-κB inhibitor BAY11-7082 (Enzo Life Sciences; 1 μM) for 1 hour as described (68), and then infected with ALV-J at an MOI of 0.1. After 48 h, supernatants were collected for IL-6 assay by ELISA, and cells were harvested for IL-6 mRNA analysis by real-time RT-PCR.
Dual-Glo luciferase assay
Splenocytes seeded on a 12-well plate were pretreated with the PI3K inhibitor LY294002 (5 μM) or equivalent DMSO for 1 h. Then the cells were transfected using Lonza Nucleofector Kits with NF-κB activity test plasmids (including a luciferase signal, provided by professor Xiaojun Wang) together with pRL-TK control plasmid (Promega) and simultaneously infected with ALV-J (MOI = 0.1) or DMEM. After another 6 h, the cells were harvested for luciferase analysis with Dual-Glo Luciferase Assay System (Promega) following manufacturer’s instructions.
VEGF-A and VEGFR-2 mRNA quantification
To test the effect of IL-6 on VEGF-A and VEGFR-2 expression, VECs pretreated with the STAT3 inhibitor stattic (Santa Cruz Biotechnology; 5 μM) or equal volume of DMSO were transfected with the IL-6 gene expression vector for 48 h. Total RNA was extracted using TRIzol and purified using RNeasy Mini kit. Real-time RT-PCR was performed using specific primers for chicken VEGF-A (forward 5′-GTC GTA CAT ATT CAG GCC ATC-3′, reverse 5′-GAT TCT TTG GTC TGC AGT CAC-3′) or VEGFR-2 (forward 5′-TAA GGC ATC CAA CCA GAC AAG-3′, reverse 5′-GTA CTA GAG TGG CGG GGA CAC-3′) and the One Step SYBR PrimeScript RT-PCR Kit II (Perfect Real Time) (TaKaRa) following the manufacturer’s instructions using the Roche LightCycler real-time PCR system. VEGF-A and VEGFR-2 gene expression was normalized to 28S rRNA and presented as fold induction relative to medium control.
Embryos inoculated with recombinant IL-6 protein (1 μg/embryo) or PBS at equal volumes were used to test the effect of IL-6 on VEGF-A and VEGFR-2 expression in vivo. At 1, 2, and 3 days after inoculation, three embryos were tested each from the control and infected groups. RNAs were isolated from the vascular tissues of the embryos and used to test mRNA expression by real-time RT-PCR as described above.
To explore whether ALV-J induced VEGF-A and VEGFR-2 expression via IL-6, VECs were transfected with IL-6 or negative control siRNA and infected with ALV-J 24 h later for an additional 24 h. The expression of VEGF-A and VEGFR-2 were then tested as described above.
SOCS3 mRNA quantification
To further study whether the STAT3 pathway was activated upon IL-6-induced VEGF-A and VEGFR-2 expression, VECs were transfected with the IL-6 gene expression vector or control vector for 48 h. Total RNA was extracted and purified as previously described. Real-time RT-PCR was performed using specific primers for chicken SOCS3 (forward 5′-TCA GCT CTA AGA GCG AGT ACC-3′, reverse 5′-GCT GAG GGT GAA GAA GTG C-3′) or VEGFR-2 (forward 5′-TAA GGC ATC CAA CCA GAC AAG-3′, reverse 5′-GTA CTA GAG TGG CGG GGA CAC-3′) and the One Step SYBR PrimeScript RT-PCR Kit II (Perfect Real Time) according to manufacturer instruction for the Roche LightCycler real-time PCR system. SOCS3 gene expression was normalized to 28S rRNA and presented as fold induction relative to medium control.
Embryos inoculated with recombinant IL-6 protein (1 μg/embryo) or equivalent volumes of PBS were also used to test the activation of the STAT3 signaling pathway. On 1, 2, and 3 days after inoculation, three embryos were tested each from the control and infected groups. RNAs were isolated from the vascular tissues of the embryos and were used to test the expression of SOCS3 mRNA by real-time RT-PCR as described above.
Statistical analysis
All experiments were performed with at least three independent replicates and the data are reported as the means ± standard deviation (SD). Differences in data were evaluated by the Student’s t test. A p value of < 0.05 was considered to be statistically significant.
Conflicts of Interest
All authors declare no competing financial interests.
Grant Support
This work was funded by the National Natural Science Foundation of China (31372437); Newton Fund Project on “China-United Kingdom International Centre for Research on Avian Diseases”; and the Heilongjiang Province Science Fund for Distinguished Young Scholars (JC2015007).
References
1. Payne LN, Brown SR, Bumstead N, Howes K, Frazier JA, Thouless ME. A novel subgroup of exogenous avian leukosis virus in chickens. J Gen Virol. 1991; 72: 801-807.
2. Payne LN, Gillespie AM, Howes K. Unsuitability of chicken sera for detection of exogenous ALV by the group-specific antigen ELISA. Vet Rec. 1993; 132: 555-557.
3. Payne LN, Gillespie AM, Howes K. Introduction of myeloid leukosis and other tumors with HPRS-103 strain of ALV. Vet Rec. 1991; 129: 447-448.
4. Payne LN, Gillespie AM, Howes K. Myeloid leukaemogenicity and transmission of the HPRS-103 strain of avian leukosis virus. Leukemia. 1992; 6: 1167-1176.
5. Fadly AM, Smith EJ. Isolation and some characteristics of a subgroup J-like avian leukosis virus associated with myeloid leukosis in meat-type chickens in the United States. Avian Dis. 1999; 43: 391-400.
6. Du Y, Cui Z, Qin A. Subgroup J avian leukosis viruses in China. China Poult. Sci. 1999; 3: 1-4.
7. Gao YL, Yun B, Qin L, Pan W, Qu Y, Liu Z, Wang Y, Qi X, Gao H, Wang X. Molecular epidemiology of avian leukosis virus subgroup J in layer flocks in China. J Clin Microbiol. 2012; 50: 953-960.
8. Gao YL, Qin LT, Pan W, Wang YQ, Le Qi X, Gao HL, Wang XM. Avian leukosis virus subgroup J in layer chickens, China. Emerg Infect Dis. 2010; 16: 1637-1638.
9. Pan W, Gao Y, Sun F, Qin L, Liu Z, Yun B, Wang Y, Qi X, Gao H, Wang X. Novel sequences of subgroup J avian leukosis viruses associated with hemangioma in Chinese layer hens. Virol J. 2011; 8: 552.
10. Poirier F, Jullien P, Dezelee P, Dambrine G, Esnault E, Benatre A, Calothy G. Role of the mitogenic property and kinase activity of p60src in tumor formation by Rous sarcoma virus. J Virol. 1984; 49: 325-332.
11. Justice J 4th, Malhotra S, Ruano M, Li Y, Zavala G, Lee N, Morqan R, Beemon K. The MET gene is a common integration target in avian leucosis virus subgroup J-induced chicken hemangiomas. J Virol. 2015; 89: 4712-4719.
12. Hayward WS, Neel BG, Astrin SM. Activation of a cellular onc gene by promoter insertion in ALV-induced lymphoid leukosis. Nature. 1981; 290: 475-480.
13. Payne GS, Bishop JM, Varmus HE. Multiple arrangements of viral DNA and an activated host oncogene in bursal lymphomas. Nature. 1982; 295: 209-214.
14. Clurman BE, Hayward WS. Multiple proto-oncogene activations in avian leukosis virus-induced lymphomas: evidence for stage-specific events. Mol Cell Biol. 1989; 9: 2657-2664.
15. Yang F, Xian RR, Li Y, Polony TS, Beemon KL. Telomerase reverse transcriptase expression elevated by avian leukosis virus integration in B cell lymphomas. Proc Natl Acad Sci USA. 2007; 104: 18952-18957.
16. Li Y, Liu X, Yang Z, Xu C, Liu D, Qin J, Dai M, Hao J, Feng M, Huang X, Tan L, Cao W, Liao M. The MYC, TERT, and ZIC1 genes are common targets of viral integration and transcriptional deregulation in avian leukosis virus subgroup J-induced myeloid leukosis. J Virol. 2014; 88: 3182-3191.
17. Chesters PM, Howes K, McKay JC, Payne LN, Venugopal K. Acutely transforming avian leukosis virus subgroup J strain 966: defective genome encodes a 72-kilodalton Gag-Myc fusion protein. J Virol. 2001; 75: 4219-4225.
18. Barr SD, Leipzig J, Shinn P, Ecker JR, Bushman FD. Integration targeting by avian sarcoma-leukosis virus and human immunodeficiency virus in the chicken genome. J Virol. 2005; 79: 12035-12044.
19. Mitchell RS, Beitzel BF, Schroder AR, Shinn P, Chen H, Berry CC, Ecker JR, Bushman FD. Retroviral DNA integration: ASLV, HIV, and MLV show distinct target site preferences. PLoS Biol. 2004; 2: E234.
20. Wang Q, Gao Y, Wang Y, Qin L, Qi X, Qu Y, Gao H, Wang X. A 205-nucleotide deletion in the 3’ untranslated region of avian leukosis virus subgroup J, currently emergent in China, contributes to its pathogenicity. J Virol. 2012; 86: 12849-12860.
21. Song G, Li Y, Jiang G. Role of VEGF/VEGFR in the pathogenesis of leukemias and as treatment targets (Review). Oncol Rep. 2012; 28: 1935-1944.
22. Kerbel RS. Tumor angiogenesis. N Engl J Med. 2008; 358: 2039-2049.
23. Ellis LM, Hicklin DJ. VEGF-targeted therapy: mechanisms of anti-tumour activity. Nat Rev Cancer. 2008; 8: 579-591.
24. Medinger M, Fischer N, Tzankov A. Vascular endothelial growth factor-related pathways in hemato-lymphoid malignancies. J Oncol. 2010; 729725.
25. Zhu Z, Hattori K, Zhang H, Jimenez X, Ludwig DL, Dias S, Kussie P, Koo H, Kim JH, Lu D, Liu M, Tejada R, Friedrich M, et al. Inhibition of human leukemia in an animal model with human antibodies directed against vascular endothelial growth factor receptor 2. Correlation between antibody affinity and biological activity. Leukemia. 2003; 17: 604-611.
26. Aggarwal BB, Sethi G, Ahn KS, Sandur SK, Pandey MK, Kunnumakkara AB, Sung B, Ichikawa H. Targeting signal-transducer-and-activator-of-transcription-3 for prevention and therapy of cancer: modern target but ancient solution. Ann N Y Acad Sci. 2006; 1091: 151-169.
27. Eriksen KW, Kaltoft K, Mikkelsen G, Nielsen M, Zhang Q, Geisler C, Nissen MH, Röpke C, Wasik MA, Odum N. Constitutive STAT3-activation in Sezary syndrome: tyrphostin AG490 inhibits STAT3-activation, interleukin-2 receptor expression and growth of leukemic Sezary cells. Leukemia. 2001; 15: 787-793.
28. Nielsen M, Nissen MH, Gerwien J, Zocca MB, Rasmussen HM, Nakajima K, Röpke C, Geisler C, Kaltoft K, Ødum N. Spontaneous interleukin-5 production in cutaneous T-cell lymphoma lines is mediated by constitutively activated Stat3. Blood. 2002; 99: 973-977.
29. Linher-Melville K, Singh G. The transcriptional responsiveness of LKB1 to STAT-mediated signaling is differentially modulated by prolactin in human breast cancer cells. BMC Cancer. 2014; 14: 415.
30. Waldner MJ, Neurath MF. Master regulator of intestinal disease: IL-6 in chronic inflammation and cancer development. Semin Immunol. 2014; 26: 75-79.
31. McGovern JL, Nguyen DX, Notley CA, Mauri C, Isenberg DA, Ehrenstein MR. Th17 cells are restrained by Treg cells via the inhibition of interleukin-6 in patients with rheumatoid arthritis responding to anti-tumor necrosis factor antibody therapy. Arthritis Rheum. 2012; 64: 3129-3138.
32. Moon YM, Lee J, Lee SY, Her YM, Ryu JG, Kim EK, Son HJ, Kwok SK, Ju JH, Yang CW, Park SH, Kim HY, Cho ML. Gene associated with retinoid-interferon-induced mortality 19 attenuates murine autoimmune arthritis by regulation of th17 and treg cells. Arthritis Rheumatol. 2014; 66: 569-578.
33. Zhang C, Zhang X, Chen XH. Inhibition of the interleukin-6 signaling pathway: a strategy to induce immune tolerance. Clin Rev Allergy Immunol. 2014; 47: 163-173.
34. Berri F, Haffar G, Le̊ VB, Sadewasser A, Paki K, Lina B, Wolff T, Riteau B. Annexin V incorporated into influenza virus particles inhibits gamma interferon signaling and promotes viral replication. J Virol. 2014; 88: 11215-11228.
35. Ouaquia L, Mrizak D, Renaud S, Moralès O, Delhem N. Control of the inflammatory response mechanisms mediated by natural and induced regulatory T-cells in HCV-, HTLV-1-, and EBV-associated cancers. Mediators Inflamm. 2014; 564296.
36. de Munnik SM, Smit MJ, Leurs R, Vischer HF. Modulation of cellular signaling by herpesvirus-encoded G protein-coupled receptors. Front Pharmacol. 2015; 6: 40.
37. Gao Y, Liu Y, Guan X, Li X, Yun B, Qi X, Wang Y, Gao H, Cui H, Liu C, Zhang Y, Wang X Gao Y. Differential expression of immune-related cytokine genes in response to J group avian leucosis virus infection in vivo. Mol Immunol. 2015; 64: 106-111.
38. Becker C, Fantini MC, Schramm C, Lehr HA, Wirtz S, Nikolaev A, Burg J, Strand S, Kiesslich R, Huber S, Ito H, Nishimoto N, Yoshizaki K, et al. TGF-beta suppresses tumor progression in colon cancer by inhibition of IL-6 trans-signaling. Immunity. 2004; 21: 491-501.
39. Waldner MJ, Wirtz S, Jefremow A, Warntjen M, Neufert C, Atreya R, Becker C, Weiqmann B, Vieth M, Rose-John S, Neurath MF. VEGF receptor signaling links inflammation and tumorigenesis in colitis-associated cancer. J Exp Med. 2010; 207: 2855-2868.
40. Mitsuyama K, Matsumoto S, Masuda J, Yamasakii H, Kuwaki K, Takedatsu H, Sata, M. Therapeutic strategies for targeting the IL-6/STAT3 cytokine signaling pathway in inflammatory bowel disease. Anticancer Res. 2007; 27: 3749-3756.
41. Culiq Z. Proinflammatory cytokine interleukin-6 in prostate carcinogenesis. Am J Clin Exp Urol. 2014; 2: 231-238.
42. Endo TA, Masuhara M, Yokouchi M, Suzuki R, Sakamoto H, Mitsui K, Matsumoto A, Tanimura S, Ohtsubo M, Misawa H, Miyazaki T, Leonor N, Taniguchi T, et al. A new protein containing an SH2 domain that inhibits JAK kinases. Nature. 1997; 387: 921-924.
43. Starr R, Willson TA, Viney EM, Murray LJ, Rayner JR, Jenkins BJ, Gonda TJ, Alexander WS, Metcalf D, Nicola NA, Hilton DJ. A family of cytokine-inducible inhibitors of signalling. Nature. 1997; 387: 917-921.
44. Alexander WS. Suppressors of cytokine signaling (SOCS) in the immune system. Nat Rev Immunol. 2002; 2: 410-416.
45. Knüpfer H, Preiss R. Significance of interleukin-6 (IL-6) in breast cancer (review). Breast Cancer Res Treat. 2007; 102: 129-135.
46. Greten FR, Eckmann L, Greten TF, Park JM, Li ZW, Egan LJ, Kagnoff MF, Karin M. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell. 2004; 118: 285-296.
47. Naugler WE, Sakurai T, Kim S, Maeda S, Kim K, Elsharkawy AM, Karin M. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science. 2007; 317: 121-124.
48. Bova CA, Manfredi JP, Swanstrom R. env genes of avian retroviruses: nucleotide sequence and molecular recombinants define host range determinants. Virology. 1986; 152: 343-354.
49. Bova-Hill C, Olsen JC, Swanstorm R. Genetic analysis of the Rousarcoma virus subgroup D env gene: mammal tropism correlates with the temperature sensitivity of gp85. J Virol. 1991; 65: 2073-2080.
50. Bova CA, Olsen JC, Swanstrom R. The avian retrovirus env gene family: molecular analysis of host range and antigenic variants. J Virol. 1988; 62: 75-83.
51. Del Cornò M, Donninelli G, Varano B, Da Sacco L, Masotti A, Gessani S. HIV-1 gp120 activates the STAT3/interleukin-6 axis in primary human monocyte-derived dendritic cells. J Virol. 2014; 88: 11045-11055.
52. Yang H, Ji X, Zhao G, Ning J, Zhao Q, Aiken C, Gronenborn AM, Zhang P, Xiong Y. Structural insight into HIV-1 capsid recognition by rhesus TRIM5α. Proc Natl Acad Sci U S A. 2012; 109: 18372-18377.
53. Grütter MG, Luban J. TRIM5 structure, HIV-1 capsid recognition, and innate immune signaling. Curr Opin Virol. 2012; 2: 142-150.
54. Weiss RA. The discovery of endogenous retroviruses. Retrovirology. 2006; 3: 67.
55. Hortling L, Suni J, Vaheri A. Localization of the major group-specific protein (p27) of avian tumor viruses by immunofluorescence in chicken cells and tissues. Intervirology. 1975; 5: 252-259.
56. Spencer JL, Gilka F. Lymphoid leukosis: detection of group specific viral antigen in chicken spleens by immunofluorescence and complement fixation. Can J Comp Med. 1982; 46: 370-375.
57. Stedman NL, Brown TP, Brooks RL Jr, Bounous DI. Heterophil function and resistance to staphylococcal challenge in broiler chickens naturally infected with avian leukosis virus subgroup J. Vet Pathol. 2001; 38: 519-527.
58. Barbour EK, Bouljihad M, Hamdar B, Sakr W, Eid A, Safieh-Garabedian B. Dynamics of protein 27 of avian leukosis virus and transforming growth factor beta2 in lymphoid leukosis susceptible and resistant broiler chicken breeding stock. Vet Res Commun. 1999; 23: 191-200.
59. Pang P, Chen FY, Tang J, Ma LF, Wang JC, Zheng SJ. Avian leukosis virus p27 inhibits tumor necrosis factor alpha expression in RAW264.7 macrophages after stimulation with lipopolysaccharide. Acta Virol. 2010; 54: 119-124.
60. Bromberg J, Wang TC. Inflammation and cancer: IL-6 and STAT3 complete the link. Cancer Cell. 2009; 15: 79-80.
61. Mathers C, Schafer X, Martinez-Sobrido L, Munger J. The human cytomegalovirus UL26 protein antagonizes NF-κB activation. J Virol. 2014; 88: 14289-14300.
62. Yamaya M, Nadine LK, Ota C, Kubo H, Makiquchi T, Nagatomi R, Nishimura H. Magnitude of influenza virus replication and cell damage is associated with interleukin-6 production in primary cultures of human tracheal epithelium. Respir Physiol Neurobiol. 2014; 202: 16-23.
63. Liu WL, Lin YH, Xiao H, Xing S, Chen H, Chi PD, Zhang G. Epstein-Barr virus infection induces indoleamine 2,3-dioxygenase expression in human monocyte-derived macrophages through p38/mitogen-activated protein kinase and NF-κB pathways: impairment in T cell functions. J Virol. 2014; 88: 6660-6671.
64. Xin M, Deng X. Nicotine inactivation of the proapoptotic function of Bax through phosphorylation. J Biol Chem. 2005; 280: 10781-10789.
65. Hofseth LJ, Hussain SP, Woqan GN, Harris CC. Nitric oxide in cancer and chemoprevention. Free Radic Biol Med. 2003; 34: 955-968.
66. Jeong SJ, Pise-Masison CA, Radonovich MF, Park HU, Brady JN. Activated AKT regulates NF-kappaB activation, p53 inhibition and cell survival in HTLV-1-transformed cells. Oncogene. 2005; 24: 6719-6728.
67. Vandermoere F, EI Yazidi-Belkoura I, Adriaenssens E, Lemoine J, Hondermarck H. The antiapoptotic effect of fibroblast growth factor-2 is mediated through nuclear factor-kappaB activation induced via interaction between Akt and IkappaB kinase-beta in breast cancer cells. Oncogene. 2005; 24: 5482-5491.
68. Mitsuyama K, Matsumoto S, Masuda J, Yamasakii H, Kuwaki K, Takedatsu H, Sata H. Therapeutic strategies for targeting the IL-6/STAT3 cytokine signaling pathway in inflammatory bowel disease. Anticancer Res. 2007; 27: 3749-3756.
69. Waldner MJ, Wirtz S, Jefremow A, Warntjen M, Neufert C, Atreya R, Becker C, Weiqmann B, Vieth M, Rose-John S, Neurath MF. VEGF receptor signaling links inflammation and tumorigenesis in colitis-associatedcancer. J Exp Med. 2010; 207: 2855-2868.
70. Brekken RA, Overholser JP, Stastny VA, Waltenberger J, Minna JD, Thorpe PE. Selective inhibition of vascular endothelial growth factor (VEGF) receptor 2 (KDR/Flk-1) activity by a monoclonal anti-VEGF antibody blocks tumor growth in mice. Cancer Res. 2000; 60: 5117-5124.
71. Ghosh AK, Secreto CR, Knox TR, Ding W, Mukhopadhyay D, Kay NE. Circulating microvesicles in B-cell chronic lymphocytic leukemia can stimulate marrow stromal cells: implications for disease progression. Blood. 2010; 115: 1755-1764.
72. Adams J, Carder PJ, Downey S, Forbes MA, MacLennan K, Allgar V, Kaufman S, Hallam S, Bricknell R, Walker JJ, Cairnduff F, Selby PJ, Perren TJ, et al. Vascular endothelial growth factor (VEGF) in breast cancer: comparison of plasma, serum and tissue VEGF and microvessel density and effects of tamoxifen. Cancer Res. 2000; 60: 2898-2905.
73. Kay NE, Bone ND, Tschumper RC, Howell KH, Geyer SM, Dewald GW, Hanson CA, Jelinek DF. B-CLL cells are capable of synthesis and secretion of both pro- and anti-angiogenic molecules. Leukemia. 2002; 16: 911-919.
74. Wang Q, Gao Y, Wang Y, Qin L, Qi X, Qu Y, Gao H, Wang X. A 205-nucleotide deletion in the 3’ untranslated region of avian leukosis virus subgroup J, currently emergent in China, contributes to its pathogenicity. J Virol. 2012; 86: 12849-12860.
75. Shah A, Verma AS, Patel KH, Noel R, Rivera-Amill V, Silverstein PS, Chaudhary S, Bhat HK, Stamatatos L, Singh DP, Buch S, Kumar A. HIV-1 gp120 induces expression of IL-6 through a nuclear factor-kappa B-dependent mechanism: suppression by gp120 specific small interfering RNA. PLoS One. 2011; 6: e21261.
76. Bollrath J, Phesse TJ, von Burstin VA, Putoczki T, Bennecke M, Bateman T, Nebelsiek T, Lundgren-May T, Canli O, Schiwitalla S, Matthews V, Schimid RM, Kirchner T, et al. gp130-mediated Stat3 activation in enterocytes regulates cell survival and cell-cycle progression during colitis-associated tumorigenesis. Cancer Cell. 2009; 15: 91-102.
77. Grivennikov S, Karin E, Terzic J, Mucida D, Yu GY, Vallabhapurapu S, Scheller J, Rose-John S, Cheroutre H, Eckmann L, Karin M. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell. 2009; 15: 103-113.
78. Thomas E, Gonzalez VD, Li Q, Modi AA, Chen W, Noureddin M, Rotman Y, Liang TJ. HCV infection induces a unique hepatic innate immune response associated with robust production of type III interferons. Gastroenterology. 2012; 142: 978-988.
79. Zou W, Restifo NP. T(H)17 cells in tumour immunity and immunotherapy. Nat Rev Immunol. 2010; 10: 248-256.
80. Karin M, Greten FR. NF-𝜅B: Linking inflammation and immunity to cancer development and progression. Nat Rev Immunol. 2005; 5: 749-759.
81. Martin A, Lillehoj HS, Kaspers B, Bacon LD. Antigen-specific T cell proliferation following coccidia infection. Poult Sci. 1993; 72: 2084-2094.
82. Fu Y, Quan R, Zhang H, Hou J, Tang J, Feng WH. Porcine reproductive and respiratory syndrome virus induces interleukin-15 through the NF-κB signaling pathway. J Virol. 2012; 86: 7625-7636.