
INTRODUCTION
DNA methylation is one of the major mechanisms for the regulation of genetic information and gene expression in vertebrates [1]. It maintains gene silencing in the nuclear genome including the repetitive DNA elements [2, 3]. In this mechanism, a methyl group is covalently added at the 5th position of carbon on the cytosine ring in CG, CHG and CHH sequence contexts [4–7]. Loss of global DNA methylation on the repetitive DNA elements and subsequent alteration in chromatin structure was the first epigenetic change demonstrated in human cancers [8, 9]. Subsequent studies have shown that numerous tumor-suppressor genes in cancers are hypermethylated and transcriptionally silent [10]. Therefore, the observation of aberrant DNA methylation is considered a hallmark of human cancer. The most studied change of DNA methylation in neoplasms is the silencing of tumor suppressor genes by hypermethylation of associated CpG island promoters, for example p16 (INK4a), BRCA1, and hMLH1 [10–12]. Indeed, aberrant DNA methylation was demonstrated to be the dominant mechanism in MDS progression to AML correlating with poor clinical outcomes [13], highlighting its relevance to oncogenesis.
DNA methylation is carried out by three different DNA (cytosine-5) methyltransferases in humans, DNMT1, DNMT3A and DNMT3B [14, 15]. Amongst these three, DNMT3A and 3B are known as de novo methyltransferases, and their roles in early developmental DNA methylation pattern establishment is well studied in mouse model systems [14, 15]. DNMT1 is popularly known as the maintenance DNA methyltransferase as it copies the original DNA methylation pattern onto the daughter strand during DNA replication [16]. DNMT1 acts on hemimethylated DNA at the replication fork with a host of accessory proteins including UHRF1, which confers specificity to the enzyme [17–19]. Indeed, UHRF1 is indispensable for maintenance DNA methylation and its null mutation results in hypomethylation of the ES genome [20]. The complex of DNMT1-UHRF1 and the clamp-loading factor PCNA are essential components of DNA methylation preservation during DNA replication. These observations suggest that there may be other protein factors including transcription factors that can influence DNA methylation by direct DNMT1 interaction. DNMT1 is essential for embryonic development and its loss results in embryonic lethality [21]. Indeed, many cancer cells show deregulation of either or both DNMT1 and UHRF1, suggesting that aberrant DNA methylation is a result of deregulation of enzymes and essential protein factors [22, 23].
Several studies have demonstrated that methylation fidelity in normal cells is tightly regulated by one or more enzyme targeting mechanisms. These mechanisms include recruitment of enzymes via specific interactions with histone tail modifications or through protein partners including chromatin remodelers and transcription factors [24–27]. Similarly, lymphoid specific helicase (LSH) protein, which belongs to the family of switch sucrose non-fermentable (SWI/SNF) chromatin remodelers, has been found to play an essential role in de novo DNA methylation in mice, strengthening the connection between chromatin remodeling and DNA methylation [28]. Transcription factors such as p53 can directly interact and cooperate with DNMT1 to selectively repress p53-repressed genes such as survivin [25]. DNMT1 also binds Retinoblastoma protein (Rb) in a complex with the transcription factor E2F1 and HDAC1 to repress transcription from promoters containing E2F-binding sites [26]. Therefore sequence specific transcription factors often participate in DNA methylation programing or reprograming.
The transcription factor LSF (Late SV40 Factor), also known as TFCP2, is involved in many biological events, including cell cycle, DNA synthesis and cell survival [29, 30]. In HCC cell lines, LSF also regulates genes involved in invasion, angiogenesis, and chemoresistance, consistent with its oncogenic and metastatic role in HCC [30]. LSF is overexpressed in human HCC cell lines when compared to normal hepatocytes. Furthermore, in more than 90% of cases of patient HCC samples, LSF expression levels show significant correlation with the stages and grades of the disease [31]. In a subsequent study, Grant et al., identified a small molecule factor quinolinone inhibitor 1 (FQI1) that effectively inhibits LSF DNA-binding activity [32]. FQI1 also dramatically displayed anti-proliferative activity in LSF overexpressing cells, including HCC cells, leading to rapid apoptosis in cell culture and inhibition of HCC growth in multiple mouse tumor models [32, 33].
Structural predictions of the LSF protein family suggest that they coevolved independently with the critical cell cycle regulator p53, as they contain a similar binding motif [34]. Based on this observation several functional hypotheses on structure-function relationships between LSF and p53 have been drawn. Since p53 can directly interact and cooperate with DNMT1 to selectively repress p53-regulated genes, we attempted to examine if LSF binds DNMT1 and accessory factor UHRF1, and if this interaction is affected by its inhibitor FQI1, leading to epigenome alterations.
RESULTS
LSF-DNMT1 complex in cells
The transcription factors of the LSF family are characterized by the possession of a distinctive DNA-binding domain that bears no clear sequence relationship to other known DNA-binding domains [35]. However, based on structural predictions, a common origin for the LSF and the p53 has been proposed based on similarities in the folding of their DNA-binding domains [34]. Since p53 recruits DNMT1 and promotes DNA methylation in a p53 dependent manner [25], we investigated if such a relationship exists between DNMT1 and LSF. We immune-precipitated human cell (HEK293T) nuclear extract with anti-LSF antibody along with an anti-IgG control and probed for DNMT1. Indeed, a full-length and a shorter form of DNMT1 were detected (Figure 1A). To confirm that it was the DNMT1 complex, the same blot was probed for UHRF1, an essential partner of DNMT1 during DNA methylation (Figure 1A). UHRF1 was observed as a co-immunoprecipitated product. This strengthened our conclusion that LSF indeed is in a complex with DNMT1 machinery in the cell. For visualization of this interaction, we also co-expressed FLAG-LSF and DsRed-DNMT1 fusions in COS-7 cells. The staining pattern of LSF within the cells was largely cytoplasmic, but a small but significant percentage of LSF were found inside the nucleus, colocalizing with DsRed-DNMT1 as was observed by a punctate yellow merged pattern with a pearson correlation coefficient of 0.3 (Figure 1B). Both DNMT1 and LSF are multi-domain proteins (Figure 1C). To determine if the interaction between DNMT1 and LSF is direct and which domains are involved in binding, we performed GST-pulldown assays. Overlapping GST-fusions representing the entire length of DNMT1 were bound to beads and incubated with a purified MBP-LSF fusion. After a thorough wash to remove non-bound LSF, the bound proteins were immunoblotted and probed for LSF. LSF binds to fragments representing the amino terminus regulatory region of DNMT1 (amino acids 1-446 and 431-836) (Figure 1D). In a reciprocal assay, overlapping GST-fusions representing the entire length of LSF bound to the beads were incubated with purified full-length DNMT1 and after a thorough wash to remove non-bound DNMT1, the bound proteins were western-blotted and probed with anti-DNMT1 antibody. DNMT1 binds to fragments representing both the carboxy terminus and DNA interaction regions of LSF (amino acids 380-502 and 65-259) (Figure 1E).
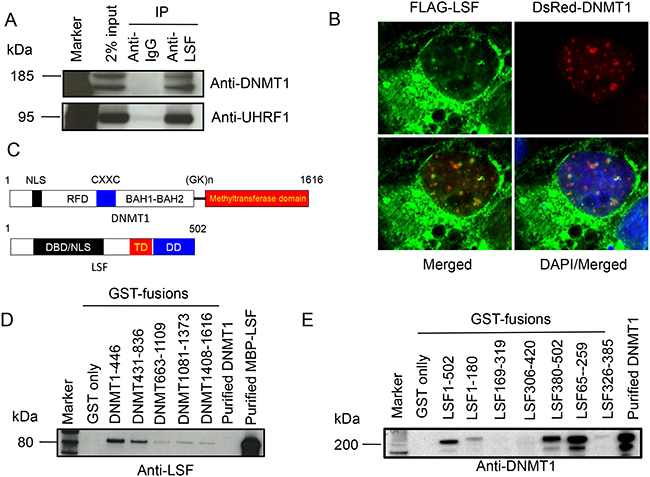
Figure 1: LSF directly binds DNMT1. A. Immunoprecipitation of endogenous LSF with DNMT1 and UHRF1 in cellular extracts. Antibodies used for the western blot are indicated on the right. Two different isoforms of DNMT1 are detected by the anti-DNMT1 N-terminus antibody. B. Colocalization of DNMT1 and LSF in COS-7 cells. Plasmids expressing FLAG-LSF and DsRed-DNMT1 (red) were transfected into the cells; the anti-FLAG antibody reveals LSF (green). The merged image indicates colocalization by the yellow punctate pattern of nuclear LSF and DNMT1. C. Schematic structure of human DNMT1 and LSF protein. The numbers indicate amino acid residues. NLS: nuclear localization signal; RFD: replication fork binding domain; CXXC: DNA binding domain of DNMT1; BAH1-BAH2: bromo-adjacent homology domains; (GK)n: GK repeats; DBD: DNA-binding domain; TD: tetramerization domain; and DD: dimerization domain. D. GST-pull down analysis of various overlapping domains of DNMT1 with purified full-length LSF, as MBP-LSF fusion protein. E. GST-pull down analysis of various overlapping domains of LSF with purified full-length DNMT1. Antibodies used for immunoblotting are indicated on the bottom of C and D.
FQI1 dissociates LSF-DNMT1 complex in vitro and in cells
We incubated purified DNMT1 with increasing amounts of His-LSF recombinant protein to determine its influence on DNA methylation (Figure 2A) and thus the biological significance of DNMT1-LSF interactions. Indeed, as the molar ratio of His-LSF to DNMT1 increased from 2:1 to 4:1, the methyltransferase activity of DNMT1 increased about two-fold (Figure 2A). However using similar reaction conditions, the presence of FQI1 inhibitor negated the methyltransferase stimulation. As controls, addition of MBP (maltose binding protein) protein alone or in the presence of 5 μM FQI1 had no effect on methyltransferase activity (Figure 2A). This result along with the GST pull-down assays suggest that LSF may activate DNA methylation by direct interaction with DNMT1, and by antagonizing this interaction, FQI1 prevents stimulation of methyltransferase activity.
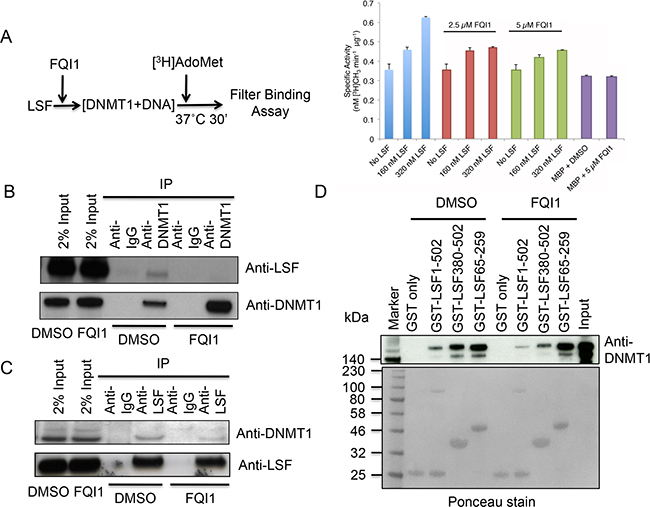
Figure 2: LSF stimulates DNMT1 and FQI1 negates methyltransferase activation. A. Schematic diagram of methyltransferase assay in the absence or presence of FQI1 (left panel). DNMT1 assay performed in the presence of the indicated amounts of LSF, plus FQI1 (2.5 μM) or FQI1 (5 μM) demonstrating the effect of FQI1 on DNA methylation. Note that addition of FQI1 alone in the DNMT1 reaction with or without control MBP protein had no effect on DNA methyltransferase activity. B. The cells were treated with DMSO or FQI1 and nuclear extract was used for immunoprecipitation of DNMT1 to reveal the degree of LSF binding. Antibodies used for western blots are indicated on the right. C. The cells were treated with DMSO or FQI1 and nuclear extract was used for immunoprecipitation of LSF to reveal the degree of DNMT1 binding. Antibodies used for western blots are indicated on the right. D. GST-pull down analysis of various overlapping domains of LSF and purified full-length DNMT1 in the presence or absence of FQI1. LSF1-502, LSF380-502, LSF65-259 represents LSF full length, LSF Ubiquitin domain, and LSF DNA binding domain respectively. Ponceau staining of the gel is shown to evaluate protein loading. Note that the level of GST-full length LSF is lower than those of the other proteins, which is reflected in the decreased amounts of associated DNMT1 with the full-length LSF. The degree of DNMT1 binding is revealed upon immunoblotting with anti-DNMT1 antibody.
We hypothesized that if LSF were an epigenetic modulator by recruitment of DNMT1, dissociation of DNMT1-LSF complex resulting from addition of FQI1 would lead to expression of DNA hypomethylation-dependent cellular genes. For validation, we performed two different experiments, testing the sensitivity of the LSF-DNMT1 interaction to FQI1, both in vivo and in vitro. To avoid possible cellular toxicity we incubated HEK293T cells with increasing concentrations of FQI1 and found 2.5 μM as the optimal concentration (data not shown). First, we treated HEK293T cells with 2.5 μM FQI1 to determine if the inhibitor has any effect on DNMT1, UHRF1 and LSF gene expression. Indeed, FQI1 had no effect on UHRF1 and LSF protein levels and small reduction of DNMT1 level (Supplementary Figure S1). To determine if a small reduction of DNMT1 has any effect on DNA methylation we performed nucleotide analysis of genomic DNA after 48 hrs of FQI1 treatment. We did not observe any difference in the global 5mC (control 3.0% vs. FQI1 treated 3.04% of total cytosine) or 5hmC content (0.03% of total 5mC) indicating that a slight reduction of DNMT1 level does not change 5mC or 5hmC levels.
Therefore, we measured DNMT1 and LSF complex formation and secondly, we also performed GST pull-down assays in the presence of 2.5 μM FQI1. Immunoprecipitation of endogenous DNMT1 in the presence of DMSO control resulted in LSF pull-down and addition of FQI1 into the cells resulted in dissociation of DNMT1 and LSF, since LSF was greatly diminished in the pull-down (Figure 2B). In a reciprocal experiment using anti-LSF antibody for immunoprecipitation in the presence of DMSO control or FQI1, a similar result was observed with severe reduction of DNMT1 in the complex (Figure 2C). These results indicate that FQI1 not only inhibits the ability of LSF to bind DNA in cells, as shown earlier [32], but that it is also a potent destabilizer of LSF-DNMT1 complex formation. GST-pull down assay in the presence of FQI1 also displayed either inhibition of complex formation or complex dissociation between LSF and DNMT1. Interestingly, FQI1 diminished association between both full length LSF and the carboxy-terminal domain of LSF with DNMT1, but not that between the DNA-binding domain of LSF and DNMT1. Full-length LSF binding with full-length DNMT1 was reduced 2 fold by FQI1, and that of the C-terminal LSF domain was reduced 2 fold by FQI1 (Figure 2D). Together, these results show that binding of FQI1 to LSF may indeed destabilize its binding to DNMT1.
FQI1 promotes aberrant DNA methylation
Since FQI1 inhibits LSF-DNMT1 complex formation, we performed reduced representation bisulfite sequencing (RRBS) to determine the methylation pattern and differences between control, DMSO-treated and FQI1-treated HEK293T genomes. Libraries were sequenced on an Illumina GAII platform. For each biological replicate, greater than 15 million of 72 bp high-quality paired-end reads (Phred score > 20, adaptor trimmed) were uniquely mapped to human genome hg19. With a minimum read coverage threshold of three, about 6.8 million average CpG sites were covered by each RRBS library, which represents about 25% and 23% of the total CpG sites, respectively, for DMSO and FQI1 treated human genome libraries.
At the single base resolution level, 9775 differentially methylated CpG sites were identified. The number of hypermethylated and hypomethylated DMR were 4666 and 5109 respectively, accounting overall for a small percentage (~2.3%) of hypomethylation across the genome (Figure 3A). We further clustered the DMR based on their distribution on genetic elements. Indeed, all DMRs were found in known genes, introns and CpG island regions including the transcription start sites, suggesting FQI1-mediated aberrant DNA methylation may have functional consequence in gene expression (Figure 3B). We also characterized the DMRs by distribution of number of CpG sites per DMR. The majority of DMR contained ~4 CpG sites, and almost all DMRs had less than 10 CpGs representing both hyper and hypomethylated sequences (Figure 3C). Further DMR width analysis showed that all DMRs are less than 400 bps and majority are ~80 bps (Figure 3D). In summary of the DMR analysis, hypermethylation was observed in the DMRs when the number of CpGs in the DMR is relatively lower; as the number of CpGs in the DMR increased, there is a trend towards hypomethylation. Also as the size of the DMR increases we observed a similar trend from hypermethylation to hypomethylation.
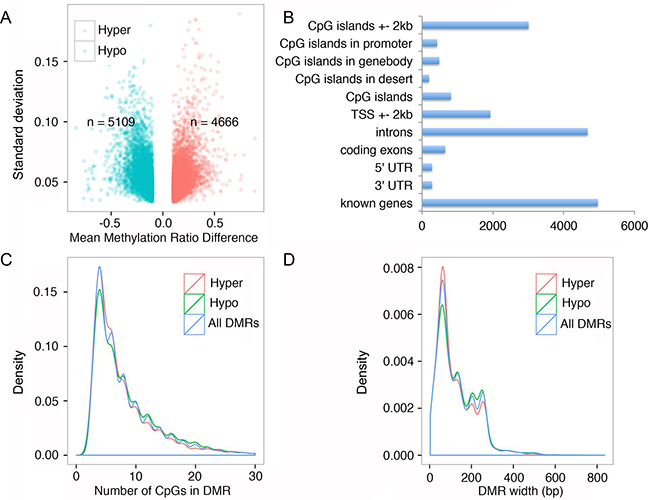
Figure 3: FQI1 treatment triggers alteration of genome wide DNA methylation. A. Volcano plot showing differentially methylated regions in the genome after HEK293T cells were treated with 2.5 μM FQI1 for 48 hrs. Green dots represent hypomethylated DMRs and red dots represent hypermethylated DMRs. Total number of hyper and hypomethylated DMRs are annotated in the plot. B. Bar graph showing the distribution of the 9775 DMRs in various regions of the genome. Each DMR is included in all appropriate categories. C. Density plot displaying the distribution of number of CpGs in DMRs. Red graph represents hypermethylated DMRs, green represents hypomethylated DMRs and blue represents all DMRs together. D. Density plot displaying the distribution of the size of DMRs. Red graph represents hypermethylated DMRs, green represents hypomethylated DMRs and blue represents all DMRs together.
FQI1 alters gene expression
LSF functions both as a transcription activator and repressor. It binds DNA regions as a homotetramer, and regulates a variety of cellular promoters. FQI1 is an antagonist to the DNA binding activity of LSF, and also promotes aberrant DNA methylation. Therefore both inhibiting binding of LSF to DNA and destabilizing its complex with DNMT1 would lead to aberrant gene expression. Therefore, transcriptional changes are of significant interest to understand the mechanisms of FQI1 cellular activity. We performed RNA-seq in triplicate with control and FQI1-treated biological samples. Clustering of samples based on Euclidean distance demonstrated good correlation within the replicates for control and treatment groups. However, there is a clear difference between control and treatment groups (Supplementary Figure S2). About 42 million of the 72-bp read pairs were mapped to hg19 for each library, and about 38 million mapped reads in each library could be assigned to a known gene. A total number of 4337 genes were found to be differentially expressed, where 2502 genes were up-regulated and 1835 genes were down-regulated in cells treated with FQI1 (Figure 4A). The regularized logarithm (rlog) transformed read counts of the top 100 genes with lowest adjusted p value were plotted on the heatmap, demonstrating clustering of control and FQI1-treated triplicates displaying differential expression (Figure 4B). Pathway analysis performed using GAGE [36] with high stringency (q value <0.01) revealed enrichment of genes involved in proteasome, spliceosome, RNA transport, protein processing in ER, and MAP kinase signaling pathways (Supplementary Table S1). FQI1 predominantly inhibited the DNA replication pathway (Supplementary Figure S1). Specific genes upregulated by FQI1 treatment included Aurora Kinase A (AURKA), suppressor APC Domain Containing 2 (SAPCD2), Kinesin Heavy Chain Member 2A (KIF2A), all of which were validated by RT-qPCR. All upregulated genes were activated within the first 12 h of FQI1 treatment (Figure 4C). Similarly among down regulated genes, MCM5, MCM6 and MCM7 were validated and repressed (Figure 4C). Therefore FQI1’s cellular influence involves both up and down regulation of gene expression and/or mRNA stability.
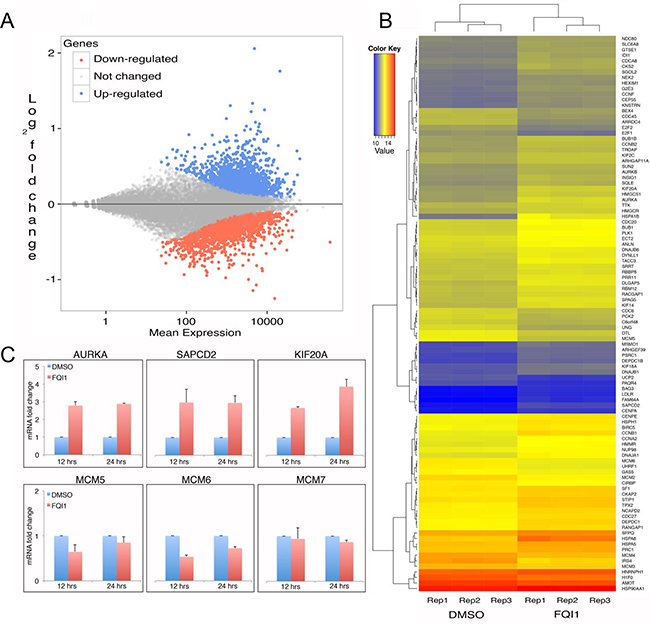
Figure 4: Transcriptome analysis of FQI1- versus vehicle-treated HEK293T cells. A. MA plot analysis of FQI1 vs DMSO control samples displaying distribution of up-regulated (blue), down-regulated (red) and unchanged (grey) gene expression. B. Heatmap showing the distribution of top 100 genes (descending order) for three replicates of DMSO and FQI1 treated samples. C. qPCR validation of genes identified with significantly altered expression from RNAseq. Top panel shows genes with increased expression and bottom panel shows genes with decreased expression.
Correlations between differentially methylated regions and gene expression
Further characterization of FQI1-mediated aberrant DNA methylation led us to identify many genic regions with either hyper or hypomethylations. RRBS genic regions were chosen for demonstration of DMRs. For example, chr19: 2,128,860 – 2,129,304, chr19: 54,677,584 – 54,678,078, and chr19: 14,590,249 – 14,590,472 were hypomethylated and chr4: 4,765,010 – 4,765,535, chr17: 73,545,489 – 73,546,127, and chr18: 13,677,234 – 13,677,609 were hypermethylated (Figure 5A). Since FQI1 promotes aberrant DNA methylation, we correlated DMR with the transcriptomic data. A driving force for this analysis was that 1678 DMRs located in TSS +- 2kb regions were associated with genes that were differentially regulated in the presence of FQI1. Therefore, the DMRs located near transcription initiation sites were clustered into hypomethylated or hypermethylated regions and then correlated with RNA-seq (log2 fold changes) data for the nearby gene. Hypermethylated DMRs were associated with no changes in gene expression (29.6%), reduced gene expression (9.1%) and elevated gene expression (9.5%). Similarly, hypomethylated DMRs represented no changes in gene expression (32.2%), reduced gene expression (9.5%) and elevated gene expression (10.2%) suggesting aberrant DNA methylation by FQI1 has a profound effect on aberrant gene expression (Figure 5B).
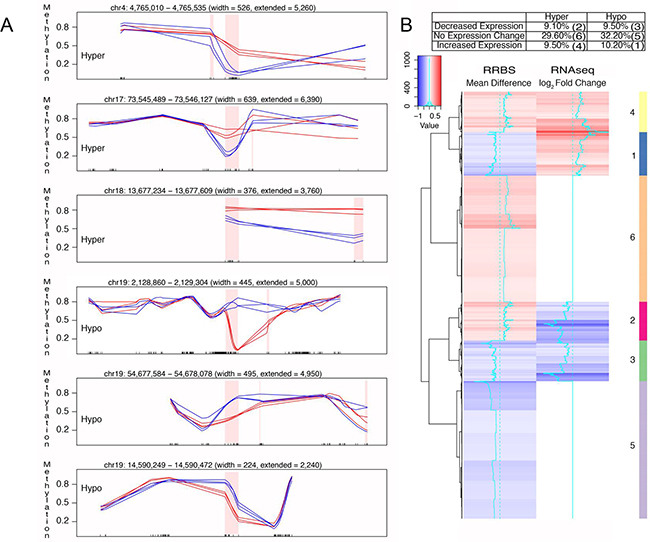
Figure 5: Correlation of differential DNA methylation and altered gene expression with FQI1 treatment. A. Tracks showing hyper and hypomethylated DMRs. Blue lines represent CpG methylation levels in DMSO control triplicates and red lines represent CpG methylation levels in FQI1 treated triplicates. Pink shaded region depicts the DMR and the ticks on the X-axis represent the location of CpGs. B. Heatmap showing the correlation between hyper and hypomethylated DMRs and their corresponding expression levels. Clustering is performed along the rows for 1678 DMRs located near the TSS of the genes represented by the RNAseq data.
Since DNA methylation is historically known as a gene repressor signal, we analyzed the previously demonstrated distribution in unperturbed HEK293T cells for H3K4me3, H3K27ac, and H3K36me3 (activation signature) and H3K4me1 and H3K9me3 (repression signature) histone marks within 6 kb around the 1678 DMRs near the transcription start sites (Supplementary Figure S3 and S4). Both hyper- and hypomethylated DMRs that exhibited transcriptional activation upon addition of FQI1 were already enriched for H3K4me3 and H3K27ac (Supplementary Figure S3 and S4). Notably, in hypomethylated DMRs the ground state H3K4me3 signature was more pronounced in gene sets in which expression was increased upon FQI1 treatment, compared to those with either a decrease or no change in gene expression coinciding with loss of nucleosome (Supplementary Figure S4). We further characterized these DMRs near transcription start sites using WebGestalt, a web-based gene set analysis tool kit to perform GO analysis. Both hyper and hypomethylated DMRs with no change in transcript levels displayed strong enrichment for metabolic pathways (e.g. FBP1, DNMT3A, NMNAT1, AK7, FBP2, B3GAT3, ADA). In the case of genes with decreased expression, we observed enrichment of G1 to S cell cycle control (e.g. POL2A, MCM2) and DNA replication (e.g. PRIM1) pathways for hypermethylated DMRs. For hypomethylated DMRs with decreased expression, GO terms for metabolic pathways (e.g. GSTZ1, ALDH5A1, NDUFS1) were enriched. Finally, for DMRs whose associated genes had increased expression upon treatment with FQI1, we observed enrichment of GO terms for pathways in cancer (e.g. PIAS4, SMAD3, TRAF4, MAP2K1) and RNA transport (e.g. NUP188, EIF3A, NUP35, RAN) in both hyper and hypomethylated DMRs (Supplementary Table S2). Thus, FQI1 treatment impacts the cell cycle and a number of growth signaling pathways, with potential to impact cancer development.
Transcription factor binding may be altered by FQI1-mediated DMRs
We performed in silico analysis for transcription factor-binding motifs in both hypermethylated and hypomethylated DMRs to better understand the effect of aberrant DNA methylation at DMRs on gene expression. DMRs with hypomethylation did not reveal any significant enrichment for transcription factor binding motifs. However, we found enrichment of motifs for STAT (Stat3), ETS (Elk4, Elk1, Fli1), Zinc Finger (Maz, Znf263, REST-NRSF), and bZIP (c-Jun-CRE, Atf1) family of transcription factors in hypermethylated DMRs (Supplementary Table S3). Several studies have demonstrated the negative correlation of CpG methylation within binding sequence with transcription factor binding, particularly for STAT3, c-Jun-CRE and ATF1 [37–40].
DISCUSSION
The transcription factor LSF is a promising protein target for chemotherapy in hepatocellular carcinoma. It is highly expressed in HCC patient samples and cell lines, and promotes oncogenesis in a rodent xenograft HCC model [31, 32]. FQI1 promotes apoptosis in LSF-overexpressing cells, including HCC cells, although primary or immortalized hepatocytes remain unaffected, demonstrating its cancer selectivity [32]. Limited data on in vitro treatment of human HCC cells with LSF inhibitors demonstrated mitotic arrest [33]. However, the molecular mechanism of this inhibitor in cells is significantly lacking. Factor quinolinone inhibitor 1 (FQI1) is a small molecule that effectively and selectively inhibits LSF cellular activity by inhibiting LSF DNA-binding activity both in vitro and in cells [32].
Here we have used human HEK293T cells as a model system to study the effect of FQI1 on its epigenome and gene transcription. HEK293T cells produce similar quantities of LSF as that in HCC cell lines and offer a more homogenous genome compared to cancer cells. Our study suggested that exposure of cells to FQI1 alter the epigenome. Indeed, DNA methylation, a major component of the epigenome showed aberrant DNA methylation and gene expression covering over 2K DMRs. As shown, FQI1 can cause both hyper- and hypomethylation resulting in either activation or repression of gene expression. One hypothesis centered on involvement of differential methylation posits that LSF recruits DNMT1 to gene regulatory regions to repress LSF target genes by traditional methylation mediated gene repression. Then, FQI1 would disrupt the LSF-DNMT1 interaction (in addition to removing LSF from the DNA), which would lead to poor DNMT1 targeting resulting in aberrant DNA methylation and altered DMR (Figure 6A). In addition, LSF target genes that are generally transcriptionally activated or repressed by LSF binding (associated with cofactors other than DNMT1) will lose LSF-mediated gene regulation in the presence of FQI1 (Figure 6B). Finally, some of the genes whose expression was significantly altered downstream of FQI1 treatment (by either mechanism in Figure 6A or 6B) are transcription factors whose binding motifs were overrepresented in DMRs (e.g. STAT3, ELK4, ATF2 and REST), suggesting that the differential gene expression could include a downstream, secondary wave of regulation.
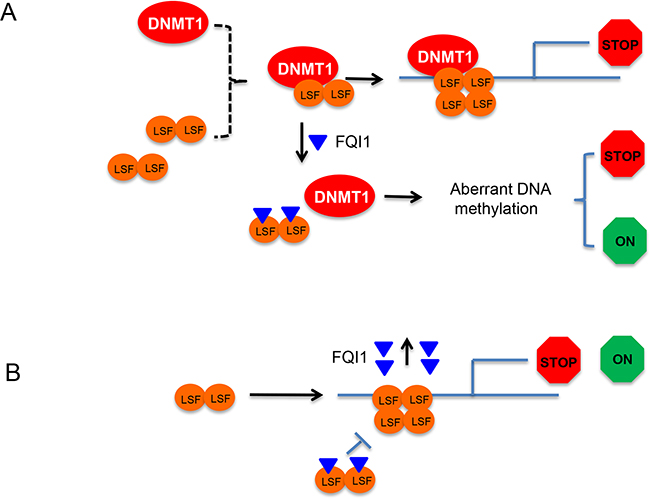
Figure 6: Working models of LSF-DNMT1 complex dissociation by FQI1 and aberrant DNA methylation. A. LSF is depicted as dimer in solution and DNMT1 as monomer. LSF and DNMT1 form a complex for sequence specific gene methylation and gene repression. FQI1 directly binds LSF and interferes with the DNMT1-LSF complex formation leading to mistargeting of DNMT1 resulting in aberrant DNA methylation and both transcriptional activation (green) and repression (red). B. FQI1 binds to LSF and inhibits its binding on DNA. LSF that is targeted for binding to DNA will dissociate FQI1 before binding as tetramer for gene regulation (both positive and negative).
Other transcription factors have been associated with aberrant gene expression patterns in cancer, and also serve as the convergence points of oncogenic signaling that are functionally altered in many cancers. Our data suggest that LSF may behave similarly. For example, the Myc oncogene, a member of the bHLHZip family of transcription factors, is well known for its contribution to cancer by protein/protein and protein/DNA interactions. Myc forms heterodimers with Max, and binds in the promoters of pro-growth and proliferation targets and thereby activates their expression. Activation of these Pol II targets is mediated by the recruitment of different chromatin modifying activities. Myc binds the corepressor Dnmt3a and associates with DNA methyltransferase activity in vivo. Indeed, Myc and Dnmt3a form a ternary complex with Miz-1, which then represses the p21Cip1 promoter [41]. Since LSF association with DNMT1 resulted in activation of DNA methylation, we speculate that interaction between both may trigger hypermethylation of CpG islands containing tumor suppressor genes. It is plausible that the interaction of LSF with the N-terminal of DNMT1 may facilitate release of the catalytic domain from an auto-inhibitory fold formed by interactions between the DNMT1 N- and C-terminal regions thus aiding DNA hypermethylation.
Small molecule inhibitors that can alter the epigenome have become increasingly important in cancer therapy [42]. Recently, small molecule inhibitors such as Decitabine have been used to treat myelodysplastic syndrome (MDS) [43]. This potent DNA methyltransferase inhibitor alters the epigenome in a rapid manner, leading to apoptosis. From the current study, we also observed FQI1 triggers signaling pathways involved in cell cycle regulation.
Recent studies underscore the extensive reprogramming of every component of the epigenetic machinery in cancer including DNA methylation, histone modifications, nucleosome positioning and non-coding RNAs. Epigenome reprogramming has a diverse effect on genome integrity and transcriptional output. A comprehensive understanding of the numerous and diverse molecular phenomena occurring in the epigenome of normal and malignant cells, and the pathways altered by small molecule drugs, will hopefully provide novel mechanistic insights into more effective epigenetic cancer treatment strategies.
MATERIALS AND METHODS
Cell culture, immunoprecipitation and immunofluorescence
HEK293T and COS7 cells were cultured in DMEM media supplemented with 10% fetal bovine serum. For FQI1 treatment, HEK293Tcells were treated with 2.5 μM of FQI1 for 24 h at 37 ˚C.
Immunoprecipitation and immunofluorescence were carried out as described previously [44, 45]. For the co-IP of DNMT1 and LSF in HEK293T cells, 1 mg of the whole cell lysate was incubated with 2 μg of anti-DNMT1 antibody (sc-20701, Santa Cruz) or anti-LSF antibody (Millipore). IP reactions were blotted with anti-DNMT1 (M0231S, New England Biolabs), anti-LSF (610818, BD) and anti-UHRF1 (612264, BD) antibodies as per the manufacturer’s dilution recommendations. Normal IgG was used as a control in all IP reactions. For the detection of DNMT1 and LSF colocalization, COS7 cells were grown on coverslips and co-transfected with DsRed-DNMT1 and 3xFLAG-LSF plasmids. DsRed-DNMT1 was visualized with an excitation wavelength of 594 nm, epitope tagged LSF was detected by mouse anti-FLAG antibody (F3165, Sigma-Aldrich) and visualized with an anti-mouse IgG coupled with Alexa Fluor 488 dye (Molecular Probes). DAPI was used for nuclear staining. Pearson’s correlation coefficient was calculated using NIH imageJ/JACoP [46].
GST-pull down assays
For GST pull-down assays, GST – LSF fragments (1-180, 169-319, 306-420, 383-503, 65-259 (DNA Binding Domain), 326-385 (Sterile Alpha Motif, or SAM, domain, [47]) amino acids) were cloned into the pGEX-5X-1 vector (GE Healthcare), overexpressed in E. coli, and GST-tagged expressed proteins were captured using Glutathione Sepharose beads (GE Healthcare). Sepharose beads containing 10 μg of fusion protein were incubated with 200 ng of recombinant baculovirus expressed DNMT1. Protein bound to the beads was resolved by SDS-PAGE. DNMT1 was visualized by immunoblotting by using Anti-DNMT1 (M0231S, New England Biolabs). For the reciprocal experiment, GST-DNMT1 fusion beads were incubated with 1 μg of purified MBP-LSF protein. LSF was visualized by immunoblotting by using Anti-LSF (610818, BD).
DNA methyltransferase assays
DNA methyltransferase assays were carried out as described previously [48]. The role of LSF and FQI1 on DNA methylation was determined by assaying the activity of DNMT1 in the presence and absence of LSF and FQI1. Methylation reactions were performed using 80 nM DNMT1, 100 ng hemimethylated substrate and 5 μM tritiated AdoMet, incubating for 30 minutes at 37˚C along with various concentrations of LSF. Samples were processed using a filter disc method and the [3H]CH3 incorporated into the DNA was determined using a liquid scintillation counter.
Genome-wide DNA methylation analysis
Genome-wide DNA methylation analysis was carried out using the Reduced Representation Bisulfite Sequencing method [49]. Genomic DNA (2 μg) isolated from HEK293T treated with FQI1 or DMSO control (biological triplicates) for 48 hrs was digested with MspI, end-repaired and dA-tailed. Methylated NEB Illumina loop adaptor was ligated to the processed fragmented DNA (E7370S, New England Biolabs) and digesting the uracil with USER enzyme opened the adaptor loop. Ligation products were size-selected for 150 to 400 bp fragments on 2% agarose gels and bisulfite converted using the EZ DNA Methylation Kit (Zymo Research). Libraries were enriched by PCR using EpiMark Hot Start Taq DNA Polymerase (New England Biolabs) and sequenced on the Illumina GAII platform with 72 bp paired-end reads. Libraries were made and sequenced using two independent replicates.
Adaptor and low quality sequences (Phred score < 20) were trimmed from sequencing reads using the trim_galore package (http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/) with the parameter of –RRBS –paired. Reads were mapped to hg19 using Bismark with Bowtie2 [50]. CpG methylation levels were calculated with uniquely mapped reads using Bismark methylation extractor with the parameter of -p –no_overlap and a minimum coverage of 3. Differential methylation analysis was carried out using the bsseq R package [51]; CpGs present in at least two replicates of each group were retained for downstream analysis. DMRs were identified containing a minimum of 3 CpGs and mean difference between the control and FQI1-treated samples of greater than 0.1 using BSmooth package [51]. DMRs were annotated by mapping the genomic coordinates to various genomic regions, i.e., ±2 kb of transcription start site (TSS), CpG islands in promoter, CpG islands in gene body, CpG islands in deserts, CpG islands, CpG island ± 2kb, 5’ UTR, coding exon, intron, 3’ UTR, and known genes using BEDTools [52]. DMRs were visualized by plotting the CpG methylation levels in a 5 kb window around the DMR using bsseq R package. Density plots for number of CpGs in DMR and the lengths of DMRs were plotted using ggplot2 R package [53].
RNA-seq analysis
Total RNA was extracted using Trizol reagent (Invitrogen) from HEK293T cells treated with either 2.5 μM of FQI1 or 0.01% DMSO for 12 hr at 37°C. RNA-seq libraries were constructed using the NEBNext® Ultra™ Directional RNA Library Prep Kit (New England Biolabs). Average insert size of libraries was 200 bp. Libraries were sequenced in 76 bp paired-end read mode on the IlluminaNextSeq 500 platform. Sequencing reads were mapped to hg19 using TopHat2 (-r 60 –library-type fr-firststrand) [54]. The number of reads mapped to each known gene was counted using htseq-count tools (-t exon -s reverse –i gene_id) and differential gene expression analyzed with DESeq2 [55, 56]. The p-value of differential expression was corrected using the Benjamini-Hochberg adjustment method. Genes with adjusted p-value < 0.01 were considered significantly differentially expressed. Sample distance was calculated using Euclidean distance on the rlog-transformed counts. Heatmaps were generated using the Z-scored-transformed rlog values of significantly changed genes (top 100). MA plot showing differential expression (log2 fold change) versus mean expression value between FQI and control was plotted. GO analysis was performed using GAGE package (Generally Applicable Geneset Enrichment for Pathway Analysis) [57].
Correlation of DNA methylation and gene expression change
In order to investigate the correlation of DNA methylation alteration and gene expression change after FQI1 treatment, we analyzed the differential methylation again and this time we focused on the ±2 kb regions flanking TSS. Only regions with more than three CpGs were kept. Only genes with significant expression changes (adj p-value < 0.05) and significant methylation changes (q-value < 0.05) were included in the correlation study. Genes were clustered based on the CpG methylation difference of their TSS ±2 kb regions and their expression level using k-means clustering.
Transcription factor binding motif analysis
Presence of transcription factor binding motifs in the DMRs (hyper and hypomethylated) associated with RNAseq data was evaluated using findMotifsGenome function of homer with masked genomic regions [58]. Motifs with a p-value of at least 0.01 were considered for analysis.
Quantitative PCR analysis of gene expression
HEK293Tcells were treated with 2.5 μM of FQI1 for 12 or 24 hr at 37°C. Total RNA was extracted using Trizol reagent (Invitrogen) from HEK293T treated FQI1, or DMSO as a control. 1 μg of total RNA was reverse transcribed to cDNA using ProtoScript II First Strand cDNA Synthesis Kit (New England Biolabs) and 40 ng cDNA was subjected to real-time PCR analysis using SYBR Green Supermix (Bio-Rad). The following primers were used: AURKA_F: GCAGATTTTGGGTGGTCAGT, AURKA_R: TCTTGAAATGAGGTCCCTGG; SAPC D2_F: GAGCAGAACCGACTCCTCAC, SAPCD2_R: GGCCCACAAGGACTAGATGA; KIF2A_F: CACCCA CCTCAACCAGAACT, KIF2A_R: AGCCAGCCAGAT CACAGAGT; MCM5_F: GACTTCATGCCCACCA TCTT, MCM5_R: TCACGTGCAGAGTGATGACA; MCM6_F: AACCAGCAACTTTCCACCAC, MCM6_R: GAAAAGTTCCGCTCACAAGC; MCM7_F: TGAGTTC GACAAGATGGCTG, MCM7_R: CCGTAGGTCAT TGTCTCGGT; expression change was calculated using the 2-ΔΔCt method.
GO analysis
GO analysis for the hypermethylated and hypomethylated DMRs was performed using WEB-based GEne SeT AnaLysis Toolkit (WebGestalt) [57]. Annotation of the DMRs was performed using homer and the gene names were used as the input for GO analysis with default parameters (minimum of 2 hits per category, hypergeometric statistics method and “BH” multiple test adjustment settings) [58]. Top 10 enriched GO terms from KEGG and WIKI pathway analysis were generated.
ChIP fragment depth analysis for histone marks
ChIP-seq data sets for H3K4me3, H3K4me1, H3K9me3, H3K27ac and H3K36me3 histone marks in HEK293T cell line were downloaded from ENCODE (ENCFF001FJF, ENCFF002AAV, ENCFF002AAX, ENCFF002ABA, and ENCFF002ABD). Tag densities for each data set was calculated using makeTagDirectory function of homer package. Further, a histogram of the tag densities for each histone mark was plotted using the annotate Peaks function of homer by extending ±3 kb around the DMRs that showed no expression change, decreased expression and increased expression in case of both hypermethylated and hypomethylated condition [58].
ACKNOWLEDGMENTS
We would like to thank Drs. Donald Comb, Rich Roberts, William Jack and Clotilde Carlow at New England Biolabs Inc. for research support and encouragement. The authors thank Dr. Lauren Brown (Boston University Center for Molecular Discovery) for the preparation of FQI1. UH research on this project was supported by Ignition Awards from Boston University and a Johnson & Johnson Clinical Innovator’s Award through Boston University. SES research is supported by the NIH (P50 GM067041 & R24 GM111625). Research performed by HGC was partly a requirement for the MCBB graduate program at Boston University and supported by NEB.
CONFLICTS OF INTEREST
The authors declare that they have no conflicts of interest.
Author contributions
Hang Gyeong Chin performed majority of experiments
V K Chaithanya Ponnaluri performed Bioinformatic analysis
Guoqiang Zhang performed Bioinformatic analysis
Pierre-Olivier Estève performed microscopy and image analysis
Scott E. Schaus provided FQI1 inhibitor and experiment planning
Ulla Hansen provided experimental planning, manuscript writing and supervision
Sriharsa Pradhan experimental planning, wrote the manuscript and supervised experimental work included in the manuscript
REFERENCES
1. Zhang G, Pradhan S. Mammalian epigenetic mechanisms. IUBMB Life. 2014; 66: 240-56. doi: 10.1002/iub.1264.
2. Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012; 13: 484-92. doi: 10.1038/nrg3230.
3. Schubeler D. Function and information content of DNA methylation. Nature. 2015; 517: 321-6. doi: 10.1038/nature14192.
4. Wu JC, Santi DV. Kinetic and catalytic mechanism of HhaI methyltransferase. J Biol Chem. 1987; 262: 4778-86.
5. Ziller MJ, Gu H, Muller F, Donaghey J, Tsai LT, Kohlbacher O, De Jager PL, Rosen ED, Bennett DA, Bernstein BE, Gnirke A, Meissner A. Charting a dynamic DNA methylation landscape of the human genome. Nature. 2013; 500: 477-81. doi: 10.1038/nature12433.
6. Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, Nery JR, Lee L, Ye Z, Ngo QM, Edsall L, Antosiewicz-Bourget J, Stewart R, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009; 462: 315-22. doi: 10.1038/nature08514.
7. Cokus SJ, Feng S, Zhang X, Chen Z, Merriman B, Haudenschild CD, Pradhan S, Nelson SF, Pellegrini M, Jacobsen SE. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature. 2008; 452: 215-9. doi: 10.1038/nature06745.
8. Ting AH, McGarvey KM, Baylin SB. The cancer epigenome--components and functional correlates. Genes Dev. 2006; 20: 3215-31. doi: 10.1101/gad.1464906.
9. Baylin SB, Chen WY. Aberrant gene silencing in tumor progression: implications for control of cancer. Cold Spring Harb Symp Quant Biol. 2005; 70: 427-33. doi: 10.1101/sqb.2005.70.010.
10. Herman JG. Hypermethylation of tumor suppressor genes in cancer. Semin Cancer Biol. 1999; 9: 359-67. doi: 10.1006/scbi.1999.0138.
11. Dobrovic A, Simpfendorfer D. Methylation of the BRCA1 gene in sporadic breast cancer. Cancer Res. 1997; 57: 3347-50.
12. Herman JG, Jen J, Merlo A, Baylin SB. Hypermethylation-associated inactivation indicates a tumor suppressor role for p15INK4B. Cancer Res. 1996; 56: 722-7.
13. Jiang Y, Dunbar A, Gondek LP, Mohan S, Rataul M, O’Keefe C, Sekeres M, Saunthararajah Y, Maciejewski JP. Aberrant DNA methylation is a dominant mechanism in MDS progression to AML. Blood. 2009; 113: 1315-25. doi: 10.1182/blood-2008-06-163246.
14. Bestor TH. Cloning of a mammalian DNA methyltransferase. Gene. 1988; 74: 9-12.
15. Okano M, Xie S, Li E. Cloning and characterization of a family of novel mammalian DNA (cytosine-5) methyltransferases. Nat Genet. 1998; 19: 219-20. doi: 10.1038/890.
16. Leonhardt H, Page AW, Weier HU, Bestor TH. A targeting sequence directs DNA methyltransferase to sites of DNA replication in mammalian nuclei. Cell. 1992; 71: 865-73.
17. Bostick M, Kim JK, Esteve PO, Clark A, Pradhan S, Jacobsen SE. UHRF1 plays a role in maintaining DNA methylation in mammalian cells. Science. 2007; 317: 1760-4. doi: 10.1126/science.1147939.
18. Sharif J, Muto M, Takebayashi S, Suetake I, Iwamatsu A, Endo TA, Shinga J, Mizutani-Koseki Y, Toyoda T, Okamura K, Tajima S, Mitsuya K, Okano M, et al. The SRA protein Np95 mediates epigenetic inheritance by recruiting Dnmt1 to methylated DNA. Nature. 2007; 450: 908-12. doi: 10.1038/nature06397.
19. Hashimoto H, Horton JR, Zhang X, Bostick M, Jacobsen SE, Cheng X. The SRA domain of UHRF1 flips 5-methylcytosine out of the DNA helix. Nature. 2008; 455: 826-9. doi: 10.1038/nature07280.
20. Feng S, Cokus SJ, Zhang X, Chen PY, Bostick M, Goll MG, Hetzel J, Jain J, Strauss SH, Halpern ME, Ukomadu C, Sadler KC, Pradhan S, et al. Conservation and divergence of methylation patterning in plants and animals. Proc Natl Acad Sci U S A. 2010; 107: 8689-94. doi: 10.1073/pnas.1002720107.
21. Li E, Bestor TH, Jaenisch R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell. 1992; 69: 915-26.
22. Pacaud R, Brocard E, Lalier L, Hervouet E, Vallette FM, Cartron PF. The DNMT1/PCNA/UHRF1 disruption induces tumorigenesis characterized by similar genetic and epigenetic signatures. Sci Rep. 2014; 4: 4230. doi: 10.1038/srep04230.
23. Mudbhary R, Hoshida Y, Chernyavskaya Y, Jacob V, Villanueva A, Fiel MI, Chen X, Kojima K, Thung S, Bronson RT, Lachenmayer A, Revill K, Alsinet C, et al. UHRF1 overexpression drives DNA hypomethylation and hepatocellular carcinoma. Cancer Cell. 2014; 25: 196-209. doi: 10.1016/j.ccr.2014.01.003.
24. Du J, Johnson LM, Jacobsen SE, Patel DJ. DNA methylation pathways and their crosstalk with histone methylation. Nat Rev Mol Cell Biol. 2015; 16: 519-32. doi: 10.1038/nrm4043.
25. Esteve PO, Chin HG, Pradhan S. Human maintenance DNA (cytosine-5)-methyltransferase and p53 modulate expression of p53-repressed promoters. Proc Natl Acad Sci U S A. 2005; 102: 1000-5. doi: 10.1073/pnas.0407729102.
26. Robertson KD, Ait-Si-Ali S, Yokochi T, Wade PA, Jones PL, Wolffe AP. DNMT1 forms a complex with Rb, E2F1 and HDAC1 and represses transcription from E2F-responsive promoters. Nat Genet. 2000; 25: 338-42. doi: 10.1038/77124.
27. Pradhan S, Kim GD. The retinoblastoma gene product interacts with maintenance human DNA (cytosine-5) methyltransferase and modulates its activity. EMBO J. 2002; 21: 779-88. doi: 10.1093/emboj/21.4.779.
28. Myant K, Stancheva I. LSH cooperates with DNA methyltransferases to repress transcription. Mol Cell Biol. 2008; 28: 215-26. doi: 10.1128/mcb.01073-07.
29. Veljkovic J, Hansen U. Lineage-specific and ubiquitous biological roles of the mammalian transcription factor LSF. Gene. 2004; 343: 23-40. doi: 10.1016/j.gene.2004.08.010.
30. Santhekadur PK, Rajasekaran D, Siddiq A, Gredler R, Chen D, Schaus SE, Hansen U, Fisher PB, Sarkar D. The transcription factor LSF: a novel oncogene for hepatocellular carcinoma. Am J Cancer Res. 2012; 2: 269-85.
31. Yoo BK, Emdad L, Gredler R, Fuller C, Dumur CI, Jones KH, Jackson-Cook C, Su ZZ, Chen D, Saxena UH, Hansen U, Fisher PB, Sarkar D. Transcription factor Late SV40 Factor (LSF) functions as an oncogene in hepatocellular carcinoma. Proc Natl Acad Sci U S A. 2010; 107: 8357-62. doi: 10.1073/pnas.1000374107.
32. Grant TJ, Bishop JA, Christadore LM, Barot G, Chin HG, Woodson S, Kavouris J, Siddiq A, Gredler R, Shen XN, Sherman J, Meehan T, Fitzgerald K, et al. Antiproliferative small-molecule inhibitors of transcription factor LSF reveal oncogene addiction to LSF in hepatocellular carcinoma. Proc Natl Acad Sci U S A. 2012; 109: 4503-8. doi: 10.1073/pnas.1121601109.
33. Rajasekaran D, Siddiq A, Willoughby JL, Biagi JM, Christadore LM, Yunes SA, Gredler R, Jariwala N, Robertson CL, Akiel MA, Shen XN, Subler MA, Windle JJ, et al. Small molecule inhibitors of Late SV40 Factor (LSF) abrogate hepatocellular carcinoma (HCC): Evaluation using an endogenous HCC model. Oncotarget. 2015; 6: 26266-77. doi: 10.18632/oncotarget.4656.
34. Kokoszynska K, Ostrowski J, Rychlewski L, Wyrwicz LS. The fold recognition of CP2 transcription factors gives new insights into the function and evolution of tumor suppressor protein p53. Cell Cycle. 2008; 7: 2907-15.
35. Traylor-Knowles N, Hansen U, Dubuc TQ, Martindale MQ, Kaufman L, Finnerty JR. The evolutionary diversification of LSF and Grainyhead transcription factors preceded the radiation of basal animal lineages. BMC Evol Biol. 2010; 10: 101. doi: 10.1186/1471-2148-10-101.
36. Luo W, Friedman MS, Shedden K, Hankenson KD, Woolf PJ. GAGE: generally applicable gene set enrichment for pathway analysis. BMC Bioinformatics. 2009; 10: 161. doi: 10.1186/1471-2105-10-161.
37. Shimozaki K, Namihira M, Nakashima K, Taga T. Stage- and site-specific DNA demethylation during neural cell development from embryonic stem cells. J Neurochem. 2005; 93: 432-9. doi: 10.1111/j.1471-4159.2005.03031.x.
38. Takizawa T, Nakashima K, Namihira M, Ochiai W, Uemura A, Yanagisawa M, Fujita N, Nakao M, Taga T. DNA methylation is a critical cell-intrinsic determinant of astrocyte differentiation in the fetal brain. Dev Cell. 2001; 1: 749-58.
39. Jones B, Chen J. Inhibition of IFN-gamma transcription by site-specific methylation during T helper cell development. EMBO J. 2006; 25: 2443-52. doi: 10.1038/sj.emboj.7601148.
40. Kuroda A, Rauch TA, Todorov I, Ku HT, Al-Abdullah IH, Kandeel F, Mullen Y, Pfeifer GP, Ferreri K. Insulin gene expression is regulated by DNA methylation. PLoS One. 2009; 4: e6953. doi: 10.1371/journal.pone.0006953.
41. Brenner C, Deplus R, Didelot C, Loriot A, Vire E, De Smet C, Gutierrez A, Danovi D, Bernard D, Boon T, Pelicci PG, Amati B, Kouzarides T, et al. Myc represses transcription through recruitment of DNA methyltransferase corepressor. EMBO J. 2005; 24: 336-46. doi: 10.1038/sj.emboj.7600509.
42. Sharma S, Kelly TK, Jones PA. Epigenetics in cancer. Carcinogenesis. 2010; 31: 27-36. doi: 10.1093/carcin/bgp220.
43. Ahuja N, Sharma AR, Baylin SB. Epigenetic Therapeutics: A New Weapon in the War Against Cancer. Annu Rev Med. 2016; 67: 73-89. doi: 10.1146/annurev-med-111314-035900.
44. Andrews NC, Faller DV. A rapid micropreparation technique for extraction of DNA-binding proteins from limiting numbers of mammalian cells. Nucleic Acids Res. 1991; 19: 2499.
45. Estève PO, Chin HG, Smallwood A, Feehery GR, Gangisetty O, Karpf AR, Carey MF, Pradhan S. Direct interaction between DNMT1 and G9a coordinates DNA and histone methylation during replication. Genes Dev. 2006; 20: 3089-103. doi: 10.1101/gad.1463706.
46. Bolte S, Cordelieres FP. A guided tour into subcellular colocalization analysis in light microscopy. J Microsc. 2006; 224: 213-32. doi: 10.1111/j.1365-2818.2006.01706.x.
47. Kim CA, Bowie JU. SAM domains: uniform structure, diversity of function. Trends Biochem Sci. 2003; 28: 625-8. doi: 10.1016/j.tibs.2003.11.001.
48. Pradhan S, Bacolla A, Wells RD, Roberts RJ. Recombinant human DNA (cytosine-5) methyltransferase. I. Expression, purification, and comparison of de novo and maintenance methylation. J Biol Chem. 1999; 274: 33002-10.
49. Meissner A, Gnirke A, Bell GW, Ramsahoye B, Lander ES, Jaenisch R. Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Res. 2005; 33: 5868-77. doi: 10.1093/nar/gki901.
50. Krueger F, Andrews SR. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics. 2011; 27: 1571-2. doi: 10.1093/bioinformatics/btr167.
51. Hansen KD, Langmead B, Irizarry RA. BSmooth: from whole genome bisulfite sequencing reads to differentially methylated regions. Genome Biol. 2012; 13: R83. doi: 10.1186/gb-2012-13-10-r83.
52. Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010; 26: 841-2. doi: 10.1093/bioinformatics/btq033.
53. Wickham H. (2009). ggplot2: elegant graphics for data analysis: Springer).
54. Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013; 14: R36. doi: 10.1186/gb-2013-14-4-r36.
55. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014; 15: 550. doi: 10.1186/s13059-014-0550-8.
56. Anders S, Pyl PT, Huber W. HTSeq-a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015; 31: 166-9. doi: 10.1093/bioinformatics/btu638.
57. Zhang B, Kirov S, Snoddy J. WebGestalt: an integrated system for exploring gene sets in various biological contexts. Nucleic Acids Res. 2005; 33: W741-8. doi: 10.1093/nar/gki475.
58. Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010; 38: 576-89. doi: 10.1016/j.molcel.2010.05.004.