
INTRODUCTION
Abnormalities in centrosome number and function are common in many cancers, indicating that loss of centrosome cycle regulation may be a major factor in tumor progression [1]. Nek2 (NIMA-related kinase 2) is a human cell cycle-dependent serine/threonine protein kinase that localizes to the centrosome [2]. Nek2 is related to the fungal protein NIMA (never in mitosis gene A), an essential mediator of mitotic entry in Aspergillus nidulans. Like NIMA the activity of Nek2 peaks prior to mitotic entry, although in contrast to NIMA, Nek2 is not essential for mitotic entry in human cells. However, Nek2 does play a key role in ensuring timely assembly of the mitotic spindle, a scaffold that is vital for accurate segregation of sister chromatids during mitosis [3]. Nek2 interacts with centrosomal proteins that assemble into a filamentous linker that holds centrosomes in close proximity throughout interphase. Phosphorylation of these proteins by Nek2 in late G2 promotes linker disassembly and loss of centrosome cohesion; this in turn allows the timely separation of centrosomes in prophase [4]. In addition, Nek2 can promote cell cycle progression through phosphorylation of motor proteins that lead to disassembly of primary cilia [5]. Overexpression of Nek2 results in premature centrosome separation, which is a cause of chromosome segregation errors, aneuploidy and chromosomal instability, common genetic abnormalities observed in tumor cells. Upregulation of Nek2 expression has been observed in many human tumors, including ovarian [6], colorectal [7], prostatic [8], hepatocellular carcinoma [9] and breast cancer [10]. Nek2 depletion in a number of tumor cell lines causes growth suppression and apoptosis, while anti-tumor activity has been reported in a range of tumor cell lines following abrogation of Nek2 activity by RNAi depletion alone, or combined with cisplatin [11–14]. Hence, inhibiting Nek2 with small-molecule kinase inhibitors has potential as a novel cancer therapy.
Increasing efforts are now being applied to the development of Nek2 inhibitors with several small molecule inhibitors disclosed (Table 1). Examples include the aminopyrazine ATP-competitive inhibitor 1 (Nek2IC50 = 0.23 μM) [15] and a benzimidazole-based series with > 200-fold selectivity for Nek2 over Plk1 (e.g. 2:Nek2 IC50 = 0.36 μM) [16]. To address the modest ligand efficiency (LE) of 2, a hybrid class of compounds was generated by combining the core aminopyrazine moiety of the initial series with side-chains from the benzimidazole series. Optimisation of this new class of inhibitors improved potency, LE and kinase selectivity against a panel of cell cycle kinases, and culminated in the identification of 3 (Nek2 IC50 = 0.022 μM) [17]. A series of ATP-competitive reversible Nek2 inhibitors was also identified via high-throughput screening. These viridin/wortmannin-like compounds exhibited only modest Nek2-inhibitory activity (e.g. 4:Nek2 IC50 = 1.9 μM), albeit with selectivity over other Nek family members. In addition, some activity was observed in cellular growth inhibition and centrosome separation assays [18]. More recently, an irreversible inhibitor (5) of Nek2 was identified via structure-guided design [19]. Interestingly, compound 5 was shown to react with the Cys22 residue of Nek2 and achieved sub-micromolar activity (Nek2 IC50 = 0.77 μM).
Table 1. Examples of reported small molecule Nek2 inhibitors
In the present study, a medium-throughput screen revealed that purines bearing 6-alkoxy substituents were ATP-competitive inhibitors of both Nek2 and CDK2 (e.g. 6: CDK2 IC50 = 0.005 μM; Nek2 IC50 = 12 μM) (Table 1). Modifications around the purine scaffold were carried out to repurpose this class of kinase inhibitor to improve the potency against Nek2, whilst reducing activity against CDK2. Whilst it is acknowledged that a significant extent of structural homology exists between kinome members, and dissecting CDK2 inhibiton from Nek2 inhibition was expected to present a significant challenge, it was considered that structural differences between the two proteins could be highlighted using appropriately designed probes. From our initial screen, it was evident that in particular, purines bearing meta- or para-substituted 2-arylamino groups that contained a basic functionality afforded improved selectivity for Nek2 over CDK2 (e.g. 7: CDK2 IC50 = 5.6 μM; Nek2 IC50 = 0.89 μM).
This paper describes extensive structure-guided design, synthesis and structure-activity relationship (SAR) studies conducted with the purine scaffold and directed towards the development of potent and selective reversible Nek2 inhibitors. These tools have provided an initial insight in to some of the key requirements for selective inhibition of Nek2 over CDK2 using purine-based inhibitors.
RESULTS AND DISCUSSION
Structure-guided inhibitor design
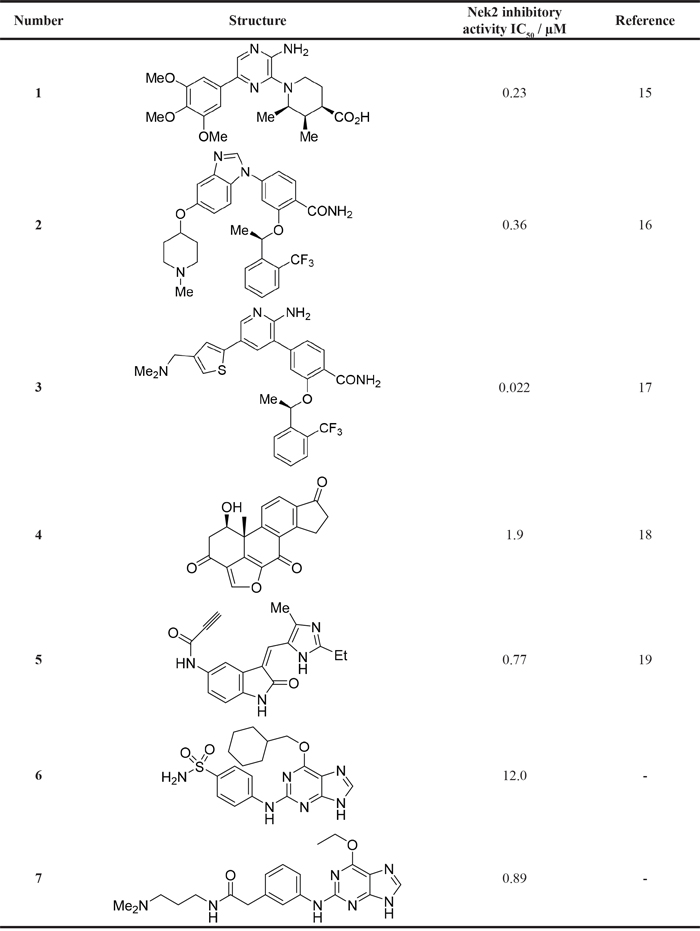
As a starting point for understanding the mode of binding of arylaminopurines to Nek2, the X-ray crystal structure of 8, identified from the initial screening, was determined in complex with Nek2 and revealed that the compound binds via a hydrogen bonding triplet between the purine N9-H, N3 and C2-NH, and the kinase hinge region residues Cys-89 and Glu-87 (Figure 1). Alkylation or removal of the participating purine nitrogen atoms would, therefore, be expected to be detrimental to activity towards Nek2 as for CDK2 and offer no basis for differentiation. However, the 6-alkoxy substituent was considered a candidate for remodeling of prototype inhibitors to differentiate between Nek2 and CDK2 inhibition. The 6-cyclohexylmethyl group occupies a lipophilic cavity near the ribose-binding pocket in CDK2 and is critical for activity [20]. A van der Waals contact may be formed between the 2-arylamino group and Gly-92. No definite interactions were observed between the amide functionality of 8 with the binding site, although the amide group is close to Asp-93 (Figure 1C), and it was considered that this may be exploitable.
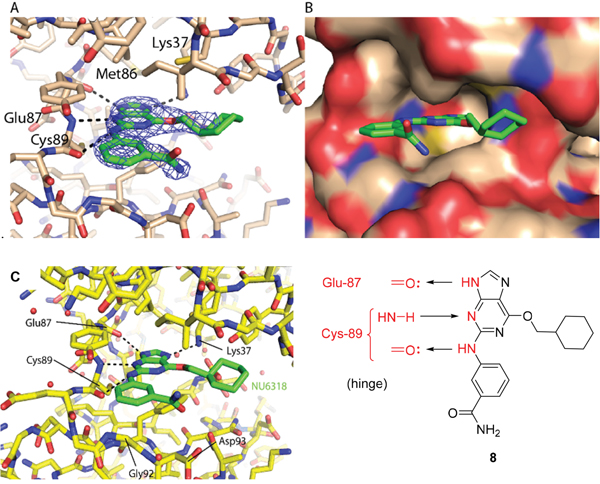
Figure 1: X-ray crystal structure of Nek2 in complex with 6-alkoxypurine inhibitor 8. A. View of compound 8 (carbon atoms coloured green) in the ATP-binding pocket of Nek2 (carbon atoms coloured beige). H-bonds are shown as dashed lines. A 2mFo-dFc electron density map is shown as a blue wire-mesh around the compound. B. View of the ATP-binding pocket of Nek2 shown as a surface. C. Crystal structure of carboxamide 8 (green) bound to the T175A Nek2 mutant (carbon atoms are coloured yellow, oxygen coloured red, and nitrogen coloured blue). Hydrogen bonds are represented as dotted lines and important residues are highlighted.
A comparison of the purines 6 and 8 in the CDK2 and Nek2 ATP-binding sites is shown in Figure 2. The aromatic ring systems of 8 in Nek2 are co-planar, whereas for 6 bound to CDK2 the 2-arylamino ring is rotated ~13° relative to the purine core due to sulfonamide interactions with Asp-86 (equivalent to Asp-93 of Nek2). Thus, interactions between the 3-benzamide moiety and the Nek2 binding site do not appear to affect the conformation of the purine. As a starting point for these studies, it was proposed that selective inhibition of Nek2 over CDK2 may be achieved through judicious modification of the purine 2-arylamino motif or the O6-substituent.
Figure 2: Structural overlay of 8-T175A-Nek2A complex (green and cyan) and the 6-T160pCDK2-cyclin A (pink and yellow). Rotation of the 2-arylamino group with respect to the purine is only observed for 6 in CDK2, but not for 8 in Nek2.
Synthesis of purine-based probes for selective Nek2 inhibition
Modifications at the 2-Arylamino Position
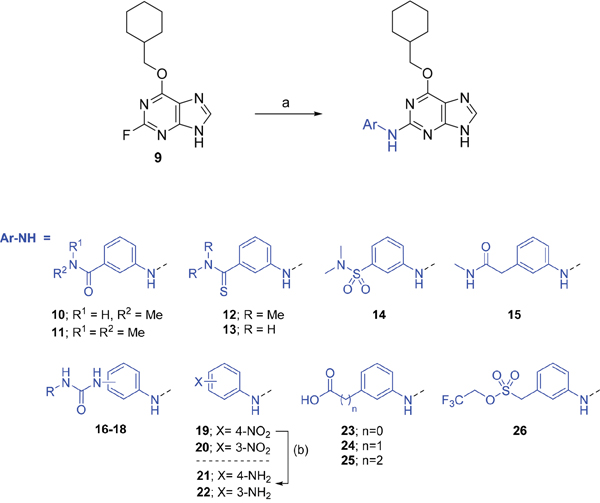
Investigation at the purine 2-position began with the synthesis of simple 2-arylamino purine derivatives containing hydrogen-bond donors/acceptors, in which the substitution pattern around the aryl ring was varied and the linker length between the ring and various functionalities was altered. From the initial screening data, several structural motifs at this position showed Nek2 inhibition, including carboxamides, sulfonamides and ureas. These served as the targets for synthesis. Initial compounds were prepared by direct nucleophilic aromatic substitution of a purine 2-fluoro substituent of 9 with a range of anilines (see supporting information for synthesis of corresponding anilines where otherwise not shown); a second approach allowed diversification of some of these simple 2-arylamino-purines.
As one of the original hits from the screen was a carboxamide derivative, it was decided to begin this study by synthesising analogues of the carboxamide to investigate how this may be modified. Carboxamides 10 and 11 and related thiocarboxamides 12 and 13 were prepared by reaction of fluoropurine 9, with either a commercially available or a synthesized (see ESI) aniline, in 2,2,2-trifluoroethanol (TFE) containing trifluoroacetic acid (TFA) [20–22] (Scheme 1). A carboxamide isostere, the thiocarboxamide group, was investigated as the sulfur atom of a thioamide is a weaker hydrogen bond acceptor than the oxygen atom of an amide, but has a stronger hydrogen bond donating ability at the NH [23]. The sulfonamide equivalent of carboxamide 11 was synthesized via a similar route affording 14. To probe the effect of sidechain homologation of compound 10, N-methyl-homocarboxamide 15 was prepared from the corresponding precursor, 3-aminophenyl-(N-methyl)-acetamide (see ESI). Compounds 16-18, comprising a urea motif bearing linkers to a terminal basic group, were also prepared according to the standard method using synthesised anilines (see ESI). Para- and meta-nitrophenylaminopurines 19 and 20 were prepared in the same manner and subsequently converted in to the amino compounds (21 and 22) by catalytic hydrogenation. Anilines 21 and 22, carboxylic acids 23 and 24 and sulfonate ester 26 were prepared principally for further elaboration of the 2-arylamino sidechain (Scheme 2).
Scheme 1: Synthesis of 2-substituted purine derivatives I.a
a Reagents and conditions: (a) Appropriate aniline, TFA, 2,2,2-trifluoroethanol, 90 °C, 18 h, 17-77%; (b) Pd/C, H2, MeOH, RT, 18 h.
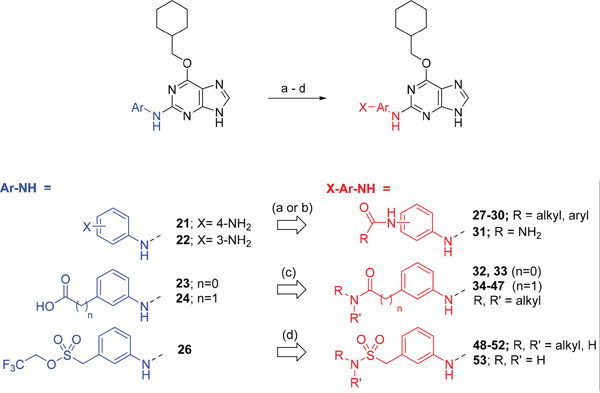
Scheme 2: Synthesis of 2-substituted purine derivatives II.a a Reagents and conditions: (a) i. RCOCl, Et3N, DMAP, THF, RT (or 70 °C as required), 18 h, 31-81%; ii. TFA, DCM, RT, 18 h, 43 - 100%; (b) i. NaOCN, TFA, DMF, RT, 18 h; ii. TFA, DCM, RT, 18 h; (c) i. CDI, DIPEA, DMF, RT, 90 min; ii. RR'NH, RT, 18 h, 20-81%; (d) i. DBU, RR'NH, THF, MW, 160 °C, 15 min; ii. (for compound 53) 52, TFA (neat), RT, 6 h, 85%.
As indicated, para- and meta-aminophenylaminopurines 21 and 22 (Scheme 1) were treated with acyl chlorides to provide the desired N-acyl aniline ‘reversed amide’ derivatives 27-30 without purine N-9 protection (Scheme 2). In addition to the desired products, the reaction also afforded an unstable acylation product at the purine N-9, which was easily removed by treatment with TFA. This allowed the investigation of the exact preferences for the amide group orientation, as well as the substitution position at the 2-arylamino group. To investigate whether a second hydrogen bond-donor group could form additional favourable interactions over that of a simple carboxamide and to quantify this in comparison with more complex urea derivatives, the parent urea e.g. 31 was prepared. Arylureas were synthesised by treatment of precursor aniline 22 with isocyanic acid (HN=C=O), generated in situ from sodium cyanate and TFA (Scheme 2) [24]. As previously observed within the reversed amides series, an undesired urea product was also formed at the purine N-9 and was cleaved by treatment with TFA.
For the synthesis of a focussed set of homocarboxamides a convergent multiple-parallel approach was undertaken (Scheme 2). Using carboxylic acids 25 and 26 a library of amides (32-47) was obtained by coupling with aliphatic or aromatic amines [25, 26]. To further understand the effect of homologation of the hydrogen bond donor-acceptor group, a series of N-2-arylmethanesulfonamides (48-52) was also synthesized from the parent 2,2,2-trifluoroethanesulfonate 26 (Scheme 2). To complete the SARs, the meta-substituted methylsulfonamide 53 was also synthesized by deprotection of the p-methoxybenzyl (PMB) group of 52.
Modifications at the O6-Alkyl Position
Deletion and contraction of the 6-cyclohexylmethyl substituent
Crystal structures of Nek2 and CDK2 suggested that the O6-cyclohexylmethyl group was more important for CDK2 inhibition than Nek2, and indicates that varying the size and conformation of the purine 6-substituent might enable discrimination between Nek2 and CDK2. Furthermore, a suitable group at this position could possibly make a favorable interaction with Lys-37 (K37), which is near the ribose binding pocket of Nek2 (Figure 3B). Although CDK2 also has a lysine residue (K33) near the ribose site (Figure 3A), its disposition and environment differs and so substituents at the purine 6-position could increase Nek2 selectivity by exhibiting a selective impact on its Lys-37.
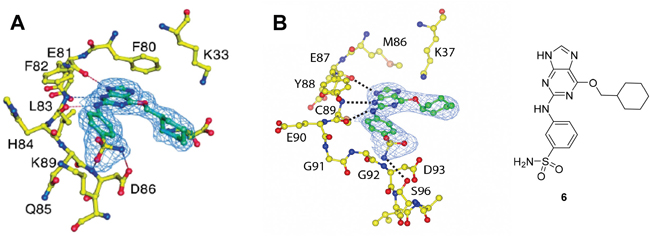
Figure 3: Binding of purine 6 to: A. active Thr 160-phosphorylated CDK2-cyclin A complex (T160pCDK2-cyclin A); and B. inactive Nek2.
Purines bearing O6-alkyl substituents, ethyl and sec-butyl (59 and 60, respectively), were prepared from the 6-chloropurine precursor (54), by 6-position substitution with alkoxides, followed by addition of anilines at the 2-position that had been found to confer Nek2 inhibition (Scheme 3). To overcome some purification problems encountered during the synthesis of purines with dimethylaminopropyl side chains, carboxylic acid 61 was synthesized and used without further purification in the preparation of 1-(3-aminopropyl)imidazole derivative 62.
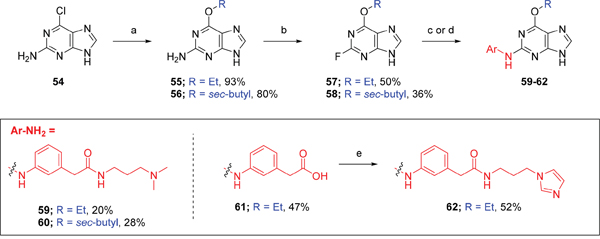
Scheme 3: Synthesis of O6-alkyl-2-arylaminopurines.a a Reagents and conditions: (a) ROH, Na, reflux, 18 h; (b) HBF4, NaNO2, H2O, 0 °C → RT, 24 h; (c) (i) 3-aminophenylacetic acid, TFA, 2,2,2-trifluoroethanol, 90 °C, 24 h, (ii) NaOH, THF/H2O,RT, 18 h; (d) TFA, 2,2,2-trifluoroethanol, 90 °C, 18 h; (e) (i) CDI, DIPEA, DMF, RT, 90 min, (ii) 1-(3-aminopropyl)imidazole, RT, 18 h.
To provide a reference point for these studies, the 6-substituent was deleted entirely. Thus, the 6-unsubstituted intermediate 64 was prepared from 2-fluoro-6-chloropurine (63) [27], by selective dehalogenation of the 6-chloro group using catalytic transfer hydrogenation [28, 29]. Coupling of 64 with the appropriate anilines gave derivatives 65 and 66, with 66 being converted to amide 67 (Scheme 4).
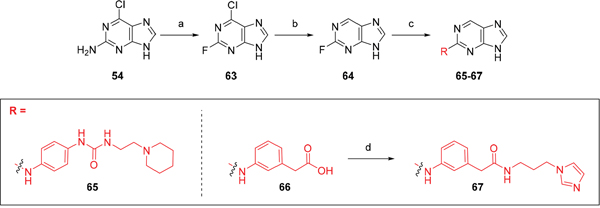
Scheme 4: Synthesis of 6-unsubstituted 2-arylaminopurines.a a Reagents and conditions: (a) HBF4, NaNO2, 0 °C to RT, 75 min, 75%; (b) Pd(OH)2, NH4OOCH, MeOH, 65 °C, 1 h, 100%; (c) (i) anilines, TFA, 2,2,2-trifluoroethanol, 90 ºC, 48 h; (ii) for 66: KOH, THF/H2O, RT, 18 h 34-45%; (d) (i) CDI, DIPEA, DMF, RT, 90 min; (ii) RNH2, RT, 18 h, 40 – 59%.
Replacement of the O6-cyclohexylmethyl substituent
Given the differences between the ribose-binding domains of Nek2 and CDK2, it was considered that potency may be improved and off-target activity reduced, if the group at C-6 could be conformationally restricted by the introduction of an alkene. This premise was explored by synthesis of (E)-6-(2-dialkylaminovinyl)purines (69-73). 6-Ethynyl- and 6-vinylpurines have been reported as substrates for conjugate addition reactions with secondary amines affording (E)-enamines or ethylamines, respectively [30, 31]. Addition of primary amines to a 6-ethynyl group afforded an inseparable mixture of (Z)- and (E)-enamines as a consequence of imine-enamine tautomerism. Reported conditions for this transformation utilized purine N-9 protection and typically required lengthy reaction times of 1-3 days at room temperature [30]. We found that the addition of amines to the 6-ethynylpurine 68 could be accelerated by microwave heating (Scheme 5). 6-Ethynylpurine 68 was prepared from N-9 THP-protected 2-fluoro-6-chloropurine utilizing Sonagashira alkynylation. Following cleavage of the N-9 hemiaminal ether by acidic hydrolysis, introduction of the 2-arylamino group and silyl group deprotection afforded the alkynyl intermediate. In all cases the subsequent conjugate addition took place smoothly in moderate-to-high yield without N-9 protection and a series of (E)-enamines (69-73) was obtained (see ESI for more examples). The configuration at the newly formed double bond was validated by 1H NMR (3J = 15.0 Hz). This methodology is applicable for the facile synthesis of enamine derivatives from a diverse set of secondary amines.
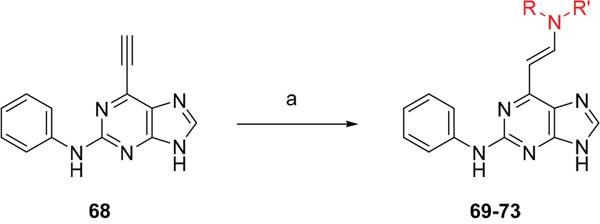
Scheme 5: Synthesis of 6-(dialkylamino)vinyl-purines.a a Reagents and conditions: (a) RR′NH, THF, microwave 100 °C, 10 min, 60 – 98% (Supplementary Table 1).
Biological evaluation of synthesized purines
Preliminary data attained following a medium-throughput screening campaign, identified purines bearing 6-alkoxy substituents (e.g. 6; CDK2 IC50 = 0.005 μM; Nek2 IC50 = 12 μM) as dual inhibitors of Nek2 and CDK2. Interestingly, deletion of the sulfonamide moiety from 6, resulted in a 200-fold reduction in CDK2 inhibitor activity as well as some reduction in Nek2 activity (2-aminophenyl-6-(cyclohexylmethoxy)-9H-purine: CDK2 IC50 = 1 μM; Nek2 IC50 = 22 μM). To gain a further understanding of the requirements for selective Nek2 inhibition, a structure-activity study was undertaken to explore the scope for modification of the purine hits at the 2- and 6-positions.
Modifications at the 2-arylamino position
We have previously reported that hydrogen bond donor-acceptor groups, such as sulfonamides at the 2-arylamino position, form important interactions (e.g. with Asp-86) at the ‘specificity surface’ of CDK2 [32]. To discriminate between the interactions required for Nek2 inhibition and CDK2 inhibition, variations of the 2-arylamino substitution were studied by synthesis of acetamide, carboxamide, sulfonamide and urea derivatives. Both 4- and 3-nitro-substituted precursors were inactive against Nek2 yet retained modest potency against CDK2 (19; CDK2 IC50 = 1.9 μM; Nek2 IC50 = > 50 μM and 20; CDK2 IC50 = 0.89 μM; Nek2 IC50 = > 50 μM, respectively). The corresponding aniline improved activity against Nek2 in both cases (21; CDK2 IC50 = 0.22 μM; Nek2 IC50 = 6.3 μM and 22; CDK2 IC50 = 0.7 μM; Nek2 IC50 = 5.9 μM, respectively), suggesting that there may be a hydrogen-bonding interaction available in the Nek2 ATP-binding domain. Activity was not enhanced by acylation of the respective aniline compounds, with N-acetyl compound 27, showing only weak activity against Nek2. Further homologation and branching of the acyl group of the 4-substituted derivatives did not enhance Nek2 inhibition (e.g. 28), and substantial loss of Nek2 inhibitory activity occurred with benzoyl (29) or isonicotinoyl (30) groups.
The 3-substituted purine 8 had modest activity against CDK2 (IC50 = 0.48 μM) and weak activity against Nek2 (IC50 = 19 μM). Addition of a methyl substituent at the amide nitrogen (compound 10) reduced potency against CDK2 but only marginally increased Nek2. Reversing the orientation of the carboxamide group (27; CDK2 IC50 = 0.88 μM; Nek2 IC50 = 8.3 μM) increased Nek2 inhibition in comparison to 8, without increasing activity against CDK2. However, unexpectedly, addition of a second methyl group to produce the dimethylamide 11 afforded a marked increase in Nek2 inhibitory activity whilst also greatly reducing CDK2 inhibition, suggesting that the amide NH hydrogen bond may not be required for Nek2 inhibition. Although tethering the dimethylamino group into a ring, as in piperidine derivative 32, further reduced CDK2 inhibitory activity, Nek2 inhibition was also abolished.
Replacing the carboxamide group by a carboxylate (23) increased Nek2 activity (IC50 = 4.3 μM) compared to 8, indicating a possible ionic interaction with the carboxylate and that one hydrogen bond donor is optimal. However, 23 still retained potency against CDK2 (IC50 = 0.99 μM). The thioamides 12 and 13 were similar in potency to their amide equivalents (11 and 8) against CDK2, but were less active against Nek2.
Overall, the SARs around the amides, benzamides and thioamides at the meta position of the 2-arylamino-O6-cyclohexylmethylpurines revealed that a hydrogen bond donor at the meta position favoured activity against CDK2 (e.g. 8, 10, 13, 23), whereas introducing a larger, bulkier substituent (32) was unfavourable for activity against both CDK2 and Nek2 (Table 2). Furthermore, the dimethylcarboxamide 11 was both the most potent Nek2 inhibitor and the compound with the greatest Nek2/CDK2 selectivity (> 10-fold) in this series. These observations could possibly be rationalised through a putative interaction with Asp-93 (Figure 4), and may also be the case with primary carboxamide 8 in Figure 1; however, no direct evidence for this supposition has yet been attained.
Table 2: Inhibition of CDK2 and Nek2 by representative 2-arylamino-O6-cyclohexylmethylpurines.a
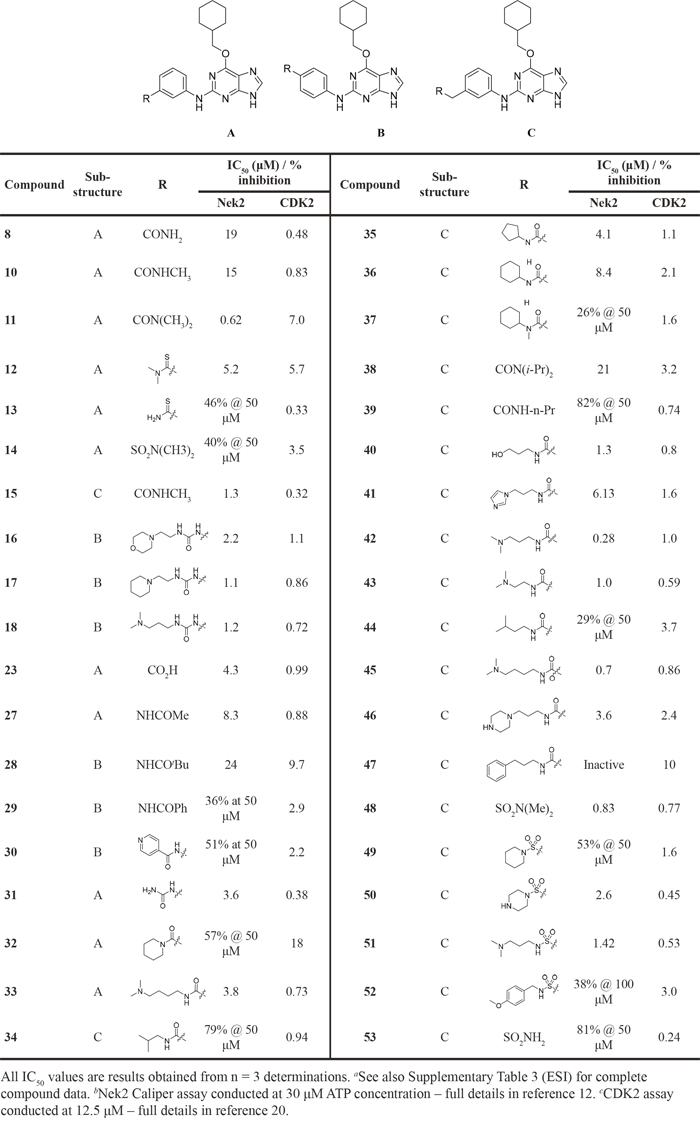
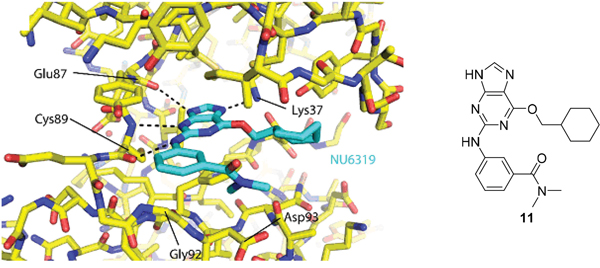
Figure 4: Crystal structure of carboxamide 11 (cyan) bound to the T175A Nek2 mutant (carbon atoms are coloured yellow, oxygen coloured red, and nitrogen coloured blue). Hydrogen bonds are represented as dotted lines and important residues are highlighted.
With the homocarboxamide series (Table 2, sub-structure C), removing the donor NH of the amide group by synthesis of di-substituted carboxamides (37 and 38) in general retained affinity for CDK2 but not Nek2. This observation was clearly shown by the matched pair of compounds 36 and 37, where a methyl group replaced a hydrogen bond donor and markedly reduced activity against Nek2 but not CDK2. Increasing the size of the di-substituted amides decreased Nek2 activity further whilst retaining modest CDK2 activity (e.g. 38), indicating that hydrophobic interactions may be particularly unfavourable for Nek2 inhibitory activity in this region. A similar decrease in potency was observed for Nek2 and CDK2 (~2-fold) when the size of the alkyl ring of cyclic secondary amides was increased (35 and 36). Compound 15 possesses the amide NH hydrogen bond donor but had a higher potency against CDK2. Replacing the methyl group of 15 with n-propyl (39) reduced activity against Nek2 whilst maintaining CDK2 inhibition. The iso-butyl analogue (34) was also less active against Nek2 than 15 but equipotent to 39, indicating that the Nek2 active site is sterically limited. In contrast, the effect on CDK2 inhibition was limited (IC50 values ranging from 0.3-0.9 μM).
Introducing a terminal hydroxy group on the propyl chain (40) was tolerated for Nek2 inhibition, and a dimethylamino group (42) at the end of the propyl side chain increased activity against Nek2. Shortening the alkyl chain of 42 from propyl to ethyl (43) did not improve Nek2 inhibition, and removal of the amino functionality of 43 to give 44 greatly reduced Nek2 inhibition, suggesting that the basicity of the tertiary amino group was required. In summary, these results indicated the possibility of an additional interaction in the Nek2 binding site for a basic moiety at the end of the propyl chain.
Selectivity for CDK2 over Nek2 was observed for the sulfonamides shown in Table 2 with the exception of 48, which was equipotent against both CDK2 and Nek2. The primary sulfonamide 53 exhibited good potency against CDK2 and some activity against Nek2. Cyclic secondary sulfonamides (e.g. 49) were essentially inactive against Nek2 except for heterocyclic derivatives (e.g. 50), which showed modest activity against Nek2. Nevertheless, all of the sulfonamides tested were more potent against CDK2 than Nek2. Inhibition of Nek2 by 50 could be attributed to the basic properties of the piperidine ring, which appears to be favourable for Nek2 activity. Compound 49 supports this observation given its lower activity against Nek2. Larger lipophilic substituents (e.g. 52) resulted in greatly decreased activity against Nek2 and variable effects on CDK2 activity. In comparison to the most potent carboxamide (42) the corresponding sulfonamide (51) was both less active (5-fold) and less selective for Nek2.
The urea moiety was also investigated as a putative non-classical isosteric replacement for the sulfonamide functional group. 6-Alkoxy-2-arylaminopurines with urea-based side-chains exhibited low-micromolar activity against Nek2, as compared to micromolar or sub-micromolar inhibition of CDK2; as exemplified by 16 (Nek2 IC50 = 2.2 μM, CDK2 IC50 = 1.1 μM). Disappointingly, primary ureas e.g. 31 were found to be 10-fold selective for CDK2 over Nek2. Elaboration of this group bysynthesis of secondary ureas e.g. N-ethylmorpholine derivative 16, enhanced activity against Nek2. Replacement of the N-ethyl-morpholino group by e.g. N-ethyl-piperidine (17) and dimethylaminopropyl (18) side-chains, did not greatly affect inhibition or selectivity profile for Nek2 or CDK2. These results suggest that whilst, potency against Nek2 may be gained from incorporation of a basic group and a donor-acceptor moiety at the 2-arylamino position, the additional hydrogen-bond acceptor in the morpholine is unnecessary or even detrimental.
Modifications at the purine 6-position
In order to improve potency against Nek2 whilst retaining the selectivity observed within some examples from the homocarboxamide series e.g. 42, alternative substitution at the 6-position was investigated (Table 3). In comparison to 42, selectivity for Nek2 over CDK2 was maintained by combining an O6-ethyl substituent with the dimethylaminopropyl homocarboxamide side chain (59;CDK2 IC50 = 5.6 μM; Nek2 IC50 = 0.89 μM), albeit with some loss of potency against both kinases. Imidazole derivative 62 exhibited 2-fold improved Nek2 inhibitory activity combined with a 2-fold reduction in CDK2 inhibition, compared with the analogous cyclohexylmethyl derivative 41. Interestingly, the O6-sec-butyl derivative with a N,N'-dimethylaminopropyl side chain 60 gained potency against CDK2 with 10-fold selectivity over Nek2.
Table 3: Nek2 and CDK2 inhibition by selected 6-unsubstituted and 6-substituted purines. a
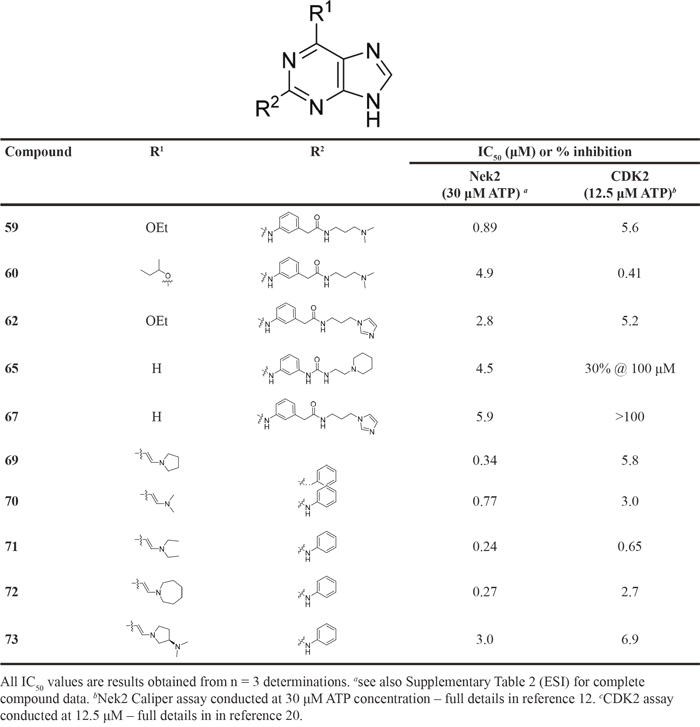
Guanine derivatives lacking the O-alkyl substituent (65 and 67) exhibited a dramatic reduction in potency against CDK2 compared with the parent 6-alkoxy compounds, yet Nek2 inhibition was maintained. The 6-unsubstituted purine derivative bearing a substituted urea side-chain (65)was approximately 4-fold less potent against Nek2 than the parent 6-alkoxypurine 17 but, importantly, gave only 30% inhibition of CDK2 at 100 μM. Activity against Nek2 was also maintained for the 6-unsubstituted imidazole homocarboxamide purine (67), and CDK2 inhibitory activity was again abolished. This result indicated that a 6-cyclohexylmethyl substituent conferred potent CDK2 inhibition but was not necessary for Nek2 inhibition. A significant improvement in selectivity was gained as a result of this modification, confirming that there are exploitable differences between the ribose-binding pockets of Nek2 and CDK2.
Having generated a series of enamines to probe the replacement of the O6-alkoxy substituent, SARs revealed that polar substituents, e.g. morpholine (see ESI), and hydrophobic alkyl substituents such as those in compounds 69-71 were tolerated. Larger cyclic secondary amines such as homopiperidine (72) were found to be sub-micromolar inhibitors of Nek2 (IC50 = 0.27 μM). However, the addition of a second basic group as in 3-dimethylaminopyrrolidine (73) reduced the inhibition of Nek2 around 10-fold as compared with 69. The CDK2 counter-screening data were largely unremarkable as the compounds 69-73 were only low-micromolar CDK2 inhibitors with the exception of the diethylamino compound 71 (IC50 = 0.65 μM). From the data obtained, it appeared that the most potent enamine-based Nek2 inhibitors were generally also more potent against CDK2. To assess the broader kinase selectivity of this series of 6-(dialkylamino)vinyl-purines, the diethylamino derivative 71 was chosen for wider screening against 24 kinases at 2 μM inhibitor concentration (Figure 5 – method as in reference 12). Compound 71 did not significantly inhibit other kinases studied with the exception of the mitotic kinases aurora A (100% inhibition at 2 μM) and Chk2 (65% at 2 μM). If 71 is representative of the entire series, this may indicate a lack of selectivity for Nek2. In a separate counter-screen, dimethylamino derivative 70 was found to inhibit weakly another mitotic kinase, Plk1 (41% at 10 μM – method as in reference 12). Cell growth inhibition studies (Figure 6) with compounds 69-73 in human tumor cell lines, including U2OS, MDA-MB-231 and HeLa cells, showed modest effects on cell viability (GI50 > 10 μM). Interestingly, the cellular activity did not reflect the relative potencies in the Nek2 kinase assay and may indicate that cell growth inhibition is due to inhibition of a combination of Nek2 and other kinases e.g. aurora A.
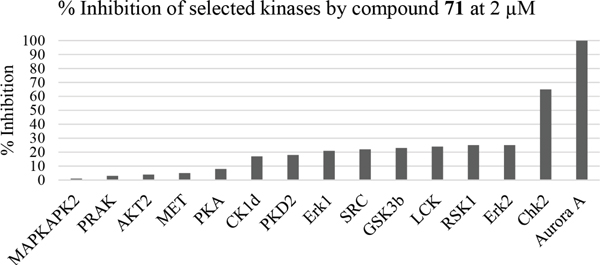
Figure 5: Counter-screening of 71 against a panel of kinases.
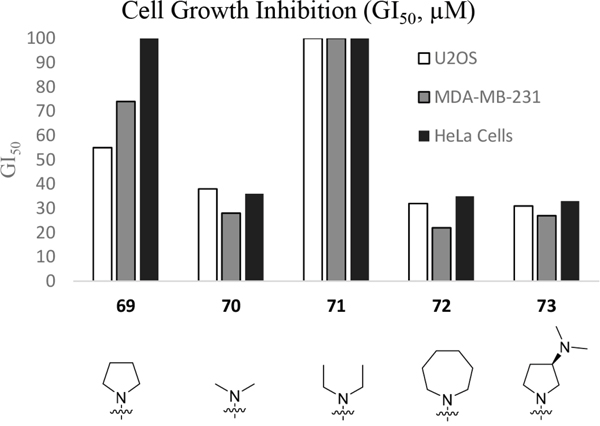
Figure 6: Growth inhibition (GI50) of selected cell-lines by enamines 69-73.
A representative enamine (70) was shown not to exhibit time-dependent Nek2 inhibition kinetics (Figure 7A (see also ESI), indicating that the compound binds reversibly to Nek2 and that the enamine is not chemically reactive within the Nek2 active site. This observation was also confirmed by kinetic studies of kinase inhibition, which showed that removal of the inhibitor by rapid dilution of the assay medium, restored kinase activity (Figure 7B) [33].
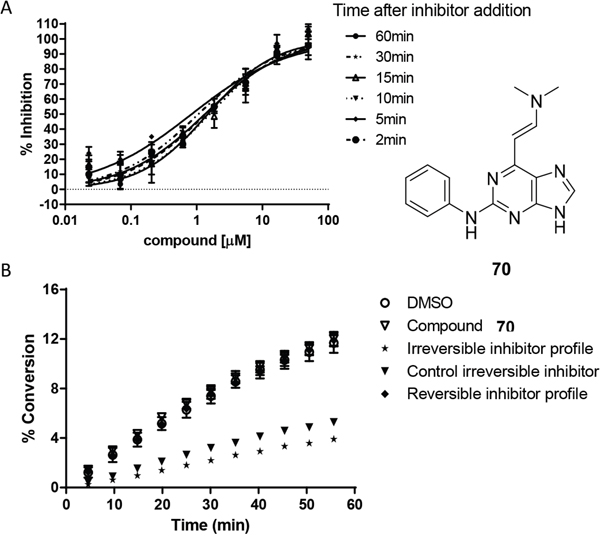
Figure 7: Mechanism of action studies with enamine 70. A. The effect of pre-incubation (2-60 min) of Nek2 with 70 on inhibitory potency. B. Reversibility studies indicating that 70 was not an irreversible inhibitor of Nek2 (Supplementary Table 4).
Structural biology studies confirmed that the 6-(2-aminovinyl)purine Nek2 inhibitors occupied a binding orientation nearly identical to that of the O6-alkylpurines. The interactions between the purine and the kinase hinge motif were clearly visible (Figure 8A) and the enamine 6-substituent was shown to point towards the hydrophobic glycine-rich loop (Figure 8B), which is consistent with the O6-alkyl purine hits from the initial screening results.
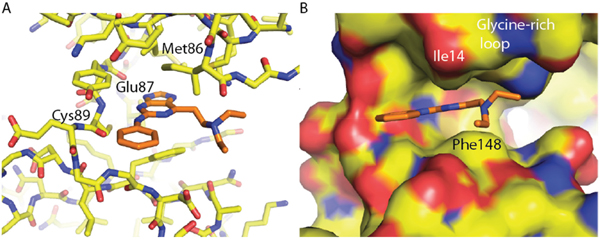
Figure 8: Crystal structures of compound 71 in complex with Nek2. A. Interaction of 6-(2-aminovinyl)purine derivative 71 (carbon atoms coloured orange) with the hinge region of Nek2. B. Orientation of the C-6 substituents towards the glycine-rich loop.
Stability of enamines
To determine whether the enamines were hydrolysed under the assay conditions, the stability of representative enamine derivative 72 was assessed in a range of assay media and pH buffers. Stock solutions of 72 in each of the various media were prepared and aliquots were extracted from each solution at various time points and subjected to HPLC analysis to measure the remaining intact enamine (Figure 9).
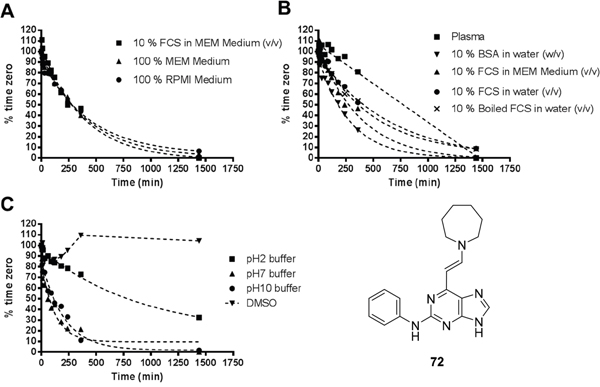
Figure 9: Stability of 72 in various assay media. BSA (bovine serum albumin) and FCS. Note: MEM (minimum essential medium) and RPMI (Roswell Park Memorial Institute) media contain no proteins or enzymes.
In two different standard assay media (MEM and RPMI, Figure 9A) enamine 72 was degraded at a similar rate. Addition of 10% foetal calf serum (FCS) to the media caused no difference in the observed stability, suggesting that breakdown was independent of the composition of the media and was non-enzymatic. The lack of enzymatic involvement in the breakdown was confirmed by incubation of 72 in protein solutions (Figure 9B). Similar rates of decomposition of 72 were observed in media containing FCS and water containing FCS, both of which contained active enzymes, compared with solutions of boiled (inactivated) FCS in water. A surprising result was that 72 was relatively stable over a period of 24 hours in human plasma, potentially reflecting the impact of reversible plasma protein binding. Whilst, the enamine 72 was stable in DMSO solutions for over 24 h and relatively stable at pH 2.2, under neutral (pH 7) and basic (pH 10.4) conditions the enamine 72 was rapidly degraded (Figure 9C), contrary to the expected profile. Together, these stability observations suggest that the enamine motif is relatively unstable under model conditions, which are similar to those used for the cell-based assays. This may explain the mostly flat SARs observed in cellular assays for this series, perhaps due to the enamine group degradation to a reactive aldehyde, which is a hydrolysis product common to all members of this series. Some notable differential activity is noted, however, and may be attributed to varying rates of enamine hydrolysis to the corresponding aldehyde more or less quickly than 72. Reduction of the enamine to the corresponding tertiary amine may provide a possible solution to stability problems.
Structural insights into Nek2 activation
To gain an insight in to the molecular mechanisms of Nek2 activation, structural biology studies were conducted using Nek2 in complex with carboxamide 11, which was one of the newly synthessied inhibitors with the greatest Nek2-selectivity profile (Table 2). In complex with purine-based ATP-competitive inhibitors, it has been previously shown that Nek2 adopts an inactive conformation characterized by a partially disordered activation loop and an outward position of the αC-helix (Figure 10A). This is similar to that observed in previously determined Nek2 structures such as the complex with ADP (Figure 10B) [34]. Here, in complex with carboxamide 11, and in contrast to previous structures, the activation loop of Nek2 has a DFG-in conformation, indicating that 11 is the first reported inhibitor of Nek2 to function in this way. The phenylalanine (Phe-160 in Nek2) of the DFG motif is one of the four regulatory (R-) spine residues, which adopt a continuous hydrophobic column in active kinases (Figure 10C) [35]. The DFG-in conformation is required to form the R-spine. The HRD motif also adopts the position expected in an active kinase structure and thus, in the presence of 11, Nek2 has a more ordered R-spine. However, one of the four R-spine residues is out of position compared to that expected in an active kinase because the αC-helix is in an αC-out position.
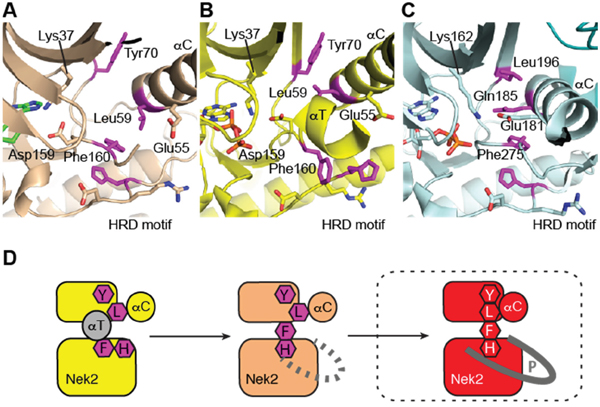
Figure 10: Purine ATP-competitive inhibitors induce a pre-active, DFG-in, αC-out conformation of Nek2. A. Crystal structure of Nek2 in complex with compound 11. B. Crystal structure of Nek2 in complex with ADP (PDB code 2W5A). C. Crystal structure of active Aurora-A (PDB code 2W5A). In panels A-C, hydrophobic R-spine residues are coloured magenta.
The conformation adopted here by Nek2 appears to be stabilized by 11. The clearest connection here is the interaction of the ligand with Lys-37, which is connected to the DFG motif through an interaction with the side chain of Asp-159. The conformations of the DFG and HRD motifs are coupled through aromatic stacking between the side chains of Phe-160 and His-139. Thus, the purine scaffold induces a specific conformation of key structural elements of Nek2 through a network of H-bond interactions.
The crystal structure of Nek2 bound to purine compounds suggests an intermediate state of a kinase poised for activity, and we propose a stepwise pathway for Nek2 activation (Figure 10D). Unphosphorylated Nek2 is in an autoinhibited state, in which the activation loop forms an α-helix (αT) that stabilizes the outwards position of the αC-helix and blocks formation of an R-spine. Refolding of αT by formation of a DFG-in conformation of the activation loop is coupled to changes in the HRD motif that results in a partially formed R-spine and a disordered activation loop. In this pre-active state the outward location of the αC-helix is destabilized and the activation loop is disordered. The kinase may have sufficient flexibility to transiently adopt an active state necessary for autophosphorylation. Nek2 autophosphorylation is expected to result in a fully-active conformation of the kinase with a fully-assembled R-spine and an ordered activation loop. In our study, the pre-active state of Nek2 was induced by ATP-competitive inhibitors based on a purine scaffold (i.e. carboxamide 11). In a physiological context, it is likely that Nek2 activation is promoted by protein-protein interactions, such as Nek2 dimerization.
In crystal structures of Nek2 bound to purine inhibitors, the region C-terminal to the αE helix and N-terminal to the HRD motif is ordered (Figure 11A), unlike other structures of Nek2 (Figure 11B). The loop structure contacts the αC-helix in the N-lobe of the protein and occupies the equivalent space as the activatory helix of TPX2 in the Aurora-A/TPX2 complex (Figure 11C). Based on these observations, we predict that this loop will fulfill a similar role in the stabilization of the activation loop in the active Nek2 structure. This loop is not present in other Neks and is indeed unique to Nek2 among human kinases (Figure 11D). Importantly, targeting this unique protein conformation with small molecules presents an attractive opportunioty for selective inhibition of Nek2 over other kinome members.
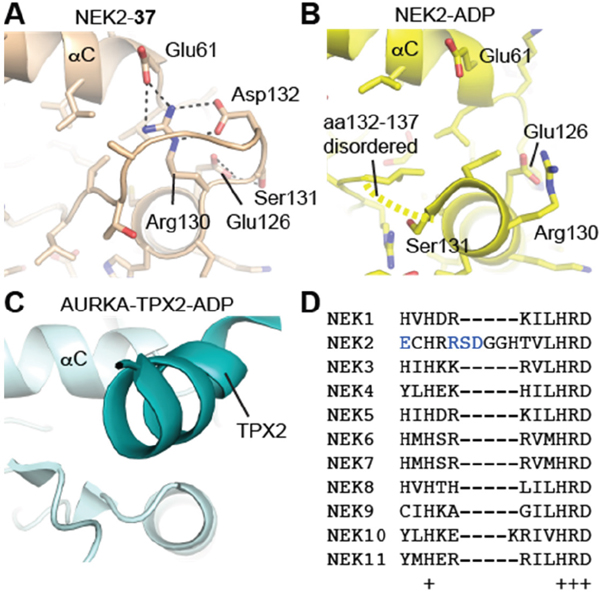
Figure 11: Nek2 has a unique insertion N-terminal to the ‘HRD’ motif. A. Crystal structure of Nek2 in complex with compound 11. B. Crystal structure of Nek2 in complex with ADP (PDB code 2W5A). C. Crystal structure of active Aurora-A (PDB code 2W5A). D. Sequence alignment of human Neks in the vicinity of the ‘HRD’ motif, showing the insert sequence that is unique to Nek2.
In conclusion, a number of key structural requirements for selective Nek2 inhibition over CDK2 have been elucidated by synthesis and evaluation of purines probes bearing informative modifications, specifically at the 2-arylamino and 6-positions. These significantly include the importance of a terminal basic group, especially the NMe2 residue, on the meta-substituent of 2-arylamino derivatives which consistently showed good potency when incorporated into (homo) sulfonamide, (homo) carboxamide and urea sidechains. It was also noteworthy that a hydrogen bond donor-acceptor group, e.g. urea, played a role in potent Nek2 inhibition, whilst sulfonamides may confer CDK2 inhibitory activity due to interactions previously reported [32]. It may also be concluded that para-substitution on the 2-arylamino ring may be preferable for CDK2 inhibition, whereas meta-substitution generally affords lower CDK2 inhibitory activities, and in some cases improved Nek2 inhibition.
Aided by structural biology, we have shown that it may be possible to obtain further selectivity by removal of the O6-cyclohexylmethyl group, which abolished CDK2 inhibition but was not essential for Nek2 inhibition. A facile and rapid synthesis of 6-(dialkylaminovinyl) purines was described and such groups at the purine 6-position conferred potent Nek2 inhibition and a degree of kinase selectivity. Subsequently, it has been found that the precursor of these enamines, 6-ethynyl-2-phenylaminopurine 68, reacts covalently with Cys-22 of Nek2, thus behaving as an irreversible inhibitor. Identification and structure-activity relationships for 2-arylamino-6-ethynylpurines as irreversible inhibitors of Nek2 kinase will be the subject of our next publication.
Crystal structures of Nek2 in complex with purines e.g. carboxamide 11 provide a snapshot of Nek2 in a conformation closer to that of an active kinase than was observed in previous Nek2 structures. This provides a potentially unique and selective molecular target for small molecule kinase inhibition. Moving forward, the development of tool compounds with improved potency and selectivity will be required in future studies to assist with target validation, cellular studies and to understand better the role of Nek2 in mitosis and cancer.
MATERIALS AND METHODS
Synthetic chemistry
All chemicals were purchased from standard suppliers. Solvents were purified and stored according to standard procedures. Melting points were obtained on a Stuart Scientific SMP3 apparatus and are uncorrected. TLC was performed with Merck 60 F254 silica gel plates. Where appropriate, compound mixtures were separated and purified using either medium pressure (‘flash’) chromatography, employing Davisil silica 40-60 μm, or using a Biotage SP4 automated chromatography system with UV monitoring at 254 and 290 nm. When using the Biotage SP4 to purify samples, the stationary phase was KP-SIL (silica), Biotage KP-NH or KP-C18 as appropriate. 12+M, 25+M or 40+M pre-packed columns were used as required. KP-NH was used for the normal-phase purification of polar organic amines. KP-C18 (18% carbon by weight) was used for the separation of polar and ionisable organic compounds, requiring a water-based eluent and lipophilic stationary phase. 1H NMR and 13C NMRspectra were recorded on a Bruker Spectrospin AC 300E spectrometer (300 MHz for 1H, 75 MHz for 13C) or a Bruker AMX (500 MHz for 1H, 126 MHz for 13C). Samples were acquired in deuterated solvents including CDCl3 and DMSO-d6. Where appropriate (compounds 69-73), reactions were carried out with microwave heating, in sealed vessels, using a Biotage Initiator reactor equipped with a ‘Sixty robot’. Samples were irradiated at 2.45 GHz, reaching temperatures from 60-250 ºC (rate of heating 2-5 °C/sec) and pressures up to 20 bars. LCMS was carried out on either a Micromass Platform instrument operating in positive and negative ion electrospray mode, employing a 50 x 4.6 mm C18 column (Supelco Discovery or Waters Symmetry) and a 15 min gradient elution of 0.05% formic acid and methanol (10-90%), or on a Finnegan LCQ instrument in positive ion mode with a Phenomenex 5μ Luna C18 column, 4.6 mm x 50 mm and an 8 min gradient of 0.1% aqueous formic acid and acetonitrile (5-98%), with a flow rate of 2 mL/min. IR spectra were recorded on a Bio-Rad FTS 3000MX diamond ATR. HRMS were measured using a Finnigan MAT 95 XP or a Finnigan MAT 900 XLT by the EPSRC National Mass Spectrometry Service Centre (Swansea).
Synthesis of 2-arylaminopurines (compounds 8, 10-26, 59-61, 65, 66). Method I. General Procedure
To a stirred suspension of the appropriate 2-fluoro-9H-purine (0.49 mmol) and the required aniline derivative (0.98 mmol) in TFE (25 mL/g of fluoropurine) was added TFA (0.19 mL, 2.46 mmol) dropwise. The resulting solution was heated under reflux for 48 h under a nitrogen atmosphere. The solvent was removed in vacuo and the residue was redissolved in EtOAc (10 mL). The solution was washed several times with saturated NaHCO3 solution (3 × 10 mL), and the aqueous extracts were combined and washed with EtOAc (10 mL). The combined organic layers were dried (Na2SO4) and the solvent was removed to give a residue that was purified as indicated.
3-(6-Cyclohexylmethoxy-9H-purin-2-ylamino)benzamide (8)
The title compound was synthesised according to Method I using 2-fluoro-6-cyclohexylmethoxypurine (9, 125 mg, 0.50 mmol) and 3-aminobenzamide (136 mg, 1.0 mmol), TFE (5 mL), and TFA (0.19 mL, 2.5 mmol). The crude product was dry-loaded onto silica (~30 mL) and purified by column chromatography (eluent: EtOAc) to give a light brown powder, (62 mg, 34%): Rf = 0.18 (MeOH-EtOAc; 1:9); mp 231-232 °C; IR (cm-1) 3350, 2922, 2851, 2160, 1577, 1541, 1118; 1H NMR (300 MHz, DMSO-d6) δ 0.85-1.70 (11H, m, cyclohexyl), 4.36 (2H, d, J = 5.4 Hz, OCH2), 7.24-7.34 (2H, m, 2 × ArH), 7.39 (1H, d, J = 8.4 Hz, ArH), 7.85 (2H, s, CONH2), 7.99 (1H, s, H-8),8.37 (1H, s, ArH), 9.43 (1H, s, ArNHAr), 12.83 (1H, s br, N9-H); 13C NMR (75 MHz, DMSO-d6) δ 25.6, 26.4, 29.6, 37.4, 71.6, 97.0, 114.4, 118.7, 120.2, 121.6, 128.4, 135.5, 141.9, 155.8, 169.0; LCMS (ES+) m/z 367 [M+H]+; HRMS (ES+) calcd for C19H23N6O2 [M+H]+ 367.1877, found 367.1875; λmax (EtOH) 273, 293 nm.
3-(6-Cyclohexylmethoxy-9H-purin-2-ylamino)-N-methylbenzamide (10)
The title compound was synthesised according to Method I using 2-fluoro-6-cyclohexylmethoxypurine (9, 75 mg, 0.30 mmol), 3-amino-N-methylbenzamide (see ESI S3; 102 mg, 0.68 mmol), TFE (3 mL), and TFA (0.11 mL, 1.5 mmol). The crude product was allowed to stand in DCM (20 mL) for 2 h at room temperature. The resulting precipitate was filtered and washed with DCM (2 × 5 mL) to give the title compound as a pale pink solid (88 mg, 77%): Rf = 0.10 (EtOAc); mp 252-254 °C; IR (cm-1) 3257, 3061, 2928, 2854, 1638, 1612, 1579, 1538, 1485, 1446, 1381, 1257; 1H NMR (300 MHz, DMSO-d6) δ 1.0–1.84 (11H, m, cyclohexyl), 2.77 (3H, d, J = 4.4 Hz, NHCH3), 4.35 (2H, d, J = 6.1 Hz, OCH2), 7.32 (2H, m, 2 × ArH), 7.85 (1H, br, CONH), 7.98 (1H, s, ArH), 8.27-8.35 (2H, m, ArH and H-8), 9.45 (1H, s, ArNHAr), 12.83 (1H, s, N9-H); LCMS (ES+) m/z 381.45 [M+H]+; HRMS (ES+) calcd for C20H25N6O2 [M+H]+ 381.2034, found 381.2034; λmax (EtOH) 273 nm.
3-(6-Cyclohexylmethoxy-9H-purin-2-ylamino)-N,N-dimethylbenzamide (11)
The title compound was synthesised according to Method I using 2-fluoro-6-cyclohexylmethoxypurine (9, 80 mg, 0.32 mmol), 3-amino-N,N-dimethylbenzamide (see ESI S4; 105 mg, 0.64 mmol), TFE (4 mL), and TFA (0.12 mL, 1.6 mmol). The crude product was dry-loaded onto silica and purified by column chromatography (EtOAc-petrol; 9:1) to give an off-white powder (68 mg, 54%): Rf = 0.09 (EtOAc); mp 120-121 °C; IR (cm-1) 3266, 2922, 2849, 1973, 1581, 1435, 1386; 1H NMR (300 MHz, DMSO-d6) δ 0.95-1.80 (11H, m, cyclohexyl), 2.95 (6H, s, N(CH3)2), 4.33 (2H, d, J = 6.3 Hz, OCH2), 6.90 (1H, d, J = 7.6 Hz, ArH), 7.31 (1H, dd, J = 7.8, 8.0 Hz, ArH), 7.77 (1H, d, J = 8.1 Hz, ArH), 7.98 (1H, s, ArH), 8.03 (1H, s, H-8), 9.45 (1H, s, ArNHAr), 12.91 (1H, s, N9-H); 13C NMR (75 MHz, DMSO-d6) δ 25.6, 26.4, 29.6, 37.3, 71.5, 114.1, 116.93, 119.3, 119.4, 128.6, 137.3, 140.2, 141.6, 155.6, 160.0, 170.8; LCMS (ES+) m/z 395 [M+H]+; HRMS (ES+) calcd for C21H27N6O2 [M+H]+ 395.2190, found 395.2189; λmax (EtOH) 272, 295 nm.
3-(6-Cyclohexylmethoxy-9H-purin-2-ylamino)-N,N-dimethylthiobenzamide (12)
The title compound was synthesised according to Method I using 2-fluoro-6-cyclohexylmethoxypurine (9, 88 mg, 0.35 mmol), 3-amino-N,N-dimethylthiobenzamide (see ESI S7; 140 mg, 0.78 mmol), TFE (3 mL), and TFA (0.13 mL, 1.8 mmol). The crude product was dry-loaded onto silica (4 mL) and purified by column chromatography (EtOAc-petrol; 7:3) followed by further purification by semi-prep HPLC (mobile phase A – eluted with 40% acetonitrile, flow-rate 12.75 mL/min, wavelength 280 nm) to give the title compound as a yellow solid (35 mg, 24%): Rf = 0.18 (EtOAc-petrol; 7:3); mp 231-232 °C (dec.); IR (cm-1) 2926, 2851, 1629, 1580, 1521, 1487, 1431, 1393, 1258, 1116; 1H NMR (300 MHz, DMSO-d6) δ 0.9-1.7 (11H, m, cyclohexyl), 3.17 (3H, s, NCH3), 3.50 (3H, s, NCH3), 4.33 (2H, d, J = 6.1 Hz, OCH2), 6.78 (1H, d, J = 7.4 Hz, ArH), 7.26 (1H, dd, J = 7.9, 7.9 Hz, ArH), 7.66 (1H, d, J = 7.7 Hz, ArH), 7.90 (1H, s, ArH), 8.01 (1H, s, H-8), 9.45 (1H, s, ArNHAr), 12.72 (1H, s br, N9-H); LCMS (ES+) m/z 411 [M+H]+; HRMS (ES+) calcd for C21H27N6OS [M+H]+ 411.1962, found 411.1959; λmax (EtOH) 236, 277 nm.
3-(6-Cyclohexylmethoxy-9H-purin-2-ylamino)thiobenzamide (13)
The title compound was synthesised according to Method I using 2-fluoro-6-cyclohexylmethoxypurine (9, 75 mg, 0.3 mmol), 3-aminothiobenzamide (see ESI S8; 92 mg, 0.6 mmol), TFE (3 mL), and TFA (0.11 mL, 1.5 mmol). The crude product was dry-loaded onto silica (4 mL) and purified by column chromatography (EtOAc-petrol; 7:3 up to EtOAc) followed by further purification by semi-prep HPLC (5 → 100% v/v acetonitrile: water: NH4OH over 25 min; flow-rate 12.75 mL/min, wavelength 280 nm) to give the title compound as a yellow solid (37 mg, 32%): Rf = 0.28 (EtOAc-petrol; 9:1); mp 148-149 °C (dec.); IR (cm-1) 3271, 3077, 2920, 2849, 2363, 2337, 1597, 1537, 1483, 1438, 1391, 1352, 1319, 1283, 1117, 973; 1H NMR (300 MHz, DMSO-d6) δ 1.00-1.90 (11H, m, cyclohexyl), 4.37 (2H, d, J = 5.8 Hz, OCH2), 7.29 (2H, d, J = 7.3 Hz, ArH), 7.79 (1H, d, J = 7.3 Hz, ArH), 7.98 (1H, s, H-8), 8.44 (1H, s, ArH), 9.43 (2H, br, NH2), 9.82 (1H, s, ArNHAr), 12.81 (1H, s br, N9-H); 13C NMR (125 MHz, DMSO-d6) δ 25.2, 26.0, 29.2, 36. 9, 71.2, 114.9, 118.2, 118.8, 120.9, 127.8, 139.0, 140.7, 140.7, 154.2, 155.3, 160.1, 201.1; LCMS (ES+) m/z 411 [M+H]+; HRMS (ES+) calcd for C19H23N6OS [M+H]+ 383.1649, found 383.1647; λmax (EtOH) 278, 295 nm.
3-(6-Cyclohexylmethoxy-9H-purin-2-ylamino)-N,N-dimethylbenzenesulfonamide (14)
The title compound was synthesised according to Method I using 2-fluoro-6-cyclohexylmethoxypurine (9,75 mg, 0.30 mmol), 3-amino-N,N-dimethylbenzenesulfonamide (see ESI S10; 136 mg, 0.68 mmol), TFE (3 mL), and TFA (0.11 mL, 1.5 mmol). After concentration in vacuo, the residual oil was extracted into DCM (10 mL) and cooled to 0 °C overnight. The resulting precipitate was collected by filtration under vacuum, washed with DCM (10 mL) and dried (Na2SO4) to give the title compound as an off-white solid (35 mg, 27%): Rf= 0.54 (EtOAc); mp 143-145 °C; IR (cm-1) 2926, 2850, 1598, 1579, 1543, 1433, 1394, 1334, 1249, 1149, 951; 1H NMR (300 MHz, DMSO-d6) δ 1.2-1.9 (11H, m, cyclohexyl), 2.64 (6H, s, N(CH3)2, 4.36 (2H, d, J = 6.2 Hz, OCH2), 7.24 (1H, d, J = 7.5 Hz, ArH), 7.52 (1H, dd, J = 8.0, 8.0 Hz, ArH), 7.96 (1H, d, J = 7.4 Hz, ArH), 8.04 (1H, s, H-8), 8.50 (1H, s, ArH), 9.77 (1H, s, ArNHAr), 12.89 (1H, s, N9-H); 13C NMR (125 MHz, DMSO-d6) δ 25.2, 26.0, 29.2, 36.9, 37.7, 71.3, 116.2, 119.1, 122.0, 129.3, 134.9, 142.0, 154.9; LCMS (ES+) m/z 431 [M+H]+; Anal. calcd for C20H26N6O3: C, 55.80; H, 6.09; N, 19.52%; found: C, 55.77; H, 6.00; N, 19.47; λmax (EtOH) 275, 293 nm.
[3-(6-Cyclohexylmethoxy-9H-purin-2-ylanilino]-N-methylacetamide (15)
The title compound was synthesised according to Method I using 2-fluoro-6-cyclohexylmethoxypurine (9, 75 mg, 0.30 mmol), 2-(3-aminophenyl)-N-methylacetamide (see ESI S12; 98 mg, 0.60 mmol), TFE (3 mL), and TFA (0.11 mL, 1.5 mmol). The crude product was adsorbed onto silica gel (~3 mL) and purified by column chromatography (eluent: 5% MeOH/ EtOAc) and HPLC to give an off-white powder (53 mg, 45%): Rf = 0.23 (MeOH-EtOAc; 1:9); mp 176-177 °C; IR (cm-1) 3267, 2921, 2850, 2362, 2160, 2013, 1587, 1499, 1396, 1346, 1239, 1119; 1H NMR (300 MHz, DMSO-d6) δ 1.0–1.8 (11H, m, cyclohexyl), 2.50 (3H, d, J = 4.6 Hz, NCH3), 4.26 (2H, d, J = 6.3 Hz, OCH2), 6.74 (1H, d, J = 7.6 Hz, ArH), 7.10 (1H, dd, J = 7.8, 7.9 Hz, ArH), 7.56 (1H, s, ArH), 7.62 (1H, d, J = 8.2 Hz, ArH), 7.85 (1H, q, J = 4.6 Hz, CONH), 7.96 (1H, s, H-8), 9.21 (1H, s, ArNHAr), 12.68 (1H, br, N9-H); 13C NMR (75 MHz, DMSO-d6) δ 25.6, 26.0, 26.4, 29.6, 37.3, 43.1, 44.5, 71.5, 117.2, 119.9, 122.0, 128.4, 134.6, 136.8, 139.8, 141.4, 156.0, 170.8; LCMS (ES+) m/z 395 [M + H]+; Anal. calcd for C21H26N6O2: C, 63.94; H, 6.64; N, 21.31%; found: C, 63.80; H, 6.85; N, 20.97; λmax (EtOH) 207.0, 271.5, 293.5 nm.
1-(4-(6-(Cyclohexylmethoxy)-9H-purin-2-ylamino)phenyl)-3-(2-morpholinoethyl)urea (16)
Following Method I, the title compound was prepared using 6-(cyclohexylmethoxy)-2-fluoro-9H-purine (9) (0.15 g, 0.59 mmol) and 1-(4-aminophenyl)-3-(2-morpholinoethyl) urea (see ESI S18; 0.31 g, 1.17 mmol) and TFA (0.27 mL, 3.5 mmol) in TFE (6.0 mL). The compound was purified by recrystallisation from EtOAc to obtain a white solid (50 mg, 17%): mp 161-163 ºC; IR (cm-1) 3134, 2849, 2812, 1634 υ(NN'C=O), 1558, 1506; 1H NMR (300 MHz, DMSO-d6) δ 1.12-1.42 (5H, m, cyclohexyl), 1.78-1.99 (6H, m, cyclohexyl), 2.54 (6H, m, N(CH2)3), 3.38 (2H, dt, J = 6.0, 12.0 Hz, CH2), 3.73 (4H, t, J = 4.5 Hz, OCH2 morpholine), 4.37 (2H, d, J = 6.0 Hz, OCH2), 7.21 (2H, d, J = 8.5 Hz, H-2' and H-6'), 7.67 (2H, d, J = 8.5 Hz, H-3' and H-5'), 7.92 (1H, s, H-8); 13C NMR (75 MHz, DMSO-d6) δ 27.3, 28.1, 31.2, 36.4, 39.1, 54.1, 59.4, 68.1, 73.3, 115.1, 121.4, 121.6, 123.0, 135.2, 137.9, 151.1, 151.9, 158.0, 159.1; LCMS (ES+) m/z 495.1 [M+H]+; λmax (EtOH) 280.0, 238.0 nm.
1-(4-(6-(Cyclohexylmethoxy)-9H-purin-2-ylamino)phenyl)-3-(2-(piperidin-1-yl)ethyl)urea (17)
Following Method I, the title compound was prepared using 6-(cyclohexylmethoxy)-2-fluoro-9H-purine (9, 0.15 g, 0.59 mmol) and 1-(4-aminophenyl)-3-(2-(piperidin-1-yl)ethyl)urea (see ESI S20; 0.30 g, 1.17 mmol) and TFA (0.27 mL, 3.5 mmol) in TFE (4 mL). The compound was purified using the Biotage SP4 chromatography (KP-NH; MeOH-EtOAc; 1:9) to obtain the desired compound as an off-white solid (0.14 g, 48%): mp 158-160 ºC; IR (cm-1) 2924, 2849, 1644 υ(NN'C=O), 1610, 1556, 1506; 1H NMR (300 MHz, DMSO-d6) δ 1.10-1.19 (5H, m, cyclohexyl), 1.48-1.55 (6H, m, CH2 piperidine), 1.76-1.84 (6H, m, cyclohexyl), 3.17 (2H, m, CH2), 2.33 (6H, m, N(CH2)3), 4.30 (2H, d, J = 6.0 Hz, OCH2), 7.27 (2H, d, J = 8.0 Hz, H-2' and H-6'), 7.62 (2H, d, J = 8.0 Hz, H-3' and H-5'), 7.95 (1H, s, H-8), 8.47 (1H, s, NH-4'), 9.09 (1H, s, NH); 13C NMR (125 MHz, DMSO-d6) δ 24.1, 25.2, 25.5, 26.0, 29.2, 36.4, 36.8, 54.0, 58.2, 70.9, 118.1, 119.2, 119.3, 128.4, 134.3, 134.9, 138.5, 154.3, 155.3, 155.7, 160.3; LCMS (ES+) m/z 493.6 [M+H]+; HRMS (ES+) calcd for C26H36N8O2 [M+H]+ 493.3034, found 493.3040; λmax (EtOH) 281.5, 238.0 nm.
1-(4-(6-(Cyclohexylmethoxy)-9H-purin-2-ylamino)phenyl)-3-(3-(dimethylamino)propyl)urea (18)
Following Method I, the title compound was prepared using 6-(cyclohexylmethoxy)-2-fluoro-9H-purine (9) (0.15 g, 0.59 mmol) and 1-(4-aminophenyl)-3-(3-(dimethylamino)propyl)urea (see ESI S22; 0.30 g, 1.17 mmol) and TFA (0.27 mL, 3.5 mmol) in TFE (4 mL). The compound was purified using the Biotage SP4 chromatography (KP-NH; MeOH-EtOAc; 1:4) to obtain title compound as an off-white solid (94 mg, 34%): mp 110-112 ºC; IR (cm-1) 2925, 2855, 2127, 2050, 1672 υ(NN'C=O), 1614, 1551, 1508; 1H NMR (300 MHz, DMSO-d6) δ 1.07-1.21 (5H, m, cyclohexyl), 1.50-1.57 (2H, quin., J = 7.0 Hz, CH2), 1.69-1.75 (6H, m, cyclohexyl), 2.12 (6H, s, N(CH3)2), 2.22 (2H, t, J = 7.0 Hz, CH2), 3.09 (2H, dt, J = 5.5, 7.0 Hz, CH2), 4.30 (2H, d, J = 6.0 Hz, OCH2), 6.15 (1H, br t, J = 5.5 Hz, NH), 7.27 (2H, d, J = 8.5 Hz, H-2' and H-6'), 7.62 (2H, d, J = 8.5 Hz, H-3' and H-5'), 7.96 (1H, s, H-8), 8.34 (1H, s, NH-4'), 9.08 (1H, s, NH); 13C NMR (125 MHz, DMSO-d6) δ 25.2, 26.0, 27.8, 29.2, 36.8, 37.5, 45.2, 56.8, 70.9, 118.2, 119.2, 134.3, 134.9, 138.9, 155.5, 155.7, 158.1; LCMS (ES+) m/z 467.7 [M+H]+; HRMS (ES+) calcd for C24H35N8O2 [M+H]+ 467.2877, found 467.2870; λmax (EtOH) 279.0, 238.0 nm.
6-(Cyclohexylmethoxy)-N-(4-nitrophenyl)-9H-purin-2-amine (19)
The title compound was prepared according to Method I using: 6-(cyclohexylmethoxy)-2-fluoro-9H-purine (9, 2.00 g, 8.0 mmol), 4-nitroaniline (2.21 g, 16.0 mmol) and TFA (3.06 mL, 40.0 mmol) in TFE (50 mL). The crude orange solid was purified by recrystallisation from EtOAc and obtained as a yellow solid (2.18 g, 74%): mp 180-181 ºC; IR (cm-1) 3390, 3331, 2936, 2862, 2758, 1728, 1591, 1522 υ(strong, br, NO2 asymmetric stretch), 1330 υ(strong, sharp, NO2 symmetric stretch); 1H NMR (300 MHz, DMSO-d6) δ 1.02-1.29 (5H, m, cyclohexyl), 1.66-1.85 (6H, m, cyclohexyl), 4.32 (2H, d, J = 6.0 Hz, OCH2), 8.04 (2H, d, J = 9.0, H-2' and H-6'), 8.18 (2H, d, J = 9.0, H-3' and H-5'), 8.21 (1H, s, H-8); 13C NMR (75 MHz, DMSO-d6) δ 21.0, 25.4, 26.5, 30.1, 71.9, 117.5, 125.5, 140.2, 148.2, 154.6; LCMS (ES+) m/z 369.3 [M+H]+; HRMS (ES+) calcd for C18H20N6O3 [M+H]+ 369.3614, found 369.3611.
6-(Cyclohexylmethoxy)-N-(3-nitrophenyl)-9H-purin-2-amine (20)
According to Method I, the title compound was prepared using: 6-(cyclohexylmethoxy)-2-fluoro-9H-purine (9) (2.00 g, 8.0 mmol), 3-nitroaniline (2.21 g, 16.0 mmol) and TFA (3.06 mL, 40.0 mmol) in TFE (50 mL). The crude orange solid was purified by column chromatography (silica; EtOAc-petrol; 4:6) and isolated as a yellow solid (1.89 g, 64%): mp 217-218 ºC; IR (cm-1) 3419, 3351, 2931, 2854, 2769, 1732, 1592, 1532 υ(strong, br, NO2 asymmetric stretch), 1351 υ(strong, sharp, NO2 symmetric stretch); 1H NMR (300 MHz, DMSO-d6) δ 1.17 (5H, m, cyclohexyl), 1.77 (6H, m, cyclohexyl), 4.38 (2H, d, J = 6.0 Hz, OCH2), 7.53 (1H, dd, J = 7.5, 8.0 Hz, H-5'), 7.73 (1H, dd, J = 1.5, 2.0 Hz, H-2'), 7.94-8.11 (2H, m, H-4', H-6'), 8.29 (1H, s, H-8), 9.10 (1H, s, N-2 H); LCMS (ES+) m/z 369.4 [M+H]+; HRMS (ES+) calcd for C18H20N6O3 [M+H]+ 369.3614, found 369.3609; λmax (EtOH) 379, 292, 271, 227.5 nm.
Reduction of aryl-nitro groups to corresponding anilines (compounds 21 and 22)
To a stirred solution of the nitroaromatic compound in sufficient anhydrous solvent as indicated was added 10% palladium on activated carbon (30% w/w). The resulting mixture was stirred under an atmosphere of H2 at room temperature for 24 h. The reaction mixture was filtered through a bed of Celite eluting with a mixture of MeOH-DCM (1:9), to afford the title compound following removal of the solvent in vacuo.
2-(4-Aminophenyl)amino-6-cyclohexylmethoxy-purine (21)
The title compound was synthesised from 6-(cyclo hexylmethoxy)-N-(4-nitrophenyl)-9H-purin-2-amine (19, 1.20 g, 3.26 mmol) with 10% palladium on activated carbon (0.36 g) in THF (100 mL) to obtain the title compound as a beige solid (1.10 g, 100%): Rf= 0.30 (MeOH-DCM; 1:9); mp 228-230 ºC; IR (cm-1) 3431 υ(NH), 3370 υ(NH2), 3234, 2923, 2834, 2359, 1613, 1584; 1H NMR (300 MHz, DMSO-d6) δ 1.02-1.29 (5H, m, cyclohexyl), 1.69-1.85 (6H, m, cyclohexyl), 4.26 (2H, d, J = 6.0 Hz, OCH2), 4.72 (2H, br s, NH2), 6.50 (2H, d, J = 8.5 Hz, H-2' and H-6'), 7.35 (2H, d, J = 8.5 Hz, H-3' and H-5'), 7.99 (1H, s, H-8), 9.01 (1H, s, NH); 13C NMR (75 MHz, DMSO-d6) δ 25.2, 26.5, 29.6, 37.2, 71.1, 113.8, 121.3, 130.2, 143.7, 156.6; LCMS (ES+) m/z 339.1 [M+H]+; HRMS (ES+) calcd for C18H22N6O [M+H]+ 389.1928, found 389.1931; λmax (EtOH) 277.0, 240.0 nm.
2-(3-Aminophenyl)amino-6-cyclohexylmethoxy-purine (22)
The title compound was synthesised from 6-(cyclo hexylmethoxy)-N-(3-nitrophenyl)-9H-purin-2-amine (20, 0.30 g, 0.82 mmol) with 10% palladium on activated carbon (90 mg) in THF (40 mL) to afford the title compound as a brown solid (0.23 g, 84%): mp 117-118 ºC (dec); IR (cm-1) 3254 υ(NH2), 2922, 2847, 1587, 1443, 1389, 1352, 1118; 1H NMR (300 MHz, DMSO-d6) δ 1.38-1.87 (5H, m, cyclohexyl), 1.72 (6H, m, cyclohexyl), 4.31 (2H, d, J = 6.0 Hz, OCH2), 4.89 (2H, br s, NH2), 6.18 (1H, dd, J = 7.5, 8.0 Hz, H-5'), 6.95 (3H, m, H-2', H-4', H-6'), 7.95 (1H, s, H-8), 8.96 (1H, s, NH); 13C NMR (125 MHz, DMSO-d6) δ 25.2, 26.0, 29.2, 36.9, 70.9, 104.7, 107.3, 107.7, 114.5, 128.5, 138.6, 141.5, 148.6, 155.7, 160.0; LCMS (ES+) m/z 339.0 [M+H]+; HRMS (ES+) calcd for C18H22N6O [M+H]+ 389.1928, found 389.1934; λmax (EtOH) 301, 271, 224.5 nm.
Synthesis of aminophenylacetic acid derivatives (23 – 25)
To a mixture of the aniline (2.25 mol. equiv.) and (9, 1 mol. equiv.) in TFE (5 mL/mmol) was added TFA (5 mol. equiv.). The mixture was boiled at reflux for 24 h and allowed to cool to room temperature. After concentration in vacuo, THF (20 mL) and NaOH aqueous solution (1 M, 15 mL) were added to the residue and the resulting mixture was stirred overnight. The pH was adjusted to around 1 with conc. HCl and the product was extracted with EtOAc (250 mL). The organic phase was separated, washed with 10 % HCl solution and dried (NaSO4). Removal of the solvent gave crude product, to which Et2O (100 mL) was added. After allowing the mixture to stand for 3 h the resulting precipitate was collected by suction filtration and washed with diethyl ether (30 mL). Recrystallisation from MeOH gave the pure product.
3-(6-Cyclohexylmethoxy-9H-purin-2-ylamino)benzoic acid (23)
The title compound was synthesised using 6-cyclohexylmethoxy-2-fluoro-9-methyl-9H-purine (9, 250 mg, 1.0 mmol), 3-aminobenzoic acid (309 mg, 2.3 mmol), TFE (5.0 mL), and TFA (0.37 mL, 5 mmol) to give an off-white solid (259 mg, 71%): Rf = 0.18 (MeOH-EtOAc; 1:9); mp 182-184 °C (dec.); IR (cm-1) 2925, 2850, 1593, 1564, 1440, 1357, 1114, 972; 1H NMR (300 MHz, DMSO-d6) δ 1.0-1.9 (11H, m, cyclohexyl), 4.36 (2H, d, J = 5.8 Hz, OCH2), 7.35 (1H, dd, J = 7.6, 7.8 Hz, ArH), 7.49 (1H, d, J = 7.3 Hz, ArH), 7.92 (1H, d, J = 7.6 Hz, ArH), 8.03 (1H, s, ArH), 8.55 (1H, s, H-8), 9.50 (1H, s, ArNHAr), 12.86 (1H, s br, N9-H); LCMS (ES+) m/z 368 [M+H]+; HRMS (ES+) calcd for C19H22N6O3 [M+H]+ 368.1717, found 368.1722; λmax (EtOH) 225, 274 nm.
[3-(6-Cyclohexylmethoxy-9H-purin-2-ylamino)phenyl]acetic acid (24)
The title compound was synthesised using 6-cyclohexylmethoxy-2-fluoro-9-methyl-9H-purine (9, 2.0 g, 8.0 mmol), 3-aminophenylacetic acid (2.7 g, 18 mmol), TFE (20 mL), and TFA (3.0 mL, 40 mmol) to give a colourless solid (1.7 g, 55%): Rf = 0.05 (MeOH-EtOAc; 0.5-9.5); mp 221-222 °C (dec.); IR (cm-1) 3434, 3118, 2923, 2851, 1603, 1493, 1417, 1261; 1H NMR (300 MHz, DMSO-d6) δ 1.0-1.9 (11H, m, cyclohexyl), 3.51 (2H, s, ArCH2), 4.33 (2H, d, J = 6.2 Hz, OCH2), 6.79 (1H, d, J = 7.3 Hz, ArH), 7.18 (1H, dd, J = 7.8, 7.9 Hz, ArH), 7.65 (1H, d, J = 8.3 Hz, ArH), 7.73 (1H, s, ArH), 8.00 (1H, s, H-8), 9.29 (1H, s, ArNHAr), 12.29 (1H, s br, CO2H), 12.78 (1H, s br, N9-H); LCMS (ES+) m/z 382 [M+H]+; λmax (EtOH) 209, 272 nm.
[3-(6-Cyclohexylmethoxy-9H-purin-2-ylamino)phenyl]propionic acid (25)
The title compound was synthesised using 6-cyclohexylmethoxy-2-fluoro-9-methyl-9H-purine (9, 300 mg, 1.2 mmol), 3-(3-aminophenyl)propionic acid (446 mg, 2.7 mmol), TFE (5.0 mL), and TFA (0.45 mL, 6.0 mmol) to give an off-white solid (225 mg, 48%): Rf = 0.18 (MeOH-EtOAc; 1:9); mp 231-232 °C (dec.); IR (cm-1) 2922, 2849, 1707, 1639, 1595, 1491, 1415, 1251, 1128, 972; 1H NMR (300 MHz, DMSO-d6) δ 1.0-1.9 (11H, m, cyclohexyl), 2.53 (2H, t, J = 7.9 Hz, CH2CO2H), 2.80 (2H, t, J = 7.7 Hz, ArCH2), 4.35 (2H, d, J = 6.3 Hz, OCH2), 6.81 (1H, d, J = 7.6 Hz, ArH), 7.17 (1H, dd, J = 7.8, 7.9 Hz, ArH), 7.58 (1H, d, J = 8.1 Hz, ArH), 7.71 (1H, s, ArH), 8.38 (1H, s, H-8), 9.41 (1H, s, ArNHAr), 12.10 (1H, s br, N9-H); 13C NMR (125 MHz, DMSO-d6) δ 21.1, 25.2, 25.9, 29.1, 30.7, 35.3, 36.8, 39.0, 71.4, 116.5, 118.5, 121.0, 128.3, 139.4, 140.7, 141.0, 154.3, 156.0, 159.0, 173.7; LCMS (ES+) m/z 396 [M+H]+; HRMS (ES+) calcd for C21H26N5O3 [M+H]+ 396.2030, found 396.2026; λmax (EtOH) 272 nm.
2,2,2-Trifluoroethyl-3-(6-cyclohexylmethoxy-9H-purin-2-ylamino)phenylmethanesulfonate (26)
ThThe title compound was synthesised according to Method I using 2-fluoro-6-cyclohexylmethoxypurine (9) (1.24 g, 5.0 mmol), 2,2,2-trifluoroethyl 3-aminophenylmethanesulfonate (S26, 2.8 g, 10 mmol), TFE (25 mL), and TFA (1.84 mL, 25 mmol). The crude product was adsorbed onto silica (~50 mL) and purified by chromatography (silica; EtOAc-petrol; 6:4) to give a viscous oil that was triturated with DCM (35 mL), to afford a pale yellow powder (1.83 g, 74%): Rf = 0.22 (EtOAc-petrol; 6:4); mp 199-200 °C; IR (cm-1) 3436, 3112, 2924, 2848, 1591, 1537, 1494, 1435, 1392, 1338, 1287, 1151, 1028; 1H NMR (300 MHz, DMSO-d6) δ 1.0-1.9 (11H, m, cyclohexyl), 4.35 (2H, d, J = 6.2 Hz, OCH2), 4.84 (2H, s, ArCH2), 4.94 (2H, q, J = 8.6 Hz, CH2CF3), 6.99 (2H, d, J = 7.6 Hz, 2 × ArH overlap), 7.31 (1H, t, J = 7.8, 8.1 Hz, ArH), 7.86-7.88 (2H, m, 2 × ArH), 8.03 (1H, s, H-8), 9.44 (1H, s, ArNHAr), 12.86 (1H, br, N9-H); 13C NMR (125 MHz, DMSO-d6) δ 25.2, 26.0, 29.2, 36.9, 55.5, 55.7, 64.9 (q, 2JC-F = 36 Hz), 71.2, 114.9, 118.8, 121.5, 122.6 (q, 1JC-F = 276 Hz), 123.2, 128.6, 139.0, 141.4, 155.3, 160.1; 19F NMR (470 MHz, DMSO-d6) δ -72.86 (t, J = 8.6 Hz, CF3); LCMS (ES+) m/z 500 [M+H]+; Anal. calcd for C21H24F3N5O4S: C, 50.50; H, 4.84; N, 14.02%; found: C, 50.80; H, 4.56; N, 13.91; λmax (EtOH) 272, 292 nm.
Synthesis of 4-N-acyl-(2—phenyl)-amino-9H-purines (compounds 27-30)
A stirred solution of the 4- (21) or 3-substituted aniline (22) (0.20 g, 0.59 mmol), 4-dimethylaminopyridine (0.07 g, 0.59 mmol) and Et3N (0.33 mL, 2.36 mmol) in THF (5 mL) was maintained at 0 ºC. To the chilled mixture was slowly added the required acyl chloride (1.77 mmol) and the reaction mixture was allowed to warm to room temperature (or heated to reflux as stated) with continued stirring for 18 h. EtOAc (20 mL) was added to the crude mixture and washed with sat. NaHCO3 solution (3 × 10 mL). The combined aqueous phase was re-extracted using EtOAc (2 × 10 mL). The combined organic extracts were washed with 0.1 M HCl (3 × 10 mL) and the acidic extracts again re-extracted with EtOAc (2 × 10 mL). The combined organic extracts were dried (Na2SO4). Products were obtained after purification as described. The N-9 acyl compound (0.10 g) was dissolved in a 1:1 mixture of DCM (2 mL) and TFA (2 mL) and stirred at room temperature for 18 h. After this time, the solvent was removed in vacuo and the trifluoroacetate salt of the purine was suspended in EtOAc (5 mL). The suspension was washed with saturated NaHCO3 solution (3 × 10 mL) and the organic phase was dried (Na2SO4). Solvents were removed under reduced pressure and the resulting solid was purified as required.
N-(3-(6-(Cyclohexylmethoxy)-9H-purin-2-ylamino)phenyl)acetamide (27)
The title compound was obtained using acetyl chloride (0.13 mL, 1.77 mmol) with N-1-(6-(cyclohexylmethoxy)-9H-purin-2-yl)benzene-1,3-diamine (22). The crude mixture was purified using chromatography (silica; EtOAc) to obtain the compound as an off-white solid (0.15 g, 60%): mp 83-85 ºC; IR (cm-1) 3294, 2923, 2852, 1971, 1734 υ(amide C=O), 1671 υ(amide C=O), 1586, 1541; 1H NMR (300 MHz, DMSO-d6) δ 1.16 (5H, m, cyclohexyl), 1.74 (6H, m, cyclohexyl), 2.04 (3H, s, CH3), 2.88 (3H, s, CH3), 4.34 (2H, d, J = 6.0 Hz, OCH2), 7.02-7.11 (1H, m, H-4'), 7.13-7.23 (1H, dd, J = 7.5, 8.0 Hz, H-5'), 7.38-7.46 (1H, m, H-6'), 8.14-8.15 (1H, m, H-2'), 8.43-8.43 (1H, s, H-8), 9.65 (1H, s, NH), 9.86 (1H, br s, CONH); 13C NMR (125 MHz, DMSO-d6) δ 24.0, 25.2, 25.9, 26.0, 29.1, 36.7, 36.8, 71.0, 71.4, 110.2, 110.5, 112.4, 113.0, 113.9, 114.3, 128.3, 137.7, 139.3, 156.0, 160.7, 168.0 (C=O), 168.2 (C=O); LCMS (ES+) m/z 423.3 [M+H]+; λmax (EtOH) 306.5, 209.0 nm. After acidolysis according to the general procedure, the title compound was isolated as an orange solid was achieved without further purification (90 mg, 100%): mp 93-95ºC; IR (cm-1) 3260, 2923, 2851, 2031, 1666 υ(amide C=O), 1591, 1537; 1H NMR (300 MHz, DMSO-d6) δ 1.04-1.32 (5H, m, cyclohexyl), 1.64-1.84 (6H, m, cyclohexyl), 2.03 (3H, s, CH3), 4.34 (2H, d, J = 6.0 Hz, OCH2), 7.07-7.18 (2H, m, H-4' and H-6'), 7.47 (1H, dd, J = 8.0, 8.5 Hz, H-5'), 7.96 (1H, m, H-2'), 7.99 (1H, s, H-8), 9.30 (1H, s, NH), 9.85 (1H, s, CONH); 13C NMR (125 MHz, DMSO-d6) δ 18.5, 24.0, 25.2, 26.0, 29.2, 36.8, 56.0, 71.0, 110.2, 112.4, 113.9, 114.7, 138.8, 139.3, 141.3, 154.2, 155.5, 160.1, 168.0 (C=O); LCMS (ES+) m/z 381.4 [M+H]+; HRMS (ES+) calcd for C20H24N6O2 [M+H]+ 381.2034, found 381.2037; λmax (EtOH) 293.5, 270.0, 230.0 nm.
N-(4-(6-(Cyclohexylmethoxy)-9H-purin-2-ylamino)phenyl)pivalamide (28)
The title compound was obtained from pivaloyl chloride (0.22 mL, 1.77 mmol) with N-1-(6-(cyclohexylmethoxy)-9H-purin-2-yl)benzene-1,4-diamine (21, 0.20 g, 0.59 mmol), DMAP (70 mg, 0.59 mmol) and triethylamine (0.33 mL, 2.36 mmol) at reflux for 18 h. The crude mixture was purified using column chromatography (silica; EtOAc-petrol; 1:1) to isolate the compound as an off-white solid (92 mg, 31%): mp 109-111 ºC; IR (cm-1) 3421, 3328, 3123, 2969, 2921, 2844, 1732 υ(amide C=O), 1658 υ(amide C=O), 1592, 1561, 1517; 1H NMR (300 MHz, DMSO-d6) δ 1.11 (9H, s, tBu), 1.16 (5H, m, cyclohexyl), 1.50 (9H, s, tBu), 1.71 (6H, m, cyclohexyl), 4.34 (2H, d, J = 6.0 Hz, OCH2), 7.55 (1H, d, J = 9.0 Hz, H-2' and H-6'), 7.67 (1H, d, J = 9.0 Hz, H-3' and H-5'), 8.43 (1H, s, H-8), 9.11 (1H, s, CONH), 9.51 (1H, s, NH); 13C NMR (125 MHz, DMSO-d6) δ 25.9, 26.0, 27.0, 27.1, 27.3, 28.6, 29.2, 36.6, 37.7, 70.9, 71.4, 118.3, 120.4, 120.7, 127.5, 128.7, 132.7, 136.7, 137.7, 155.5, 176.0 (C=O), 176.4 (C=O); LCMS (ES+) m/z 507.5 [M+H]+; λmax (EtOH) 312.0; 266.5, 207.5 nm. After acidolysis according to the general procedure, the title compound was isolated as a pale pink solid and was used without further purification (83 mg, 100%): mp 164-166ºC; IR (cm-1) 2921, 2850, 2161, 1622 υ(amide C=O), 1589, 1508; 1H NMR (300 MHz, DMSO-d6) δ 0.92-1.48 (14H, m, tBu and cyclohexyl), 1.70-1.84 (6H, m, cyclohexyl), 4.30.4.34 (2H, d, J = 6.0 Hz, OCH2), 7.50 (2H, d, J = 7.0 Hz, H-2' and H-6'), 7.60 (2H, d, J = 7.0 Hz, H-3' and H-5'), 7.95 (1H, s, H-8), 9.15 (1H, s, NH), 9.22 (1H, s, CONH); LCMS (ES+) m/z 423.5 [M+H]+; HRMS (ES+) calcd for C23H30N6O [M+H]+ 423.2503, found 423.2509; λmax (EtOH) 369.5, 300.0, 291.5, 284.0, 207.0 nm.
N-(4-(6-(Cyclohexylmethoxy)-9H-purin-2-ylamino)phenyl)benzamide (29)
The title compound was obtained from benzoyl chloride (0.21 mL, 1.77 mmol) with N-1-(6-(cyclohexylmethoxy)-9H-purin-2-yl)benzene-1,4-diamine (21, 0.20 g, 0.59 mmol), DMAP (70 mg, 0.59 mmol) and triethylamine (0.33 mL, 2.36 mmol). The crude mixture was purified using chromatography (silica; EtOAc-petrol; 1:4) to obtain the compound as a white solid (0.26 g, 81%): mp 206-208 ºC; IR (cm-1) 3301, 3139, 2920, 2845, 1701 υ(amide C=O), 1634 υ(amide C=O), 1549, 1506; 1H NMR (300 MHz, DMSO-d6) δ 1.00-1.39 (5H, s, cyclohexyl), 1.60-1.95 (6H, s, cyclohexyl), 4.35 (2H, d, J = 6.0 Hz, OCH2), 7.19-7.29 (2H, d, J = 8.0 Hz, H-2' and H-6'), 7.38-7.46 (2H, d, J = 8.0 Hz, H-3' and H-5'), 7.49-7.69 (5H, m, Ph), 7.80-7.88 (1H, t, J = 7.5, Hz, Ph), 7.89-7.99 (4H, m, Ph), 8.43 (1H, s, H-8), 9.52 (1H, s, NH); 13C NMR (125 MHz, DMSO-d6) δ 25.2, 26.0, 29.2, 36.8, 71.3, 115.4, 118.0, 120.5, 127.5, 128.3, 128.6, 130.3, 131.3, 132.0, 132.7, 132.9, 135.2, 136.2, 139.2, 152.7, 155.6, 160.6, 165.1 (C=O), 166.5 (C=O); LCMS (ES+) m/z 546.3 [M+H]+; λmax (EtOH) 315.0, 229.5 nm. After acidolysis according to the general procedure, the title compound was isolated as a beige solid was isolated and was used without further purification (81 mg, 100%): mp 260-262ºC; IR (cm-1) 3300, 3139, 2920, 2845, 1634 υ(amide C=O), 1553, 1506; 1H NMR (300 MHz, DMSO-d6) δ 0.99-1.37 (5H, m, cyclohexyl), 1.58-1.96 (6H, m, cyclohexyl), 4.27-4.40 (2H, d, J = 6.0 Hz, OCH2), 7.48-7.60 (3H, m, phenyl), 7.62-7.72 (2H, d, J = 9.0 Hz, H-2' and H-6'), 7.73-7.83 (2H, d, J = 9.0 Hz, H-3' and H-5'), 7.91-8.05 (3H, m, phenyl and H-8), 9.28 (1H, s, NH), 10.14 (1H, s, CONH), 12.68-12.93 (1H, br s, N9-H); 13C NMR (125 MHz, DMSO-d6) δ 25.2, 26.0, 29.2, 36.8, 71.0, 118.5, 120.8, 127.5, 128.3, 131.3, 132.5, 135.1, 137.1, 155.5, 165.0 (C=O); LCMS (ES+) m/z 443.5 [M+H]+; HRMS (ES+) calcd for C25H26N6O2 [M+H]+ 443.2190, found 443.2196; λmax (EtOH) 315.0 nm.
N-[4-(6-Cyclohexylmethoxy-9H-purin-2-ylamino)phenyl]isonicotinamide (30)
The title compound was obtained from isonicotinoyl chloride (0.32 g, 1.77 mmol) with N-1-(6-(cyclohexylmethoxy)-9H-purin-2-yl)benzene-1,4-diamine (21, 0.2 g, 1.77 mmol), DMAP (70 mg, 0.59 mmol) and triethylamine (0.33 mL, 2.36 mmol). The crude mixture was washed with saturated NaHCO3 solution (20 mL) and beige solid collected by filtration and washed with MeOH (20 mL) (0.14 g, 43%): mp 248-250 ºC (dec.); IR (cm-1) 3339, 2927, 2851, 1664 υ(amide C=O), 1622, 1587, 1544, 1516; 1H NMR (300 MHz, DMSO-d6) δ 1.30-1.07 (5H, m, cyclohexyl), 1.83-1.71 (6 H, m, cyclohexyl), 4.32 (2H, d, J = 6.0 Hz, OCH2), 7.83 (2H, d, J = 8.5 Hz, H-2' and H-6'), 7.87 (2H, d, J = 5.5 Hz, pyridyl), 7.64 (2H, d, J = 8.5 Hz, H-3' and H-5'), 8.78 (2H, d, J = 5.5 Hz, pyridyl), 9.05 (1H, s, NH), 10.46-10.33 (1H, s, CONH); 13C NMR (125 MHz, DMSO-d6) δ 25.2, 26.0, 29.3, 36.9, 70.8, 118.1, 120.9, 121.5, 131.4, 138.1, 142.1, 150.2, 154.7, 157.4 (C=O), 159.3 (C=O), 163.3; LCMS (ES+) m/z 444.4 [M+H]+; HRMS (ES+) calcd for C24H25N7O 444.2142 [M+H]+, found 444.2143; λmax(EtOH) 315.5, 271.0, 206.0 nm.
Synthesis of urea-substituted 2-arylaminopurines (e.g. 31 – see ESI for further example)
To a stirred suspension of the required N-(6-cyclo hexylmethoxy-9H-purin-2-yl)benzene-diamine (21 or 22) (0.20 g, 0.59 mmol) and NaOCN (80 mg, 1.18 mmol) in a mixture of DCM (10 mL) and DMF (3 mL) was added TFA (9 μL, 1.18 mmol) over 2 min. The mixture was stirred under N2 for 18 h. Solvents were removed under reduced pressure and the residue was dissolved in DCM (25 ml) and extracted with 0.1 M HCl (3 × 20 mL). The organic extract was dried using (Na2SO4) and the filtrate was concentrated to give a beige solid as crude mixture of mono- and di-urea compounds. The mixture was dissolved in a 1:1 mixture of DCM (2 mL) and TFA (2 mL) and stirred at room temperature for 18 h. After this time, the solvent was removed and the trifluoroacetate salt of the purine was suspended in EtOAc (5 mL). The suspension was washed with saturated NaHCO3 solution (3 × 10 mL) and the organic phase was dried (Na2SO4). Solvents were removed under reduced pressure and the resulting solid was used without further purification.
1-(3-(6-(Cyclohexylmethoxy)-9H-purin-2-ylamino)phenyl) urea (31)
Using N-(6-cyclohexylmethoxy-9H-purin-2-yl)benzene-1,3-diamine (22) the product was obtained as a off-white solid (0.14 g, 64%) was isolated without further purification: mp193-194 ºC; IR (cm-1) 3285 υ(NH2), 3159, 2919, 2849, 1674 υ(NN'C=O), 1620, 1589, 1541; 1H NMR (300 MHz, DMSO-d6) δ 1.17 (5H, m, cyclohexyl), 1.74 (6H, m, cyclohexyl), 4.33 (2H, d, J = 6.0 Hz, OCH2), 7.05 (2H, m, H-4' and H-6'), 7.43 (1H, dd, J = 7.5, 8.0 Hz, H-5'), 7.67 (1H, s, H-2'), 7.95 (1H, s, H-8), 8.46 (1H, s, NH-4'), 9.21 (1H, s, NH), 12.77 (1H, s, NH-9); 13C NMR (75 MHz, DMSO-d6) δ 14.4, 25.6, 26.4, 29.6, 37.3, 71.4, 109.7, 128.5, 138.9, 140.9, 141.7, 156.1, 156.3; LCMS (ES+) m/z 382.3 [M+H]+; HRMS (ES+) calcd for C19H23N7O2 [M+H]+ 382.4311, found 382.4312; λmax (EtOH) 295.0, 272.0, 229.0 nm.
Synthesis of amide derivatives (compounds 32-47)
To the appropriate carboxylic acid (23-24) (1 mol. equiv.) and DIPEA (2 mol. equiv.) in DMF (3 mL/mmol) was added carbonyldiimidazole (2 mol. equiv.) and the resulting mixture was stirred for 1.5 h at room temperature. The appropriate amine (4 mol. equiv.) was added and the reaction was stirred overnight. Solvents were removed and the residue was extracted into EtOAc or THF depending on solubility. The extract was washed with saturated aqueous NaHCO3 and dried (Na2SO4). The solvent was removed to afford the crude product, which was purified as indicated by either by chromatography on silica, or by using the Biotage SP4 purification system.
[3-(6-Cyclohexylmethoxy-9H-purin-2-ylamino)phenyl]piperidin-1-ylmethanone (32)
The title compound was prepared using 23 (50 mg, 0.14 mmol), carbonyldiimidazole (45 mg, 0.28 mmol), DIPEA (50 μL, 0.28 mmol), and piperidine (55 μL, 0.56 mmol) in DMF (2 mL). The crude product was purified by chromatography on silica using EtOAc-petrol (9:1) as eluent to give an off-white powder (33 mg, 54%): Rf = 0.48 (EtOAc); mp 133-135 °C; IR (cm-1) 2923, 2850, 1581, 1539, 1437, 1390, 1348, 1276, 1209, 1114, 975; 1H NMR (300 MHz, DMSO-d6) δ 1.1-1.9 (17H, m, cyclohexyl and piperidyl), 3.34 (br, overlap with H2O, NCH2), 3.58 (2H, s, NCH2), 4.34 (2H, d, J = 6.3 Hz, OCH2), 6.87 (1H, d, J = 7.5 Hz, ArH), 7.31 (1H, dd, J = 7.8, 7.9 Hz, ArH), 7.79 (1H, d, J = 8.3 Hz, ArH), 7.94 (1H, s, ArH), 8.03 (1H, s, H-8), 9.46 (1H, s, ArNHAr), 12.87 (1H, s br, N9-H); LCMS (ES+) m/z 435.42 [M+H]+; Anal. calcd for C24H30N6O2: C, 66.34; H, 6.96; N, 19.34%; found: C, 66.41; H, 7.07; N, 19.04; λmax (EtOH) 273, 293 nm.
3-(6-Cyclohexylmethoxy-9H-purin-2-ylamino)-N-(4-dimethylaminobutyl)benzamide (33)
The title compound was prepared using 23 (80 mg, 0.22 mmol), carbonyldiimidazole (71 mg, 0.44 mmol), DIPEA (79 μL, 0.44 mmol), and N,N-dimethyl-1,4-butanediamine (128 mg, 1.1 mmol) in DMF (3 mL). The crude product was purified using a Biotage SP4 purification system (12 + M KP-NH Si cartridge; MeOH-EtOAc; 1:4) to give a colourless powder (53 mg, 51%): Rf = 0.67 (NH2-modified silica – MeOH-EtOAc; 1:4); mp 125-127 °C; IR (cm-1) 3076, 2920, 2849, 2363, 2337, 1597, 1537, 1483, 1438, 1391, 1352, 1283, 1117, 973; 1H NMR (300 MHz, DMSO-d6) δ 1.00-1.90 (15H, m, cyclohexyl and CH2CH2CH2CH2), 2.11 (6H, s, 2 × CH3), 2.21 (2H, t, J = 6.9 Hz, Me2NCH2), 3.25 (2H overlap with H2O, m, CH2CH2NHCO), 4.35 (2H, d, J = 6.1 Hz, OCH2), 7.29-7.34 (2H, m, 2 × ArH), 7.85 (1H, m, ArH), 8.01 (1H, s, H-8), 8.31 (1H, s, ArH), 8.38 (1H, t, J = 5.3 Hz, CONH), 9.40 (1H, s, ArNHAr); LCMS (ES+) m/z 466.50 [M+H]+; HRMS (ES+) calcd for C25H36N7O2 [M+H]+ 466.2925, found 466.2920; λmax (EtOH) 273, 293 nm.
2-[3-(6-Cyclohexylmethoxy-9H-purin-2-anilino)]-N-isobutylacetamide (34)
The title compound was prepared using 24 (60 mg, 0.16 mmol), carbonyldiimidazole (50 mg, 0.31 mmol), DIPEA (56 μL, 0.31 mmol), and isobutylamine (64 μL, 0.63 mmol) in DMF (2 mL). The crude product was purified by chromatography (silica; EtOAc-petrol; 9:1) to give a white powder (35 mg, 52%): Rf = 0.59 (EtOAc); mp 144-145 °C; IR (cm-1) 3280, 3091, 2925, 2853, 1643, 1586, 1437, 1344, 1256, 1161, 974; 1H NMR (300 MHz, DMSO-d6) δ 0.82 (6H, d, J = 6.7 Hz, CH(CH3)2), 1.0–2.0 (11H, m, cyclohexyl), 1.69 (1H, m, CH2CH(CH3)2), 2.88 (2H, t, J = 6.5 Hz, CH2NHCO), 3.37 (2H, s, ArCH2), 4.34 (2H, d, J = 6.3 Hz, OCH2), 6.82 (1H, d, J = 7.6 Hz, ArH), 7.17 (1H, dd, J = 7.8, 7.9 Hz, ArH), 7.64 (1H, s, ArH), 7.71 (1H, d, J = 8.2 Hz, ArH), 7.97 (1H, t, J = 5.6 Hz, CONH), 8.15 (1H, s, H-8), 9.26 (1H, s, ArNHAr), 12.76 (1H, br, N9-H); LCMS (ES+) m/z 437 [M+H]+; HRMS (ES+) calcd for C24H33N6O2 [M+H]+ 437.2660, found 437.2659; λmax (EtOH) 214, 272, 291 nm.
2-[3-(6-Cyclohexylmethoxy-9H-purin-2-ylanilino]-N-cyclopentylacetamide (35)
The title compound was prepared using 24 (60 mg, 0.16 mmol), carbonyldiimidazole (50 mg, 0.31 mmol), DIPEA (56 μL, 0.31 mmol), and cyclopentylamine (62 μL, 0.63 mmol) in DMF (2 mL). The crude product was purified by chromatography (silica; EtOAc-petrol; 9:1) to give a white powder (14 mg, 20%): Rf = 0.57 (EtOAc); mp 228-229 °C; IR (cm-1) 3277, 2925, 2851, 1641, 1587, 1537, 1441, 1394, 1251; 1H NMR (300 MHz, DMSO-d6) δ 1.0–1.9 (19H, m, cyclohexyl and cyclopentyl), 3.17 (2H, s, ArCH2), 3.97 (1H, m, cyclopentyl), 4.34 (2H, d, J = 6.7 Hz, OCH2), 6.80 (1H, d, J = 7.3 Hz, ArH), 7.18 (1H, dd, J = 7.4, 7.6 Hz, ArH), 7.60 (1H, s, ArH), 7.71 (1H, d, J = 7.7 Hz, ArH), 8.01 (1H, d, J = 7.6 Hz, CONH), 9.27 (1H, s, ArNHAr), 12.79 (1H, br, N9-H); LCMS (ES+) m/z 449 [M+H]+; HRMS (ES+) calcd for C25H33N6O2 [M+H]+ 449.2660, found 449.2664; λmax (EtOH) 216, 272 nm.
2-[3-(6-Cyclohexylmethoxy-9H-purin-2-ylanilino]-N-cyclohexylacetamide (36)
The title compound was prepared using 24 (60 mg, 0.16 mmol), carbonyldiimidazole (50 mg, 0.31 mmol), DIPEA (56 μL, 0.31 mmol), and cyclohexylamine (72 μL, 0.63 mmol) in DMF (2 mL). The crude product was purified by chromatography (silica; EtOAc-petrol; 9:1) to give a white powder (17 mg, 24%): Rf = 0.64 (EtOAc); mp 226-227 °C; IR (cm-1) 3272, 2922, 2850, 2159, 1639, 1585, 1537, 1442, 1346, 1253, 1126; 1H NMR (300 MHz, DMSO-d6) δ 1.0–1.9 (21H, m, cyclohexyl), 3.35 (2H, s, ArCH2 overlap with H2O), 3.50 (1H, m, cyclohexyl 3′ CH), 4.34 (2H, d, J = 6.2 Hz, OCH2), 6.81 (1H, d, J = 7.4 Hz, ArH), 7.17 (1H, dd, J = 7.6, 8.1 Hz, ArH), 7.59 (1H, s, ArH), 7.71 (1H, d, J = 8.5 ArH), 7.89 (1H, d, J = 7.7 Hz, CONH), 7.96 (1H, s, H-8), 9.26 (1H, s, ArNHAr), 12.76 (1H, br, N9-H); LCMS (ES+) m/z 463 [M+H]+; HRMS (ES+) calcd for C26H35N6O2 [M+H]+ 463.2816, found 463.2815; λmax (EtOH) 215, 272 nm.
[3-(6-Cyclohexylmethoxy-9H-purin-2-ylanilino]-N-methyl-N-cyclohexylacetamide (37)
The title compound was prepared using 24 (50 mg, 0.13 mmol), carbonyldiimidazole (42 mg, 0.26 mmol), DIPEA (43 μL, 0.26 mmol), and N-methylcyclohexylamine (69 μL, 0.52 mmol) in DMF (2 mL). The crude product was purified by chromatography (silica; EtOAc-petrol; 9:1) to give a white powder (14 mg, 23%): Rf = 0.67 (EtOAc); mp 134-135 °C; IR (cm-1) 2922, 2850, 2157, 1583, 1539, 1489, 1437, 1390, 1205, 1123; 1H NMR (300 MHz, DMSO-d6) δ 1.0–1.9 (21H, m, cyclohexyl), 2.72 (3H, s, NCH3), 3.65 (2H, s, ArCH2), 4.27 (1H, m, cyclohexyl 3′ CH), 4.33 (2H, d, J = 5.9 Hz, OCH2), 6.76 (1H, d, J = 7.8 Hz, ArH), 7.18 (1H, dd, J = 7.8, 8.0 Hz, ArH), 7.62 (1H, s, ArH), 7.67-7.72 (2H, m, ArH and H-8), 9.27 (1H, s, ArNHAr), 12.79 (1H, br, N9-H); LCMS (ES+) m/z 477 [M+H]+; HRMS (ES+) calcd for C18H22N5O2 [M+H]+ 477.2973, found 477.2972; λmax (EtOH) 216, 272, 291 nm.
[3-(6-Cyclohexylmethoxy-9H-purin-2-ylanilino]-N, N-diisopropylacetamide (38)
The title compound was prepared using 24 (60 mg, 0.16 mmol), carbonyldiimidazole (50 mg, 0.31 mmol), DIPEA (56 μL, 0.31 mmol), and diisopropylamine (89 μL, 0.63 mmol) in DMF (2 mL). The crude product was purified by chromatography (silica; EtOAc-petrol; 9:1) to give a white powder (35 mg, 49%): Rf = 0.71 (EtOAc); mp 126-127 °C; IR (cm-1) 2925, 2851, 2160, 1586, 1441, 1342, 1211, 1118; 1H NMR (300 MHz, DMSO-d6) δ 1.0–1.9 (11H, m, cyclohexyl), 1.05 (6H, d, J = 6.5 Hz, CH(CH3)2), 1.40 (6H, d, J = 6.7 Hz, CH(CH3)2), 3.41 (1H, m, 3′ i-propyl CH), 3.66 (2H, s, ArCH2), 4.00 (1H, m, 3′ i-propyl CH), 4.30 (2H, d, J = 6.2 Hz, OCH2), 6.82 (1H, d, J = 7.8 Hz, ArH), 7.18 (1H, dd, J = 7.9, 8.3 Hz, ArH), 7.47 (1H, m, ArH), 7.59 (1H, s, ArH), 7.64 (1H, s, H-8), 11.65 (1H, br, N9-H); LCMS (ES+) m/z 465 [M+H]+; HRMS (ES+) calcd for C26H37N6O2 [M+H]+ 465.2973, found 465.2976; λmax (EtOH) 214, 272, 292 nm.
[3-(6-Cyclohexylmethoxy-9H-purin-2-ylanilino]-N-propylacetamide (39)
The title compound was prepared using 24 (50 mg, 0.13 mmol), carbonyldiimidazole (42 mg, 0.26 mmol), DIPEA (43 μL, 0.26 mmol), and n-propylamine (42 μL, 0.52 mmol) in DMF (2 mL). The crude product was purified by chromatography (silica; MeOH-EtOAc; 0.5:9.5) to give a white powder (13 mg, 24%): Rf = 0.23 (MeOH-EtOAc; 0.5:9.5); mp 141-142 °C (dec.); IR (cm-1) 3275, 3081, 2929, 2853, 1648, 1450, 1358, 1256; 1H NMR (300 MHz, DMSO-d6) δ 0.81 (3H, t, J = 7.8 Hz, CH2CH3), 1.0–1.81 (13H, m, cyclohexyl and CH3CH2), 2.99 (2H, m, CH2CH2NH), 3.49 (2H, s, ArCH2), 4.32 (2H, d, J = 6.2 Hz, OCH2), 6.79 (1H, d, J = 7.3 Hz, ArH), 7.16 (1H, dd, J = 7.6, 7.8 Hz, ArH), 7.62 (1H, s, ArH), 7.97 (1H, d, J = 8.26 Hz, ArH), 7.97 (2H, br, CONH and H-8), 9.24 (1H, s, ArNHAr), 12.81 (1H, s br, N9-H); LCMS (ES+) m/z 423 [M+H]+; λmax (EtOH) 272 nm.
2-[3-(6-Cyclohexylmethoxy-9H-purin-2-ylamino]-N-(3-hydroxy-propyl)phenylacetamide (40)
The title compound was prepared using 24 (60 mg, 0.16 mmol), carbonyldiimidazole (50 mg, 0.31 mmol), DIPEA (56 μL, 0.31 mmol), and 3-amino-1-propanol (48 μL, 0.63 mmol) in DMF (2 mL). The crude product was purified by chromatography (silica; MeOH-EtOAc; 0.5:9.5) to give a white powder (24 mg, 34%): Rf = 0.08 (EtOAc); mp 132-133 °C; IR (cm-1) 3267, 2920, 2845, 1641, 1587, 1494, 1436, 1256; 1H NMR (300 MHz, DMSO-d6) δ 1.0–1.9 (11H, m, cyclohexyl), 1.55 (2H, m, CH2CH2CH2), 3.11 (2H, m, CH2NH), 3.36-3.42 (4H, m, ArCH2, CH2OH), 4.34 (2H, d, J = 6.5 Hz, OCH2), 6.81 (1H, d, J = 7.3 Hz, ArH), 7.18 (1H, dd, J = 7.7, 8.3 Hz, ArH), 7.63 (1H, s, ArH), 7.71 (1H, d, J = 8.9 Hz, ArH), 8.01 (2H, br m, H-8 and CONH), 9.27 (1H, s, ArNHAr), 12.89 (1H, br, N9-H); LCMS (ES+) m/z 439 [M+H]+; HRMS (ES+) calcd for C23H31N6O3 [M+H]+ 439.2452, found 439.2451; λmax (EtOH) 272, 291 nm.
2-[3-(6-Cyclohexylmethoxy-9H-purin-2-ylamino)phenyl]-N-(3-imidazol-1-yl-propyl)acetamide (41)
The title compound was prepared using 24 (50 mg, 0.13 mmol), carbonyldiimidazole (42 mg, 0.26 mmol), DIPEA (43 μL, 0.26 mmol), and N-(3-aminopropyl)imidazole (62 μL, 0.52 mmol) in DMF (2 mL). The crude product was purified using a Biotage SP4 purification system (12 + M KP-NH silica cartridge; MeOH-EtOAc; 1.5:8.5) to give a white powder (15 mg, 24%): Rf = 0.03 (MeOH-EtOAc; 1:4); mp 192-193 °C; IR (cm-1) 3272, 3199, 2923, 2843, 1616, 1548, 1434, 1381, 1349, 1220, 1122; 1H NMR (300 MHz, DMSO-d6) δ 1.0–1.9 (13H, m, cyclohexyl and CH2CH2CH2), 3.02 (2H, m, CH2NH), 3.40 (2H, s, ArCH2CONH), 3.92 (2H, t, J = 6.9, CH2), 4.32 (2H, d, J = 6.5 Hz, OCH2), 6.81 (1H, d, J = 7.6 Hz, ArH), 6.85 (1H, s, imidazole-H), 7.13 (1H, s, imidazole-H), 7.17 (1H, dd, J = 7.8, 8.0 Hz, ArH), 7.57 (1H, s, imidazole-H), 7.63 (1H, s. ArH), 7.71 (1H, d, J = 7.8 Hz, ArH), 8.1 (1H, s, H-8), 9.27 (1H, s, ArNHAr); LCMS (ES+) m/z 489 [M+H]+; HRMS (ES+) calcd for C26H33N8O2 [M+H]+ 489.2721, found 489.2724; λmax (EtOH) 215, 273, 292 nm.
2-[3-(6-Cyclohexylmethoxy-9H-purin-2-ylamino)phenyl]-N-(3-dimethylamino-propyl)acetamide (42)
The title compound was prepared using 24 (50 mg, 0.13 mmol), carbonyldiimidazole (42 mg, 0.26 mmol), DIPEA (43 μL, 0.26 mmol), and N,N-dimethyl-1,3-propanediamine (67 μL, 0.52 mmol) in DMF (2 mL). The crude product was purified using a Biotage SP4 purification system (12 + M KP-NH silica cartridge; MeOH-EtOAc; 1.5:8.5) to give a white powder (15 mg, 24%): Rf = 0.03 (MeOH-EtOAc; 1:4); mp 96-97 °C; IR (cm-1) 2924, 2850, 2157, 1587, 1537, 1442, 1352, 1249, 1119; 1H NMR (300 MHz, DMSO-d6) δ 1.03 (2H, d, J = 6.6 Hz, CH2CH2CH2), 1.0–1.9 (11H, m, cyclohexyl), 2.11 (6H, s, N(CH3)2), 2.19 (2H, m, CH2N(CH3)2), 3.25-3.40 (overlap with H2O, CH2NH and ArCH2), 4.34 (2H, d, J = 6.3 Hz, OCH2), 6.80 (1H, d, J = 7.6 Hz, ArH), 7.16 (1H, dd, J = 7.8, 7.9 Hz, ArH), 7.63 (1H, s, ArH), 7.70 (1H, br, CONH), 7.81 (1H, d, J = 7.9 Hz, ArH), 7.95 (1H, s, H-8), 9.17 (1H, s, ArNHAr); LCMS (ES+) m/z 466 [M+H]+; HRMS (ES+) calcd for C25H36N7O2 [M+H]+ 466.2925, found 466.2929; λmax (EtOH) 215, 272, 292 nm.
2-[3-(6-Cyclohexylmethoxy-9H-purin-2-ylamino)phenyl]-N-(2-dimethylaminoethyl)acetamide (43)
The title compound was prepared using 24 (75 mg, 0.20 mmol), carbonyldiimidazole (65 mg, 0.40 mmol), DIPEA (72 μL, 0.40 mmol), and N,N-dimethylethylenediamine (71 mg, 0.80 mmol) in DMF (3 mL). The crude product was purified using a Biotage SP4 purification system (12 + M KP-NH silica cartridge; MeOH-EtOAc; 1:9) to give a colourless powder (56 mg, 62%): Rf = 0.10 (NH2-modified silica; MeOH-EtOAc; 1:9); mp 161-162 °C; IR (cm-1) 3249, 2918, 2845, 1587, 1541, 1438, 1396, 1346, 1292, 1240, 1116; 1H NMR (300 MHz, DMSO-d6) δ 1.0–1.84 (11H, m, cyclohexyl), 2.12 (6H, s, N(CH3)2), 2.27 (2H, t, J = 6.7 Hz, CH2N(Me)2), 3.13 (2H, m, CH2NHCO), 3.34 (ArCH2, overlap with H2O), 4.33 (2H, d, J = 6.2 Hz, OCH2), 6.81 (1H, d, J = 7.4 Hz, ArH), 7.17 (1H, t, J = 7.8, 7.8 Hz, ArH), 7.64 (1H, s, ArH), 7.69 (1H, d, J = 8.0 Hz, ArH), 7.93 (1H, br t, J = 5.2 Hz, CONH), 7.98 (1H, s, H-8), 9.27 (1H, s, ArNHAr), 12.80 (1H, s, N9-H); 13C NMR (125 MHz, DMSO-d6) δ 25.2, 26.0, 29.2, 36.9, 42.6, 45.2, 58.2, 71.0, 116.6, 119.3, 121.4, 128.1, 136.5, 138.8, 141.0, 155.5, 170.0; LCMS (ES+) m/z 452.65 [M+H]+; HRMS (ES+) calcd for C24H34N7O2 [M+H]+ 452.2768, found 452.2766; λmax (EtOH) 272 nm.
2-[3-(6-Cyclohexylmethoxy-9H-purin-2-ylamino)phenyl]-N-(3-methylbutyl)acetamide (44)
The title compound was prepared using 24 (60 mg, 0.16 mmol), carbonyldiimidazole (52 mg, 0.32 mmol), DIPEA (57 μL, 0.32 mmol), and isopentylamine (74 μL, 0.64 mmol) in DMF (3 mL). The crude product was purified by chromatography (silica; MeOH-EtOAc; 0.5:9.5) to give an off-white solid (34 mg, 48%): Rf = 0.45 (MeOH-EtOAc; 1:9); mp 204-205 °C; IR (cm-1) 3326, 3232, 3137, 3061, 2920, 2851, 2162, 2023, 1643, 1593, 1530, 1487, 1440, 1390, 1354, 1282, 1240, 1170, 1127; 1H NMR (300 MHz, DMSO-d6) δ 0.84 (6H, d, J = 6.6 Hz, (CH3)2CH), 1.0–1.85 (11H, m, cyclohexyl), 1.28 (2H, m, CHCH2CH2), 1.56 (1H, m, (CH3)2CHCH2), 3.05 (2H, m, CH2CH2NH), 3.35 (ArCH2, overlap with H2O), 4.33 (2H, d, J = 6.2 Hz, OCH2), 6.80 (1H, d, J = 7.6 Hz, ArH), 7.17 (1H, dd, J = 7.8, 7.8 Hz, ArH), 7.63 (1H, s, ArH), 7.68 (1H, d, J = 8.1 Hz, ArH), 7.95 (1H, br t, J = 5.3 Hz, CONH), 8.00 (1H, s, H-8), 9.26 (1H, s, ArNHAr), 12.83 (1H, s, N9-H); LCMS (ES+) m/z 451 [M+H]+; HRMS (ES+) calcd for C25H35N6O2 [M+H]+ 451.2816, found 451.2820; Anal. calcd for C25H34N6O2: C, 66.64; H, 7.61; N, 18.66%; found: C, 66.44; H, 7.68; N, 18.60; λmax (EtOH) 267 nm.
2-[3-(6-Cyclohexylmethoxy-9H-purin-2-ylamino)phenyl]-N-(4-dimethylaminobutyl)acetamide (45)
The title compound was prepared using 24 (75 mg, 0.20 mmol), carbonyldiimidazole (65 mg, 0.40 mmol), DIPEA (72 μL, 0.40 mmol), and N,N-dimethyl-1,4-butanediamine (93 mg, 0.80 mmol) in DMF (3 mL). The crude product was purified using a Biotage SP4 purification system (12 + M KP-NH silica cartridge; MeOH-EtOAc; 1.5:8.5) to give a colourless powder (61 mg, 64%): Rf = 0.12 (NH2-modified silica; MeOH-EtOAc: 1:9); mp 125-127 °C; IR (cm-1) 3490, 3276, 3052, 2927, 2852, 2779, 1606, 1558, 1495, 1448, 1400, 1354, 1309, 1246, 1217; 1H NMR (300 MHz, DMSO-d6) δ 1.0–1.84 (15H, m, cyclohexyl and CH2CH2CH2CH2), 2.14 (2H, t, J = 5.7 Hz, CH2N(Me)2), 3.04 (2H, m, CH2NHCO), 3.34 (ArCH2, overlap with H2O), 4.33 (2H, d, J = 5.8 Hz, OCH2), 6.81 (1H, d, J = 7.4 Hz, ArH), 7.17 (1H, dd, J = 7.8, 7.9 Hz, ArH), 7.64 (1H, s, ArH), 7.69 (1H, d, J = 8.0 Hz, ArH), 8.00 (2H, br, CONH and H-8), 9.27 (1H, s, ArNHAr), 12.82 (1H, s, N9-H); 13C NMR (125 MHz, DMSO-d6) δ 24.4, 25.2, 26.0, 27.0, 29.2, 36.8, 38.5, 42.8, 45.1, 58.7, 71.0, 116.5, 119.2, 121.3, 128.1, 136.6, 141.0, 155.4, 169.8; LCMS (ES+) m/z 480.71 [M+H]+; HRMS (ES+) calcd for C26H38N7O2 [M+H]+ 480.3081, found 480.3084; λmax (EtOH) 272 nm.
2-[3-(6-Cyclohexylmethoxy-9H-purin-2-ylamino)phenyl]-N-[3-(4-methylpiperazin-1- yl)propyl]acetamide (46)
The title compound was prepared using 24 (75 mg, 0.20 mmol), carbonyldiimidazole (65 mg, 0.40 mmol), DIPEA (72 μL, 0.40 mmol), and 1-(3-aminopropyl)-4-methylpiperazine (126 mg, 0.80 mmol) in DMF (3 mL). The crude product was purified using a Biotage SP4 purification system (12 + M KP-NH silica cartridge; MeOH-EtOAc; 1:4) to give a colourless powder (72 mg, 69%): Rf = 0.09 (NH2-modified silica; MeOH-EtOAc; 1:9); mp 157-158 °C; IR (cm-1) 3614, 3273, 3203, 3108, 3039, 2923, 2848, 2802, 1639, 1606, 1585, 1495, 1451, 1394, 1354, 1131, 1216, 1127; 1H NMR (300 MHz, DMSO-d6) δ 1.0–1.85 (13H, m, cyclohexyl and NCH2CH2CH2N), 2.11 (3H, s, NCH3), 2.22 (8H, br t, J = 7.2 Hz, 2 × NCH2CH2N), 3.05 (2H, m, CH2CH2NHCO), 3.34 (ArCH2, overlap with H2O), 4.33 (2H, d, J = 6.1 Hz, OCH2), 6.80 (1H, d, J = 7.5 Hz, ArH), 7.17 (1H, dd, J = 7.8, 7.8 Hz, ArH), 7.64 (1H, s, ArH), 7.68 (1H, d, J = 8.0 Hz, ArH), 7.97 (2H, br, CONH and H-8), 9.28 (1H, s, ArNHAr), 12.79 (1H, s, N9-H); 13C NMR (125 MHz, DMSO-d6) δ 25.2, 26.0, 26.3, 29.2, 36.8, 37.1, 42.8, 45.7, 52.6, 54.7, 55.4, 71.0, 116.6, 119.2, 121.4, 128.1, 136.5, 141.0, 155.4, 169.9; HRMS (ES+) calcd for C28H41N8O2 [M+H]+ 521.3347, found 521.3352; λmax (EtOH) 272 nm.
2-[3-(6-Cyclohexylmethoxy-9H-purin-2-ylamino)phenyl]-N-(3-phenylpropyl)acetamide (47)
The title compound was prepared using 24 (75 mg, 0.20 mmol), carbonyldiimidazole (65 mg, 0.40 mmol), DIPEA (72 μL, 0.40 mmol), and 3-phenylpropylamine (108 mg, 0.80 mmol) in DMF (3 mL). The crude product was purified by chromatography (silica; EtOAc) to give an off-white solid (53 mg, 53%): Rf = 0.46 (MeOH-EtOAc; 1:9); mp 154-155 °C; IR (cm-1) 3273, 3207, 3126, 3080, 2921, 2850, 1616, 1585, 1547, 1489, 1444, 1382, 1350, 1283, 1218, 1178, 1119, 976; 1H NMR (300 MHz, DMSO-d6) δ 1.0-1.85 (13H, m, cyclohexyland CH2CH2CH2), 2.55 (2H, t, J = 7.6 Hz, CH2Ph), 3.06 (2H, m, CH2NHCO), 3.36 (2H, s, ArCH2CONH), 4.32 (2H, d, J = 6.2 Hz, OCH2), 6.81 (1H, d, J = 7.5 Hz, ArH), 7.13-7.29 (6H, m, ArH and PhH), 7.67 (1H, s, ArH), 7.70 (1H, d, J = 8.3 Hz, ArH), 7.95 (1H, s, H-8), 8.06 (1H, t br, J = 5.5 Hz, CONH), 9.18 (1H, s, ArNHAr); 13C NMR (125 MHz, DMSO-d6) δ 25.2, 26.0, 29.2, 30.9, 32.5, 36.8, 38.2, 68.3, 71.0, 116.6, 121.4, 125.7, 128.1, 128.2, 128.2, 136.6, 141.7, 155.5, 170.0; LCMS (ES+) m/z 499.54 [M+H]+; HRMS (ES+) calcd for C29H35N6O2 [M+H]+ 499.2816, found 499.2820; λmax (EtOH) 272 nm.
Synthesis of 2-arylaminopurine derivatives (48-53)
2,2,2-Trifluoroethyl-3-(6-cyclohexylmethoxy-9H-purin-2-ylamino)phenylmethanesulfonate (26, 1 mol. equiv.),1,8-diazabicyclo[5.4.0]undec-7-ene (3 mol. equiv.), and the appropriate amine (2.5 mol. equiv.) were heated under microwave conditions in anhydrous THF (2 mL) for 15 min at 160 °C. After removal of the solvent, the white solid was extracted with EtOAc or THF and washed with saturated aqueous NaHCO3 solution and brine. The filtrate was concentrated to give a residue that was purified by medium pressure chromatography or using the Biotage SP4 purification system.
[3-(6-Cyclohexylmethoxy-9H-purin-2-ylamino)-phenyl]-N,N-dimethylmethanesulfonamide (48)
The product from the reaction of 2,2,2-trifluoroethyl-3-(6-cyclohexylmethoxy-9H-purin-2-ylamino) phenylmethanesulfonate (26) (46 mg, 0.09 mmol), 1,8-diazabicyclo[5.4.0]undec-7-ene (42 μl, 0.18 mmol), and dimethylamine (12 μL, 0.18 mmol), was purified by chromatography (silica; EtOAc-petrol; 8:2) to give the title compound as an off-white solid (16 mg, 40%): Rf = 0.14 (EtOAc); mp 132-133 °C; IR (cm-1) 2922, 2849, 2158, 2027, 1968, 1588, 1440, 1309, 1118; 1H NMR (300 MHz, DMSO-d6) δ 1.0–1.8 (11H, m, cyclohexyl), 2.75 (6H, s, N(CH3)2), 4.34 (2H, s, SO2CH2), 4.36 (2H, d, J = 6.2 Hz, OCH2), 6.96 (1H, d, J = 7.6 Hz, ArH), 7.27 (1H, dd, J = 7.8, 7.9 Hz, ArH), 7.82 (1H, d, J = 7.6 Hz, ArH), 7.86 (1H, s, ArH), 8.02 (1H, s, H8), 9.40 (1H, s, ArNHAr), 12.78 (1H, br, N9-H); 13C NMR (75 MHz, DMSO-d6) δ 25.57, 26.37, 29.62, 37.74, 39.36, 49.55, 54.77, 71.52, 118.68, 121.09, 123.60, 128.62, 130.05, 140.26, 141.74, 155.77; LCMS (ES+) m/z 445 [M+H]+; Found: C, 56.79; H, 6.31; N, 18.61. C21H28N6O3S requires C, 56.74; H, 6.35; N, 18.91%; λmax (EtOH) 212.5, 272.5, 294 nm.
1-(3-(6-Cyclohexylmethoxy-9H-purin-2-ylamino)phenylmethanesulfonyl)piperidine (49)
The product from the reaction of 2,2,2-trifluoroethyl-3-(6-cyclohexylmethoxy-9H-purin-2-ylamino) phenylmethanesulfonate (26) (75 mg, 0.15 mmol),1,8-diazabicyclo[5.4.0]undec-7-ene (68 μL, 0.45 mmol), and piperidine (37 μL, 0.38 mmol) was purified by chromatography (silica; EtOAc-MeOH; 9.5:0.5) to give the title compound as a white solid (54 mg, 75%): Rf = 0.26 (EtOAc); mp 217-218 °C; IR (cm-1) 2922, 2853, 2362, 2338, 1589, 1541, 1437, 1396, 1350, 1310, 1242, 1152, 1121, 1067, 1045; 1H NMR (300 MHz, DMSO-d6) δ 1.0–1.8 (11H, m, cyclohexyl), 1.47 (6H, m, piperidyl CH), 3.12 (4H, br m, 2 × piperidyl CH2), 4.28 (2H, s, ArCH2), 4.35 (2H, d, J = 6.2 Hz, OCH2), 6.94 (1H, d, J = 7.5 Hz, ArH), 7.25 (1H, dd, J = 7.9, 8.0 Hz, ArH), 7.81 (1H, d, J = 8.5 Hz, ArH), 7.88 (1H, s, ArH), 8.02 (1H, s, H-8), 9.42 (1H, s, ArNHAr), 12.76 (1H, br, N9-H); LCMS (ES+) m/z 485.18 [M+H]+; Anal. calcd for C24H32N6O3S: C, 59.48; H, 6.66; N, 17.34%; found: C, 59.22; H, 6.56; N, 17.16; λmax (EtOH) 272, 293 nm.
1-(3-(6-Cyclohexylmethoxy-9H-purin-2-ylamino)phenylmethanesulfonyl)piperazine (50)
The product from the reaction of 2,2,2-trifluoroethyl-3-(6-cyclohexylmethoxy-9H-purin-2-ylamino)phenylmethanesulfonate (26) (75 mg, 0.15 mmol), 1,8-diazabicyclo[5.4.0]undec-7-ene (68 μL, 0.45 mmol), and piperazine (45 mg, 0.53 mmol) was purified using Biotage SP4 (12 + M KP-NH silica cartridge; EtOAc-MeOH; 9.5:0.5) to give the title compound as a colourless solid (66 mg, 91%): Rf = 0.73 (NH2-modified silica; EtOAc-MeOH; 8:2); mp 127-128 °C; IR (cm-1) 2922, 2853, 2362, 2338, 1589, 1541, 1437, 1396, 1350, 1310, 1242, 1152, 1121, 1067, 1045; 1H NMR (300 MHz, DMSO-d6) δ 1.0–1.9 (11H, m, cyclohexyl), 3.11 (4H, br, 2 × piperazinyl CH2), 3.16 (4H, br, 2 × piperazinyl CH2), 4.33 (2H, s, ArCH2), 4.35 (2H, d, J = 6.7 Hz, OCH2), 6.95 (1H, d, J = 7.5 Hz, ArH), 7.26 (1H, dd, J = 7.8, 7.9 Hz, ArH), 7.62 (1H, d, J = 8.0 Hz, ArH), 7.91 (1H, s, ArH), 7.97 (1H, s, H-8), 9.34 (1H, s, ArNHAr), 12.77 (1H, br, N9-H); LCMS (ES+) m/z 486 [M+H]+; HRMS (ES+) calcd for C23H32N7O3S [M+H]+ 484.2136, found 484.2145; λmax (EtOH) 272, 293 nm.
1-[3-(6-Cyclohexylmethoxy-9H-purin-2-ylamino)phenyl]-N-(3-dimethylaminopropyl)methanesulfonamide (51)
The product from the reaction of 2,2,2-trifluoroethyl-3-(6-cyclohexylmethoxy-9H-purin-2-ylamino)phenylmethanesulfonate (26) (60 mg, 0.12 mmol),1,8-diazabicyclo[5.4.0]undec-7-ene (55 μL, 0.36 mmol), and N,N-dimethylpropane-1,3-diamine (38 μL, 0.30 mmol) was purified using the Biotage SP4 (12 + M KP-NH silica cartridge; EtOAc-MeOH; 8.5:1.5) to give the title compound as a white solid (55 mg, 92%): Rf = 0.13 (NH2-modified silica; EtOAc-MeOH; 9:1); mp 167-168 °C; IR (cm-1) 2924, 2850, 1589, 1541, 1489, 1441, 1390, 1356, 1308, 1243, 1214, 1146, 1121; 1H NMR (300 MHz, DMSO-d6) δ 1.00-1.90 (13H, m, cyclohexyl and NCH2CH2CH2NH), 2.07 (6H, s, 2 × CH3), 2.19 (2H, t, J = 6.9 Hz, Me2NCH2), 2.95 (2H, t, J = 6.7 Hz, CH2CH2NH), 4.23 (2H, s, ArCH2), 4.35 (2H, d, J = 5.8 Hz, OCH2), 6.92 (1H, d, J = 5.8 Hz, ArH), 7.26 (1H, dd, J = 7.8, 7.8 Hz, ArH), 7.83 (1H, d, J = 7.7 Hz, ArH), 7.84 (1H, s, ArH), 8.00 (1H, s, H-8), 9.35 (1H, s, ArNHAr); LCMS (ES+) m/z 502.36 [M+H]+; HRMS (ES+) calcd for C24H36N7O3S [M+H]+ 502.2595, found 502.2600; Anal. calcd for C24H35N7O3S: C, 57.46; H, 7.03; N, 19.55%; found: C, 57.76; H, 7.15; N, 19.67%; λmax (EtOH) 272, 293 nm.
1-[3-(6-Cyclohexylmethoxy-9H-purin-2-ylamino)phenyl]-N-(4-methoxybenzyl)methanesulfonamide (52)
The product from the reaction of2,2,2-trifluoroethyl-3-(6-cyclohexylmethoxy-9H-purin-2-ylamino)phenylmethanesulfonate (26) (150 mg, 0.3 mmol),1,8-diazabicyclo[5.4.0]undec-7-ene (136 μL, 0.9 mmol), and 4-methoxybenzylamine (97 μL, 0.75 mmol) was purified by chromatography (silica; EtOAc-petrol; 9:1) to give the title compound as a white solid (143 mg, 89%): Rf = 0.37 (EtOAc); mp 118-119 °C; IR (cm-1) 2922, 2850, 2361, 2338, 2026, 1589, 1541, 1506, 1444, 1395, 1354, 1303, 1244, 1121, 1030; 1H NMR (300 MHz, DMSO-d6) δ 1.00-1.90 (11H, m, cyclohexyl), 3.70 (3H, s, OCH3), 4.05 (2H, d, J = 9.0 Hz, ArCH2NH), 4.22 (2H, s, ArCH2SO2NH), 4.35 (2H, d, J = 6.1 Hz, OCH2), 6.86–6.90 (3H, m, 3 × ArH), 7.23 (2H, d, J = 7.0 Hz, ArH), 7.26 (1H, dd, J = 7.6, 7.9 Hz, ArH), 7.57 (1H, t, J = 6.0 Hz, SO2NH), 7.81-7.85 (2H, m, 2 × ArH), 7.99 (1H, s, H-8), 9.37 (1H, s, ArNHAr), 12.77 (1H, s br, N9-H); LCMS (ES+) m/z 537.40 [M+H]+; HRMS (ES-) calcd for C27H31N6O4S [M-H]- 535.2133, found 535.2143; λmax (EtOH) 272 nm.
[3-(6-Cyclohexylmethoxy-9H-purin-2-ylamino)phenyl]methanesulfonamide (53)
[3-(6-Cyclohexylmethoxy-9H-purin-2-ylamino)phenyl]-N-(4-methoxybenzyl)methanesulfonamide (52) (80 mg, 0.15 mmol) was stirred in neat TFA (2 mL) for 6 h. Upon completion of the reaction, the TFA was removed in vacuo and the residual solid was extracted into EtOAc (50 mL). The organic layer was washed with aqueous NaHCO3 (50 mL), dried (Na2SO4) and concentrated to give a white solid. Purification by chromatography (silica; EtOAc) afforded the title compound as a white powder (53 mg, 85%): Rf = 0.50 (EtOAc); mp 134-135 °C; IR (cm-1) 3344, 2924, 2852, 2361, 2338, 1719, 1599, 1542, 1488, 1449, 1393, 1336, 1256, 1156, 1123, 1046; 1H NMR (300 MHz, DMSO-d6) δ 1.0–1.9 (11H, m, cyclohexyl), 4.20 (2H, s, ArCH2SO2NH2), 4.34 (2H, d, J = 6.0 Hz, OCH2), 6.85 (2H, s br, SO2NH2), 6.93 (1H, d, J = 7.3 Hz, ArH), 7.26 (1H, dd, J = 7.8, 7.9 Hz, ArH), 7.75 (1H, s, ArH), 7.85 (1H, d, J = 8.3 Hz, ArH), 7.97 (1H, s, H-8), 9.37 (1H, s, ArNH), 12.77 (1H, br, N9-H); LCMS (ES+) m/z 417 [M+H]+; HRMS (ES-) calcd for C19H23N6O3S [M-H]- 417.1703, found 417.1702; λmax (EtOH) 272, 292 nm.
Synthesis of 2-amino-6-alkoxypurines (55 and 56)
Sodium (5 mol. equiv.) was cautiously dissolved in the appropriate alcohol (3.4 mL/mmol), and to the resulting solution was added 2-amino-6-chloropurine (54, 1 mol. equiv). The mixture was stirred at reflux for 18 h and cooled to room temperature. The alcohol was removed in vacuo and water (100 mL) was added to the residual solid. The resulting solution was neutralised with AcOH. Purification was achieved either by cooling the mixture to ~ 3 °C and collecting the product by filtration under vacuum, or by extracting the product into EtOAc (3 × 100 mL). The combined extracts were dried (Na2SO4) and the solvent was removed to give the 6-alkoxy-2-aminopurine that was used directly in subsequent steps.
O6-Ethylguanine (55)
The title compound was prepared using 2-amino-6-chloropurine (3.0 g, 18 mmol) and sodium (1.2 g, 53 mmol) in EtOH (80 mL) to give a colourless solid (2.9 g, 93%): mp 280-281 °C (dec); 1H NMR (300 MHz, DMSO-d6) δ 1.35 (3H, t, J = 7.0 Hz, CH2CH3), 4.43 (2H, q, J = 7.0 Hz, OCH2), 6.20 (2H, s, NH2), 7.81 (1H, s, H-8), 12.41 (1H, br, N9-H).
(±)-O6-sec-Butylguanine (56)
The title compound was prepared using 2-amino-6-chloropurine (1.5 g, 8.9 mmol), sodium (0.60 g, 27 mmol), butan-2-ol (25 mL) and THF (15 mL) to give a colourless powder (1.46 g, 80%): mp 84-85 °C; 1H NMR (300 MHz, DMSO-d6) δ 0.91 (3H, t, J = 7.4 Hz, CH3CH2), 1.30 (3H, d, J = 6.3 Hz, CHCH3), 1.62 (2H, m, CH2CH3), 5.32 (1H, m, OCH), 6.20 (2H, s, 2-NH2), 7.79 (1H, s, H-8), 12.29 (1H, br, N9-H).
Synthesis of 6-alkoxy-2-fluoropurines (57 and 58)
To a stirred solution of aqueous HBF4 (48%, 20 mol. equiv.) at 0 °C was added the appropriate 2-amino-6-alkoxypurine (1 mol. equiv.). To the resulting solution was added NaNO2 (2 mol. equiv.) in water (1 mL/mmol of NaNO2) dropwise, ensuring that the reaction temperature did not exceed 10 °C. The reaction mixture was allowed to warm to room temperature and was stirred for 24 h before being neutralised slowly with saturated Na2CO3 solution. The resulting precipitate was collected via filtration and washed with water. The solid was stirred for 1 h in EtOAc (4 × 150 mL) and filtered. The combined filtrates were concentrated under reduced pressure to yield the product, which was used directly.
6-Ethoxy-2-fluoropurine (57)
The title compound was synthesised using O6-ethylguanine (55) (2.5 g, 14 mmol), 48% aqueous HBF4 (25 mL, 280 mmol) and NaNO2 (1.9 g, 28 mmol) to give a colourless solid (1.26 g, 50%): mp 220-222 °C; 1H NMR (300 MHz, DMSO-d6) δ 1.41 (3H, t, J = 7.0 Hz, CH2CH3), 4.57 (2H, q, J = 7.0 Hz, OCH2), 8.39 (1H, s, H-8), 13.40 (1H, br, N9-H).
(±)-6-sec-Butoxy-2-fluoropurine (58)
The title compound was synthesised using O6-sec-butylguanine (56) (1.0 g, 4.8 mmol), 48% aqueous HBF4 (10 mL, 45 mmol) and NaNO2 (0.67 g, 9.7 mmol) to give a colourless solid (369 mg, 36%): mp 189-190 °C (dec); 1H NMR (300 MHz, DMSO-d6) δ 0.92 (3H, t, J = 7.4 Hz, CH3CH2), 1.35 (3H, d, J = 6.3 Hz, CHCH3), 1.71 (2H, m, CH2CH3), 5.32 (1H, m, OCH), 8.38 (1H, s, H-8), 13.34 (1H, br, N9-H); 19F NMR (470 MHz, DMSO-d6) δ -51.94 (s, ArF).
N-(3-Dimethylaminopropyl)-2-[3-(6-ethoxy-9H-purin-2-ylamino)phenyl]acetamide (59)
The title compound was synthesised according to Method I using 6-ethoxy-2-fluoropurine (57) (100 mg, 0.55 mmol), 2-(3-aminophenyl)-N-(3-dimethylaminopropyl)acetamide (see ESI S49; 291 mg, 1.2 mmol), TFE (4 mL), and TFA (0.20 mL, 2.8 mmol). The crude product was purified using the Biotage SP4 purification system (25 + M KP-NH Si cartridge; MeOH-EtOAc; 1:9 to 3:7) to give a semi-pure compound that was further purified by semi-prep HPLC (5 → 100% v/v acetonitrile: water: NH4OH over 25 min; flow-rate 12.75 mL/min, wavelength 280 nm) to give the title compound as a brown gum (43 mg, 20%):Rf = 0.06 (NH2–modified silica; MeOH-EtOAcl 1:9); mp 178-179 °C; IR (cm-1) 3290, 2937, 2762, 1641, 1602, 1571, 1534, 1493, 1433, 1381, 1336, 1317, 1251, 1163, 1122, 1019;1H NMR (300 MHz, DMSO-d6) δ 1.42 (3H, t, J = 7.1 Hz, CH3CH2O), 1.52 (2H, qn, J = 6.9 Hz, NCH2CH2CH2NH), 2.07 (6H, s, 2 × CH3), 2.18 (2H, t, J = 7.1 Hz, Me2NCH2), 3.06 (2H, m, CH2CH2NHCO), 3.34 (ArCH2, overlap with H2O), 4.57 (2H, q, J = 7.0 Hz, OCH2), 6.81 (1H, d, J = 7.6 Hz, ArH), 7.18 (1H, dd, J = 7.7, 7.8 Hz, ArH), 7.65 (2H, m, 2 × ArH), 8.00 (2H, br, CONH and H-8), 9.28 (1H, s, ArNHAr), 12.81 (1H, br, N9-H); LCMS (ES+) m/z 398.54 [M+H]+;λmax (EtOH) 272 nm.
(±)-2-[3-(6-sec-Butoxy-9H-purin-2-ylamino)phenyl]-N-(3-dimethylaminopropyl)acetamide (60)
The title compound was synthesised according to Method I using 6-sec-butoxy-2-fluoropurine (58) (75 mg, 0.36 mmol), 2-(3-aminophenyl)-N-(3-dimethylaminopropyl)acetamide (see ESI S49; 190 mg, 0.81 mmol), TFE (3 mL), and TFA (0.13 mL, 1.8 mmol). The crude product was purified using the Biotage SP4 purification system (25 + M KP-NH Si cartridge; EtOAc → MeOH-EtOAc; 1.5:8.5) to give a semi-pure compound that was further purified by semi-prep HPLC (5 → 100% v/v acetonitrile: water: NH4OH over 25 min; flow-rate 12.75 mL/min, wavelength 280 nm) to give the title compound as a white solid (43 mg, 28%): Rf = 0.08 (NH2-modified silica; MeOH-EtOAc; 1:9); mp 97-98 °C; IR (cm-1) 3283, 2970, 2935, 2777, 2164, 1587, 1538, 1493, 1438, 1373, 1313, 1245, 1215, 1165, 1114; 1H NMR (300 MHz, DMSO-d6) δ 0.95 (3H, t, J = 7.4 Hz, CH3CH2CH(CH3)O), 1.37 (3H, d, J = 6.2 Hz, OCH(CH3)), 1.51 (2H, qn, J = 7.0 Hz, NCH2CH2CH2N), 1.65-1.84 (2H, m, CH3CH2CH), 2.05 (6H, s, 2 × NCH3), 2.16 (2H, t, J = 7.1 Hz, Me2NCH2), 3.06 (2H, m, CH2CH2NHCO), 3.34 (ArCH2, overlap with H2O), 5.42 (1H, m, OCH), 6.80 (1H, d, J = 7.5 Hz, ArH), 7.18 (1H, dd, J = 7.8, 7.8 Hz, ArH), 7.62 (1H, s, ArH), 7.67 (1H, d, J = 7.7 Hz, ArH), 7.98 (1H, s, H-8), 9.24 (1H, s, ArNHAr), 12.78 (1H, br, N9-H);LCMS (ES+) m/z 426.46 [M+H]+; HRMS (ES+) calcd for C22H32N7O2 [M+H]+ 426.2612, found 426.2613; λmax (EtOH) 272 nm.
[3-(6-Ethoxy-9H-purin-2-ylamino)phenyl]acetic acid (61)
6-Ethoxy-2-fluoropurine (57) (0.90 g, 5.0 mmol), 3-aminophenylacetic acid (1.87 g, 12.4 mmol) were combined in TFE (7 mL), to which TFA (1.8 mL, 25 mmol) was added and the reaction mixture was heated to reflux for 24 h. After concentration in vacuo, THF (20 mL) and NaOH aqueous solution (1 M, 15 mL) were added to the residue and the resulting mixture was stirred overnight. The pH was adjusted to 1 with conc. HCl and the product was extracted with EtOAc (250 mL). The organic phase was separated, washed with 10 % HCl solution and dried (NaSO4). Removal of the solvent gave crude product, to which Et2O (100 mL) was added. After allowing the mixture to stand for 3 h the resulting precipitate was collected by filtration under vacuum and washed with Et2O (30 mL). Recrystallisation from MeOH gave the pure product as a light brown powder (729 mg, 47%): mp 205-206 °C; 1H NMR (300 MHz, DMSO-d6) δ 1.42 (3H, t, J = 7.0 Hz, CH3CH2O), 3.50 (2H, s, ArCH2,), 4.57 (2H, q, J = 6.9 Hz, OCH2), 6.81 (1H, d, J = 7.2 Hz, ArH), 7.20 (1H, dd, J = 7.7, 7.8 Hz, ArH), 7.65 (1H, d, J = 8.1 Hz, ArH), 7.75 (1H, s, ArH), 7.90 (1H, s, H-8), 9.29 (1H, s, ArNHAr), 12.78 (1H, br, N9-H); LCMS (ES+) m/z 314.21 [M+H]+.
2-[3-(6-Ethoxy-9H-purin-2-ylamino)-phenyl]-N-(3-imidazol-1-ylpropyl)acetamide (62)
Carboxylic acid 61 (80 mg, 0.26 mmol) was treated with carbonyldiimidazole (84 mg, 0.52 mmol) and DIPEA (93 μL, 0.52 mmol) in DMF (3 mL) and stirred for 1.5 h at room temperature. N-(3-aminopropyl)imidazole (130 mg, 0.80 mmol) was added in one portion and stirring was continued overnight. Solvents were removed and the residue was extracted into EtOAc. The extract was washed with saturated aqueous NaHCO3 and dried (Na2SO4). The solvent was removed to afford the crude product, which was purified using a Biotage SP4 purification system (12 + M KP-NH silica cartridge; EtOAc → 15% MeOH/ EtOAc), followed by purification by semi-prep HPLC (5 → 100% v/v acetonitrile: water: NH4OH over 25 min; flow-rate 12.75 mL/min, wavelength 280 nm) to give an off-white solid (57 mg, 52%): Rf = 0.55 (NH2-modified silica; MeOH-EtOAc; 3:7); mp 176-177 °C; IR (cm-1) 3268, 2953, 2926, 2362, 2337, 1636, 1578, 1539, 1493, 1436, 1379, 1315, 1251, 1165, 1110, 1079; 1H NMR (300 MHz, DMSO-d6) δ 1.42 (3H, t, J = 7.0 Hz, CH3CH2O), 1.83 (2H, m, CH2CH2CH2NH), 3.01 (2H, m, CH2CH2NHCO), 3.38 (ArCH2, overlap with H2O), 3.94 (2H, t, J = 6.8 Hz, ImCH2), 4.57 (2H, q, J = 7.1 Hz, OCH2), 6.83 (1H, d, J = 7.6 Hz, ArH), 6.86 (1H, s, imidazole-H), 7.13 (1H, s, imidazole-H), 7.19 (1H, dd. J = 7.7, 7.7 Hz, ArH), 7.58 (1H, s, imidazole-H), 7.65-7.71 (2H, m, 2 × ArH), 8.00 (1H, s, H-8), 8.07 (1H, t, J = 5.1 Hz, CONH), 9.27 (1H, s, ArNHAr); LCMS (ES+) m/z 421.34 [M+H]+; HRMS (ES+) calcd for C21H25N8O2 [M+H]+ 421.2095, found 421.2099; λmax (EtOH) 272, 292 nm.
6-Chloro-2-fluoro-9H-purine (63) [36]
To a stirred solution of HBF4 (48% aqueous, 120 mL) at 0 ºC, was added 2-amino-6-chloropurine (6.0 g, 35.0 mmol). Over 20 min, a solution of NaNO2 (4.9 g, 70.0 mmol) in water (200 mL) was added dropwise, ensuring the temperature remained close to 0 ºC. The pale yellow solution was raised to room temperature and stirred for 18 h. The resulting solution was neutralised to pH 7 in an ice bath at 0 °C, by addition of Na2CO3 (6.00 g) in water (200 mL). Solvents were removed in vacuo and the residual solid was redissolved in MeOH (100 mL) and adsorbed onto silica (250 mL). The crude material was purified by chromatography (silica; MeOH-DCM; 1:9) to afford the title compound as a white crystalline solid (4.52 g, 75%): mp 158-159 °C (Lit.,32 mp 161-162 °C); IR (cm-1) 2964, 2785, 1735, 1581;1H NMR (300 MHz, DMSO-d6) δ 8.72 (1H, s, H-8), 14.12 (1H, br s, N9-H); LCMS (ES+) m/z 172.6 [M+H]+; λmax (EtOH) 393 nm.
2-Fluoro-9H-purine (64) [37, 38]
To a stirred suspension of 6-chloro-2-fluoropurine (63) (0.30 g, 1.74 mmol) and palladium hydroxide on carbon (0.30 g) in MeOH (15 mL) was added ammonium formate (0.34 g, 5.35 mmol). The suspension was heated under reflux for 1h before filtering through a pad of Celite, eluting with MeOH (20 mL). Removal of volatiles under reduced pressure yielded the desired compound as a white solid (240 mg, 100%): mp 219 °C (dec.) (lit. [37], decomposed at 216 °C); 1H NMR (300 MHz, DMSO-d6) δ 8.60 (1H, s, H-8), 9.01 (1H, s, H-6), 13.9 (1H, s, NH-9); LCMS (ES+) m/z 139.2 [M+H]+.
1-(4-((9H-purin-2-yl)amino)phenyl)-3-(2-(piperidin-1-yl)ethyl)urea (65)
Synthesis of the title compound was achieved according to Method I using 2-fluoro-9H-purine (64) (79 mg, 0.57 mmol) and 1-(4-aminophenyl)-3-(2-(piperidin-1-yl)ethyl)urea (see ESI; S20) (0.30 g, 1.15 mmol) with TFA (220 μL, 2.88 mmol) in TFE (4 mL). The product was isolated using the Biotage SP4 system (12 + M KP-NH; MeOH-EtOAc; 1:9) as a pale orange solid (95 mg, 45%): mp 96-98 °C; IR (cm-1) 3296, 3151, 2920, 2850, 1704, 1629 υ(NN'C=O), 1581, 1547, 1512; 1H NMR (300 MHz, DMSO-d6) δ 1.45 (6H, m, CH2), 2.33 (6H, m, N(CH2)3), 3.17 (2H, m, CH2), 6.02 (1H, m, NH), 7.28 (2H, d, J = 8.5 Hz, H-2' and H-6'), 7.63 (2H, d, J = 8.5 Hz, H-3' and H-5'), 8.18 (1H, s, H-8), 8.52 (1H, s, H-6), 8.76 (1 H, s, NH-2), 9.28 (1H, s, NH-4´); 13C NMR (125 MHz, DMSO-d6) δ 24.1, 25.5, 36.4, 54.0, 58.2, 118.1, 119.2, 134.3, 134.9, 155.3, 156.8; LCMS (ES+) m/z 381.4 [M+H]+; HRMS (ES+) calcd for C19H24N8O [M+H]+ 381.2146, found 381.2142; λmax (EtOH) 334.0, 280.0, 244.0 nm.
[3-(9H-Purin-2-ylamino)phenyl]acetic acid (66)
To a stirred mixture of 2-fluoro-9H-purine (64) (700 mg, 5.1 mmol), 3-aminophenylacetic acid (1.72 g, 11.4 mmol) in TFE (10 mL) was added TFA (1.9 mL, 25 mmol) and the reaction mixture was heated to reflux for 24 h. The reaction mixture was cooled to room temperature and then concentrated in vacuo to give a brown oil. The oil was redissolved in THF (10 mL) and a solution of KOH (2 g) in water (10 mL) was added, and the resulting solution was stirred overnight. THF was removed and the aqueous layer was adjusted to pH 6 with 2 M HCl. The resulting precipitate was collected by filtration under vacuum and washed with water (30 mL), Et2O (15 mL) and MeOH (15 mL). The resulting solid was dried to give the title compound as a light brown powder (462 mg, 34%): mp > 250 °C; 1H NMR (DMSO-d6) δ 3.51 (2H, s, ArCH2), 6.82 (1H, d, J = 7.5 Hz, ArH), 7.21 (1H, dd, J = 7.8, 7.9 Hz, ArH), 7.64 (1H, s, ArH), 7.79 (1H, d, J = 8.2 Hz, ArH), 8.24 (1H, s, H-8), 8.81 (1H, s, H-6), 9.51 (1H, s, ArNH); LCMS (ES+) m/z 270.20 [M+H]+.
N-(3-Imidazol-1-yl-propyl)-2-[3-(9H-purin-2-ylamino)phenyl]acetamide (67)
Carboxylic acid 66 (100 mg, 0.37 mmol) was stirred with carbonyldiimidazole (120 mg, 0.74 mmol) and DIPEA (133 μL, 0.74 mmol) in DMF (3 mL) at room temperature for 1.5 h. Following this, N-(3-aminopropyl)imidazole (185 mg, 1.48 mmol) was added in one portion and stirring was continued overnight. Solvents were removed and the residue was extracted into EtOAc. The extract was washed with saturated aqueous NaHCO3 and dried (Na2SO4). The solvent was removed to afford the crude product, which was purified using a Biotage SP4 purification system (12 + M KP-NH silica cartridge; EtOAc → MeOH-EtOAc; 1:4) to give a light brown solid (55 mg, 40%): Rf = 0.14 (NH2-modified silica; MeOH-EtOAc; 1:4); mp 121-122 °C; IR (cm-1) 3252, 2923, 2362, 2337, 1580, 1537, 1394, 1333, 1282, 1214, 1180, 1105, 1080; 1H NMR (300 MHz, DMSO-d6) δ 1.84 (2H, m, CH2CH2CH2), 3.02 (2H, m, CH2NH), 3.38 (2H, s, ArCH2CONH), 3.94 (2H, t, J = 6.8 Hz, CH2CH2CH2NHCO), 6.81-6.88 (2H, m, ArH and imidazole-H), 7.14 (1H, s, imidazole-H), 7.20 (1H, dd, J = 7.8, 7.9 Hz, ArH), 7.59 (2H, s and d, J = 7.4 Hz, imidazole-H and ArH), 7.78 (1H, d, J = 7.9 Hz, ArH), 8.10 (1H, t br, J = 5.3 Hz, CONH), 8.23 (1H, s, H-8), 8.79 (1H, s, H-6), 9.46 (1H, s, ArNHAr); LCMS (ES+) m/z 375.39 [M+H]+; HRMS (ES+) calcd for C19H21N8O [M+H]+ 375.1687, found 375.1689; λmax (EtOH) 272, 330 nm.
Synthesis of 2-arylamino-6-(dialkylaminovinyl)-purines (69-73)
A solution of 6-ethynyl-2-phenylaminopurine (68) (50 mg, 0.21 mmol) and the required secondary amine (20.0 eq.) in anhydrous THF (2 mL) was subjected to microwave heating at 100 °C for 10 minutes in a sealed nitrogen flushed Biotage microwave vial. The cooled solution was partitioned between EtOAc (20 mL) and saturated NaHCO3 solution (20 mL). The organic extract was concentrated in vacuo to a yellow/orange syrup which was isolated using the Biotage SP4 purification system (12 + M KP-NH; MeOH-DCM; 0.5:9.5).
(E)-N-Phenyl-6-(2-(pyrrolidin-1-yl)vinyl)-9H-purin-2-amine (69)
The title compound was prepared by reaction of pyrrolidine (355 μL, 4.23 mmol). Yellow solid (60 mg, 93%): mp 138-140 °C; IR (cm-1) 3040, 2957, 1358, 2921, 2852, 1630, 1559; 1H NMR (500 MHz, DMSO-d6) δ 1.94 (4H, m, CH2), 3.36 (4H, m, CH2), 5.32-5.35 (1H, d, J = 15.0 Hz, alkene CH), 6.85-6.87 (1H, t, J = 10.1 Hz, H-4'), 7.22-7.26 (2H, dd, J = 9.9, 10.1 Hz, H-3' and H-5'), 8.82-8.84 (2H, d, J = 9.9 Hz, H-2' and H-6'), 7.91 (1H, s, H-8), 8.54-8.57 (1H, d, J = 15.0 Hz, alkene CH), 8.97(1H, br s, NH), 12.52 (1H, br s, NH-9); LCMS (ES+) m/z 307.3 [M+H]+; HRMS (ES+) calcd for C17H18N6 [M+H]+ 307.1671, found 307.1666; λmax (EtOH) 362.5, 282.5, 254.5 nm.
(E)-6-(2-(Dimethylamino)vinyl)-N-phenyl-9H-purin-2-amine (70)
The title compound was prepared by reaction dimethylamine solution 2.0 M in THF (2.13 mL, 4.23 mmol). Yellow solid (63%): mp 129-131 °C; IR (cm-1) 2946, 2922, 2853, 1562, 1525; 1H NMR (500 MHz, DMSO-d6) δ 2.99 (6H, s, NCH3), 5.15-5.18 (1H, d, J = 15.0 Hz, alkene CH), 6.63-6.65 (1H, t, J = 10.0 Hz, H-4'), 7.00-7.04 (2H, dd, J = 9.9, 10.0 Hz, H-3' and H-5'), 7.60-7.62 (2H, d, J = 9.9 Hz, H-2' and H-6'), 7.70 (1H, s, H-8), 8.08-8.11 (1H, d, J = 15.0 Hz, alkene CH), 8.75(1H, br s, NH), 12.30 (1H, br s, NH-9); 13C NMR (125 MHz, DMSO-d6) δ 84.5, 91.0, 117.9, 119.8, 122.0, 128.2, 138.1, 141.9, 149.6, 151.5, 156.0, 156.6; LCMS (ES+) m/z 281.2 [M+H]+; HRMS (ES+) calcd for C15H16N6 [M+H]+ 281.1508, found 281.1509; λmax (EtOH) 358.0, 281.0, 253.0 nm.
(E)-6-(2-(Diethylamino)vinyl)-N-phenyl-9H-purin-2-amine (71)
The title compound was prepared by reaction of diethylamine (442 μL, 4.23 mmol). Yellow solid (39 mg, 60%): mp 127-129 °C; IR (cm-1) 2970, 2911, 1630, 1559; 1H NMR (500 MHz, DMSO-d6) δ 1.11-1.13 (6H, t, J = 5.0 Hz, CH3), 3.21-3.24 (4H, q, J = 5.0 Hz, NCH2), 5.52-5.53 (1H, d, J = 15.0 Hz, alkene CH), 6.96-7.00 (1H, dd, J = 10.1 Hz, H-4'), 7.19 (1H, s, H-8), 7.21-7.25 (2H, dd, J = 9.8, 10.1 Hz, H-3' and H-5'), 7.40 (1H, br s, NH-2), 7.44-7.46 (2H, d, J = 9.8 Hz, H-2' and H-6'), 8.18-8.21 (1H, d, J = 15.0 Hz, alkene CH), 13.05 (1H, br s, NH-9); 13C NMR (125 MHz, DMSO-d6) δ 29.7, 22.2, 121.3, 122.8, 123.0, 129.3, 137.7, 140.16, 148.3, 151.1, 156.0, 158.3; LCMS (ES+) m/z 309.3 [M+H]+; HRMS (ES+) calcd for C17H20N6 [M+H]+ 309.1824, found 309.1822; λmax (EtOH) 359.0, 279.5, 253.5 nm.
(E)-6-(2-(Azepan-1-yl)vinyl)-N-phenyl-9H-purin-2-amine (72)
The title compound was prepared by reaction of homopiperadine (480 μL, 4.23 mmol). Yellow solid (69 mg, 98%): mp 135-137 °C; IR (cm-1) 3030, 2921, 2850, 1629, 1559; 1H NMR (500 MHz, CDCl3) δ 1.49 (4H, m, CH2), 1.64-1.71 (4H, m, CH2), 3.28-3.38 (4H, m, CH2), 5.49-5.52 (1H, d, J = 15.0 Hz, alkene CH), 6.95-6.99 (1H, t, J = 10.5 Hz, H-4'), 7.19 (1H, s, H-8), 7.22-7.26 (2H, dd, J = 9.5, 10.5 Hz, H-3' and H-5'), 7.32 (1H, br s, NH), 7.44-7.46 (2H, d, J = 9.5 Hz, H-2' and H-6'), 8.23-8.26 (1H, d, J = 15.0 Hz, alkene CH), 12.89 (1H, br s, NH-9); 13C NMR (125 MHz, CDCl3) δ 25.9, 26.9, 28.2, 30.5, 48.1, 56.0, 121.2, 122.8, 123.0, 129.3, 137.6, 140.1, 149.8, 151.0, 156.0, 158.3; LCMS (ES+) m/z 335.3 [M+H]+; HRMS (ES+) calcd for C19H22N6 [M+H]+ 335.1975, found 335.1979;λmax (EtOH) 360.0, 238.0, 254.5 nm.
(R, E)-6-(2-(3-(Dimethylamino)pyrrolidin-1-yl)vinyl)-N-phenyl-9H-purin-2-amine (73)
The title compound was prepared by reaction of (R)-3-dimethylaminopyrrolidine (540 μL, 4.23 mmol). Yellow solid (68 mg, 93%): mp 136-138 °C; IR (cm-1) 2954, 2778, 2118, 1625, 1561, 1528; 1H NMR (500 MHz, CDCl3) δ 1.75-1.83 (1H, m, pyrrolidine H-3''), 2.08 (1H, m, pyrrolidine CH), 2.20 (6H, s, N(CH3)2), 2.70 (1H, m, pyrollidine CH), 3.49-3.50 (4H, m, CH2), 5.40 (1H, d, J = 15.0 Hz, alkene CH), 6.79-7.00 (1H, t, J = 8.0 Hz, H-4'), 7.23-7.26 (2H, dd, J = 7.5, 8.0 Hz, H-3' and H-5'), 7.45-7.46 (2H, d, J = 7.5 Hz, H-2' and H-6'), 8.31 (1H, d, J = 15.0 Hz, alkene CH); 13C NMR (125 MHz, DMSO-d6) δ 14.2, 29.7, 30.2, 44.3, 65.2, 76.8, 77.1, 77.3, 91.8, 121.2, 122.7, 123.0, 129.3, 137.7, 140.0, 146.2, 151.2, 155.9, 155.8; LCMS (ES+) m/z 350.3 [M+H]+; HRMS (ES+) calcd for C19H23N7 [M+H]+ 350.2085, found 350.2088; λmax (EtOH) 263.0, 282.0, 361.0 nm.
Kinase inhibition conter-screening assays
Assays were conducted using ProfilerPro kinase selectivity assay kit 1 (Caliper Life Sciences). The ATP concentration used was that of the apparent ATP KM of each individual kinase and inhibitors were incubated at a single concentration of 2 μM, giving percentage inhibition values. Briefly, the protocol (provided with the kit on purchase) involved thawing of frozen enzyme and peptide/ATP plates before reconstitution of the enzymes in buffer solution. Compound solutions were added to the enzyme plate, mixed and pre-incubated at 28 °C for 15 min before transferring the peptide/ATP solution to each well. The reaction mixture was incubated at 28 °C for a further 90 min before addition of stop-buffer solution. The complete assay reactions were read using the Caliper EZ Reader II. For more information see: reference 16 and http://www.perkinelmer.co.uk/product/ez-reader-ship-level-122919 (accessed via internet on 05/07/2016).
Cellular growth inhibition assays
CellTiter-Blue Assay for Growth Inhibition. U2OS human osteosarcoma cells (American Type Culture Collection, Manassas, Virginia, United States) were grown in McCoy’s 5A medium supplemented with 1.5 mM L-glutamine, 25 mM HEPES, 2% penicillin/streptomycin (Invitrogen, Paisley, United Kingdom) and 10% (v/v) foetal calf serum (FCS) (Biosera, Ringmer, East Sussex, United Kingdom). MDA-MB-231 human breast cancer cells (American Type Culture Collection, Manassas, Virginia, United States) were grown in RPMI 1640 medium (Invitrogen) supplemented with 2 mM L-glutamine, 25 mM HEPES, 2% penicillin/streptomycin and 10% (v/v) FCS. HeLa cells were grown in Dulbecco′s Modified Eagle Medium (D-MEM) (Invitrogen) supplemented with 2% penicillin/streptomycin and 10% (v/v) FCS. All three cell lines were maintained in a humidified atmosphere of 5% CO2 at 37°C. The medium was aspirated and the cells were washed with PBS (Invitrogen), trypsinized (Internal supply, 0.25% versene trypsin with EDTA), neutralized and counted. Cells were seeded into 384-well clear tissue culture treated microtiter plates (Corning B.V. Life Sciences, Amsterdam, The Netherlands) at 200 cells per well in a 45 μL volume of the respective media. Columns 1 and 24 had no cells added and were plated with 45 μL of media alone. Cells were incubated at 37°C / 5% CO2. At 24 hours after plating, compounds were three-fold serially diluted in large volume V-shape 384-well microplates (Greiner Bio-One, Stonehouse, Gloucestershire, United Kingdom) using an Evolution plate handling system (PerkinElmer Life Sciences, Waltham, Massachusetts, USA). Then 5 μL of diluted test compounds, etoposide as positive control (Sigma-Aldrich, Gillingham, Dorset, United Kingdom), or DMSO at 1% v/v final concentration (Fisher Scientific, Loughborough, Leicestershire, United Kingdom) were added to the wells using a MiniTrack V plate handling system (PerkinElmer Life Sciences). There were four replicates of each compound concentration, 32 replicates of DMSO wells, and 32 replicates of wells containing no cells. Test compounds were screened at final concentrations of 100 μM, 33.33 μM, 11.11 μM, 3.70 μM, 1.23 μM, 0.41 μM, 0.14 μM, and 0.05 μM. Etoposide was screened at final concentrations of 10 μM, 3.33 μM, 1.11 μM, 0.37 μM, 0.12 μM, 0.041 μM, 0.014 μM, and 0.005 μM. After 92 hours, 5 μL of CellTiter-Blue Reagent (Promega, Southampton, United Kingdom) was added to the cells using a Multidrop dispenser (Thermo Electron, Basingstoke, Hants, United Kingdom) and incubated for 4 hours in a humidified atmosphere of 5% CO2 at 37°C. After the incubation, the plates were placed at room temperature for 40 minutes before fluorescence was recorded (Ex = 560/ Em = 590) on an EnVision 2103 plate reader (PerkinElmer Life Sciences). Data were plotted as percentage of DMSO control against compound concentration using GraphPad Prism 5 Software. The 50% growth inhibition (GI50) was calculated as the compound concentration required to reduce the cell number by 50% compared with the DMSO control.
Nek2 inhibition reversibility assay
The reversible nature of Nek2 inhibition by enamine 70 was measured using a kinase inhibition-reversibility assay. The assay mixture initially contained the ‘peptide 11’ substrate (5-FAM-KKLNRTLSVA-COOH; Caliper Life Sciences) and Nek2 protein (Invitrogen) at a concentration 100-fold that of the standard inhibitory activity assay. This solution in reaction buffer was pre-incubated with inhibitor 70 at a concentration ten-fold its IC50, calculated to afford approximately 91% inhibition of Nek2. After incubation of the enzyme with compound 70 for 30 min, the system was diluted 100- fold into a buffer solution containing both peptide-11 and ATP at the concentrations of the standard assay (details given in reference 12), affording a very low concentration of inhibitor (0.1 × IC50) with respect to other substrates. Following rapid 1:100 dilution, a reaction progress curve was generated by monitoring for levels of the phosphorylated substrate as described in reference 16 using the Caliper EZ Reader II instrument at different times throughout the duration of the reaction.
HPLC analysis of enamine stability
Solutions of 10 μM 72 were prepared in100% MEM media, 100% RPMI media, 10% (v/v) FCS in MEM, 10% BSA in water (v/v), 10 % FCS (v/v) in water, or 10 % (v/v) boiled FCS in water, DMSO and phosphate buffered water at pH 2, pH 7 and pH 10 by adding 10 μL of the 1 mM working stock of 72 in MeCN to 990 μL of each medium. A 50 μL aliquot was removed directly and immediately extracted as described below. At 5, 10, 15, 30, 60, 90, 120, 180, 240, 360 and 1440 minutes after addition of the drug further aliquots of 50 μL were taken and extracted as below. At each time point the 50 μL of the test solution was added to 50 μL of acetonitrile (MeCN). Samples were mixed for 10 seconds on a vortex mixer and centrifuged at 15700 rcf for 5 minutes in a microcentrifuge. The supernatant solution from each sample was placed into an HPLC vial for analysis. Samples were analysed by reverse phase HPLC with PDA detection.
ACKNOWLEDGMENTS
This research was supported by a grant from Cancer Research UK (Grant Reference C2115/A21421). The EPSRC Mass Spectrometry Service at the University of Wales (Swansea) is also gratefully acknowledged. AMF also acknowledges the support of Worldwide Cancer Research, The Wellcome Trust and BBSRC for funding.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
Accession codes
Atomic coordinates and structure factors for Nek2/inhibitor complexes have been deposited in the PDB, accession codes are 5M51 (compound 8), 5M53 (compound 11), 5M55 (compound 71) and 5M57 (compound 6) (Supplementary Table 5).
Authors’ contributions
Christopher R. Coxon – Synthesis and characterization of compounds and manuscript preparation.
Christopher Wong – Synthesis and characterization of compounds.
Richard Bayliss – Refined the structure of Nek2 in complex with compound 71 and analysed all protein-ligand structures.
Kathy Boxall – Design, conduct and interpretation of Nek2 inhibition experiments.
Katherine H Carr – Refine the structure of Nek2 in complex with compound 11.
Andrew M. Fry – Project conception and manuscript preparation.
Ian R. Hardcastle – Experimental design and interpretation.
Christopher J. Matheson – Synthesis and characterization of compounds.
David R. Newell – Project conception, experimental design and interpretation, data analysis and manuscript preparation.
Mangaleswaran Sivaprakasam – Synthesis and characterization of compounds.
Huw Thomas – Design, conduct and interpretation of compound 72 stability experiments.
David Turner – Synthesis and characterization of compounds.
Sharon Yeoh – Refined the structure of Nek2 in complex with compound 8.
Lan Z. Wang – Design, conduct and interpretation of CDK2 inhibition experiments.
Roger J. Griffin – Project conception, experimental design and interpretation.
Bernard T. Golding – Project conception, experimental design and interpretation, data analysis and manuscript preparation.
Céline Cano – Project conception, experimental design and interpretation, data analysis and manuscript preparation (submitting author).
REFERENCES
1. Chan JY. A clinical overview of centrosome amplification in human cancers. Int. J. Biol. Sci. 2011; 7: 1122-1144.
2. Fry AM, Meraldi P and Nigg EA. A centrosomal function for the human Nek2 protein kinase, a member of the NIMA family of cell cycle regulators. EMBO J. 1998; 17: 470-481.
3. Fry AM. The Nek2 protein kinase: a novel regulator of centrosome structure. Oncogene. 2002; 21: 6184-6194.
4. Mardin BR and Schiebel E. Breaking the ties that bind: new advances in centrosome biology. J Cell Biol. 2012; 197: 11-18.
5. Kim S, Lee K, Choi JH, Ringstad N and Dynlacht BD, Nek2 activation of Kif24 ensures cilium disassembly during the cell cycle. Nat. commun. 2015; 6: 8087.
6. Liu X, Gao Y, Lu Y, Zhang J, Li L and Yin F. Upregulation of Nek2 is associated with drug resistance in ovarian cancer. Oncol Rep. 2014; 31: 745-754.
7. Neal CP, Fry AM, Moreman C, McGregor A, Garcea G, Berry DP and Manson MM. Overexpression of the Nek2 kinase in colorectal cancer correlates with beta-catenin relocalization and shortened cancer-specific survival. J Surg Oncol. 2014; 110: 828-838.
8. Zeng YR, Han ZD, Wang C, Cai C, Huang YQ, Luo HW, Liu ZZ, Zhuo YJ, Dai QS, Zhao HB and Liang YX. Overexpression of NIMA-related kinase 2 is associated with progression and poor prognosis of prostate cancer. BMC Urol. 2015; 1: 90.
9. Wubetu GY, Morine Y, Teraoku H, Yoshikawa M, Ishikawa D, Yamada S, Ikemoto T, Saito UU, Imura S and Shimada M. High Nek2 Expression Is a Predictor of Tumor Recurrence in Hepatocellular Carcinoma Patients After Hepatectomy. Anticancer Res. 2016; 36: 757-762.
10. Hayward DG, Clarke RB, Faragher AJ, Pillai MR., Hagan IM, Fry AM. The Centrosomal Kinase Nek2 Displays Elevated Levels of Protein Expression in Human Breast Cancer. Cancer Res. 2004; 64: 7370-7376.
11. Zhou W, Yang Y, Xia J, Wang H, Salama ME, Xiong W, Xu H, Shetty S, Chen T, Zeng Z and Shi L. Nek2 induces drug resistance mainly through activation of efflux drug pumps and is associated with poor prognosis in myeloma and other cancers. Cancer Cell. 2013; 23: 48-62.
12. Cappello P, Blaser H, Gorrini C, Lin DCC, Elia AJ, Wakeham A, Haider S, Boutros PC, Mason JM, Miller NA and Youngson B. Role of Nek2 on centrosome duplication and aneuploidy in breast cancer cells. Oncogene. 2014; 33: 2375-2384.
13. Kokuryo T, Senga T, Yokoyama Y, Nagino M, Nimura Y, Hamaguch, M. Nek2 as an Effective Target for Inhibition of Tumorigenic Growth and Peritoneal Dissemination of Cholangiocarcinoma. Cancer Res. 2007; 67: 9637-9642.
14. Suzuki K, Kokuryo T, Senga T, Yokoyama Y, Nagino M, Hamaguchi M. Novel combination treatment for colorectal cancer using Nek2 siRNA and cisplatin. Cancer Sci. 2010; 101: 1163-1169.
15. Whelligan DK, Solanki S, Taylor D, Thomson DW, Cheung KMJ, Boxall K, Mas-Droux C, Barillari C, Burns S, Grummitt CG, Collins I, van Montfort RLM, Aherne GW, et al. Aminopyrazine Inhibitors Binding to an Unusual Inactive Conformation of the Mitotic Kinase Nek2: SAR and Structural Characterization. J Med Chem. 2010; 53: 7682-7698.
16. Solanki S, Innocenti P, Mas-Droux C, Boxall K, Barillari C, van Montfort RLM, Aherne GW, Bayliss R, Hoelder S. Benzimidazole Inhibitors Induce a DFG-Out Conformation of Never in Mitosis Gene A-Related Kinase 2 (Nek2) without Binding to the Back Pocket and Reveal a Nonlinear Structure−Activity Relationship. J Med Chem. 2011; 54: 1626-1639.
17. Innocenti P, Cheung KMJ, Solanki S, Mas-Droux C, Rowan F, Yeoh S, Boxall K, Westlake M, Pickard L, Hardy T, Baxter JE, Aherne GW, Bayliss R, Fry AM, Hoelder S. Design of Potent and Selective Hybrid Inhibitors of the Mitotic Kinase Nek2: Structure–Activity Relationship, Structural Biology, and Cellular Activity. J Med Chem. 2012; 55: 3228-3241.
18. Hayward DG, Newbatt Y, Pickard L, Byrne E, Mao G, Burns S, Sahota NK, Workman P, Collins I, Aherne W, Fry AM. Identification by High-Throughput Screening of Viridin Analogs as Biochemical and Cell-Based Inhibitors of the Cell Cycle–Regulated Nek2 Kinase. J Biomol Screening 2010; 15: 918-927.
19. Henise JC, Taunton J. Irreversible Nek2 Kinase Inhibitors with Cellular Activity. J Med Chem. 2011; 54: 4133-4146.
20. Arris CE, Boyle FT, Calvert AH, Curtin NJ, Endicott JA, Garman EF, Gibson AE, Golding BT, Grant S, Griffin RJ and Jewsbury P, Identification of Novel Purine and Pyrimidine Cyclin-Dependent Kinase Inhibitors with Distinct Molecular Interactions and Tumor Cell Growth Inhibition Profiles. J Med Chem. 2000; 43: 2797-2804.
21. Whitfield HJ, Griffin RJ, Hardcastle IR, Henderson A, Meneyrol J, Mesguiche V, Sayle KL, Golding BT. Facilitation of addition-elimination reactions in pyrimidines and purines using TFA in trifluoroethanol. Chem Commun. 2003; 2802-2803.
22. Carbain B, Coxon CR, Lebraud H, Elliott KJ, Matheson CJ, Meschini E, Roberts AR, Turner DM, Wong C, Cano C and Griffin RJ. Trifluoroacetic Acid in 2, 2, 2-Trifluoroethanol Facilitates SNAr Reactions of Heterocycles with Arylamines. Chem Eur J. 2014; 20: 2311-2317..
23. Lee HJ, Choi YS, Lee KB, Park J and Yoon CJ. Hydrogen bonding abilities of thioamide. J Chem Phys A. 2002; 106: 7010-7017.
24. Loev B and Kormendy MF. An Improved Synthesis of Carbamates. J Org Chem. 1963; 28: 3421-3426.
25. Jencks WP, Oakenfull DG and Salvesen K. Reactions of acetylimidazole and acetylimidazolium ion with nucleophilic reagents. Mechanisms of catalysis. J Am Chem. Soc. 1971; 93: 188-194.
26. Woodman EK, Chaffey JGK, Hopes PA, Hose DRJ, Gilday JP. N,N'-Carbonyldiimidazole-Mediated Amide Coupling: Significant Rate Enhancement Achieved by Acid Catalysis with Imidazole·HCl. Org. Process Res Dev. 2009; 13: 106-113.
27. Beach JW, Kim HO, Jeong LS, Nampalli S, Islam Q, Ahn SK, Babu JR, Chu CK. A Highly Stereoselective Synthesis of Anti-HIV 2',3'-dideoxy- and 2',3'-didehydro-2',3'-dideoxynucleosides. J Org Chem. 1992; 57: 3887-3894.
28. Baumgarten HE, Petersen JM. Reactions of Amines. V. Synthesis of α-Amino Ketones.1,2 J Am Chem Soc. 1960; 82: 463-468.
29. Kos NJ, Jongejan H and Van Der Plas HC. Deamination, Involving Ring Opening, in Reactions of 1-Aminopurinium Mesitylenesulfonates with MeOHic Ammonia. Tetrahedron. 1987; 43: 4841-4848.
30. Kuchar M, Pohl R, Votruba I, Hocek M. Synthesis of Purines Bearing Functionalized C-Substituents by the Conjugate Addition of Nucleophiles to 6-Vinylpurines and 6-Ethynylpurines. Eur J Org Chem. 2006; 5083-5098.
31. Kuchar M, Hocek M, Pohl R, Votruba I, Shih IH, Mabery E and Mackman R, Synthesis, cytostatic, and antiviral activity of novel 6-[2-(dialkylamino)ethyl]-, 6-(2-alkoxyethyl)-, 6-[2- 60 (alkylsulfanyl)ethyl]-, and 6-[2-(dialkylamino)vinyl]purine Nucleosides. Bioorg Med Chem. 2008; 16: 1400–1424.
32. Hardcastle IR, Arris CE, Bentley J, Boyle T, Chen Y, Curtin NJ, Endicott JA, Gibson AE, Golding BT, Griffin RJ, Jewsbury P, Menyerol J, Mesguiche V, et al. N2-Substituted O6-Cyclohexylmethylguanine Derivatives: Potent Inhibitors of Cyclin-Dependent Kinases 1 and 2. J Med Chem. 2004; 47: 3710-3722.
33. Copeland RA. Evaluation of Enzyme Inhibitors in Drug Discovery, 2005; Wiley & Sons, Hoboken, New Jersey.
34. Westwood I, Cheary DM, Baxter JE, Richards MW, van Montfort RL, Fry AM, Bayliss R. Insights into the conformational variability and regulation of human Nek2 kinase. J Mol Biol. 2009; 386: 476-485.
35. Kornev AP, Taylor SS, Ten Eyck LF. A helix scaffold for the assembly of active protein kinases. Proc Natl Acad Sci USA. 2008; 105: 14377-14382.
36. Kwak EL, Sordella R, Bell DW, Godin-Heymann N, Okimoto RA, Brannigan BW, Harris PL, Driscoll DR, Fidias P, Lynch TJ, Rabindran SK, McGinnis JP, Wissner A, et al. Irreversible inhibitors of the EGF receptor may circumvent acquired resistance to gefitinib. Proc Natl Acad Sci USA. 2005; 102: 7665-7670.
37. Tsou HR, Overbeek-Klumpers EG, Hallett WA, Reich MF, Floyd MB, Johnson BD, Michalak RS, Nilakantan R, Discafani C, Golas J and Rabindran SK. Optimization of 6,7-Disubstituted-4-(arylamino)quinoline-3-carbonitriles as Orally Active, Irreversible Inhibitors of Human Epidermal Growth Factor, Receptor-2 Kinase Activity. J Med Chem. 2005; 48: 1107-1131.
38. Wissner A, Overbeek E, Reich MF, Floyd MB, Johnson BD, Mamuya N, Rosfjord EC, Discafani C, Davis R, Shi X and Rabindran SK. Synthesis and Structure-Activity Relationships of 6,7-Disubstituted 4-Anilinoquinoline-3-carbonitriles. The Design of an Orally Active, Irreversible Inhibitor of the Tyrosine Kinase Activity of the Epidermal Growth Factor Receptor (EGFR) and the Human Epidermal Growth Factor Receptor-2 (HER-2). J Med Chem. 2003; 46: 49-63.