
INTRODUCTION
Breast cancer is the most common malignancy detected in women [1]. Surgical removal of the cancer followed by adjuvant therapy represents the standard of care. Accurate prediction of recurrence risk is of vital importance for tailoring adjuvant therapy for individual breast cancer patients. Conventional pathological parameters, such as histological grade, tumor size, and presence of lymph node metastasis, are not accurate enough to select subsets of patients who are at sufficiently low risk of recurrence to be spared from extensive adjuvant therapy without compromising prognosis. Accumulating evidence exists that additional molecular testing can help to better select patients who would benefit most from adjuvant therapy, at the same time sparing those who would derive little or no advantage from treatment [2–4].
The p16 tumor suppressor, encoded by the CDKN2A gene at 9p21, has been discussed as a prognostic factor in breast cancer for more than a decade. p16 inhibits cyclin-dependent kinases (CDKs) 4 and 6 at the G1 to S phase transition, thus preventing phosphorylation of the retinoblastoma (RB1) protein. Insufficiently phosphorylated RB1 leads to sequestered E2F in an incompetent RB1/E2F complex, preventing E2F from triggering cell cycle progression (reviewed in [5]). p16 plays a pivotal role in various tumor types including cancers of colon, skin, and gallbladder (reviewed in [6]). In breast cancer, studies on 10–314 patients suggested a role of p16 overexpression in tumor progression [7–10], metastasis [10], and clinical outcome [9, 10]. It is, thus, remarkable that deletion of the chromosomal region 9p21, potentially leading to reduced p16 expression, belongs to the most frequent deletions in breast cancers occurring in 11% to 65% [11–17]. Two of these studies with 39 and 166 cancers even described a link between 9p21 deletions and unfavorable tumor phenotype [15, 16].
To evaluate the potential role of both p16 expression and 9p21 deletion as prognostic features we investigated a cohort of more than 2,100 breast cancers employing immunohistochemistry (IHC) and fluorescence in-situ hybridization (FISH).
RESULTS
Prevalence of p16 expression and 9p21 deletion in breast cancer
To estimate the baseline level of p16 expression we analyzed 20 normal breast tissues. In normal breast epithelium p16 immunostaining was usually negative or limited to a small fraction of cells (< 5%) and appeared less intense (mostly weak to moderate) as compared to p16 positive cancer cells. P16 immunostaining was observed in 747 of 1,684 (44.4%) analyzable cancers. If present, p16 staining was typically found in all tumor cells, and was considered weak in 25.6%, moderate in 7.1% and strong in 12.7% of cancers. Representative images of immunostainings are shown in Figure 1A–1D. 9p21 was successfully analyzed by FISH in 1,089 (49.6%) arrayed cancer samples. 9p21 deletions were found in 167 (15.3%) interpretable breast cancers, including 13.6% heterozygous and 1.7% homozygous deletions (Figure 1E–1G).
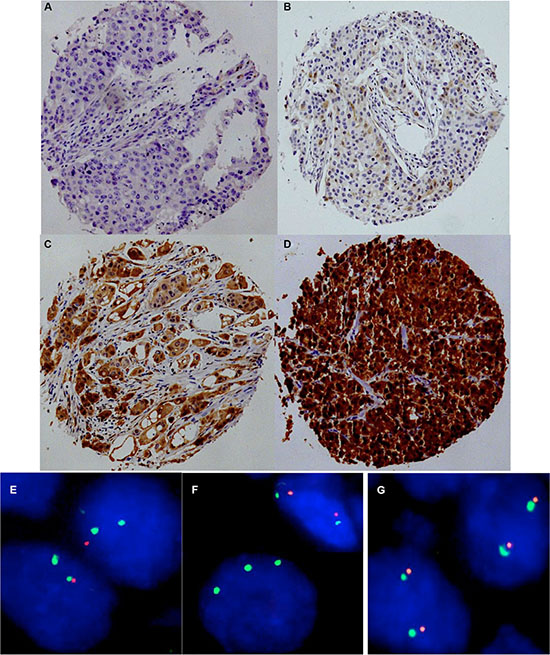
Figure 1: Representative images of p16 immunostaining with (A) negative, (B) weak, (C) moderate, and (D) strong staining and examples of FISH findings using the 9p21 deletion probe. (E) Heterozygous deletion as indicated by the lack of one orange 9p21 signal and two green centromere 9 signals, (F) Homozygous deletion as indicated by the complete lack of orange 9p21 signals in the tumor cell, (G) Normal 9p21 copy numbers as indicated by two orange 9p21 signals and two green centromere 9 signals.
Association of p16 expression and 9p21 deletion to breast cancer phenotype
Strong p16 expression was tightly linked to adverse tumor features, including histopathological grade (p < 0.0001), advanced tumor stage (p = 0.0003), and hormone receptor (ER/PR) negativity (p < 0.0001 each) in all breast cancers and in the largest subset of cancers of No Special Type (NST; p ≤ 0.0010). Also 9p21 deletion was significantly associated with unfavorable tumor features, including histological grade (p < 0.0001), presence of lymph node metastases (p = 0.0063), ER/PR negativity (p < 0.0001 for ER and p = 0.0006 for PR), and amplifications of HER2 (p = 0.0078) in all breast cancers. These associations held also true in the subset of NST cancers p < 0.05). All results are summarized in Table 1.
Table 1: Clinico-pathological association of 9p21 deletion (FISH) and p16 expression (IHC)
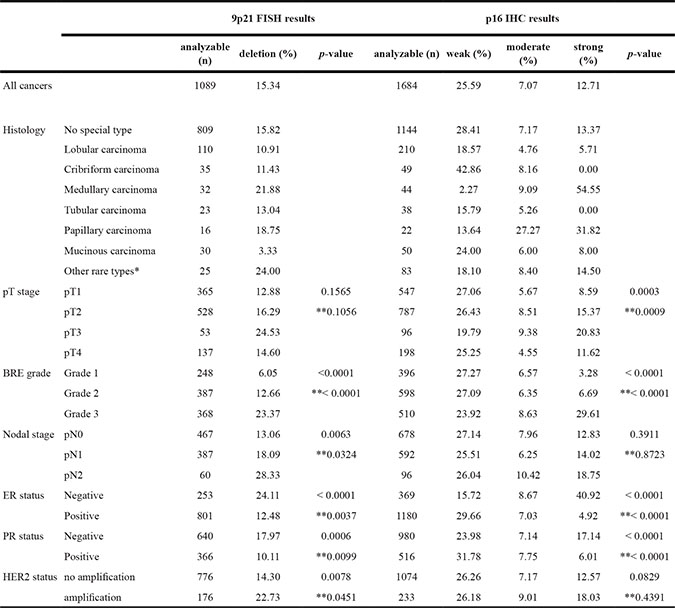
* including adenoid-cystic carcinoma, apocrine carcinoma, atyp medullary carcinoma, carcinosarcoma, clear cell carcinoma, histiocytic carcinoma, lipid rich carcinoma, lipid rich or histiocytoic carcinoma, metaplastic carcinoma, neuroendocrine carcinoma, signet ring carcinoma, and small cell carcinoma. ** in subgroup of cancers of No Special Type
Association of p16 expression and 9p21 deletion with cell proliferation
Data on the tumor cell proliferation, as determined by immunohistochemical analysis of the Ki67 antigen, were available from a previous study using the same TMA [18]. Strong p16 staining as well as 9p21 deletions were tightly associated with a high Ki67 labeling index (LI) if all cancers were jointly analyzed (p < 0.0001 each). These associations were independent from the histological grade because they held also true in the subsets of cancers with identical grade. All results are summarized in Table 2.
Table 2: Association between 9p21 deletion or p16 expression and Ki67-labeling index
9p21 FISH |
p16 IHC |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
normal |
deletion |
negative |
weak |
moderate |
strong |
|||||
analyzable (n) |
Ki67LI |
Ki67LI |
p-value |
analyzable (n) |
Ki67LI |
Ki67LI |
Ki67LI |
Ki67LI |
p-value |
|
All cancers |
947 |
28.70 ±0.52 |
36.37 ±1.23 |
p < 0.0001 |
1425 |
24.63 ±0.50 |
27.38 ±0.72 |
29.16 ±1.33 |
41.48 ±0.99 |
p <0.0001 |
Grad 1 |
211 |
19.42 ± 0.72 |
25.36 ± 3.07 |
p = 0.0610 |
337 |
18.26 ± 0.69 |
17.47 ± 1.08 |
20.00 ± 2.12 |
24.27 ± 3.07 |
p = 0.1729 |
Grad 2 |
343 |
25.63 ± 0.66 |
32.58 ± 1.69 |
p = 0.0001 |
535 |
23.87 ± 0.67 |
25.80 ± 1.00 |
25.97 ± 1.96 |
29.00 ± 1.96 |
p = 0.0494 |
Grad 3 |
319 |
40.20 ± 0.96 |
40.23 ± 1.73 |
p = 0.9907 |
443 |
34.25 ± 1.12 |
36.84 ± 1.34 |
35.23 ± 2.21 |
46.00 ± 1.19 |
p < 0.0001 |
Prognostic relevance of p16 expression and 9p21 deletion
Data on raw survival were available from 1,635 cancers with interpretable p16 IHC results and from 1,087 cancers with interpretable 9p21 FISH results. Strong p16 staining was linked to shortened overall survival if all cancers were jointly analyzed (p = 0.0038, Figure 2A), as well as in the subsets of NST cancers (p = 0.0048, Figure 2B), and in the subset of cancers with nodal metastases (p < 0.0001, Figure 2D). No association was found between p16 expression and overall survival in the subset of triple negative cancers (p = 0.9411, Figure 2E). No unequivocal association was found between 9p21 deletion and overall survival, neither in all cancers (p = 0.0720) nor in subsets of NST (p = 0.2478), nodal negative (p = 0.1469), nodal positive (p = 0.0130), or triple negative cancers (p = 0.7141, Figure 2F–2J). In a multivariate analysis including KI67LI, hormone receptor status, HER2 status, pT stage, BRE grade and nodal stage, p16 expression predicted overall survival independently from these parameters in 800 analyzable cancers (p = 0.0304, Table 3).
Table 3: Multivariate analysis (Cox regression) including pathological and molecular parameters in addition to p16 expression in all cancers
Parameter |
RR |
95%CI |
p-Value |
|---|---|---|---|
KI67LI |
1.3 |
0.6–2.7 |
0.4918 |
ER status |
|||
positive vs negative |
0.8 |
0.6–1.2 |
0.3189 |
PR status |
|||
positive vs negative |
0.6 |
0.4–0.8 |
0.0005 |
HER2 status |
|||
positive vs negative |
1.1 |
0.8–1.5 |
0.6347 |
pT stage |
|||
2 vs 1 |
1.2 |
0.8–1.6 |
0.0007 |
3 vs 2 |
1.2 |
0.7–1.7 |
|
4 vs 3 |
1.7 |
1.0–2.8 |
|
pN stage |
|||
N+ vs N0 |
2.9 |
2.2–3.9 |
< 0.0001 |
BRE grade |
|||
2 vs 1 |
1.3 |
0.8–1.9 |
< 0.0001 |
3 vs 2 |
2.3 |
1.7–3.1 |
|
p16 IHC |
|||
weak vs negative |
1.1 |
0.8–1.5 |
0.0145 |
moderate vs weak |
1.5 |
0.9–2.2 |
|
strong vs moderate |
0.5 |
0.3–0.7 |
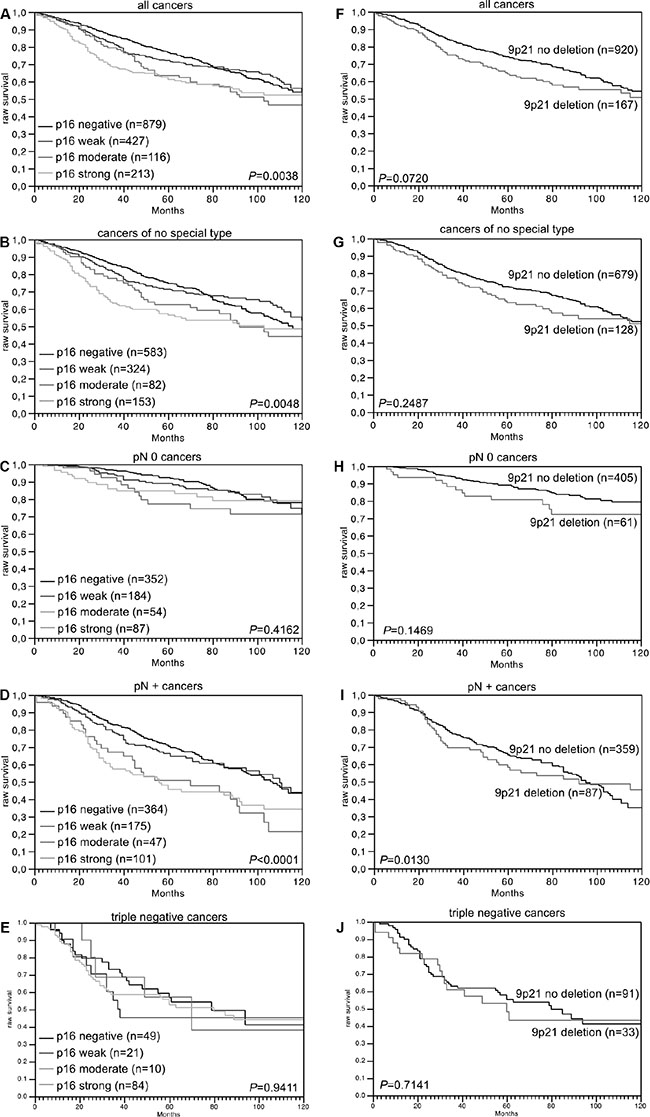
Figure 2: Association between p16 expression and raw survival in (A) all cancers, (B) no special type cancers, (C) nodal negative cancers, (D) nodal positive cancers, and (E) triple negative cancers. Association between 9p21 deletion and raw survival in (F) all cancers, (G) no special type cancers, (H) nodal negative cancers, (I) nodal positive cancers, and (J) triple negative cancers.
Relationship between p16 expression and 9p21 deletion
In all (100%) of 19 cancers with homozygous 9p21 deletion p16 immunostaining was completely absent. No significant difference of p16 expression was found between 138 cancers with heterozygous 9p21 deletions and 866 cancers without 9p21 deletion. p16 expression was even slightly higher in deleted than in undeleted cancers (p = 0.0256, Figure 3).
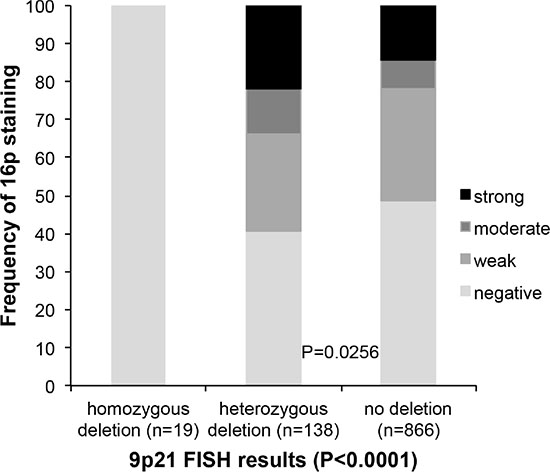
Figure 3: Association between 9p21 deletion (FISH) and p16 expression (IHC).
DISCUSSION
The results of our study on 2,100 breast cancers identify that p16 overexpression and 9p21 deletion are largely independent of each other while both features are linked to aggressive breast cancer phenotype.
Our IHC analysis shows that p16 is up regulated in a fraction of tumors as compared to normal breast epithelium. Under the selected staining conditions, positive p16 immunostaining was found in about 50% of breast cancers, including more than 10% of cancers with high intensity p16 staining. Earlier immunohistochemistry studies using TMAs and large sections examining 20–119 breast cancers reported frequencies of p16 positivity ranging from 56% to 89% (TMA) and 21% to 51% (large sections) [8, 9, 19–23]. Such discrepancies are common if a protein is analyzed by immunohistochemistry in a large number of studies. They are typically attributable to the usage of different antibodies, laboratory protocols, and scoring criteria. However, the comparably high fraction of p16 positive cases in our study using a single 0.6 mm TMA spot per cancer and earlier studies on conventional large sections argues against substantially heterogeneity of p16 expression in breast cancer.
Despite of its well-known role as a tumor suppressor gene, p16 overexpression - but not expression loss - was linked to adverse tumor parameters, including advanced stage, high grade and shorter survival independently from known prognosticators, including pT stage, nodal stage, BRE grade, hormone receptor state, HER2 and cell proliferation. Most of these associations are in line with earlier IHC studies examining 10, 60, 82 and 314 breast tumors that had already suggested associations between p16 overexpression and adverse features of breast cancer such as high grade [7–10], nodal stage [10] and poor patient prognosis [9, 10]. That p16 expression lacked prognostic relevance in the subset of triple negative cancers underscores the particularly poor prognosis of these cancers [24].
Finding a close association between p16 positivity and accelerated cell proliferation fits well to the specific role of p16 for cell cycle regulation. p16 is up regulated in G1 phase of each cell cycle and has an exceptionally long half-life time as compared to other cell cycle regulators (reviewed in [5, 25]). In slowly proliferating cells with a doubling time greater than the p16 dismantling period, it can, thus, be expected that p16 is completely cleared from the cell between two mitoses. Accordingly, we observed scattered p16 staining only in a low fraction of normal breast epithelial cells, which corresponds to the fraction of proliferating cells (< 3%). In contrast, rapidly proliferating cancers showed uniformly strong accumulation of p16 in virtually all cells, indicating that p16 clearance was not accomplished between two mitoses.
Another aim of our study was to clarify the relationship between 9p deletion and p16 expression, both of which have been suggested as markers for breast cancer progression. As expected, cancers harboring complete (i.e., homozygous) 9p deletion lacked p16 expression, which indirectly validates our experimental approaches both for FISH and IHC. That no difference of p16 expression was seen between cancers with and without heterozygous 9p deletion demonstrates that breast cancer cells are able to fully compensate for loss of one p16 (CDKN2A) allele, either by increased transcriptional activation of the remaining allele or by increased stabilization of p16 protein or mRNA (reviewed in [5, 25]). Our findings are in line with published data from the TCGA project showing no significant differences in p16 expression between cancers with partial (heterozygous) deletion and normal copy numbers (http://www.cbioportal.org [26, 27]). These results, therefore, demonstrate that heterozygous 9p deletion is not a relevant mechanism for altering p16 expression in breast cancer. We conclude from these observations that p16 may not represent the main target gene of 9p deletions in breast cancer. Complete p16 inactivation occurs in only 1.5% of 9p deleted cancers by homozygous deletion and inactivating p16 mutations – that might accompany some of the 9p deletions – occur in only 0.5 to 7% of breast cancers [12, 28–31]).
Finding 9p deletions in 15% of cancers fits well to earlier work reporting deletions of 9p in 6–25% by classical or array CGH in cohorts of 39–98 analyzed breast cancers [12, 15, 32, 33]. Higher rates of 9p deletions were only found in studies selecting for metastatic breast cancers (41% of 34) [34] or studies employing less quantitative loss of heterozygosity (LOH, 11–65%) analyses on 12–171 cases [11, 13, 14, 17]. These assays are influenced by ploidy changes and admixture of normal cells, which inevitably affect the assay specificity and sensitivity. In contrast, FISH allows for precise gene copy number determination in individual cells, rendering it independent from the purity of cancer tissues or presence of aneusomy. FISH is thus considered the gold standard for gene copy number analysis.
9p deletions were linked to features of aggressive breast cancer such as high-grade and a strong trend towards reduced survival was found. Studies using classical CGH have demonstrated that 9p deletions typically comprise large portions of 9p or even the entire chromosome arm [32, 33]. The example of p16 shows that not all effected genes must necessarily become down-regulated in 9p deleted cancers, but it seems likely that many genes will be. These might include genes with tumor suppressive properties, such as CDKN2B (9p21) [35], SH3GL2 (9p22) [36], PTPRD (9p23) [37], and DOCK8 (9p24) [38]. Such genes may, either alone or in concert contribute to tumor progression when affected by 9p deletion. A cooperative effect of genes hit by large deletions has been demonstrated for large 8p deletions in liver cancer [39].
In conclusion, the results of our study show that strong p16 overexpression occurs in 10% of breast cancers and is linked to a fraction of aggressive and rapidly proliferating breast cancers with poor prognosis.
MATERIALS AND METHODS
Breast cancer tissue microarray (TMA)
A pre-existing tissue microarray (TMA) was used for this study [18]. The TMA contained 2,197 human breast cancer samples from paraffin-embedded tissue specimens fixed in 4% neutral buffered formalin. From each patient one 0.6 mm core was taken from a representative cancer tissue block. All tissues were distributed among 6 TMA blocks, each containing 263 to 522 tumor samples. Consecutive breast cancer samples collected between 1984 and 2000 were used for this study. The median patient’s age was 63 (range 26–101) years. The use of the specimens and data for research purposes was approved by the Ethics Committee of the Basel University Hospital. Survival data were either obtained from the cancer registry of Basel or collected from the patients attending physicians. Raw survival data were available from 1,982 patients (713 patients with and 1,508 without event). The mean follow-up time was 63 months (range 1–176 months). Tumor size and nodal status were obtained from the primary pathology reports. All slides from the tumors were reviewed by specialized pathologists to define the histologic grade according to Elston and Ellis [40] and the histologic tumor tumor type. Four μm sections of the TMA blocks were transferred to an adhesive coated slide system (Instrumedics Inc., Hackensack, New Jersey) for FISH and IHC analysis. Molecular data used in this study were available from previously published studies. These included amplification data obtained by FISH for HER2 amplification as well as IHC data on estrogen receptor (ER) and progesterone receptor (PR) expression as well as Ki67 labeling index (Ki67 LI) [18, 41].
Fluorescence in-situ hybridization
Four micrometer TMA sections were used for FISH. For proteolytic slide pretreatment, a commercial kit was used (paraffin pretreatment reagent kit; Abbott, Wiesbaden, Germany). TMA sections were deparaffinized, air-dried, and dehydrated in 70%, 85%, and 100% ethanol, followed by denaturation for 5 min at 74°C in 70% formamid 2x SSC solution. The commercial Vysis CDKN2A / CEP 9 FISH probe kit (#04N61–020; Abbott, Wiesbaden, Germany) was used for detection of the 9p21 status. Hybridization was performed overnight at 37°C in a humidified chamber. Slides were subsequently washed and counterstained with 0.2 μmol/L 4′-6-diamidino-2-phenylindole in antifade solution. Stained slides were manually interpreted with an epifluorescence microscope by expert scientists (MK, CÖ, BT, KH and MR). The predominant FISH signal numbers were recorded in each tissue spot, and a consensus result was generated in case of unequivocal findings. Presence of fewer CDKN2A signals than centromere 9 probe signals in at least 60% tumor nuclei was considered a heterozygous deletion. Complete absence of CDKN2A signals in all tumor cells, but presence of centromere 9 and CDKN2A signals in adjacent normal cells, was considered a homozygous deletion. Tissue spots lacking any detectable CDKN2A signals in all (tumor and normal cells) or lack of any normal cells as an internal control for successful hybridization of the CDKN2A probe were excluded from analysis. These thresholds were based on our previous study analyzing PTEN deletions on a prostate cancer TMA where our approach resulted in a 100% concordance with array comparative genomic hybridization (CGH) data [42].
Immunohistochemical analysis
Freshly cut TMA sections were immunostained on one day and in one experiment. Slides were deparaffinized, rehydrated, washed in DAKO buffer (K8002) and transferred to a DAKO Link 48 autostainer device. The immunohistochemical staining of p16 was performed with the commercially available CINtec p16 Histology Kit (Cat.# 725–4713, Ventana Medical Systems Inc., Arizona, USA) according to the manufacturer’s instructions. p16 staining was typically nuclear and cytoplasmic. The staining intensity (0, 1+, 2+, and 3+) and the fraction of positive tumor cells were separately recorded for each tissue spot by a pathologist (PL). A final score was then built from these 2 parameters according to the following score as previously described [43, 44]. Negative scores had complete absence of staining, weak scores had staining intensity of 1+ in ≤ 70% of tumor cells or staining intensity of 2+ in ≤ 30% of tumor cells; moderate scores had staining intensity of 1+ in > 70% of tumor cells, staining intensity of 2+ in > 30% but in ≤ 70% of tumor cells or staining intensity of 3+ in ≤ 30% of tumor cells; strong scores had staining intensity of 2+ in > 70% of tumor cells or staining intensity of 3+ in > 30% of tumor cells.
Statistics
Statistical calculations were performed with JMP 9 software (SAS Institute Inc., NC, USA). Contingency table analysis and Chi square test were used to study the relationship between IHC and FISH results and clinicopathological variables. Kaplan–Meier plots were used to estimate overall survival and the statistical significance was determined by the log rank test. The log-Rank test was applied to test the significance of differences between stratified survival functions. Cox proportional hazards regression analysis was performed to test the statistical independence and significance between pathological and molecular variables.
ACKNOWLEDGMENTS
We thank Janett Lütgens, Sünje Seekamp, and Inge Brandt for excellent technical assistance.
CONFLICTS OF INTEREST
We certify that there is no actual or potential conflicts of interest in relation to this article.
FUNDING
We certify that there are no sources of any support for the work, received in the form of grants, equipment, and/or drugs in relation to this article.
REFERENCES
1. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics.CA-cancer J Clin. 2011; 61:69–90.
2. Giuliano AE, Hunt KK, Ballman KV, Beitsch PD, Whitworth PW, Blumencranz PW, Leitch AM, Saha S, McCall LM, Morrow M. Axillary dissection vs no axillary dissection in women with invasive breast cancer and sentinel node metastasis: a randomized clinical trial. Jama. 2011; 305:569–575.
3. McVeigh TP, Hughes LM, Miller N, Sheehan M, Keane M, Sweeney KJ, Kerin MJ. The impact of Oncotype DX testing on breast cancer management and chemotherapy prescribing patterns in a tertiary referral centre. Eur J Cancer. 2014; 50:2763–2770.
4. Naoi Y, Noguchi S. Multi-gene classifiers for prediction of recurrence in breast cancer patients. Breast cancer. 2015.
5. Li J, Poi MJ, Tsai MD. Regulatory mechanisms of tumor suppressor P16(INK4A) and their relevance to cancer. Biochemistry. 2011; 50:5566–5582.
6. Romagosa C, Simonetti S, Lopez-Vicente L, Mazo A, Lleonart ME, Castellvi J, Ramon y Cajal S. p16(Ink4a) overexpression in cancer: a tumor suppressor gene associated with senescence and high-grade tumors. Oncogene. 2011; 30:2087–2097.
7. Milde-Langosch K, Bamberger AM, Rieck G, Kelp B, Loning T. Overexpression of the p16 cell cycle inhibitor in breast cancer is associated with a more malignant phenotype. Breast Cancer Res Treat. 2001; 67:61–70.
8. Witkiewicz AK, Rivadeneira DB, Ertel A, Kline J, Hyslop T, Schwartz GF, Fortina P, Knudsen ES. Association of RB/p16-pathway perturbations with DCIS recurrence: dependence on tumor versus tissue microenvironment. The Am J Pathol. 2011; 179:1171–1178.
9. Karray-Chouayekh S, Baccouche S, Khabir A, Sellami-Boudawara T, Daoud J, Frikha M, Jlidi R, Gargouri A, Mokdad-Gargouri R. Prognostic significance of p16INK4a/p53 in Tunisian patients with breast carcinoma. Acta histochemica. 2011; 113:508–513.
10. Hui R, Macmillan RD, Kenny FS, Musgrove EA, Blamey RW, Nicholson RI, Robertson JF, Sutherland RL. INK4a gene expression and methylation in primary breast cancer: overexpression of p16INK4a messenger RNA is a marker of poor prognosis. Clin Cancer Res. 2000; 6:2777–2787.
11. Vieira de Oliveira SF, Oliveira MM, Urban CA, de Lima RS, Cavalli IJ, Ribeiro EM. Lack of association between LOH in the 9p region and clinicopathologic parameters in primary breast cancer. Cancer Genet Cytogenet. 2010; 200:23–27.
12. Haverty PM, Fridlyand J, Li L, Getz G, Beroukhim R, Lohr S, Wu TD, Cavet G, Zhang Z, Chant J. High-resolution genomic and expression analyses of copy number alterations in breast tumors. Gene Chromosomes Cancer. 2008; 47:530–542.
13. Silva J, Silva JM, Dominguez G, Garcia JM, Cantos B, Rodriguez R, Larrondo FJ, Provencio M, Espana P, BonillaF. Concomitant expression of p16INK4a and p14ARF in primary breast cancer and analysis of inactivation mechanisms. J Pathol. 2003; 199:289–297.
14. Brenner AJ, Aldaz CM. Chromosome 9p allelic loss and p16/CDKN2 in breast cancer and evidence of p16 inactivation in immortal breast epithelial cells. Cancer Res. 1995; 55:2892–2895.
15. Schwendel A, Richard F, Langreck H, Kaufmann O, Lage H, Winzer KJ, Petersen I, Dietel M. Chromosome alterations in breast carcinomas: frequent involvement of DNA losses including chromosomes 4q and 21q. Br J Canc. 1998; 78:806–811.
16. Loo LW, Ton C, Wang YW, Grove DI, Bouzek H, Vartanian N, Lin MG, Yuan X, Lawton TL, Daling JR, Malone KE, Li CI, Hsu L, et al. Differential patterns of allelic loss in estrogen receptor-positive infiltrating lobular and ductal breast cancer. Gene Chromosomes Cancer. 2008; 47:1049–1066.
17. Cairns P, Polascik TJ, Eby Y, Tokino K, Califano J, Merlo A, Mao L, Herath J, Jenkins R, Westra W, et al. Frequency of homozygous deletion at p16/CDKN2 in primary human tumours. Nat Gen. 1995; 11:210–212.
18. Ruiz C, Seibt S, Al Kuraya K, Siraj AK, Mirlacher M, Schraml P, Maurer R, Spichtin H, Torhorst J, Popovska S, Simon R, Sauter G. Tissue microarrays for comparing molecular features with proliferation activity in breast cancer. Int J Cancer. 2006; 118:2190–2194.
19. Stefansson OA, Jonasson JG, Olafsdottir K, Bjarnason H, Th Johannsson O, Bodvarsdottir SK, Valgeirsdottir S, Eyfjord JE. Genomic and phenotypic analysis of BRCA2 mutated breast cancers reveals co-occurring changes linked to progression. Breast Cancer Res. 2011; 13:R95.
20. Bohn OL, Fuertes-Camilo M, Navarro L, Saldivar J, Sanchez-Sosa S. p16INK4a expression in basal-like breast carcinoma. Int J Clin Exp Patho. 2010; 3:600–607.
21. Geradts J, Kratzke RA, Niehans GA, Lincoln CE. Immunohistochemical detection of the cyclin-dependent kinase inhibitor 2/multiple tumor suppressor gene 1 (CDKN2/MTS1) product p16INK4A in archival human solid tumors: correlation with retinoblastoma protein expression. Cancer Res. 1995; 55:6006–6011.
22. Herschkowitz JI, He X, Fan C, Perou CM. The functional loss of the retinoblastoma tumour suppressor is a common event in basal-like and luminal B breast carcinomas. Breast Cancer Res. 2008; 10:R75.
23. Geradts J, Wilson PA. High frequency of aberrant p16(INK4A) expression in human breast cancer. Am J Pathol 1996; 149:15–20.
24. Hennigs A, Riedel F, Gondos A, Sinn P, SchirmacherP, Marme F, Jager D, Kauczor HU, Stieber A, Lindel K, Debus J, Golatta M, Schutz F, et al. Prognosis of breast cancer molecular subtypes in routine clinical care: A large prospective cohort study. BMC cancer. 2016; 16:734.
25. Ruas M, Peters G. The p16INK4a/CDKN2A tumor suppressor and its relatives. Biochim Biophys acta. 1998; 1378:F115–177.
26. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012; 2:401–404.
27. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, Cerami E, Sander C, Schultz N. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013; 6:pl1.
28. Kurian AW, Hare EE, Mills MA, Kingham KE, McPhersonL, Whittemore AS, McGuire V, Ladabaum U, Kobayashi Y, Lincoln SE, Cargill M, Ford JM. Clinical evaluation of a multiple-gene sequencing panel for hereditary cancer risk assessment. J Clin Oncol. 2014; 32:2001–2009.
29. Monnerat C, Chompret A, Kannengiesser C, Avril MF, Janin N, Spatz A, Guinebretiere JM, Marian C, Barrois M, Boitier F, Lenoir GM, Bressac-de Paillerets B. BRCA1, BRCA2, TP53, and CDKN2A germline mutations in patients with breast cancer and cutaneous melanoma. Fam Cancer. 2007; 6:453–461.
30. Debniak T, Cybulski C, Gorski B, Huzarski T, Byrski T, Gronwald J, Jakubowska A, Kowalska E, Oszurek O, Narod SA, Lubinski J. CDKN2A-positive breast cancers in young women from Poland. Breast Cancer Res Treat. 2007; 103:355–359.
31. Lubinski J, Korzen M, Gorski B, Cybulski C, Debniak T, Jakubowska A, Jaworska K, Wokolorczyk D, Medrek K, Matyjasik J, Huzarski T, Byrski T, Gronwald J, et al. Genetic contribution to all cancers: the first demonstration using the model of breast cancers from Poland stratified by age at diagnosis and tumour pathology. Breast Cancer Res Treat. 2009; 114:121–126.
32. Jong YJ, Li LH, Tsou MH, Chen YJ, Cheng SH, Wang-Wuu S, Tsai SF, Chen CM, Huang AT, Hsu MT, Lin CH. Chromosomal comparative genomic hybridization abnormalities in early- and late-onset human breast cancers: correlation with disease progression and TP53 mutations. Cancer Genet Cytogenet. 2004; 148:55–65.
33. Seute A, Sinn HP, Schlenk RF, Emig R, Wallwiener D, Grischke EM, Hohaus S, Dohner H, Haas R, Bentz M. Clinical relevance of genomic aberrations in homogeneously treated high-risk stage II/III breast cancer patients. Int J Cancer. 2001; 93:80–84.
34. Climent J, Martinez-Climent JA, Blesa D, Garcia-Barchino MJ, Saez R, Sanchez-Izquierdo D, Azagra P, Lluch A, Garcia-Conde J. Genomic loss of 18p predicts an adverse clinical outcome in patients with high-risk breast cancer. Clin Cancer Res. 2002; 8:3863–3869.
35. Erickson S, Sangfelt O, Heyman M, Castro J, Einhorn S, Grander D. Involvement of the Ink4 proteins p16 and p15 in T-lymphocyte senescence. Oncogene. 1998; 17:595–602.
36. Sinha S, Chunder N, Mukherjee N, Alam N, Roy A, Roychoudhury S, Kumar Panda C. Frequent deletion and methylation in SH3GL2 and CDKN2A loci are associated with early- and late-onset breast carcinoma. Ann Surg Oncol. 2008; 15:1070–1080.
37. Ortiz B, White JR, Wu WH, Chan TA. Deletion of Ptprd and Cdkn2a cooperate to accelerate tumorigenesis. Oncotarget. 2014; 5:6976–6982. doi: 10.18632/oncotarget.2106.
38. Kang JU, Koo SH, Kwon KC, Park JW. Frequent silence of chromosome 9p, homozygous DOCK8, DMRT1 and DMRT3 deletion at 9p24.3 in squamous cell carcinoma of the lung. Int J Oncol. 2010; 37:327–335.
39. Xue W, Kitzing T, Roessler S, Zuber J, Krasnitz A, Schultz N, Revill K, Weissmueller S, Rappaport AR, Simon J, Zhang J, Luo W, Hicks J, et al. A cluster of cooperating tumor-suppressor gene candidates in chromosomal deletions. Proc Natl Acad Sci USA. 2012; 109:8212–8217.
40. Elston CW, Ellis IO. Pathological prognostic factors in breast cancer. I. The value of histological grade in breast cancer: experience from a large study with long-term follow-up. Histopathology. 1991; 19:403–410.
41. Al Kuraya K, Simon R, Sauter G. Tissue microarrays for high-throughput molecular pathology. Ann Saudi Med. 2004; 24:169–174.
42. Krohn A, Diedler T, Burkhardt L, Mayer PS, De Silva C, Meyer-Kornblum M, Kotschau D, Tennstedt P, Huang J, Gerhauser C, Mader M, Kurtz S, Sirma H, et al. Genomic deletion of PTEN is associated with tumor progression and early PSA recurrence in ERG fusion-positive and fusion-negative prostate cancer. Am J Pathol. 2012; 181:401–412.
43. Simon R, Mirlacher M, Sauter G. Immunohistochemical analysis of tissue microarrays. Methods Mol Biol. 2010; 664:113–126.
44. Minner S, Wittmer C, Graefen M, Salomon G, Steuber T, Haese A, Huland H, Bokemeyer C, Yekebas E, Dierlamm J, Balabanov S, Kilic E, Wilczak W, et al. High level PSMA expression is associated with early PSA recurrence in surgically treated prostate cancer. Prostate. 2011; 71:281–288.