
INTRODUCTION
Of all the malignant bone tumors, chondrosarcoma occurs as an in situ secondary incidence [1]. Currently, the treatment of chondrosarcoma involves the use of chemotherapy or radiation therapy, but its management is a complicated challenge because of its unresponsive nature [2]. Clinically, chondrosarcoma possesses a poor prognosis which lack an effective adjuvant treatment so that surgical resection is the major therapy for this mesenchymal malignancy [3]. Therefore, exploring a novel and rare side-effect strategy might be critical for the treatment of chondrosarcoma.
Reactive oxygen species (ROS) are originated with the metabolism of oxygen exhaustion. Aerobic respiration produces adenosine triphosphate (ATP) and other harmful superoxide anion radical (O2−), which can then form other ROS such as highly reactive hydroxyl radicals and hydrogen peroxide (H2O2) [4, 5]. As excess ROS or antioxidant depletion leads to disruption of balance from aerobic respiration, oxidative stress would occur. Accumulating evidence demonstrates that chemotherapy may be selectively toxic to tumor cells owing to increasing stressed cells over limitation and oxidant stress [6, 7]. In addition, activation of the mitochondria-dependent apoptosis signaling triggered ROS signaling through the apoptotic signaling proteins, such as BH3 interacting-domain death agonist (Bid), B-cell lymphoma-extra large (Bcl-XL), B cell lymphoma-2 homologous antagonist/killer (Bak), B cell lymphoma-2 associated-X protein (Bax), or B cell lymphoma-2 (Bcl-2) with permeabilization and cell death of mitochondrial membrane [4, 8]. Nevertheless, involvement of ROS and mitochondrial dependent signalings in chondrosarcoma needs to be further clarified.
As central organelle, the endoplasmic reticulum (ER) is responsible for lipid synthesis and protein folding, modification, and maturation. Due to the broken ER function, ER stress derives from various toxic distractions including protein misfolding, hypoxia, and Ca2+ overload [9–11]. Accumulating evidence indicates that ER stress plays an important role in the apoptosis regulation and connected with calcium-dependent signaling pathways and unfolded protein response [12, 13]. In addition, glucose-regulated proteins (GRPs), the primary glycoproteins, play a critical role in the ER including GRP78 and GRP94 against oxidative injury and regulate ribozyme approaches [14–16]. The induction of GRPs for antiapoptotic function may cause drug resistance and cancer development [17, 18].
Benzofuran appears structurally like natural products and functions as human protein kinase inhibitors [19]. Recently, benzofuran has been reported the role of antiproliferative activity in tumors especially against p53-independent malignant tumors [20]. The roles of benzofuran derivative in chondrosarcoma remain largely unknown. Therefore, in this study we synthesized a brand-new benzofuran derivative, 2-amino-3-(2-chlorophenyl)-6-(4-dimethylaminophenyl)benzofuran-4-yl acetate (ACDB), and evaluated the antitumor role of ACDB in response to human chondrosarcoma cells. We attempt to investigate ACDB antitumor activity and explore the mechanism by which it induces chondrosarcoma apoptosis.
RESULTS
ACDB enhanced human chondrosarcoma cells apoptosis
For the cytotoxic investigation of ACDB, we first examined its effects on the survival between human chondrosarcoma cell lines and normal chondrocytes with the MTT assay. Both chondrosarcoma cell lines and normal chondrocytes were treated with ACDB (3, 10, 30 μM) that triggered cell apoptosis of JJ012 and SW1353 cell lines with half-maximal inhibitory concentration (IC50) values of 4.9 and 19.1 μM, respectively (Figure 1B). The role of ACDB in anticancer activities was further performed using clonogenic assays (Figure 1C), which is connected with prior in vivo tumorigenicity assays in nude mice [27]. While the JJ012 cells formed clones in the untreated control wells (Figure 1D), treatment with ACDB (3, 10, 30 μM) induced a dose-dependent inhibition of clonogenicity, and the quantitative results containing normal condrocytes are shown in Figure 1D. Interestingly, ACDB led to the cell viability of normal chondrocytes without impairs in response to MTT and clonogenic assays (Figure 1B and 1D, dashed line). Furthermore, JJ012 cells treated with ACDB significantly induced the condensation of chromatin as exhibited by the DAPI staining (Figure 1E).
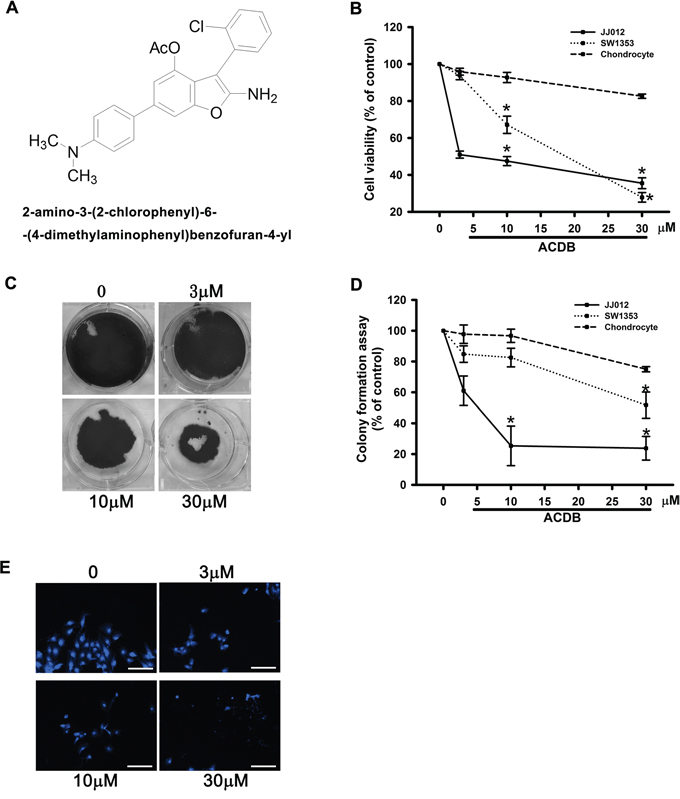
Figure 1: The effect of ACDB on cell viability and formation and colony assay in human chondrosarcoma cells. A. Novel structure of benzofuran derivative, 2-amino-3-(2-chlorophenyl)-6-(4-dimethylaminophenyl)benzofuran-4-yl acetate; ACDB. B. Cells were incubated with various concentrations of ACDB for 48 h, and the cell viability was examined by MTT assay (n=6). C. For the colony-forming assay, the clonogenic assay was performed as described in Materials and Methods section. D. The quantitative data of colony-forming assay. E. JJ012 cells were incubated with ACDB for 48 h, and stained with DAPI (scale bar = 5 μm). Results are expressed as the means ± SEM of four independent experiments. * P < 0.05 as compared with control group.
Next, we investigated if ACDB resulted in cell apoptosis of chondrosarcoma cells via various apoptotic assays. Treating cells with ACDB enhanced a dose-dependent production of cell death, causing sub G1 phase ascent of the cell cycle (Figure 2A). Thus, ACDB failed to affect sub G1 phase on normal chondrocytes (Supplementary Figure S1). To detect the cell death by using double-stained Annexin V/PI, a high proportion of Annexin V+ labeling was detected in the ACDB-treated cells compared to the control group (Figure 2B); quantitative data of the sub G1 phase and Annexin V/PI assay are shown in the Figure 2C and 2D, suggesting that ACDB probably induced late cell death during programmed cell death. In addition, those stimulated with ACDB exhibited a high increase TUNEL fluorescent intensity compared to JJ012 cells without treatment (Figure 2E). Then, we examined the DNA fragmentation, which provides information about the type of cell death and the pathways activated in the dying cells, and is a hallmark of apoptosis in biochemistry [21, 22]. As shown in Figure 2F, ACDB promoted DNA fragmentation in a dose-dependent increase (~2.2 to 38 fold) in JJ012 cells. Above results demonstrate that ACDB largely enhanced chondrosarcoma cell apoptosis through various different levels.
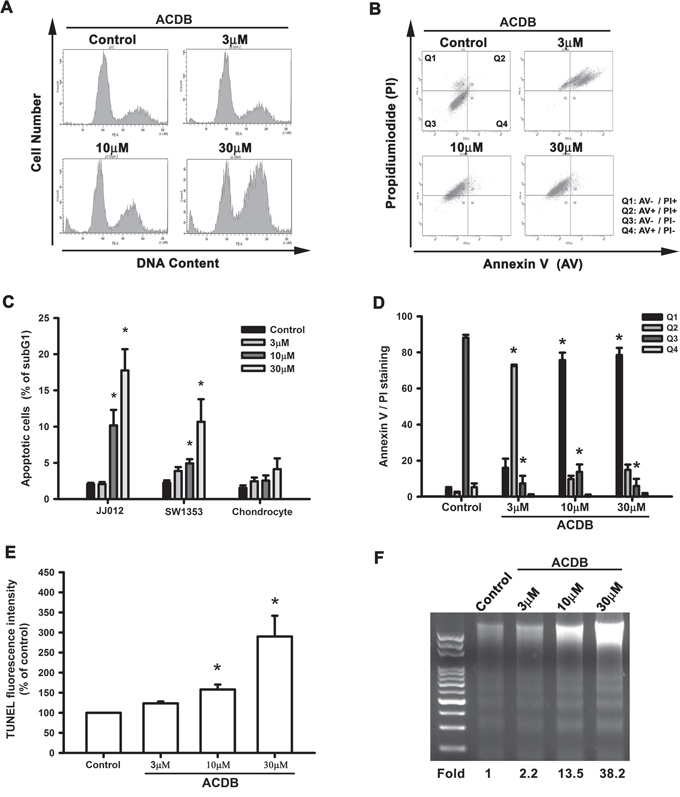
Figure 2: ACDB induces apoptosis in human chondrosarcoma cells. A and C. Cells were treated with vehicle or ACDB for 48 h. The cell cycle analysis (PI staining) was examined and quantified by flow cytometry. B and D. JJ012 cells were treated with vehicle or ACDB for 48 h. The percentage of apoptotic cells was analyzed by flow cytometry of Annexin V/PI double staining, and the results were quantified. E. Cells were treated with vehicle or ACDB for 48 h. The TUNEL positive cells were examined by flow cytometry (n = 4). F. For measuring apoptosis, the DNA fragmentation assay was performed as described in Materials and Methods section. Results are expressed as the means ± SEM of four independent experiments. * P < 0.05 as compared with control group.
Both reactive oxygen species and mitochondrial dysfunction contribute to cell apoptosis of ACDB in chondrosarcoma cells
ROS generation has been reported to play a crucial role of various anticancer agents in the proapoptotic activities [23, 24]. Therefore, we continued to examine whether ROS accumulation is involved in the ACDB-increased cell death. After treatment with ACDB, the JJ012 cells showed increased intracellular O2− and H2O2 levels, detected using the DHE- and H2DCFDA-based FACS assays, respectively (Figure 3A). Next, we further continued to explore the mitochondrial dysfunction by using a specific membrane potential dye. As shown in Figure 3B, mitochondria membrane potential was significantly decreased in a concentration-dependent trend after ACDB exposure to chondrosarcoma cells. The expression of the mitochondrial membrane-dependent family proteins was measured. Once mitochondrial dysfunction occurred, ACDB increased protein levels of Bak (12.9 fold), Bid (4.23 fold) and Bax (1.92 fold) and decrease Bcl-XL (0.24 fold) and Bcl-2 (0.45 fold) protein expression in response to chondrosarcoma cell apoptosis (Figure 3C). In numerous abnormal situations, the production of ROS derived from a critical enzyme, nicotinamide adenine dinucleotide phosphate (NADPH) oxidase [25]. Pretreatment of cells with DPI, catalase, or NAC abrogated the ACDB-induced ROS production and cell apoptosis (Figure 3D), suggesting that ACDB-induced cell apoptosis involves ROS release in chondrosarcoma. Above results indicate that ACDB enhanced chondrosarcoma cells apoptosis via ROS and mitochondrial dysfunction.
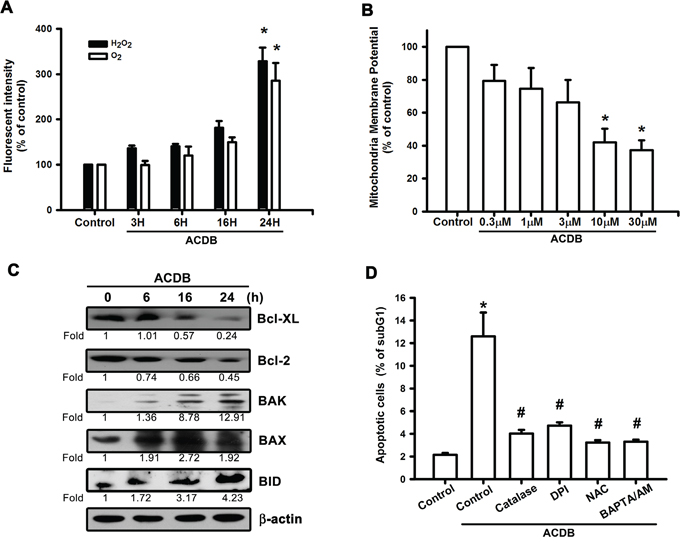
Figure 3: ACDB induces ROS production and mitochondrial dysfunction in human chondrosarcoma cells. A. JJ012 cells were incubated with ACDB for different time intervals, the production of O2− and H2O2 were examined by flow cytometry. B. JJ012 cells were incubated with various concentration of ACDB for 48 h, the mitochondrial membrane potential was examined by flow cytometry. C. JJ012 cells were incubated with ACDB for different time intervals, the Bax, Bak, BID, Bcl-2 and Bcl-XL expression were examined by Western blot analysis. D. JJ012 cells were pre-treated for 30 min with catalase (10,000 U/ml), NAC (4 μM), DPI (10 μM) or BAPTA-AM (10 μM) followed by stimulation with ACDB for 48 h, the percentage of apoptotic cells was analyzed by flow cytometry. Results are expressed as the means ± SEM of four independent experiments. *P < 0.05 as compared with control group; #P < 0.05 compared with the ACDB-treated group.
ACDB caused calcium accumulation and ER stress
A previous report demonstrated that mitochondrial dysfunction may lead to pancreatic β-cell apoptosis through ER stress and GRP regulation [26]. For the involvement of GRP in chondrosarcoma cells apoptosis, the levels of the GRP78 and GRP94 were investigated that ACDB induced their mRNA expression (Figure 4A) and promoter-luciferase activity (Figure 4B). The accumulation of ER stress originated from the depletion of luminal ER calcium stores [27], and Ca2+ mobilization was investigated in response to ACDB-induced cell apoptosis. After treated with ACDB in JJ012 cells, the Ca2+ level was significantly increased compared to control group (Figure 4C), and the BAPTA-AM, the Ca2+ chelator, decreased the ACDB-induced cell apoptosis (Figure 3D), suggesting that Ca2+ production is involved in the ACDB-promoted chondrosarcoma cell apoptosis.
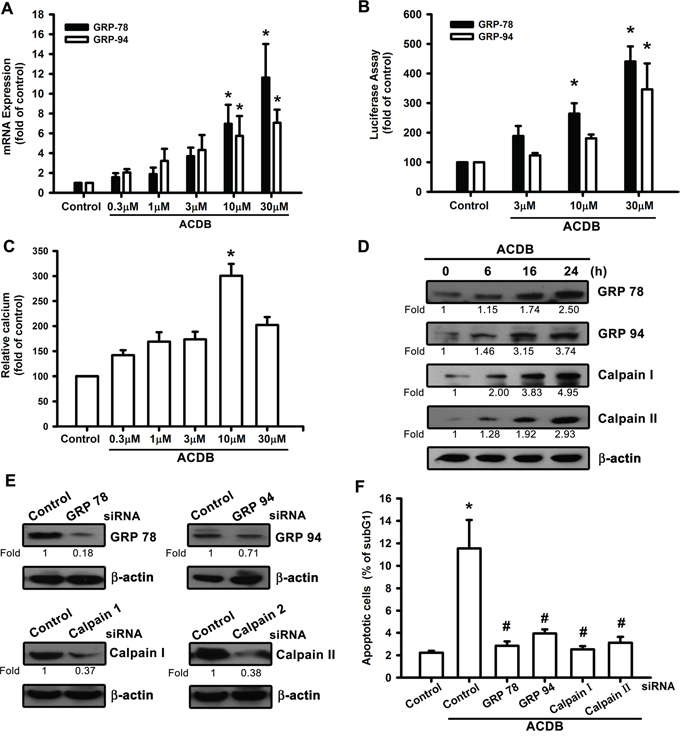
Figure 4: ER stress-related proteins activation are involved in ACDB-mediated cell apoptosis in human chondrosarcoma cells. A and B. JJ012 cells were incubated with ACDB, and mRNA expression and GRP promoter were examined by qPCR and luciferase assay, respectively. C. JJ012 cells were incubated with ACDB for different concentration. The Ca2+ flux was examined by flow cytometry. D. JJ012 cells were incubated with ACDB for different time intervals, the GRP78, GRP94, calpain I and calpain II expression were examined by Western blot analysis. E. Cells were transfected with siRNA against GRP78, GRP94, calpain I, calpain II or control for 24 h before incubated with ACDB. GRP78, GRP94, calpain I and calpain II expression was examined by Western blot analysis. F. Cells were transfected with siRNA against GRP78, GRP94, calpain I, calpain II, or control for 24 h and then stimulated with ACDB for 48 h, and the percentage of apoptotic cells was analyzed by flow cytometry of PI-stained cells. Results are expressed as the means ± SEM of four independent experiments. *P < 0.05 as compared with control group; #P < 0.05 compared with the ACDB-treated group.
In addition, calpains are critical enzymes in the intracellular signaling cascades and potential mediators of calcium-induced apoptosis [28]. ACDB markedly increased protein levels of GRPs and calpains in a time-dependent manner (~2.5 to 4.95 fold) (Figure 4D). To further substantiate the functional roles of GRPs and calpains in ACDB-induced apoptotic cell death, we examined its effects on siRNA against GRP-78, GRP-94, calpain I, and calpain II. Transfection of chondrosarcoma cells for 12-16 h with these siRNAs not only reduced the protein expression (~0.71 to 0.18 fold) (Figure 4E), but it also antagonized the ACDB-induced cell apoptosis (Figure 4F). Therefore, above results indicate that ACDB-induced apoptosis results in ER stress including GRP and calpain activation through mitochondrial dysfunction.
ACDB increased caspase-dependent signaling in chondrosarcoma cells
A previous apoptosis-associated study reported that caspase activation derived from ubiquitous calpains within ER stress [29]. We continued to clarify the apoptotic function of ACDB in chondrosarcoma that caspase-3/7 activity and protein expression (~1.26 to 7.45 fold) were elevated after ACDB exposure in JJ012 cells (Figure 5A and 5B, upper panel). Furthermore, treatment with ACDB increased the expression of the upstream protein caspase-9 (10.89 fold) and the cytosolic fraction of cytochrome C (2.04 fold) in JJ012 cells (Figure 5B) as well as cleaved-PARP expression (5.40 fold) (Figure 5B). On the other hand, we continued to determine the involvement of caspase with the specific caspase-3 inhibitor, z-DEVD-FMK, and caspase-9 inhibitor, z-LEHD-FMK [30]. JJ012 cells pretreated with z-DEVD-FMK and z-LEHD-FMK largely diminished ACDB-enhanced cell apoptosis, as shown by the cell cycle assay (Figure 5C). Therefore, our data suggest that caspase-3/7, caspase-9, and cleaved-PARP activation is involved in the ACDB-mediated human chondrosarcoma cell apoptosis.
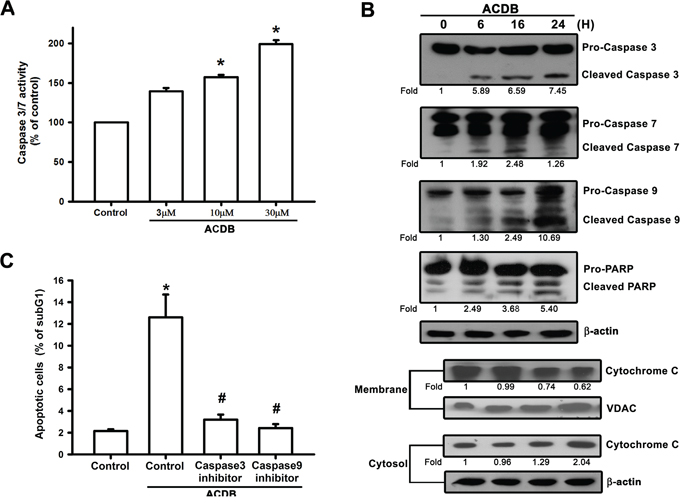
Figure 5: ACDB induces the activation of caspase in human chondrosarcoma cells. A. JJ012 cells were incubated with ACDB for 24 h. Caspase-3/7 activities were examined by caspase activity kit. B. JJ012 cells were incubated with ACDB for different time intervals. Levels of PARP, cytochrome C, caspase-3, caspase-7 and caspase-9 expressions were examined by Western blot analysis. Results are expressed as the means ± SEM of four independent experiments. C. Cells were pretreated for 30 min with z-DEVD-FMK (caspase 3 inhibitor) or z-LEHD-FMK (caspase 9 inhibitor), followed by stimulation with ACDB for 24 h. The percentage of apoptotic cells was the analyzed by flow cytometry of PI-stained cells. *P < 0.05 as compared with control group; #P < 0.05 compared with the ACDB-treated group.
ACDB retarded tumor growth in a murine xenograft model
According to the ACDB-enhanced apoptotic effect in vitro, we continued to exhibit the antitumor activities of ACDB in vivo. We performed mouse xenograft model in vivo followed by treatment with ACDB for 20 days. Mice were sacrificed at the end of the treatment and the tumors were collected.
ACDB significantly diminished xenograft tumor size and weights (Figure 6A and 6B) and tumor volume and body weights were estimated once two days (Figure 6C and 6D) compared to the control group, indicating that ACDB produced a dose-dependent inhibition of tumor growth without toxicity. Furthermore, the ex vivo tumor sections analysis showed that the protein levels of Bax, Bcl-2, GRP-94, and calpain I increased (~1.2 to 3.01 fold) or decreased (~0.55 to 0.04 fold) significantly in the ACDB-stimulated group than the control group (Figure 6E). In addition, an increase in TUNEL+ cells was observed in tumors of the ACDB-treated mice compared with that of tumors from the vehicle-treated mice (Figure 6F). Above results indicated that ACDB treatment strongly suppressed JJ012 xenograft tumor growth in vivo through regulation Bax, Bcl-2, GRP-94, and calpain I signaling (Supplementary Figure S2).
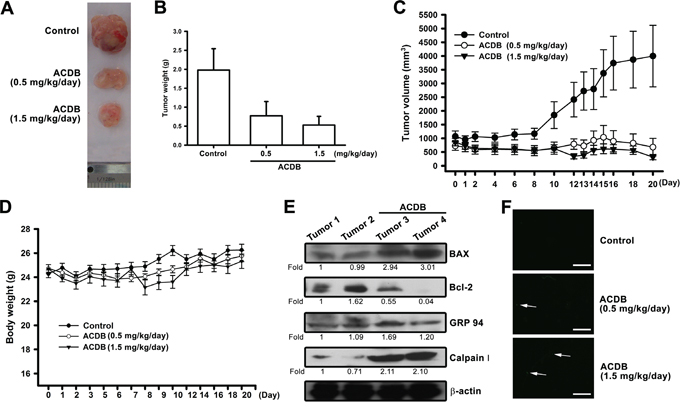
Figure 6: ACDB inhibits tumor growth in nude mice. A and B. Mice were injected subcutaneously with JJ012 tumor cells. After the tumors reached 1000 mm3 in size, ACDB (0.5 or 1.5 mg/kg) or vehicle was administered daily for 3 weeks. C and D. Mean tumor volume and body weight were measured at the indicated number of days after implantation (n=10). E. Western blot analysis determined protein levels of BAX, Bcl-2, GRP94, and calpain I from the tumors with and without ACDB treatment. F. TUNEL assay in tissues from chondrosarcoma cells xenografts. Apoptosis happened only in approximately 1% of tumor tissue in the control group, while ACDB-treated tumors show marked green staining of fragmented nuclei, indicative of apoptosis (scale bar = 50 μm).
DISCUSSION
In contrast to other mesenchymal malignancies such as osteosarcoma and Ewing’s sarcoma that have demonstrated a thrilling increase in the long-term survival of patients with the approach of effective chemotherapy, chondrosarcoma continues to possess a poor prognosis due to lack of promising adjuvant therapies [31]. Therefore, to explore a novel therapeutic agent targeting the malignant survival of chondrosarcoma would be crucial for ameliorating the prognosis of patients. Benzofuran derivative has recently been reported to show anticancer activity in breast and cervical cancer [32, 33]. However, the antitumor activity of benzofuran derivative in chondrosarcoma cells are mostly unknown.
Therefore, in this study, we synthesized a new 2-amino-3-(2-chlorophenyl)-6-(4-dimethylaminophenyl)benzofuran-4-yl acetate, ACDB as an attractive lead compound with potential anticancer activity against human chondrosarcoma cells and good pharmacological properties. We first discovered that this benzofuran derivative induced a significant cell death in JJ012 than SW1353 cell line. Although both JJ012 and SW1353 are both grade II chondrosarcoma cells, however, the sensitivity from ACDB treatment in these two cell lines requires to be further discussed in the future. The chondrocytes appeared to have a greater resistance to apoptosis-inducing compounds than the other cell lines, and ACDB showed greater anticancer potential against human chondrosarcoma cells. Moreover, the metabolic rates of the primary chondrocytes and chondrosarcoma are different, which may explain their differential sensitivities to ACDB treatment.
It has been reported that several anticancer agents induced an enhancement of oxidative stress and connected with the apoptotic effects [34, 35]. ROS leads to cell apoptosis through various mechanism including initiation of mitochondrial permeability transition pore opening, the release of pro-apoptotic factors, and activation of caspase cascade [36]. The O2− radical has also been shown to regulate Bcl-2-related protein expression, and the inhibition of O2− by ρ-benzoquinone, the O2− scavenger, prevents tumor apoptosis [37]. In this study, we found that ACDB triggered oxidative stress via activation of the O2− and H2O2 production, and the role of NADPH oxidase was further investigated in ACDB-induced cell apoptosis. We used catalase (H2O2 scavenging enzyme), NAC (a direct scavenger of ROS), and DPI (an NADPH oxidase inhibitor) to determine ACDB-increased cell apoptosis, indicating that ROS accumulation contributes to ACDB-induced cell death of human chondrosarcoma.
The mitochondria-dependent apoptosis has been described as a pivot in the downstream signaling cascade of ROS during apoptotic process [38–40]. As our results of mitochondrial dysfunction did not occur until the cell was treated with ACDB for 48 h. Thus, the relationship between ROS and mitochondrial dysfunction needs to be further analysis in the future. Some important events have been noted in apoptosis involving mitochondrial dysfunction [41]. Cell apoptosis triggered various function of Bcl-2 family members that reveals the permeabilization of mitochondrial outer-membrane and relieves subsequently cytochrome C into the mitochondrial intermembrane space and cytosol [42]. In this study, we demonstrated that ACDB reduced the mitochondrial membrane potential and increased the release of cytochrome C. Nevertheless, the balance between anti- and pro-apoptotic proteins, which regulates the mitochondria-dependent apoptosis mediated by the Bcl-2 family members modulates cell life-and-death decisions [43]. We discovered that ACDB treatment significantly increased Bax, Bak, and Bid expression while it decreased that of Bcl-2 and Bcl-XL, suggesting that variations in the ratio of pro- and anti-apoptotic Bcl-2 family proteins might contribute to the apoptotic activity we observed. In agreement with these observations, we noted that the mitochondrial dysfunction might be engaged in the ACDB-increased apoptosis of human chondrosarcoma cells.
Calcium has been reported to play an important role in activating apoptosis [44], and release of calcium stores from the ER can activate caspase and contribute to apoptosis [45]. Under a variety of stressful conditions, the accumulation of unfolded or misfolded proteins in the ER leads to the onset of ER stress [46]. Our study found that ACDB-mediated cell apoptosis not only enhances cytosolic calcium levels but also i by the chelator, BAPTA-AM, in human chondrosarcoma cells. This suggests that ACDB induced apoptotic cell apoptosis via the activation of ER stress in human chondrosarcoma cells.
The upregulation of GRP78, which is another major indicator of ER stress, is believed to increase the buffering capacity of the body against stressful insults initiated by the ER [47]. Moreover, the roles of GRP94 in protein folding, calcium binding, targeting to endoplasmic-reticulum-associated protein degradation, and protective functions have been understood and observed in resistance to radiation in cervical cancer [48, 49]. Notably, we demonstrated in this study that ACDB increased GRP protein and mRNA expression and promoter activity, which was revealed by the GRP siRNA-induced antagonism of ACDB-mediated cell apoptosis potentiation. Similar functions have been reported with the observation that mitochondrial dysfunction induces ER stress and GRP94 protein expression in apoptosis of pancreatic β-cells [26, 50], and curcumin induces apoptosis of human lung carcinoma cells through GRP78 upregulation [51]. Therefore, these results suggest that ACDB increased GRP94 expression through mitochondrial dysfunction, and GRP78 transcription activity, which may play a pro-apoptotic role in the observed apoptosis.
Two kinds of cysteine proteases containing calpains and caspases regulate pathological cell apoptosis [52]. Although both caspase and calpain share their death-related substrates to regulate cells apoptosis [51], the effects of the ACDB on these two proteases families remain unclear. Our results indicate that calpains, caspase-3/7, and caspase-9 activation is involved in the ACDB-mediated human chondrosarcoma cell apoptosis. Collectively, above findings indicate that ACDB induced apoptotic cell death through ER stress and caspase-dependent pathways in human chondrosarcoma cells.
In conclusion, our findings demonstrate that the novel benzofuran derivative, ACDB, triggered the apoptosis of human chondrosarcoma cells in vitro and in vivo. Furthermore, ACDB-mediated chondrosarcoma apoptosis through the ROS release led to mitochondrial dysfunction and likely included caspase-regulated mechanisms. ACDB also regulated ER stress, GPR78 activation, and Ca2+ release, which subsequently triggered calpains, caspase cascade, ultimately leading to apoptosis. Although we have previously published a similar pathway in the treatment of chondrosarcoma using another compound, trichodermin [41], the effects of these two on chondrosarcoma need to be further analyzed in the future. Our proposed mechanisms of this antitumor activity remain to be further investigated, and the molecular basis of these observed effects may provide help during the development of novel therapeutic strategies in chondrosarcoma.
MATERIALS AND METHODS
Materials
ACDB (Figure 1A) was composed of benzofuran and other chemicals at the Graduate Institute of Pharmaceutical Chemistry, China Medical University (Taichung, Taiwan). α-minimum essential medium (MEM), Dulbecco’s modified Eagle’s medium (DMEM), fetal bovine serum (FBS) and other cell culture reagents were obtained from Gibco-BRL Life technologies (Grand Island, NY, USA). Rabbit polyclonal antibodies specific for poly adenosine diphosphate (ADP) ribose polymerase (PARP), GRP78, GRP94, calpain I, calpain II, Bid, Bcl-XL, Bak, Bax, Bcl-2, caspase-9, caspase-3, caspase-7, and anti- rabbit and anti-mouse IgG-conjugated peroxidase were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Mouse monoclonal antibodies specific for both cytochrome C and mitochondria control protein, voltage-dependent anion channels (VDAC), were obtained from Abcam (Cambridge, MA, USA). The ON-TARGET smart pool small interfering RNA (siRNA) against GRP78, GRP94, calpain I, calpain II, and control were obtained from Dharmacon (Lafayette, CO, USA). The Trizol, Lipofectamine 2000, and MMLV RT kit were purchased from Invitrogen (Carlsbad, CA, USA). 4',6-diamidino-2-phenylindole (DAPI), 3-[4, 5-dimethylthiazol-2-yl]-2, 5-diphenyltetrazolium bromide (MTT), 5,5’,6,6’-tetrachloro-1,1’,3,3’-tetraethyl benzimidazolylcarbocyanine iodide (JC-1), Fluo-3-pentaacetoxyMethyl ester (Fluo 3/AM), dihydroethidium (DHE), and dichlorodihydrofluorescein diacetate (H2DCFDA), 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrakis(acetoxymethyl ester) (BAPTA-AM), catalase, diphenyleneiodonium (DPI), catalase or N-acetylcysteine (NAC), and other chemicals were purchased from Sigma-Aldrich (St. Louis, MO, USA).
Synthesized method of 2-amino-3-(2-chlorophenyl)-6-(4-dimethylaminophenyl)benzofuran-4-yl acetate (ACDB)
To the stirring solution of 5-[4-(dimethylamino)phenyl]cyclohexane-1,3-dione (0.346 g, 1.5 mmole) and 2-chloro-beta-nitrostyrene (0.187 g, 1 mmole) in tetrahydrofuran (50 mL) was added triethylamine (0.028 ml, 0.2 mmole) at room temperature for 9 h, and then added additional triethylamine (0.28 ml, 2 mmole), acetic anhydride (0.189 ml, 2 mmole) and dimethylaminopyridine (0.024 g, 0.2 mmole) for another 18 h. After the reacted mixture was concentrated in vaccum, the residue was subjected to column chromatography on silica gel with eluent of n-hexane:CH2Cl2 = 1:4 to afford the Rf = 0.23 fraction. The fraction was concentrated and recrystallization from acetonitril to obtain the pure purple-needle crystal ACDB (0.094 g, 23%). mp 201.1-201.7 °C; 1H NMR (500 MHz, DMSO-d6) δ 7.53-7.55 (m, 1 H, ArH), 7.48 (d, J = 8.9 Hz, 2 H, 2’’, 6’’-H), 7.42 (d, J = 1.3 Hz, 1 H, 5-H), 7.34-7.40 (m, 2 H, ArH), 7.30-7.33 (m, 1 H, ArH), 6.99 (d, J = 1.3 Hz, 1 H, 7-H), 6.77 (d, J = 8.9 Hz, 2 H, 3’’, 5’’-H), 6.34 (brs, 2 H, NH2), 2.92 (s, 6 H, 2×CH3), 1.54 (s, 3 H, CH3); 13C NMR (100 MHz, DMSO-d6) δ 168.9, 156.9, 151.2, 149.9, 141.0, 135.5, 133.8, 132.5, 131.5, 129.5, 129.0, 128.1, 127.2, 127.2, 127.2, 122.4, 114.4, 113.2, 113.2, 104.5, 86.2, 40.5, 40.5, 20.0; EIMS m/z 420.2 (M+). HPLC purity 99.40% (λmax 324 nm).
Cell culture
The human JJ012 cell line was kindly offered by Dr. Sean P Scully [53]. The human SW1353 cell line was purchased from the Bioresource Collection and Research Center in Taiwan. The cell was maintained in a humidified 37 °C incubator with α- MEM/DMEM and 10% FBS containing 100 μg/ml streptomycin and 100 units/ml penicillin. Human primary chondrocytes were cultured as described previously [54]. Primary cultures of human chondrocytes were isolated from articular cartilage as previously described [41]. The cells were grown in plastic cell culture dishes in 95% air–5% CO2 with DMEM that was supplemented with 20 mM 4-(2-hydroxyethyl)-1-piperazine-ethanesulphonic acid (HEPES), 10% FBS, 2 mM-glutamine, penicillin (100 U/mL), and streptomycin (100 mg/mL).
MTT assay
MTT assay was used to determine cell viability as described previously [41]. After treating with ACDB (3, 10, 30 μM) for 48 h, cells were incubated with MTT, and then analyzed the absorbance at 550 nm.
DAPI staining
After treating with ACDB (3, 10, 30 μM) for 48 h, cells were fixed in a 70% ethanol solution, and then stained with a DNA-binding fluorescent dye, DAPI. Nuclear morphology was observed under fluorescence microscopy.
Colony assay
For extended outcome of ACDB on chondrosarcoma, JJ012 cells (1,000/well) were stimulated with ACDB for 24 h. After formed colonies for 7 days, cells were marked with crystal violet and then dissolved in acetic acid to measure the absorbance at 550 nm.
DNA fraction
In DNA laddering assay, the procedure was described previously [55]. Briefly, treated JJ012 cells were extracted the DNA with a mixture of isoamyl alcohol, chloroform, and phenol (1:24:25). The isolated DNA was analyzed electrophoretically on a 2% agarose gel and visualized under ultraviolet transillumination.
Flow cytometric analysis
Apoptotic analysis was performed with flow cytometry through cell cycle and Annexin V-FITC kit as described previously [41]. JJ012 was treated with ACDB or vehicle in a dose-dependent concentrations and fixed in 70% chilled ethanol overnight. After washed with phosphate-citric acid buffer, cells were stained in the dark with propidium iodide (PI) buffer followed by fluorescence-activated cell sorting (FACS) and the Cellquest program (Becton Dickinson; Lincoln Park, NJ, USA). Double staining was measured according to Annexin V-FITC kit manufacturer (BD Biosciences, San Jose, CA).
Mitochondrial membrane potential detection
Mitochondrial membrane potential activity was detected by the fluorescent probe, JC-1, as described previously [41]. JJ012 was stimulated with ACDB in a dose-dependent concentrations followed by incubation with JC-1 (10 mg/ml) for 30 min at 37°C in the dark. Cells were then re-suspended in specific solution and analyzed by FACS and the Cellquest program (Becton Dickinson, USA).
Measurement of calcium concentration
Accumulating changes of calcium levels were measured as described previously [44]. Cells were plated with a density of 2x105 cells/well and treated with ACDB for the indicated concentrations. Cells were collected and re-suspended in Fluo 3/AM (3 mg/ml) for 30 min at 37°C in the dark followed by analyzed with FACS and the Cellquest program (Becton Dickinson, USA).
ROS assay with flow cytometry
Expression of intracellular O2− and H2O2 were analyzed spectrofluorimetrically by oxidation of specific probes: DHE and H2DCFDA, which used 488 nm wavelengths. JJ012 was seeded 5x105 cells/well in 6-well plates and treated with ACDB for specified time intervals. Cells were harvested, washed twice with chilled PBS, and stained with DHE (10 mM) or H2DCFDA (10mM) for 10 min in the dark. The fluorescent intensity of the samples were immediately measured by using the flow cytometry.
Immunoblot analysis
The cellular lysates were collected as described previously [56]. Sample were separated into SDS–PAGE electrophoresis followed by transferred to Immobilon polyvinyldifluoride membranes (Millipore; Billerica, MA, USA). After protein blocking, the blots were incubated with specific primary antibodies followed by specific horseradish peroxidase-conjugated secondary antibodies. The blots were enhanced with chemiluminescence and visualized by using Fujifilm LAS-3000 chemiluminescence detection system (Fujifilm; Tokyo, Japan).
Caspase activity assay
Caspase activity was determined according to caspase activity assay kit manufacturer (Abcam; Cambridge, MA, USA). Briefly, the cell lysates were collected and treated with peptide substrate, and then p-nitroaniline releasing from immunocomplexes was measured at 405 nm.
In vivo mouse xenograft model
3-5 week male nude mice (BALB/c nu/nu) were obtained from the National Laboratory Animal Center (Taipei, Taiwan) and maintained in specific pathogen-free conditions. All protocols and guidelines were approved by Animal Care Committee of China Medical Taiwan University. JJ012 1x107 cells in a volume of 160 μl were injected subcutaneously into the flanks of each mice. Once the tumors developed approximately 1000 mm3, mice were randomly divided into three groups (10 mice/group): the control group and the treatment groups, and treated with either different dose of ACDB or vehicle once daily for 20 days. The implanted tumor was measured three times a week. Tumor volume was calculated using the formula V=(LW 2)/2: where V, is the volume (mm3); L, the biggest diameter (mm); W, is the smallest diameter (mm).
ACDB-induced tumor apoptosis in vivo was detected by terminal deoxynucleotidyl transferase (TdT) deoxyuridine 5'-triphosphate (dUTP) nick-end labeling (TUNEL) assay as described previously [41]. TUNEL assay was measured according to the Apoptotic TUNEL assay kit (Trevigen, USA) manufacturer.
Statistics
All quantified results are given as the means ± SEM by three experiments at least. Statistical comparison of two groups was performed the Student’s t-test. Statistical comparisons of more than two groups were used one-way analysis of variance (ANOVA) with Bonferroni’s post-hoc test. In all cases, p < 0.05 was defined statistically significant.
ACKNOWLEDGMENTS
This work was supported by grants from the Ministry of Science and Technology of Taiwan (NSC 102-2632-B-039-001-MY3; MOST 103-2628-B-039-002-MY3) and China Medical University (CMU103-AISA-03). We thank Dr. Kazutoshi Mori for providing the GRP78 and GRP94 luciferase plasmids.
CONFLICTS OF INTEREST
All authors declare that none has any financial or personal relationships with other people or organizations that could inappropriately influence their work.
REFERENCES
1. Liau CT. (2010). Clinical oncology.
2. Su CM, Fong YC, Tang CH. An overview of current and future treatment options for chondrosarcoma. Expert Opinion on Orphan Drugs. 2014; 2:217-227.
3. Gelderblom H, Hogendoorn PC, Dijkstra SD, van Rijswijk CS, Krol AD, Taminiau AH, Bovee JV. The clinical approach towards chondrosarcoma. Oncologist. 2008; 13:320-329.
4. Feig DI, Reid TM, Loeb LA. Reactive oxygen species in tumorigenesis. Cancer Res. 1994; 54:1890s-1894s.
5. Schumacker PT. Reactive oxygen species in cancer cells: live by the sword, die by the sword. Cancer Cell. 2006; 10:175-176.
6. Ham YM, Lim JH, Na HK, Choi JS, Park BD, Yim H, Lee SK. Ginsenoside-Rh2-induced mitochondrial depolarization and apoptosis are associated with reactive oxygen species- and Ca2+-mediated c-Jun NH2-terminal kinase 1 activation in HeLa cells. J Pharmacol Exp Ther. 2006; 319:1276-1285.
7. Moungjaroen J, Nimmannit U, Callery PS, Wang L, Azad N, Lipipun V, Chanvorachote P, Rojanasakul Y. Reactive oxygen species mediate caspase activation and apoptosis induced by lipoic acid in human lung epithelial cancer cells through Bcl-2 down-regulation. J Pharmacol Exp Ther. 2006; 319:1062-1069.
8. Roos WP, Kaina B. DNA damage-induced cell death by apoptosis. Trends Mol Med. 2006; 12:440-450.
9. Yung HW, Korolchuk S, Tolkovsky AM, Charnock-Jones DS, Burton GJ. Endoplasmic reticulum stress exacerbates ischemia-reperfusion-induced apoptosis through attenuation of Akt protein synthesis in human choriocarcinoma cells. FASEB J. 2007; 21:872-884.
10. Abcouwer SF, Marjon PL, Loper RK, Vander Jagt DL. Response of VEGF expression to amino acid deprivation and inducers of endoplasmic reticulum stress. Invest Ophthalmol Vis Sci. 2002; 43:2791-2798.
11. Soboloff J, Berger SA. Sustained ER Ca2+ depletion suppresses protein synthesis and induces activation-enhanced cell death in mast cells. J Biol Chem. 2002; 277:13812-13820.
12. Feldman DE, Chauhan V, Koong AC. The unfolded protein response: a novel component of the hypoxic stress response in tumors. Mol Cancer Res. 2005; 3:597-605.
13. Moenner M, Pluquet O, Bouchecareilh M, Chevet E. Integrated endoplasmic reticulum stress responses in cancer. Cancer Res. 2007; 67:10631-10634.
14. Lee AS. The glucose-regulated proteins: stress induction and clinical applications. Trends Biochem Sci. 2001; 26:504-510.
15. Little E, Ramakrishnan M, Roy B, Gazit G, Lee AS. The glucose-regulated proteins (GRP78 and GRP94): functions, gene regulation, and applications. Crit Rev Eukaryot Gene Expr. 1994; 4:1-18.
16. Xu D, Cui S, Sun Y, Bao G, Li W, Liu W, Zhu X, Fan J, Wang Y, Cui Z. Overexpression of glucose-regulated protein 94 after spinal cord injury in rats. J Neurol Sci. 2011; 309:141-147.
17. Fu Y, Lee AS. Glucose regulated proteins in cancer progression, drug resistance and immunotherapy. Cancer Biol Ther. 2006; 5:741-744.
18. Tomida A, Yun J, Tsuruo T. Glucose-regulated stresses induce resistance to camptothecin in human cancer cells. Int J Cancer. 1996; 68:391-396.
19. Kwiecien H, Goszczynska A, Rokosz P. Benzofuran Small Molecules as Potential Inhibitors of Human Protein Kinases. A Review. Current pharmaceutical design. 2016; 22:879-894.
20. Yang YR, Wei JL, Mo XF, Yuan ZW, Wang JL, Zhang C, Xie YY, You QD, Sun HP. Discovery and optimization of new benzofuran derivatives against p53-independent malignant cancer cells through inhibition of HIF-1 pathway. Bioorg Med Chem Lett. 2016; 26:2713-2718.
21. Matassov D, Kagan T, Leblanc J, Sikorska M, Zakeri Z. Measurement of apoptosis by DNA fragmentation. Methods Mol Biol. 2004; 282:1-17.
22. Brown DG, Sun XM, Cohen GM. Dexamethasone-induced apoptosis involves cleavage of DNA to large fragments prior to internucleosomal fragmentation. J Biol Chem. 1993; 268:3037-3039.
23. Kim BC, Kim HG, Lee SA, Lim S, Park EH, Kim SJ, Lim CJ. Genipin-induced apoptosis in hepatoma cells is mediated by reactive oxygen species/c-Jun NH2-terminal kinase-dependent activation of mitochondrial pathway. Biochem Pharmacol. 2005; 70:1398-1407.
24. Hou CH, Fong YC, Chen JT, Liu JF, Lin MS, Chang CS, Tang CH. The novel isoflavone 7-hydroxy-3',4'-benzoisoflavone induces cell apoptosis in human osteosarcoma cells. Cancer Lett. 2008; 271:117-128.
25. Chowdhury AK, Watkins T, Parinandi NL, Saatian B, Kleinberg ME, Usatyuk PV, Natarajan V. Src-mediated tyrosine phosphorylation of p47phox in hyperoxia-induced activation of NADPH oxidase and generation of reactive oxygen species in lung endothelial cells. J Biol Chem. 2005; 280:20700-20711.
26. Lee JW, Kim WH, Yeo J, Jung MH. ER stress is implicated in mitochondrial dysfunction-induced apoptosis of pancreatic beta cells. Mol Cells. 2010; 30:545-549.
27. Benali-Furet NL, Chami M, Houel L, De Giorgi F, Vernejoul F, Lagorce D, Buscail L, Bartenschlager R, Ichas F, Rizzuto R, Paterlini-Brechot P. Hepatitis C virus core triggers apoptosis in liver cells by inducing ER stress and ER calcium depletion. Oncogene. 2005; 24:4921-4933.
28. Saito K, Elce JS, Hamos JE, Nixon RA. Widespread activation of calcium-activated neutral proteinase (calpain) in the brain in Alzheimer disease: a potential molecular basis for neuronal degeneration. Proc Natl Acad Sci U S A. 1993; 90:2628-2632.
29. Orrenius S, Zhivotovsky B, Nicotera P. Regulation of cell death: the calcium-apoptosis link. Nat Rev Mol Cell Biol. 2003; 4:552-565.
30. Kim SM, Park HS, Jun DY, Woo HJ, Woo MH, Yang CH, Kim YH. Mollugin induces apoptosis in human Jurkat T cells through endoplasmic reticulum stress-mediated activation of JNK and caspase-12 and subsequent activation of mitochondria-dependent caspase cascade regulated by Bcl-xL. Toxicol Appl Pharmacol. 2009; 241:210-220.
31. Fong YC, Yang WH, Hsu SF, Hsu HC, Tseng KF, Hsu CJ, Lee CY, Scully SP. 2-methoxyestradiol induces apoptosis and cell cycle arrest in human chondrosarcoma cells. J Orthop Res. 2007; 25:1106-1114.
32. Leow ML, Chin HL, Yu PS, Pasunooti KK, Tay RX, Zhang D, Yoon HS, Liu XW. Benzofuran-based estrogen receptor alpha modulators as anti-cancer therapeutics: in silico and experimental studies. Current medicinal chemistry. 2013; 20:2820-2837.
33. Chen H, Zeng X, Gao C, Ming P, Zhang J, Guo C, Zhou L, Lu Y, Wang L, Huang L, He X, Mei L. A new arylbenzofuran derivative functions as an anti-tumour agent by inducing DNA damage and inhibiting PARP activity. Scientific reports. 2015; 5:10893.
34. Haga N, Fujita N, Tsuruo T. Involvement of mitochondrial aggregation in arsenic trioxide (As2O3)-induced apoptosis in human glioblastoma cells. Cancer Sci. 2005; 96:825-833.
35. Kallio A, Zheng A, Dahllund J, Heiskanen KM, Harkonen P. Role of mitochondria in tamoxifen-induced rapid death of MCF-7 breast cancer cells. Apoptosis. 2005; 10:1395-1410.
36. Lemasters JJ, Theruvath TP, Zhong Z, Nieminen AL. Mitochondrial calcium and the permeability transition in cell death. Biochim Biophys Acta. 2009; 1787:1395-1401.
37. Wenzel U, Nickel A, Kuntz S, Daniel H. Ascorbic acid suppresses drug-induced apoptosis in human colon cancer cells by scavenging mitochondrial superoxide anions. Carcinogenesis. 2004; 25:703-712.
38. Zu K, Hawthorn L, Ip C. Up-regulation of c-Jun-NH2-kinase pathway contributes to the induction of mitochondria-mediated apoptosis by alpha-tocopheryl succinate in human prostate cancer cells. Mol Cancer Ther. 2005; 4:43-50.
39. Iwamaru A, Iwado E, Kondo S, Newman RA, Vera B, Rodriguez AD, Kondo Y. Eupalmerin acetate, a novel anticancer agent from Caribbean gorgonian octocorals, induces apoptosis in malignant glioma cells via the c-Jun NH2-terminal kinase pathway. Mol Cancer Ther. 2007; 6:184-192.
40. Aliev G, Palacios HH, Gasimov E, Obrenovich ME, Morales L, Leszek J, Bragin V, Solis Herrera A, Gokhman D. Oxidative Stress Induced Mitochondrial Failure and Vascular Hypoperfusion as a Key Initiator for the Development of Alzheimer Disease. Pharmaceuticals. 2010; 3:158-187.
41. Su CM, Wang SW, Lee TH, Tzeng WP, Hsiao CJ, Liu SC, Tang CH. Trichodermin induces cell apoptosis through mitochondrial dysfunction and endoplasmic reticulum stress in human chondrosarcoma cells. Toxicol Appl Pharmacol. 2013; 272:335-344.
42. Chan DC. Mitochondria: dynamic organelles in disease, aging, and development. Cell. 2006; 125:1241-1252.
43. Hess CJ, Berkhof J, Denkers F, Ossenkoppele GJ, Schouten JP, Oudejans JJ, Waisfisz Q, Schuurhuis GJ. Activated intrinsic apoptosis pathway is a key related prognostic parameter in acute myeloid leukemia. J Clin Oncol. 2007; 25:1209-1215.
44. Bianchi L, Gerstbrein B, Frokjaer-Jensen C, Royal DC, Mukherjee G, Royal MA, Xue J, Schafer WR, Driscoll M. The neurotoxic MEC-4(d) DEG/ENaC sodium channel conducts calcium: implications for necrosis initiation. Nat Neurosci. 2004; 7:1337-1344.
45. Verkhratsky A. Physiology and pathophysiology of the calcium store in the endoplasmic reticulum of neurons. Physiol Rev. 2005; 85:201-279.
46. Kaufman RJ. Stress signaling from the lumen of the endoplasmic reticulum: coordination of gene transcriptional and translational controls. Genes Dev. 1999; 13:1211-1233.
47. Quinones QJ, de Ridder GG, Pizzo SV. GRP78: a chaperone with diverse roles beyond the endoplasmic reticulum. Histol Histopathol. 2008; 23:1409-1416.
48. Eletto D, Dersh D, Argon Y. GRP94 in ER quality control and stress responses. Semin Cell Dev Biol. 2010; 21:479-485.
49. Kubota H, Suzuki T, Lu J, Takahashi S, Sugita K, Sekiya S, Suzuki N. Increased expression of GRP94 protein is associated with decreased sensitivity to X-rays in cervical cancer cell lines. Int J Radiat Biol. 2005; 81:701-709.
50. Fonseca SG, Burcin M, Gromada J, Urano F. Endoplasmic reticulum stress in beta-cells and development of diabetes. Curr Opin Pharmacol. 2009; 9:763-770.
51. Fettucciari K, Fetriconi I, Mannucci R, Nicoletti I, Bartoli A, Coaccioli S, Marconi P. Group B Streptococcus induces macrophage apoptosis by calpain activation. J Immunol. 2006; 176:7542-7556.
52. Tan Y, Wu C, De Veyra T, Greer PA. Ubiquitous calpains promote both apoptosis and survival signals in response to different cell death stimuli. J Biol Chem. 2006; 281:17689-17698.
53. Temple HT, Scully SP, Aboulafia AJ. Benign bone tumors. Instructional course lectures. 2002; 51:429-439.
54. Su CM, Hsu CJ, Tsai CH, Huang CY, Wang SW, Tang CH. Resistin Promotes Angiogenesis in Endothelial Progenitor Cells Through Inhibition of MicroRNA206: Potential Implications for Rheumatoid Arthritis. Stem Cells. 2015; 33:2243-2255.
55. Duke RC, Chervenak R, Cohen JJ. Endogenous endonuclease-induced DNA fragmentation: an early event in cell-mediated cytolysis. Proc Natl Acad Sci U S A. 1983; 80:6361-6365.
56. Su CM, Chiang YC, Huang CY, Hsu CJ, Fong YC, Tang CH. Osteopontin Promotes Oncostatin M Production in Human Osteoblasts: Implication of Rheumatoid Arthritis Therapy. J Immunol. 2015; 195:3355-3364.