
INTRODUCTION
Most cancer deaths are largely caused by metastases rather than their primary tumors [1, 2]. Therefore, investigating the potential metastatic mechanisms is essential to stop the progression of cancer. Morphological changes of epithelial cells into mesenchymal cells, termed epithelial-mesenchymal transition (EMT), can be observed in invasive cancer development and also during normal processes such as embryogenesis [3]. These changes include losses of polarity and cell-cell adhesion as well as the ability of mesenchymal cell to migrate away from the point of origin [4]. Loss of the cell-cell adhesion molecule, E-cadherin, a hallmark of EMT [5], is controlled by specific transcription regulators such as Snail, Slug, ZEB1/2, and Twist that can induce mesenchymal characteristics, including expression of vimentin, α-smooth muscle actin, fibronectin, and N-cadherin [6, 7]. The process of EMT induced by transforming growth factor-β (TGF-β) is well-established as a critical mechanism of tumor progression [8–10]. Binding of TGF-β to its receptor leads to activation of the transcription factors, Smad2/3, that can form complexes with Smad4 in the cytoplasm and then translocate into the nucleus where the factors can induce transcription of target genes [11].
Recent studies have indicated a prominent role of paracrine epidermal growth factor (EGF) in driving breast cancer metastasis. Aberrant EGF and its receptor EGFR (ErbB-1) signaling has been extensively described as a major cause of progression and metastasis of breast cancer [12–14]. These factors can activate extracellular signal-regulated kinase 1/2 (ERK1/2) or phosphoinositide-3 kinase/Akt (PI3K/Akt) that can potently induce EMT in cancer cells [6, 15–18]. Overexpression or activation of EGFR in mesenchymal and stem-cell featured breast cancer, such as MDA-MB-231 cell lines has recently been identified as a highly predictive marker for poor clinical outcomes [19–21]. EMT processes of these tumors can result in the development of resistance to EGFR-targeted therapies [22–24].
Therefore, the objective of this study was to determine the mechanism of EGF-induced EMT through activating Smad2/3 in MCF-7 and MDA-MB-231 breast cancer cells. A deeper understanding of the molecular mechanisms controlling EMT may help provide new targets for the treatment of metastatic breast cancer.
RESULTS
EGF activates Smad2/3 in MCF-7 cells
We examined the effect of EGF on the activation of Smad2/3, two molecules that mediate the TGF-β signaling pathway. As shown in Figures 1A-1B, the level of phospho-Smad2/3 reached its maximum levels at a concentration of 30 ng/ml and at 8 h after EGF treatment, without affecting the total expression level of Smad2/3 in MCF-7 cells. TGF-β1 usually induces phosphorylation of Smad2/3 within a few minutes of stimulation. Here, we found that EGF prolonged the phosphorylation of Smad2/3 compared to TGF-β1.
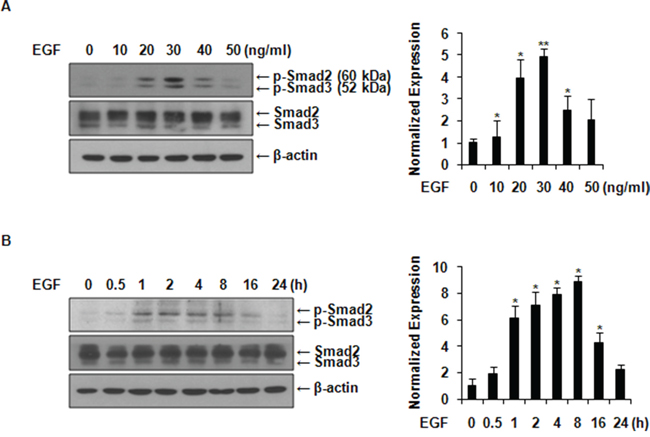
Figure 1: EGF activates Smad2/3 in MCF-7 cells. A. After serum starvation with serum-free medium, cells were treated with increasing concentrations of EGF for 8 h. Immunoblots were incubated with anti-phospho-Smad2/3 and anti-Smad2/3 antibodies. B. Cells were incubated with 30 ng/ml of EGF for the indicated time after serum starvation. The phosphorylation levels of Smad2/3 and total Smad2/3 were analyzed by western blotting at indicated time points. Phospho-Smad2/3 expression was normalized with total Smad2/3. All data represent mean ± SD of three independent experiments with similar results. P value was calculated compared to untreated Ctrl. *P < 0.05 and **P < 0.01.
EGF induces the expression of Snail and EMT markers in MCF-7 cells
As shown in Figure 2A, the expression levels of Snail, vimentin, and fibronectin were increased after EGF treatment, while the expression level of E-cadherin was decreased at 72 h. We further examined the expression of E-cadherin by immunofluorescence staining and found that the E-cadherin expression level was decreased in EGF-treated cells compared to its expression level control cells (Figure 2B). MCF-7 cells kept tight cell–cell adhesion and cell polarities before EGF treatment. However, after EGF treatment, cells scattered and lost cell–cell contacts, resulting in elongated cell shapes similar to the fibroblast-like morphologies of mesenchymal cells (Figure 2C). These results suggested that EGF could upregulate the expression of Snail, vimentin, and fibronectin, while suppressing E-cadherin expression, thus inducing EMT in MCF-7 cells.
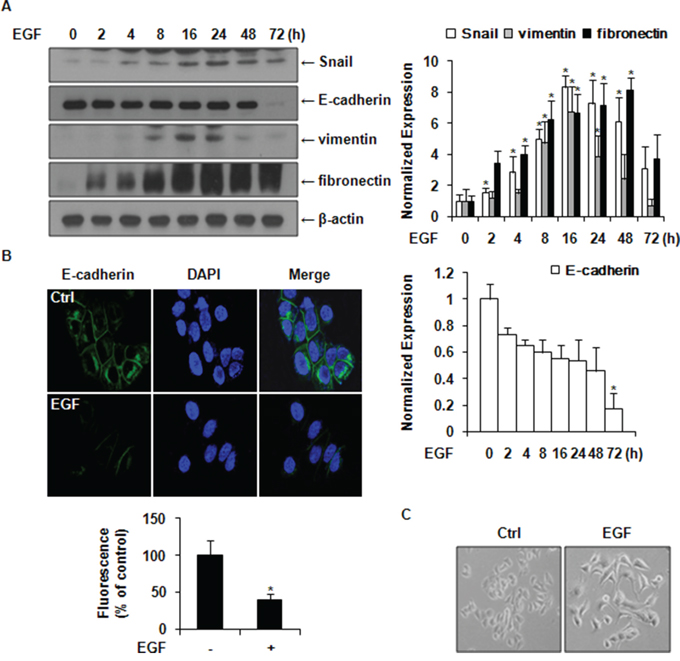
Figure 2: EGF induces the expression of Snail and EMT markers in MCF-7 cells. A. Cells were incubated with 30 ng/ml of EGF for the indicated times after serum starvation. The expression levels of Snail, vimentin, fibronectin, and E-cadherin were determined by western blotting. Protein expression levels were normalized against the level of β-actin. Data represent mean ± SD of three independent experiments with similar results. P value was calculated compared to untreated Ctrl of MCF-7 cells. *P < 0.05. B. Immunofluorescence staining of E-cadherin protein. Cells were treated with or without 30 ng/ml of EGF for 48 h. Green color represents the staining of E-cadherin. Blue color represents nuclear DNA staining by DAPI (magnification, ×400). Results were presented as a relative percentage to untreated Ctrl (defined as 100%). Data represent mean ± SD of three independent experiments in triplicates. P value was calculated compared to untreated Ctrl. *P < 0.05. C. The morphology of MCF-7 cells with or without treatment with 30 ng/ml of EGF for 24 h using phase contrast microscopy.
EGF induces activation of Smad2/3 and expression of EMT markers via ERK1/2 signaling pathway
First, we found that EGF activated ERK1/2 and Akt (data not shown) signal molecules in a time-dependent manner in MCF-7 cells (Figure 3A). Among these intracellular signal mechanisms, subsequent experiments were carried out focusing on the ERK1/2 pathway. PI3k/Akt pathway will be addressed in more details later in the discussion section. Smad2/3 phosphorylation and expression levels of Snail, vimentin, and fibronectin were inhibited by pretreatment with PD98059 prior to EGF stimulation (Figure 3B). These results suggested that EGF-induced phosphorylation of Smad2/3 and the expression of Snail, fibronectin, and vimentin via the ERK1/2 signaling pathway in MCF-7 cells.
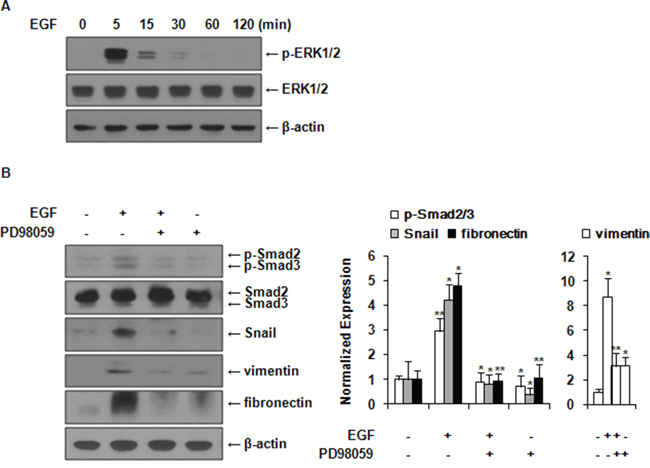
Figure 3: EGF induces activation of Smad2/3 and expression of EMT markers via ERK1/2 signaling pathway. A. MCF-7 cells were treated with 30 ng/ml of EGF and the expression levels of phospho-ERK1/2 and ERK1/2 were examined by western blot. B. Cells were pretreated with vehicle or 10 μM of PD98059 for 1 h prior to EGF treatment. The expression levels of phospho-Smad2/3, Smad2/3, Snail, vimentin, and fibronectin were examined by western blotting. Phospho-Smad2/3 expression was normalized to total Smad2/3. Protein expressions were normalized to the level of β-actin. All data represent mean ± SD of three independent experiments with similar results. P value was calculated compared to untreated Ctrl. *P < 0.05 and **P < 0.01.
EGF induces nuclear co-localization of phospho-Smad2/3 and Snail and the migration of MCF-7 cells
To investigate the possible involvement of Smad2/3 in EGF-induced Snail gene expression, MCF-7 cells were pretreated with two known inhibitors (PD169316 and SB203580) of Smad2 phosphorylation [25, 26]. SB203580 has a higher inhibitory effect on MCF-7 cells than PD169316 [27]. As shown in Figure 4A, EGF treatment for 24 h induced nuclear co-localization of phospho-Smad2/3 and Snail in MCF-7 cells. Such nucleus translocation was inhibited by pretreatment with PD98059 and SB203580 before EGF stimulation, indicating that EGF induced the expression of Snail through activating ERK1/2-Smad2/3 signaling. The mean percentages of fluorescence of phospho-Smad2/3 and Snail are shown in Figure 4A. In vitro wound healing assays revealed that pretreatment with PD98059 and SB203580 inhibited cell migration of MCF-7 cells in the presence of EGF (Figure 4B). Collectively, these data suggested that EGF induced EMT and cancer cell migration through ERK1/2-phospho-Smad2/3-Snail signaling pathway.
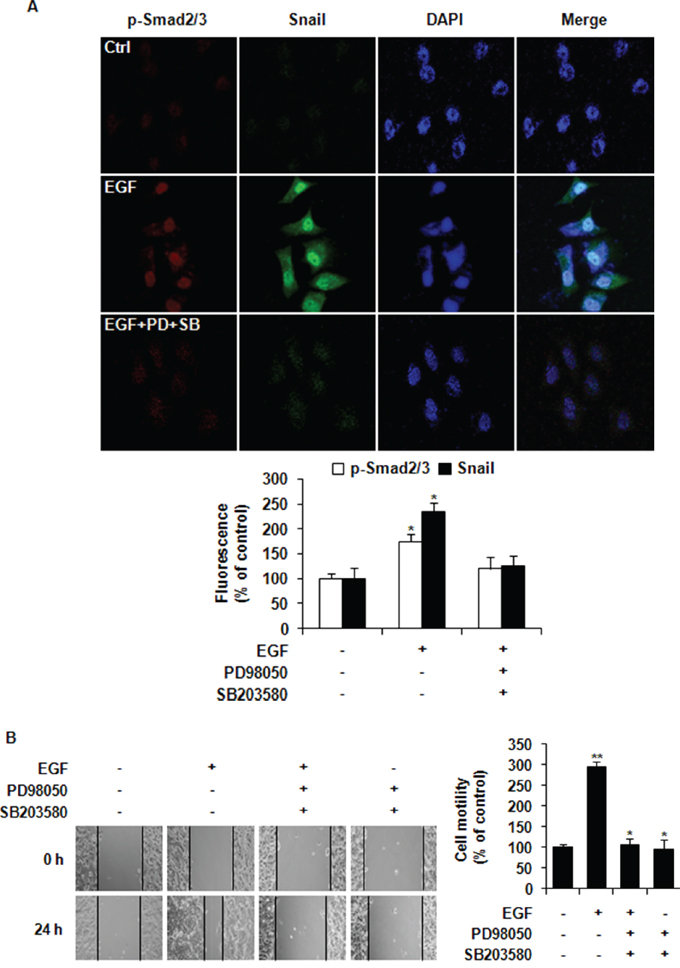
Figure 4: EGF induces nuclear co-localization of phospho-Smad2/3 and Snail and the migration of MCF-7 cells. A. Cells were pretreated with vehicle or 10 μM of PD98059 (PD) and SB203580 (SB) prior to stimulation with 30 ng/ml of EGF. After incubation for 24 h, phospho-Smad2/3 (red) and Snail (green) were observed under a confocal laser scanning microscope. Nuclear DNA was stained with DAPI (blue; magnification, ×200). The fluorescence intensities of p-Smad2/3 and Snail as relative percentages compared to untreated Ctrl were presented. B. Cell motility was assessed by wound healing assays. A scratch was made across confluent monolayers using a plastic tip. Cells were then pretreated with 10 μM of PD98059 and SB203580 prior to stimulation with EGF. Migrated cells were monitored using a light microscope with ×200 magnification. Relative wound closure percentage were presented at 24 h time point compared to that at 0 h for each treatment group. All data represent mean ± SD of three independent experiments in triplicates. P value was calculated compared to untreated Ctrl. *P < 0.05 and **P < 0.01.
Knockdown of Smad2/3 expression suppresses EGF-induced expression of Snail, vimentin, and fibronectin and the invasion of MCF-7 cells
Since Smad2 phosphorylation inhibitors could also block the phosphorylation of p38 MAP kinase, the role of Smad2 was further explored by a specific genetic approach using RNA interference (siRNA). MCF-7 cells were transfected with control or Smad2/3 siRNA. As shown in Figure 5A, EGF increased the expression levels of Snail, vimentin, and fibronectin in control siRNA-transfected cells (Ctrl siR) compared to those in untreated control cells (untreated ctrl). Such increases were suppressed in Smad2/3 siRNA-transfected cells (Smad2/3 siR). Taken together, these results suggested that Smad2/3 activation plays an important role in the expression of Snail and the induction of EMT by EGF in MCF-7 cells. Cell invasion assays revealed that knockdown of Smad2/3 by siRNA transfection inhibited cell invasion of MCF-7 cells stimulated by EGF in the matrigel-coated chamber (Figure 5B). These data suggested that EGF can stimulate cancer cell invasion through inducing EMT via ERK1/2-phospho-Smad2/3-Snail signaling pathway.
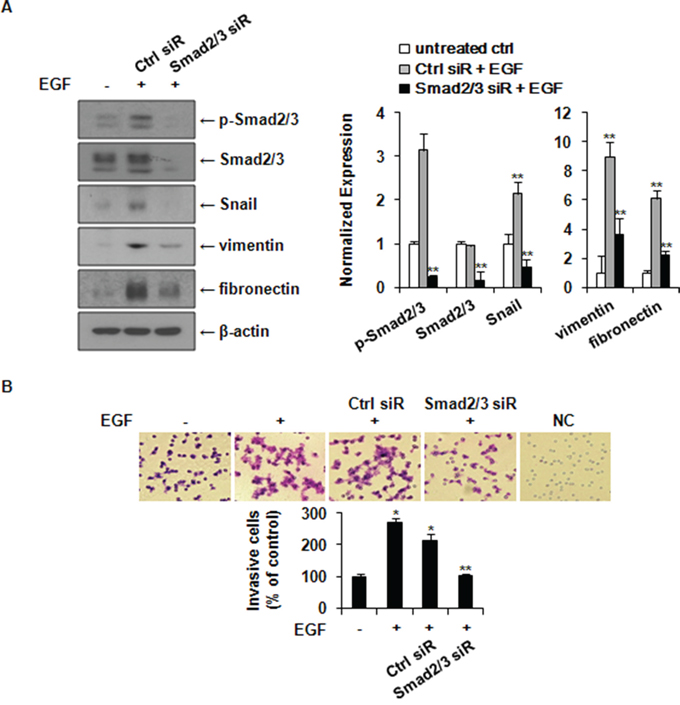
Figure 5: Knockdown of Smad2/3 expression suppresses EGF-induced expression of Snail, fibronectin, and vimentin and the invasion of MCF-7 cells. A. Cells were treated with 30 ng/ml of EGF for 24 h following transfection with control or Smad2/3 siRNA. The expression levels of phospho-Smad2/3, Smad2/3, Snail, vimentin and fibronectin were analyzed by western blotting. Protein expressions were normalized to the level of β-actin. Data represent mean ± SD of three independent experiments with similar results. P value was calculated compared to untreated Ctrl. **P < 0.01. B. A matrigel invasion assay was used to quantify cell invasion. After 24 h of transfection, cells were seeded into upper chambers and incubated for 48 h in the presence of 30 ng/ml of EGF. Cells that invaded into the lower surface were photographed under a light microscope with ×200 magnification. The relative percentages of invasive cells compared to untreated Ctrl were presented. Data represent mean ± SD of three independent experiments in triplicates. P value was calculated compared to untreated Ctrl. *P < 0.05 and **P < 0.01. NC: negative control.
EGF induces EMT through ERK1/2-phospho-Smad2/3-Snail signaling pathway in MDA-MB-231 breast cancer cells
A hallmark of EMT is the loss of E-cadherin expression. Immunofluorescence staining revealed that the expression of E-cadherin was decreased in EGF-treated cells at 48 h post treatment compared to that in the control cells (Figure 6A). EGF-induced nuclear translocation of phospho-Smad2/3 and Snail was also inhibited by pretreatment with PD98059 and SB203580 before EGF stimulation. These results indicate that EGF induced the expression of Snail through activating the ERK1/2-Smad2/3 signaling axis (Figure 6B). Knockdown of Smad2/3 expression suppressed EGF-induced expressions of Snail, vimentin, and fibronectin and the invasion of MDA-MB-231 cells (Figure 6C-6D). Therefore, EGF could induce EMT and cancer cell invasion in MDA-MB-231 cells through the ERK1/2-phospho-Smad2/3-Snail signaling pathway.
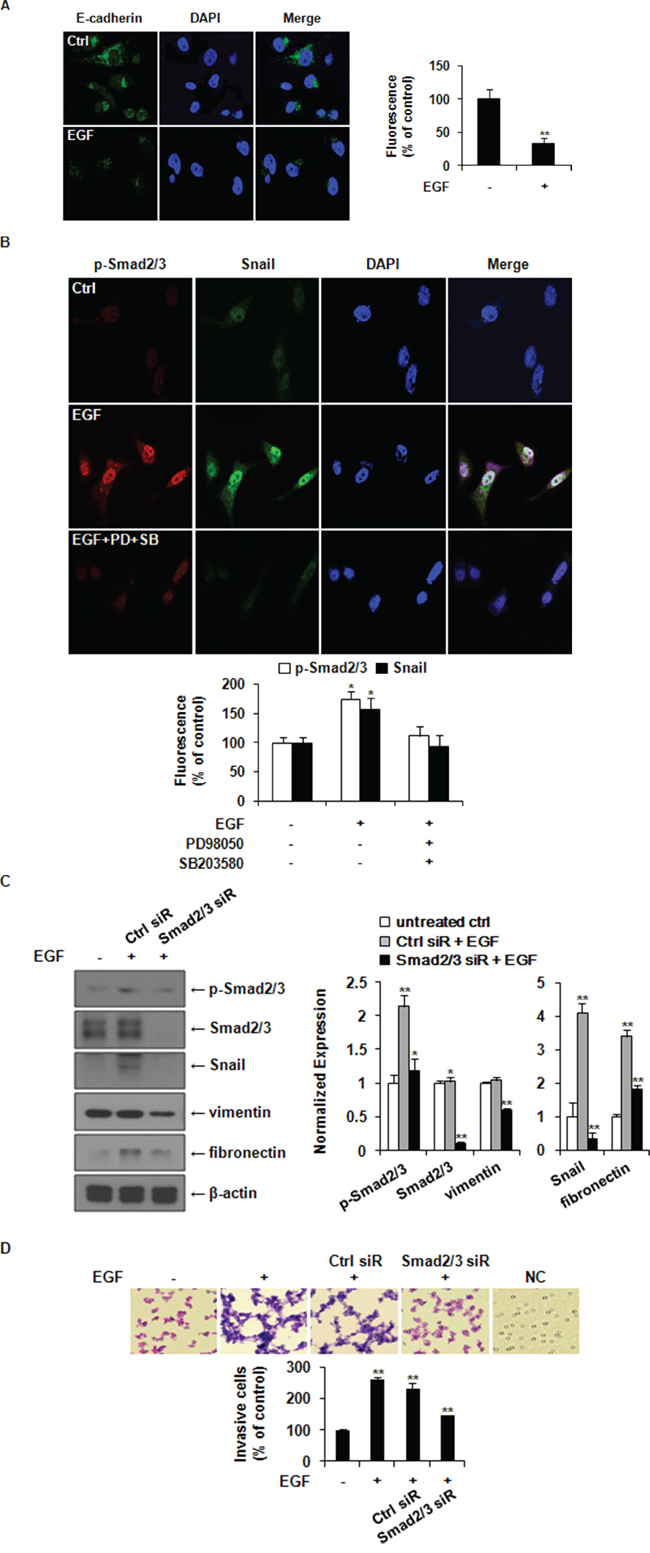
Figure 6: EGF induces EMT through ERK1/2-phospho-Smad2/3-Snail signaling pathway in MDA-MB-231 breast cancer cells. A. Immunofluorescence staining of E-cadherin expression, cells were treated with or without 30 ng/ml of EGF for 48 h. Green color represents staining of E-cadherin. Blue color represents nuclear DNA staining by DAPI (magnification, ×400). Results were presented as a relative percentages to untreated Ctrl (defined as 100%). B. Immunofluorescence analyses for nuclear co-localization of phospho-Smad2/3 and Snail. Cells were pretreated with vehicle or 10 μM of PD98059 (PD) and SB203580 (SB) for 1 h prior to stimulation with 30 ng/ml of EGF. After 24 h of incubation, p-Smad2/3 (red) and Snail (green) were observed using a confocal laser scanning microscope. Nuclear DNA was stained with DAPI (blue; magnification, ×200). The fluorescence intensities of p-Smad2/3 and Snail as relative percentages compared to untreated Ctrl were presented. C. After transfection with control or Smad2/3 siRNA, the expression levels of phospho-Smad2/3, Smad2/3, Snail, vimentin, and fibronectin were analyzed by western blotting. Protein expression levels were normalized to the level of β-actin. D. A matrigel invasion assays for cell invasion. The relative percentages of invasive cells compared to untreated Ctrl were presented. All data represent mean ± SD of three independent experiments. P value was calculated compared to untreated Ctrl. *P < 0.05 and **P < 0.01. NC: negative control.
DISCUSSION
Breast cancer is the most common cause of death among women worldwide. Most of these deaths are due to metastatic disease [28]. EMT is an important preliminary step in metastasis and invasion [29–32]. Many molecules and signaling pathways that can cause cancer invasion and metastasis are still poorly understood. A novel insight regarding the molecular mechanisms of breast cancer metastasis is that EGF-induced EMT can occur via the Smad2/3-Snail signaling pathway in MCF-7 breast cancer cells. Smads can integrate multiple signaling pathways and directly regulate the expression of target genes in TGF-β-activated cells [33].
It could be seen that activation of Smad2/3 was involved in EMT induced by EGF using a phospho-Smad2 inhibitor (SB203580) and Smad2/3 siRNA transfection. Although SB203580 is known as an inhibitor of p38 MAP kinase, some breast cancer studies using MDA-MB-231 cells have reported that the p38 MAP kinase pathway appears to be a major component of Smad-independent signaling by TGF-β [33, 34]. We clearly identified ‘biological’ EMT of EGF-stimulated breast cancer cells by using wound healing or a matrigel invasion assay. These results suggest that acquisition of the mesenchymal phenotype and motility of MCF-7 cells can be triggered through the ERK1/2-phospho-Smads2/3-Snail signaling pathway.
It has been reported that the activation of ERK1/2 by EGF and EGFR is implicated in the transcription of E-cadherin repressors, such as Snail and Slug [9, 17, 35]. The role of the Snail family zinc finger proteins in EMT and cancer has been particularly highlighted in several publications [32, 36]. On the other hand, activation of PI3K/Akt pathway may occur more via HER2 (ErbB-2) than via EGFR [37, 38]. PI3K/Akt pathway plays an important role in cell migration [39] possibly due to increased production of matrix metalloproteinases such as MMP-2 and MMP-9 caused by EGFR [15, 16].
To further explore our findings of a role for Smad2/3 activation in the EGF-induced EMT process, we examined MDA-MB-231 cells that have an extremely high expression of p-Smad2/3 as a useful positive control. EGF also induced EMT and invasion of MDA-MB-231 breast cancer cells through ERK1/2-phospho-Smad2/3-Snail signaling pathway. EMT is significantly associated with these types of breast carcinoma, particularly with increased expression of mesenchymal cytoskeletal proteins such as vimentin. This is consistent with our results as shown in Figure 6C. These may explain the aggressive behavior of mesenchymal stem-like type of MDA-MB-231 breast cancer cells with high tendency to invade and metastasize [40].
In conclusion, EGF-induced EMT via phospho-Smad2/3-Snail pathway was a crucial event during the migration and invasion of MCF-7 and MDA-MB-231 breast cancer cells. Our findings could be useful for developing novel therapeutics against metastatic breast cancers.
MATERIALS AND METHODS
Cell lines and reagents
Human breast cancer cell lines MCF-7 and MDA-MB-231 were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA). MCF-7 cells were cultured in RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin, and 100 μg/ml streptomycin (GIBCO, Grand Island, NY, USA). MDA-MB-231 cells were cultured in Leibovitz’s L-15 medium (GIBCO) supplemented with 10% FBS. Both cell lines were maintained at 37°C in a 5% CO2 humidified incubator. Epidermal growth factor (EGF) was purchased from Sigma (St Louis, MO, USA). Aliquots of the growth factor were stored at –80°C. ERK1/2 inhibitor (PD98059) and the phospho-Smad2 pharmacological inhibitor (SB203580) were purchased from Calbiochem (San Diego, CA, USA). These inhibitors were dissolved in DMSO before use.
Western blotting
Protein lysates were prepared with a radioimmunoprecipitation assay (RIPA) buffer (20 mM Tris-HCl pH 7.5, 2 mM EDTA, 150 mM NaCl, 1 mM sodium vanadate, 10 mM NaF, 2.5 mM sodium pyrophosphate, 1% sodium deoxycholate, 0.1% SDS, 1% NP-40) supplemented with a protease inhibitor cocktail (Roche, Mannheim, Germany). Protein concentrations were determined using a BCA protein assay kit (Thermo Scientific, Rockford, IL, USA). Protein samples (30 μg) were resolved by SDS-PAGE and transferred onto PVDF membranes (Bio-Rad Laboratories, Hercules, CA, USA). Blocked membranes with skim milk were incubated with primary antibodies. Washed membranes were incubated with horseradish peroxidase-conjugated anti-mouse or anti-rabbit secondary antibody and developed with an ECL plus western blot detection system reagent (GE Healthcare Biosciences, Piscataway, NJ, USA). Anti-phospho-Smad2 (Ser465/467)/Smad3 (Ser423/425) and anti-Smad2/3 antibodies were purchased from Cell Signaling Technology Inc. (Beverly, MO, USA). Anti-Snail antibody was obtained from Abcam Ltd. (Cambridge, UK). Anti-E-cadherin and anti-vimentin antibodies were purchased from BD Pharmingen (San Diego, CA, USA). Anti-fibronectin antibody was obtained from Millipore (Billerica, MA, USA). A monoclonal anti-β-actin antibody was obtained from Sigma to evaluate loading. Protein levels were quantified using ImageJ software (NIH, Bethesda, MD, USA).
Immunofluorescence assay
Nuclear translocation of p-Smad2 and Snail was examined by immunofluorescence studies. Approximately 2x104 cells/well were seeded onto a 2-well Lab-Tek II chamber slide (NUNC, Rochester, NY, USA). Serum-starved cells were incubated with EGF and specific inhibitors. Cells were washed three times with phosphate buffered saline (PBS) and then fixed with 4% paraformaldehyde for 10 min. Cells were washed three times with PBS and then permeabilized with 0.1% Triton X-100 for 20 min. After washing with PBS, the slides were blocked with 3% bovine serum albumin for 1 h at room temperature and incubated with rabbit polyclonal anti-Snail (1:500) and goat polyclonal anti-phospho-Smad2 (Ser465/467)/Smad3 (Ser423/425) (1:100) (Santa Cruz Biotechnology Inc., Dallas, TX, USA) primary antibodies. After overnight incubation at 4°C followed by three washes, cells were incubated with Alexa Fluor 488 anti-rabbit IgG and Alexa Fluor 594 anti-goat IgG secondary antibodies (Invitrogen, Grand Island, NY, USA). Washed cells were mounted with mounting medium plus DAPI (VECTOR Lab. Inc., Burlingame, CA, USA) and observed with ZEISS LSM700 confocal laser scanning microscope (Carl Zeiss, Thornwood, NY, USA). The expression of E-cadherin was determined using DyLight 488 anti-mouse IgG secondary antibody (VECTOR Lab. Inc.).
Wound healing assay
For the wound healing assay, cells were seeded in 12-well plates and grown to confluence. After serum starvation, a confluent monolayer of cells was scratched with a plastic tip and cells were incubated with EGF with indicated inhibitors for 24 h after washing with PBS to remove detached cells. Cell migration into the wounded area was monitored at indicated time points using a light microscope (Olympus BX51 Tokyo, Japan). Quantification of relative wound closure was determined using an image analysis program. Three independent experiments were performed in triplicates.
Matrigel invasion assay
For the cell invasion assay, serum-free medium (500 μl) treated with or without EGF was added into the lower chambers of a 24 transwell plate (8.0 μm pore size, Corning, NY, USA). Cells transfected with control (Ctrl), or Smad2 siRNA (2 × 105 cells in 200 μl medium) were seeded into the upper chamber coated with a matrigel (Corning, Bedford, MA, USA). After 48 h of incubation, non-migrating cells were removed with a cotton swab. Cells at the bottom of the membrane surface were stained with a Diff-Quick Staining kit (Biochemical Sciences, Swedesboro, NJ, USA). Invaded cells were photographed randomly with a microscope and quantified using an ImageJ software program (NIH, Bethesda, MD, USA). Three independent experiments were performed in triplicates.
Small interfering RNA (siRNA) transfection
Cells were grown to confluence in a 60×15 mm culture dish (Nunc, Roskilde, Denmark) and transfected with Smad2/3 siRNA or control (Ctrl) siRNA (Santa Cruz Biotechnology Inc., Dallas, TX, USA) at 60 pmol using a siRNA transfection reagent (Santa Cruz Biotechnology Inc.) according to the manufacturer’s instructions. After incubation for 6 h, the culture medium was replaced with the standard culture medium. Transfected cells were treated with EGF the next day and used in subsequent evaluations.
Statistical analysis
All the data were presented as the mean ± standard deviation of three independent experiments. Comparisons between two groups were performed using a Student’s t-test. *P < 0.05 and **P < 0.01 were considered as statistically significant.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
GRANT SUPPORT
This research was supported by a grant (HI14C3405) of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI) funded by the Ministry of Health & Welfare (MOHW), Republic of Korea.
REFERENCES
1. Gupta GP, Massagué J. Cancer metastasis: building a framework. Cell. 2006; 127:679-695.
2. Wiercinska E, Naber HP, Pardali E, van der Pluijm G, van Dam H, ten Dijke P. The TGF-β/Smad pathway induces breast cancer cell invasion through the up-regulation of matrix metalloproteinase 2 and 9 in a spheroid invasion model system. Breast cancer research and treatment. 2011; 128:657-666.
3. Thiery JP, Sleeman JP. Complex networks orchestrate epithelial–mesenchymal transitions. Nature Reviews Molecular Cell Biology. 2006; 7:131-142.
4. Hay ED. The mesenchymal cell, its role in the embryo, and the remarkable signaling mechanisms that create it. Developmental dynamics. 2005; 233:706-720.
5. Kang Y, Massagué J. Epithelial-mesenchymal transitions: twist in development and metastasis. Cell. 2004; 118:277-279.
6. Thiery JP. Epithelial–mesenchymal transitions in tumour progression. Nature Reviews Cancer. 2002; 2:442-454.
7. Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C, Savagner P, Gitelman I, Richardson A, Weinberg RA. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell. 2004; 117:927-939.
8. Zavadil J, Böttinger EP. TGF-β and epithelial-to-mesenchymal transitions. Oncogene. 2005; 24:5764-5774.
9. Moustakas A, Heldin CH. Signaling networks guiding epithelial–mesenchymal transitions during embryogenesis and cancer progression. Cancer science. 2007; 98:1512-1520.
10. Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. The Journal of clinical investigation. 2009; 119:1420.
11. TSANG KJ, TSANG D, BROWN TN, CROWE DL. A novel dominant negative Smad2 mutation in a TGFβ resistant human carcinoma cell line. Anticancer research. 2002; 22:13-19.
12. Lo H-W, Hsu S-C, Xia W, Cao X, Shih J-Y, Wei Y, Abbruzzese JL, Hortobagyi GN, Hung M-C. Epidermal growth factor receptor cooperates with signal transducer and activator of transcription 3 to induce epithelial-mesenchymal transition in cancer cells via up-regulation of TWIST gene expression. Cancer research. 2007; 67:9066-9076.
13. Olsen DA, Bechmann T, Østergaard B, Wamberg PA, Jakobsen EH, Brandslund I. Increased concentrations of growth factors and activation of the EGFR system in breast cancer. Clinical Chemistry and Laboratory Medicine. 2012; 50:1809-1818.
14. Navolanic PM, Steelman LS, McCubrey JA. EGFR family signaling and its association with breast cancer development and resistance to chemotherapy (Review). International journal of oncology. 2003; 22:237-252.
15. Ellerbroek SM, Halbleib JM, Benavidez M, Warmka JK, Wattenberg EV, Stack MS, Hudson LG. Phosphatidylinositol 3-kinase activity in epidermal growth factor-stimulated matrix metalloproteinase-9 production and cell surface association. Cancer research. 2001; 61:1855-1861.
16. Kondapaka SB, Fridman R, Reddy KB. Epidermal growth factor and amphiregulin up-regulate matrix metalloproteinase-9 (MMP-9) in human breast cancer cells. International journal of cancer. 1997; 70:722-726.
17. Christiansen JJ, Rajasekaran AK. Reassessing epithelial to mesenchymal transition as a prerequisite for carcinoma invasion and metastasis. Cancer research. 2006; 66:8319-8326.
18. Grünert S, Jechlinger M, Beug H. Diverse cellular and molecular mechanisms contribute to epithelial plasticity and metastasis. Nature Reviews Molecular Cell Biology. 2003; 4:657-665.
19. Tischkowitz M, Brunet J-S, Bégin LR, Huntsman DG, Cheang MC, Akslen LA, Nielsen TO, Foulkes WD. Use of immunohistochemical markers can refine prognosis in triple negative breast cancer. BMC cancer. 2007; 7:134.
20. Carey L, Winer E, Viale G, Cameron D, Gianni L. Triple-negative breast cancer: disease entity or title of convenience? Nature reviews Clinical oncology. 2010; 7:683-692.
21. Jeong H, Kim J, Lee Y, Seo JH, Hong SR, Kim A. Neuregulin-1 induces cancer stem cell characteristics in breast cancer cell lines. Oncology reports. 2014; 32:1218-1224.
22. Thomson S, Buck E, Petti F, Griffin G, Brown E, Ramnarine N, Iwata KK, Gibson N, Haley JD. Epithelial to mesenchymal transition is a determinant of sensitivity of non–small-cell lung carcinoma cell lines and xenografts to epidermal growth factor receptor inhibition. Cancer research. 2005; 65:9455-9462.
23. Buck E, Eyzaguirre A, Barr S, Thompson S, Sennello R, Young D, Iwata KK, Gibson NW, Cagnoni P, Haley JD. Loss of homotypic cell adhesion by epithelial-mesenchymal transition or mutation limits sensitivity to epidermal growth factor receptor inhibition. Molecular cancer therapeutics. 2007; 6:532-541.
24. Barr S, Thomson S, Buck E, Russo S, Petti F, Sujka-Kwok I, Eyzaguirre A, Gibson NW, Miglarese M, Epstein D. Bypassing cellular EGF receptor dependence through epithelial-to-mesenchymal-like transitions. Clinical & experimental metastasis. 2008; 25:685-693.
25. Qureshi HY, Ricci G, Zafarullah M. Smad signaling pathway is a pivotal component of tissue inhibitor of metalloproteinases-3 regulation by transforming growth factor beta in human chondrocytes. Biochimica et Biophysica Acta (BBA)-Molecular Cell Research. 2008; 1783:1605-1612.
26. Fu Y, O’Connor LM, Shepherd TG, Nachtigal MW. The p38 MAPK inhibitor, PD169316, inhibits transforming growth factor β-induced Smad signaling in human ovarian cancer cells. Biochemical and biophysical research communications. 2003; 310:391-397.
27. Kim J, Jeong H, Lee Y, Kim C, Kim H, Kim A. HRG-β1-driven ErbB3 signaling induces epithelial–mesenchymal transition in breast cancer cells. BMC cancer. 2013; 13:383.
28. Parkin D, Whelan S, Ferlay J, Teppo L, Thomas D. Cancer incidence in five continents Vol. VIII. IARC scientific publications. 2002; 155.
29. Thompson EW, Newgreen DF. Carcinoma invasion and metastasis: a role for epithelial-mesenchymal transition? Cancer research. 2005; 65:5991-5995.
30. Garber K. Epithelial-to-mesenchymal transition is important to metastasis, but questions remain. Journal of the National Cancer Institute. 2008; 100:232-239.
31. Thompson EW, Torri J, Sabol M, Sommers CL, Byers S, Valverius EM, Martin GR, Lippman ME, Stampfer MR, Dickson RB. Oncogene-induced basement membrane invasiveness in human mammary epithelial cells. Clinical & experimental metastasis. 1994; 12:181-194.
32. Yang J, Weinberg RA. Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Developmental cell. 2008; 14:818-829.
33. Käkönen S-M, Selander KS, Chirgwin JM, Yin JJ, Burns S, Rankin WA, Grubbs BG, Dallas M, Cui Y, Guise TA. Transforming growth factor-β stimulates parathyroid hormone-related protein and osteolytic metastases via Smad and mitogen-activated protein kinase signaling pathways. Journal of Biological Chemistry. 2002; 277:24571-24578.
34. Lindemann RK, Ballschmieter P, Nordheim A, Dittmer J. Transforming growth factor β regulates parathyroid hormone-related protein expression in MDA-MB-231 breast cancer cells through a novel Smad/Ets synergism. Journal of Biological Chemistry. 2001; 276:46661-46670.
35. Conacci-Sorrell M, Simcha I, Ben-Yedidia T, Blechman J, Savagner P, Ben-Ze'ev A. Autoregulation of E-cadherin expression by cadherin–cadherin interactions the roles of β-catenin signaling, Slug, and MAPK. The Journal of cell biology. 2003; 163:847-857.
36. Nieto MA. The snail superfamily of zinc-finger transcription factors. Nature Reviews Molecular Cell Biology. 2002; 3:155-166.
37. She Q-B, Chandarlapaty S, Ye Q, Lobo J, Haskell KM, Leander KR, DeFeo-Jones D, Huber HE, Rosen N. Breast tumor cells with PI3K mutation or HER2 amplification are selectively addicted to Akt signaling. PloS one. 2008; 3:e3065-e3065.
38. Brachmann SM, Hofmann I, Schnell C, Fritsch C, Wee S, Lane H, Wang S, Garcia-Echeverria C, Maira S-M. Specific apoptosis induction by the dual PI3K/mTor inhibitor NVP-BEZ235 in HER2 amplified and PIK3CA mutant breast cancer cells. Proceedings of the National Academy of Sciences. 2009; 106:22299-22304.
39. Holbro T, Civenni G, Hynes NE. The ErbB receptors and their role in cancer progression. Experimental cell research. 2003; 284:99-110.
40. Jeong H, Ryu Yj, An J, Lee Y, Kim A. Epithelial–mesenchymal transition in breast cancer correlates with high histological grade and triple-negative phenotype. Histopathology. 2012; 60:E87-E95.