
INTRODUCTION
The great majority (~80%) of patients with chronic lymphocytic leukemia (CLL) carry one of the chromosomal aberrations routinely analyzed by fluorescence in situ hybridization (FISH), namely deletions of 13q [del(13q)], 11q [del(11q)] and 17p [del(17p)] as well as trisomy 12. These aberrations are associated with distinct clinical outcome [1, 2]. In particular, del(17p) cases have the worst clinical outcome and the shortest overall survival (OS). Of note, it has been described that the remaining TP53 allele in del(17p) cases is generally mutated, although some CLL patients may harbor isolated TP53 mutations [3]. Taking into account both TP53 deletions and mutations, the rate of TP53 aberrant CLL cases peaks at 10% at diagnosis [4, 5] and may even exceed 40% at disease progression, mainly in patients previously treated with chemotherapy [6]. In addition, genomic complexity detected by chromosome banding analysis (CBA) or genomic microarrays also predicting rapid disease progression, is correlated to TP53 disruption in a significant proportion of patients [7–9].
TP53 aberrations (TP53abs, namely del(17p) and/or TP53 mutations) are associated with low response rates to standard chemoimmunotherapy [10]. Newly introduced signaling inhibitors represent a major advance for this group of patients, showing an improved overall response and progression-free survival even in refractory CLL cases [11–13].
Nonetheless, some CLL cases with TP53abs remain asymptomatic for extended periods of time, pointing to multiple associated genetic factors underlying the heterogeneity of this group of patients [14, 15].
Alterations of chromosome 8, and particularly losses of 8p (8p−, affecting several regions, from 8p11 to 8p23) and gains of 8q (8q+, usually involving 8q24, where MYC is located), have been suggested as new prognostic markers in CLL, even for patients harboring TP53abs. Although initial microarray studies identified chromosome 8 alterations in around 5% of the general CLL population, subsequent analyses detected an increased frequency in patients with del(17p), ranging from 28 to 80%; 8p− and 8q+ alterations were frequently concomitant and displayed an independent correlation with a shorter OS [16–21].
However, due to the small size of the evaluated cohorts, definitive conclusions could not be drawn regarding the actual impact of chromosome 8 abnormalities in CLL patients harboring TP53abs. In the present study we sought to obtain more insight into this issue by analyzing a series of 101 CLL patients with TP53abs, especially focusing on the potential prognostic value of 8p losses and 8q gains in this poor prognostic group of patients.
RESULTS
Patients
A total of 101 patients carrying TP53 abnormalities were included in this study: 92 cases with del(17p) and 9 additional cases with TP53 mutations. Demographic, clinical and biological data for the entire cohort are summarized in Table 1.
Table 1: Baseline characteristics of patients at diagnosis and last follow-up
Patients characteristics (n =101) |
|
|---|---|
Age at diagnosis |
64 (42–87) |
Male |
69 (68.3%) |
Diagnosis |
|
MBL* |
6 (5.9%) |
CLL |
95 (94.1%) |
Binet stage** (n =90) |
|
A |
57 (63.3%) |
B |
23 (25.6%) |
C |
10 (11.1%) |
B-symptoms (n = 67) |
6 (9%) |
Adenopathies (n = 71) |
37 (52.1%) |
Splenomegaly (n =68) |
12 (17.6%) |
Hepatomegaly (n =68) |
6 (8.8%) |
Absolute white blood cell count (× 109/L) (n = 71) |
20 (3.8–372) |
Absolute lymphocyte count (× 109/L) (n =68) |
15 (1–369) |
Hemoglobin (g/dL) (n = 68) |
13.8 (7–18) |
Platelets (×109/L) (n = 68) |
196 (2–356) |
Lactate dehydrogenase (IU/L) (n = 64) |
335 (180–959) |
Beta-2 microglobulin (mg/L) (n = 59) |
2.4 (1–8.4) |
Unmutated IGHV (n =32) |
27 (84.4%) |
Mutated NOTCH1 (n = 19) |
2 (10.5%) |
Last follow-up |
|
Treated patients (n =97) |
81 (83.5%) |
Time to first treatment (months, 95% CI) (n = 96) |
23 (14–33) |
Deaths |
66 (65.3%) |
Overall survival (months, 95% CI) (n = 99) |
88 (67–108) |
Follow-up (months) |
62 (0–201) |
Values are given as median (range) or number (%). Hemoglobin is expressed as mean (range).
*Although 6 patients were diagnosed as MBL (monoclonal B-cell lymphocytosis), all of them had already progressed to CLL at the time of study. **Some centers only provided information regarding the Binet stage without the physical examination and the analytical parameters.
Chromosome 8 alterations
Overall, 23/101 cases (22.8%) displayed chromosome 8 alterations. In detail, 11/75 patients (14.7%) showed 8p−, while 18/101 cases (17.8%) had 8q+ with different alteration patterns (Table 2). In 6/75 patients (8%) both abnormalities were concomitant; baseline characteristics of patients with concomitant 8p− and 8q+ are shown in Supplementary Table S1. Four of the cases with concomitant 8p− and 8q+ also presented two altered clones with shared FISH patterns (Table 2).
Table 2: Chromosome banding analysis and FISH results in patients with alterations of chromosome 8
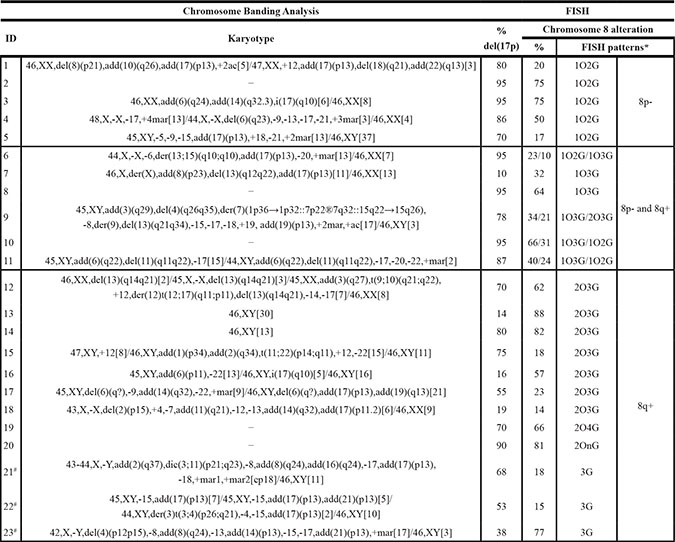
*O: LPL (8p21) signal in orange, G: MYC (8q24) signal in green.
#Only the MYC probe was analyzed.
In 12/23 abnormal patients, chromosome 8 alterations were identified prior to treatment initiation either at diagnosis or within the following 12 months (de novo). In contrast, in the remaining 11 cases chromosome 8 analysis was performed later during the disease course (n = 10/11) or after treatment administration (n = 8/11) (median time to chromosome 8 analysis: 36 months, range: 9–132). In three of the latter cases, retrospective FISH analysis of stored diagnostic material disclosed the presence of chromosome 8 alterations without del(17p); in one of these three cases, a different clonal distribution of the abnormal chromosome 8 clones was observed between the two time points (Supplementary Figure S1). Altogether, chromosome 8 alterations could be considered de novo in 65.2% of cases (15/23). Unfortunately, no previous cytogenetic studies from the remaining patients (8/23) were available to elucidate the acquisition pattern of chromosome 8 and TP53abs.
For the purpose of this study, two types of comparisons were established: patients with 8p– vs. those with normal 8p (N-8p) and patients with 8q+ vs. those with normal 8q (N-8q).
Other cytogenetic abnormalities
At the time of chromosome 8 analysis by FISH, 48.3% of patients presented del(13q), 12.9% trisomy 12 and 5.7% del(11q). Among the 66 patients with available CBA, 53 (80.3%) presented an abnormal karyotype and 31 (47%) showed a complex karyotype (CK).
When comparing cases with 8p− or 8q+ versus those with N-8p or N-8q, respectively, no differences regarding del(13q), trisomy 12 and del(11q) frequencies were detected. Both the 8p− and the 8q+ groups carried a higher median percentage of 17p-deleted cells compared to cases with normal chromosome 8, although differences were only significant for the 8p− vs. N-8p comparison (87% vs. 50%, P = 0.001; 70% vs. 48%, P = 0.07). As for CBA, both the 8p− and the 8q+ group displayed a higher median number of chromosomal alterations (7 vs. 3, P = 0.041 and 6.5 vs. 2, P = 0.001 respectively), therefore displaying a higher frequency of CK (100% vs. 45.9%, P = 0.006 and 85.7% vs. 36.5%, P = 0.002 respectively) compared to N-8p and N-8q cases.
Demographic and baseline characteristics
When comparing 8p− vs. N-8p and 8q+ vs. N-8q cases the only statistically important differences concerned female prevalence (63.6% vs. 31.3%, P = 0.049) and lower hemoglobin levels (12.8 vs. 14 g/dL, P = 0.018) in the 8p− group.
Therapy
Overall, with a median follow up of 62 months (0–201), 81/97 cases (83.5%) had received treatment. Of note, in 34/93 cases (36.6%), therapy was administered prior to chromosome 8 testing. In 17/23 cases with 8p− and/or 8q+ (73.9%), these alterations were observed before treatment, whereas in the remaining six patients therapy-related acquisition could not be formally excluded as no relevant sample from the time of diagnosis was available for testing.
Among treatment-naïve cases, both 8p− and 8q+ groups exhibited a shorter time to first treatment (TTFT; 11 vs. 45 months for 8p− vs. N-8p and 15 vs. 45 months for 8q+ vs. N-8q) although differences did not reach statistical significance.
Survival analysis
With a median follow-up of 62 months (range: 0–201), 35 patients (34.6%) remained alive. Ten-year survival analysis revealed a significant shorter median OS for both 8p− and 8q+ patients compared with N-8p and N-8q group respectively (41 vs. 94 months; P = 0.002 and 38 vs. 92 months; P = 0.012 respectively) (Figure 1, Table 3). Interestingly, even though the number of cases with concomitant 8p− and 8q+ was low (n = 6), they exhibited a significant shorter OS compared to patients with isolated chromosome 8 alterations (26 vs. 88 months, P = 0.016). An elevated percentage of 17p-deleted cells also was related to inferior outcome, with the optimal cut-off being ≥80% of deleted cells (P < 0.001).
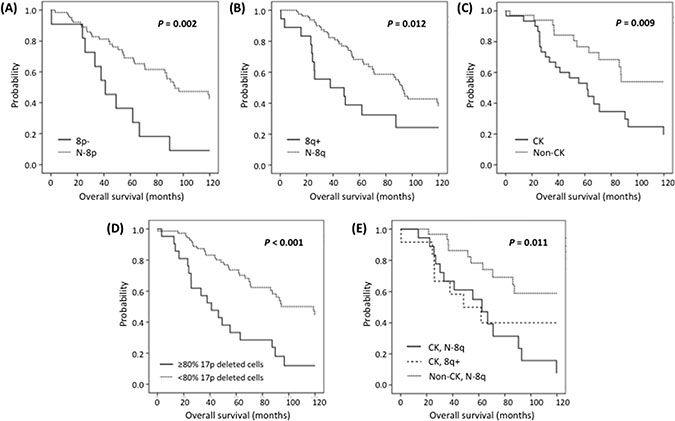
Figure 1: Kaplan-Meier plots for ten-year OS in patients carrying (A) 8p−, (B) 8q+, (C) CK, (D) ≥80% 17p deleted cells and (E) CK with 8q+ or N-8q.
Table 3: Univariate and multivariate analysis on ten-year overall survival
Variable |
Univariate |
Multivariate |
|||
|---|---|---|---|---|---|
Median OS (95% CI) in months |
P-value |
Hazard ratio (95% CI) |
P-value |
||
Loss of 8p by FISH |
8p− |
41 (24–58) |
0.002 |
NA |
NA |
N-8p |
94 (60–127) |
||||
Gain of 8q by FISH |
8q+ |
38 (0–85) |
0.012 |
1.23 (0.52–2.90) |
0.642 |
N-8q |
92 (84–100) |
||||
Complex karyotype |
Yes |
62 (40–83) |
0.009 |
2.47 (1.11–5.49) |
0.027 |
No |
NR |
||||
Percentage of cells with 17p deletion |
≥ 80% |
41 (21–61) |
< 0.001 |
2.21 (1.02–4.80) |
0.046 |
< 80% |
94 (60–129) |
||||
NA, not assessed; NR, not reached.
Regarding chromosomal abnormalities detected by CBA, patients with CK exhibited a shorter OS (P = 0.009), which was independent of the presence or absence of 8q+ (48 vs. 62 months, P = 0.538) (Figure 1). Similar results were obtained when cases analyzed after treatment were excluded from the statistical analysis (Supplementary Figure S2). On multivariate analysis including 8q+, ≥ 80% of 17p-deleted cells and CK only ≥ 80% of 17p-deleted cells (P = 0.046, HR = 2.2) and CK (P = 0.027, HR = 2.5) retained independent significance (Table 3). Multivariate analysis including 8p− cases could not be performed due to the low number of patients with available data from both CBA and 8p status.
DISCUSSION
Mounting evidence suggests that within CLL patients with TP53abs additional genetic features may influence the clinical outcome [17, 22, 23]. More specifically, alterations in chromosome 8 are more frequently detected in CLL patients with TP53abs and have been proposed as potentially prognostic markers in this aggressive group. Nevertheless, large studies focusing on the characterization and clinical outcome of this subset of patients are lacking. In order to address this issue, we retrospectively analyzed 101 CLL patients with TP53abs from our collaborating institutions. The frequency of 8p− was 14.7%, whereas 8q+ was detected in 17.8% of patients, being concomitant in 30% of cases with chromosome 8 alterations. To the best of our knowledge, this is the largest cohort of patients with TP53abs confirming the enrichment of chromosome 8 abnormalities in this disease subgroup. The frequencies reported herein are lower than those in previous studies, which, however, concerned significantly smaller cohorts, thus raising the possibility of selection bias [17, 19, 20].
The high percentage of de novo chromosome 8 alterations reported here suggests that 8p− and 8q+ can occur early in the course of the disease. This is in agreement with earlier reports of 8p− and 8q+ in treatment-naïve patients [20, 21]. Indeed, subsequent whole-exome sequencing studies analyzing clonal heterogeneity also suggested 8p− and 8q+ as early events during CLL development, whereas TP53 aberrations seem to appear later in the course of the disease [24].
Regarding the clinical characteristics of patients with alterations in chromosome 8 at diagnosis, no significant differences could be identified between groups except for gender distribution and hemoglobin levels. As for the genetic characteristics, we found a higher percentage of 17p-deleted nuclei in patients with 8p− or 8q+. Furthermore, in accordance with previous reports, a higher rate of CK was associated with both chromosome 8 abnormalities (8p− and 8q+) [25, 26]. Concerning clinical outcome, even though both 8p− and 8q+ exhibited a shorter TTFT, differences did not reach statistical significance. Nonetheless, a decreased OS was observed for both 8p− and 8q+, with concomitant 8p− and 8q+ cases exhibiting the worst outcome. Concerning patients with isolated 8p− or 8q+, given the small number of cases, we could not statistically assess their potential impact on survival. Our findings indicate that co-existence of 8p− and 8q+ may have a potential clinical impact in CLL and should be better evaluated in future research. However, further studies in larger patient cohorts are needed to discern the clinical consequences of isolated 8p− or 8q+.
Karyotypic complexity has been reported to be significantly associated with TP53abs [7, 25, 27] and to have a negative impact on survival even within this aggressive subset of patients [8, 23, 27]. Our present study confirms and significantly extends previous reports highlighting the impact of genetic instability in the pathogenesis of the disease. Indeed, although 89% of cases with 8p− and/or 8q+ showed a CK, in the multivariate analysis only karyotypic complexity and the percentage of 17p-deleted nuclei maintained the statistical significance. This finding suggests that CK rather than chromosome 8 alterations is responsible for the dismal clinical evolution of these patients. Chromosome 8 alterations could reflect underlying genomic instability leading to karyotypic complexity and could suggest the implication of different genes such as MYC in 8q+ cases and tumor suppressors such as TNFRSF10A/B (8p21) in 8p− cases [20, 28]. Notably, however, almost 40% of cases with CK did not show 8p− or 8q+ but displayed the same poor outcome as those with chromosome 8 alterations and CK.
In conclusion, our results highlight the association of chromosome 8 abnormalities with CK amongst CLL patients with TP53abs, while also revealing that CK can further aggravate the prognosis of this aggressive subgroup. These findings further underscore the relevance of chromosome banding analysis, which has the potential to refine prognosis even amongst cases with altered TP53.
MATERIALS AND METHODS
Patient selection
For the purpose of this study, we evaluated CLL patients with TP53abs and available Carnoy fixed tumoral cells from 17 collaborating institutions in Spain and Greece (Supplementary Figure S3). All patients met the iwCLL diagnostic criteria [29]. A total of 101 patients carrying TP53 abnormalities were identified: 92 cases with del(17p) detected by FISH and 9 additional cases with TP53 mutations identified by Sanger sequencing. Of note, six patients had been diagnosed of CLL-like monoclonal B cell lymphocytosis (MBL) but had already progressed to CLL at the time of the study. Treatment initiation and response assessment followed standard criteria [29, 30]. The study was performed in accordance with national and international guidelines (Professional Code of Conduct, Declaration of Helsinki) and approved by the Ethics Committee of Hospital del Mar, Barcelona (2013/5093/I).
Fluorescence in situ hybridization and chromosome banding analysis
Chromosome 8 alterations were assessed in the same Carnoy fixed tumoral cells (peripheral blood or bone marrow) which were used for the detection of TP53abs. Several samples were obtained from Parc de Salut MAR Biobank (MARBiobanc), Barcelona. The median time from diagnosis to study of chromosome 8 abnormalities was 15 months (range: 0–145) and 36.6% of the patients had received treatment prior to the analysis. Commercial FISH probes covering LPL (8p21) and MYC (8q24) genes (Abbott Molecular, Abbott Park, IL) were used in 75 and 101 patients, respectively. Moreover, data from routine FISH (D13S319, CEP12 and ATM) and CBA (72 h + TPA and/or CpG + IL2) were also available in 91 and 66 cases, respectively. Complex karyotypes (CK) were defined as the presence of three or more numerical/structural chromosomal abnormalities.
Statistical analysis
Chi-square or Fisher exact tests were employed for discrete variables, while comparisons of continuous variables were assessed by the Mann–Whitney test. TTFT was defined as the time from diagnosis to the beginning of treatment and OS as the time from diagnosis to death or last follow-up. Both parameters were evaluated using Kaplan-Meier plots within the first ten years of follow-up. The effect of different covariates was assessed employing the log-rank test. Multivariate analysis according to the Cox proportional hazards regression model was used to evaluate the maintenance of their independent predictive value. Statistical analyses were performed using SPSS v.22 software (SPSS Inc, Chicago, IL, USA). P-values below 0.05 were considered statistically significant. The optimal cut-off value for the percentage of cells harboring del(17p) was assessed using Maximally Selected Rank Statistics in R.
ACKNOWLEDGMENTS AND GRANT SUPPORT
The authors want to thank Marta Salido, Carme Melero, Ana Batlle, Mª Ángeles Piñán, Alicia Rodríguez and George Papaioannou for their contribution to the study providing clinical or laboratory data, Sergi Mojal for statistical review, and Grupo Cooperativo Español de Citogenética Hematológica (GCECGH) and Grupo Español de Leucemia Linfática Crónica (GELLC). This work has been supported by the following grants: PI11/01621, PI15/00437, RD09/0076/00036, RD12/0036/0044, RD12/0036/0069 and PT13/0010/0005 FEDER, Instituto de Salud Carlos III, Spanish Ministry of Economy and Competitiveness; 2014/SGR585 from Generalitat de Catalunya; “Xarxa de Bancs de tumors” sponsored by Pla Director d’Oncologia de Catalunya (XBTC) and Fundació La Caixa; the Swedish Cancer Society, the Swedish Research Council, the Lion’s Cancer Research Foundation, and Selander’s Foundation, Uppsala; H2020 “AEGLE, An analytics framework for integrated and personalized healthcare services in Europe” by the EU.
CONFLICTS OF INTEREST
The authors made no disclosure of conflicts of interest.
Authors’ contributions
A.P. and B.E. designed the research study. G.B., A.P. and B.E. provided patients data, analyzed the data and wrote the manuscript. P.B., A.X. and K.S. provided patients information, performed data analysis and critical discussion of the results. A.A., M.D.G., R.C., M.R.R., M.O., M.J.C., E.L., M.T.V., J.G., C.M.L., A.V., J.C., A.A., E.G., E.A., E.S., J.M.H., F.O., D.R., A.F., D.I., M.G., F.B. and P.A. provided patient data. All authors read the last version of the manuscript.
REFERENCES
1. Döhner H, Stilgenbauer S, Benner A, Leupolt E, Kröber A, Bullinger L, Döhner K, Bentz M, Lichter P. Genomic aberrations and survival in chronic lymphocytic leukemia. N Engl J Med. 2000; 343:1910–1916.
2. Van Dyke D, Werner L, Rassenti L, Neuberg D, Ghia E, Heerema N, Dal Cin P, Dell Aquila M, Sreekantaiah C, Greaves A, Kipps T, Kay N. The Dohner fluorescence in situ hybridization prognostic classification of chronic lymphocytic leukaemia (CLL): the CLL Research Consortium experience. Br J Haematol. 2016; 173:105–113.
3. Zenz T, Vollmer D, Trbusek M, Smardova J, Benner A, Soussi T, Helfrich H, Heuberger M, Hoth P, Fuge M, Denzel T, Häbe S, Malcikova J et al. TP53 mutation profile in chronic lymphocytic leukemia: evidence for a disease specific profile from a comprehensive analysis of 268 mutations. Leukemia. 2010; 24:2072–2079.
4. Baliakas P, Hadzidimitriou A, Sutton L, Rossi D, Minga E, Villamor N, Larrayoz M, Kminkova J, Agathangelidis A, Davis Z, Tausch E, Stalika E, Kantorova B et al. Recurrent mutations refine prognosis in chronic lymphocytic leukemia. Leukemia. 2015; 29:329–336.
5. Nadeu F, Delgado J, Royo C, Baumann T, Stankovic T, Pinyol M, Jares P, Navarro A, Martin-Garcia D, Bea S, Salaverria I, Oldreive C, Aymerich M et al. Clinical impact of clonal and subclonal TP53, SF3B1, BIRC3, NOTCH1, and ATM mutations in chronic lymphocytic leukemia. Blood. 2016; 127:2122–2130.
6. Zenz T, Habe S, Denzel T, Mohr J, Winkler D, Buhler A, Sarno A, Groner S, Mertens D, Busch R, Hallek M, Dohner H, Stilgenbauer S. Detailed analysis of p53 pathway defects in fludarabine-refractory chronic lymphocytic leukemia (CLL): dissecting the contribution of 17p deletion, TP53 mutation, p53-p21 dysfunction, and miR34a in a prospective clinical trial. Blood. 2009; 114:2589–2597.
7. Dicker F, Herholz H, Schnittger S, Nakao A, Patten N, Wu L, Kern W, Haferlach T, Haferlach C. The detection of TP53 mutations in chronic lymphocytic leukemia independently predicts rapid disease progression and is highly correlated with a complex aberrant karyotype. Leukemia. 2009; 23:117–124.
8. Baliakas P, Iskas M, Gardiner A, Davis Z, Plevova K, Nguyen-Khac F, Malcikova J, Anagnostopoulos A, Glide S, Mould S, Stepanovska K, Brejcha M, Belessi C et al. Chromosomal translocations and karyotype complexity in chronic lymphocytic leukemia: A systematic reappraisal of classic cytogenetic data. Am J Hematol. 2014; 89:249–255.
9. Puiggros A, Puigdecanet E, Salido M, Ferrer A, Abella E, Gimeno E, Nonell L, Herranz M, Galván A, Rodríguez-Rivera M, Melero C, Pairet S, Bellosillo B et al. Genomic arrays in chronic lymphocytic leukemia routine clinical practice: are we ready to substitute conventional cytogenetics and fluorescence in situ hybridization techniques? Leuk Lymphoma. 2013; 54:986–995.
10. Jain N, O’Brien S. Initial treatment of CLL: integrating biology and functional status. Blood. 2015; 126:463–470.
11. Furman R, Sharman J, Coutre S, Cheson B, Pagel J, Hillmen P, Barrientos J, Zelenetz A, Kipps T, Flinn I, Ghia P, Eradat H, Ervin T et al. Idelalisib and rituximab in relapsed chronic lymphocytic leukemia. N Engl J Med. 2014; 370:997–1007.
12. Byrd J, Furman R, Coutre S, Burger J, Blum K, Coleman M, Wierda W, Jones J, Zhao W, Heerema N, Johnson A, Shaw Y, Bilotti E et al. Three-year follow-up of treatment-naive and previously treated patients with CLL and SLL receiving single-agent ibrutinib. Blood. 2015; 125:2497–2506.
13. Farooqui M, Valdez J, Martyr S, Aue G, Saba N, Niemann C, Herman S, Tian X, Marti G, Soto S, Hughes T, Jones J, Lipsky A et al. Ibrutinib for previously untreated and relapsed or refractory chronic lymphocytic leukaemia with TP53 aberrations: a phase 2, single-arm trial. Lancet Oncol. 2015; 16:169–176.
14. Tam C, Shanafelt T, Wierda W, Abruzzo L, Van Dyke D, O’Brien S, Ferrajoli A, Lerner S, Lynn A, Kay N, Keating M. De novo deletion 17p13.1 chronic lymphocytic leukemia shows significant clinical heterogeneity: the M. D. Anderson and Mayo Clinic experience. Blood. 2009; 114:957–964.
15. Best O, Gardiner A, Davis Z, Tracy I, Ibbotson R, Majid A, Dyer M, Oscier D. A subset of Binet stage A CLL patients with TP53 abnormalities and mutated IGHV genes have stable disease. Leukemia. 2009; 23:212–214.
16. Tyybäkinoja A, Vilpo J, Knuutila S. High-resolution oligonucleotide array-CGH pinpoints genes involved in cryptic losses in chronic lymphocytic leukemia. Cytogenet Genome Res. 2007; 118:8–12.
17. Forconi F, Rinaldi A, Kwee I, Sozzi E, Raspadori D, Rancoita P, Scandurra M, Rossi D, Deambrogi C, Capello D, Zucca E, Marconi D, Bomben R et al. Genome-wide DNA analysis identifies recurrent imbalances predicting outcome in chronic lymphocytic leukaemia with 17p deletion. Br J Haematol. 2008; 143:532–536.
18. O’Malley D, Giudice C, Chang A, Chang D, Barry T, Hibbard M, Chen R, Chen S. Comparison of array comparative genomic hybridization (aCGH) to FISH and cytogenetics in prognostic evaluation of chronic lymphocytic leukemia. Int J Lab Hematol. 2011; 33:238–244.
19. Rinaldi A, Mian M, Kwee I, Rossi D, Deambrogi C, Mensah A, Forconi F, Spina V, Cencini E, Drandi D, Ladetto M, Santachiara R, Marasca R et al. Genome-wide DNA profiling better defines the prognosis of chronic lymphocytic leukaemia. Br J Haematol. 2011; 154:590–599.
20. Brown J, Hanna M, Tesar B, Werner L, Pochet N, Asara J, Wang Y, dal Cin P, Fernandes S, Thompson C, MacConaill L, Wu C, Van de Peer Y et al. Integrative genomic analysis implicates gain of PIK3CA at 3q26 and MYC at 8q24 in chronic lymphocytic leukemia. Clin Cancer Res. 2012; 18:3791–3802.
21. Houldsworth J, Guttapalli A, Thodima V, Yan X, Mendiratta G, Zielonka T, Nanjangud G, Chen W, Patil S, Mato A, Brown J, Rai K, Chiorazzi N, et al. Genomic imbalance defines three prognostic groups for risk stratification of patients with chronic lymphocytic leukemia. Leuk Lymphoma. 2013; 55:920–928.
22. Delgado J, Espinet B, Oliveira A, Abrisqueta P, de la Serna J, Collado R, Loscertales J, Lopez M, Hernandez-Rivas J, Ferra C, Ramirez A, Roncero J, Lopez C et al. Chronic lymphocytic leukaemia with 17p deletion: a retrospective analysis of prognostic factors and therapy results. Br J Haematol. 2012; 157:67–74.
23. Delgado J, Salaverria I, Baumann T, Martinez-Trillos A, Lee E, Jimenez L, Navarro A, Royo C, Santacruz R, Lopez C, Payer A, Colado E, Gonzalez M et al. Genomic complexity and IGHV mutational status are key predictors of outcome of chronic lymphocytic leukemia patients with TP53 disruption. Haematologica. 2014; 99: e231-e234.
24. Landau D, Tausch E, Taylor-Weiner A, Stewart C, Reiter J, Bahlo J, Kluth S, Bozic I, Lawrence M, Böttcher S, Carter S, Cibulskis K, Mertens D et al. Mutations driving CLL and their evolution in progression and relapse. Nature. 2015; 526:525–530.
25. Haferlach C, Dicker F, Schnittger S, Kern W, Haferlach T. Comprehensive genetic characterization of CLL: a study on 506 cases analysed with chromosome banding analysis, interphase FISH, IgVH status and immunophenotyping. Leukemia. 2007; 21:2442–2451.
26. Travella A, Ripollés L, Aventin A, Rodríguez A, Bezares R, Caballín M, Slavutsky I. Structural alterations in chronic lymphocytic leukaemia. Cytogenetic and FISH analysis. Hematol Oncol. 2013; 31:79–87.
27. Jaglowski S, Ruppert A, Heerema N, Bingman A, Flynn J, Grever M, Jones J, Elder P, Devine S, Byrd J, Andritsos L. Complex karyotype predicts for inferior outcomes following reduced-intensity conditioning allogeneic transplant for chronic lymphocytic leukaemia. Br J Haematol. 2012; 159:82–87.
28. Rubio-Moscardo F, Blesa D, Mestre C, Siebert R, Balasas T, Benito A, Rosenwald A, Climent J, Martinez JI, Schilhabel M, Karran EL, Gesk S, Esteller M et al. Characterization of 8p21.3 chromosomal deletions in B-cell lymphoma: TRAIL-R1 and TRAIL-R2 as candidate dosage-dependent tumor suppressor genes. Blood. 2005; 106:3214–3222.
29. Hallek M, Cheson B, Catovsky D, Caligaris-Cappio F, Dighiero G, Dohner H, Hillmen P, Keating M, Montserrat E, Rai K, Kipps T. Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: a report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute-Working Group 1996 guidelines. Blood. 2008; 111:5446–5456.
30. Cheson BD, Bennett JM, Grever M, Kay N, Keating MJ, O’Brien S, Rai KR. National Cancer Institute-sponsored Working Group guidelines for chronic lymphocytic leukemia: revised guidelines for diagnosis and treatment. Blood. 1996; 87:4990–4997.