
INTRODUCTION
Chemotherapy has been the mainstay of cancer treatment for the past several decades. Yet, there is little evidence for progress in reducing cancer mortality, because most of the patients ultimately relapse. The recent discovery of cancer stem/tumor-initiating cells (jointly CSCs) has changed our view of carcinogenesis and chemoresistance [1]. In contrast to other tumor cells, CSCs represent a subpopulation that possesses the ability to self-renew and differentiate, properties that render these cells particularly resistant to therapy. As such, it is conceivable that the relapse after remission is likely due to the inability of current chemotherapeutic drugs to kill CSCs. Despite the tumor bulk elimination, CSCs are capable of reproducing the entire tumor. Thus, strategies aiming at targeting CSCs represent rational approaches for cancer prevention and treatment. Plant-derived small molecules that exhibit potent antitumor activity but low toxicity and limited side effects could be promising new treatment modalities.
Therapeutic values of ginseng (the rhizome of Panax ginseng), a key ingredient in Chinese traditional medicine, have been recognized for >1,000 years [2]. It is now one of the most popular alternative medicines widely consumed worldwide and appears in the pharmacopeias of several countries, including the United States and Europe and employed for cancer, diabetes, and cardiovascular concerns. In recent years, ginseng has gained considered attention for the prevention and/or treatment of cancers in epidemiological, basic, and clinical research. This is supported by the findings on its ability to reduce the incidence of carcinogen-induced tumors in mice and to inhibit the growth of tumor cells in vitro [3]. A dose-dependent decrease in the risk of cancer as a regular consumption of ginseng in both prospective and case-control studies and a better survival rate and a greater quality of life are also noted [4]. Notably, such effects are not observed in patients using other Chinese traditional medicine [5]. This opens up the exciting possibility of ginseng as an important drug, emphasizing the importance to uncover the exact component(s) in the ginseng extract that contribute to the antitumor function and evaluate its pharmacological action.
Emerging evidence shows that nonpeptide small molecules, such as saponins, exhibit potent cytotoxicity, with great potential to be developed as chemotherapeutic agents [6]. A notable saponin isolated from P. quinquefolius and notoginseng is ginsenoside-Rb1, which constitutes 0.37-0.5% of ginseng extract [7, 8]. Most (~70%) orally administered Rb1 is metabolized by intestinal bacteria to its final derivative 20-O-β-D-glucopyranosyl-20(S)-protopanaxadiol (also called compound K) [9]. Compound K is reported to be easily absorbed and sustained longer in the human body. Nevertheless, the key targets of ginsenoside Rb1 and its metabolite compound K have not been explored. Nor is it clear about the molecular mechanisms.
In this study, we show for the first time that Rb1 and its metabolite compound K specifically target the formation and expansion of CSCs. We further provide evidence that Rb1 and compound K can chemosensitize CSCs to clinical anticancer drugs cisplatin and paclitaxel, inducing a synergistic cytotoxicity via Wnt/β-catenin signaling and epithelial-to-mesenchymal transition (EMT) regulation, both attractive targets for cancer treatment.
RESULTS
Cytotoxic and anti-proliferative effects of Rb1 and its metabolite compound K on ovarian CSCs
Ovarian cancer is a highly chemoresistant cancer that is rapidly fatal and most patients will develop tumor recurrence and succumb to chemoresistant disease. Thus, it provides an excellent model to identify the mechanisms required for this drug resistance. Using a functional enrichment strategy based on the self-renewal ability of CSCs to grow as nonadherent spheres under stem-cell-selective condition which recapitulates advanced stages of ovarian carcinoma cells in malignant ascites, we have successfully identified CSCs in ovarian cancer cell lines and cancerous ovarian tissues [10]. Here we first investigated the possible cytotoxic effect of Rb1 and its metabolite compound K on SKOV-3 and HEYA8 CSCs. Figure 1A shows dose-dependent effect of Rb1 and compound K on both line-derived CSCs on tumor sphere formation and growth. In addition, the spheres formed upon Rb1 or compound K treatment were smaller compared with the control (Figure 1). Both Rb1 and compound K suppressed tumor cell survival (Figure 1A). The LC50 (the concentration that leads to 50% survival) were 250 nM for Rb1 and 100 nM compound K in SKOV-3 and 230 nM for Rb1 and 125 nM for compound K in HEYA8, respectively. A clinically relevant dose of imatinib also gave similar results (Figure 1B) [10]. In accordance with this, trypan blue exclusion assay showed a 1.5-fold increase in cell death for Rb1 and compound K in SKOV-3 and a 1.4- and 2-fold increase in HEYA8, respectively (Figure 2A). Furthermore, Rb1 and compound K displayed a significant 5.9- and 9.6-fold in SKOV-3 and 1.6 and 2.9- fold in HEYA8 cells increase in apoptosis as revealed by the expression of the active (cleaved) caspase 3 (Figure 2B), suggesting that apoptosis may account for this loss of cell viability. Subsequently, CSCs were treated with 250 nM Rb1 and 125 nM compound K for different periods of time (0, 24, and 48 h). We evaluated CSC marker expression in response to Rb1 or compound K treatment. Three different markers of ovarian CSCs, Bmi-1, Nanog, and Oct4, were tested. Rb1 or compound K depleted all these markers expression in a time-dependent manner, with the maximal effects observed 48 hours following treatment (Figure 3A), reflecting Rb1- and compound K-dependent inhibition of CSC self-renewal and growth of chemotherapy-resistant CSCs.
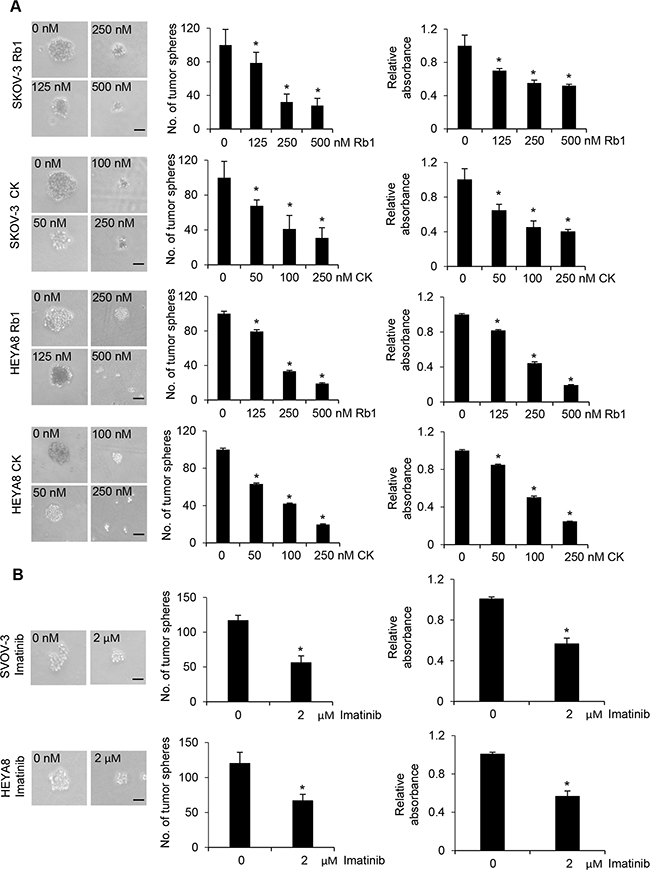
Figure 1: Rb1 and its metabolite compound K inhibit self-renewal and growth of CSCs. A. The number of tumor spheres generated were photographed (left) and counted (right). In parallel experiments, cell viability was determined by MTT assay. The absorbance of wells not exposed to Rb1 or compound K (CK) treatment was arbitrarily set as 1, and cell growth after Rb1 or CK treatment was expressed as the fold changes compared to the control. B. Cell viability was determined after imatinib treatment. Bar, 50 μm. Experiments were repeated three times, and data are shown as mean ± SD. *, P < 0.05 vs control.
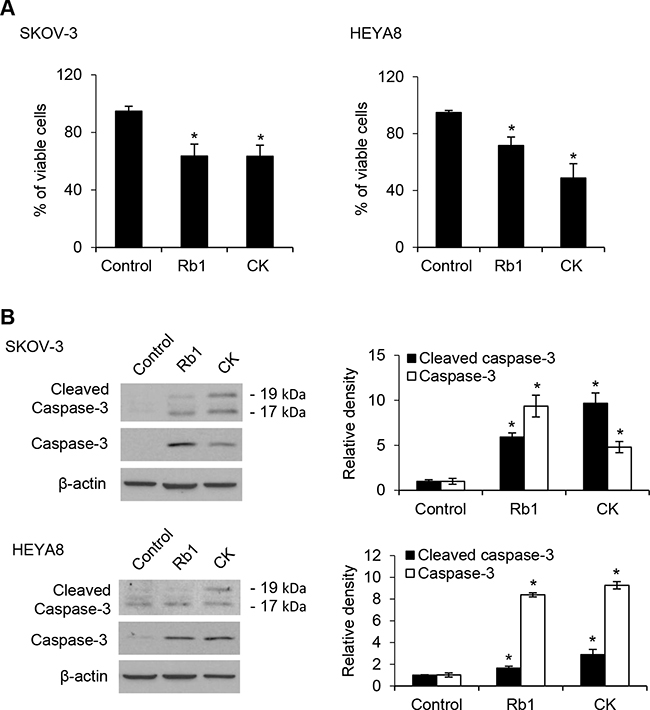
Figure 2: Rb1 and its metabolite compound K induce apoptosis of CSCs. A. Viable spheres were counted using trypan blue after Rb1 or CK treatment. B. The expression of caspase-3 and the active (cleaved) proapoptotic gene caspase 3 were determined by Western blot after Rb1 or compound K treatment. Experiments were repeated three times, and data are shown as mean ± SD. *, P < 0.05 vs control.
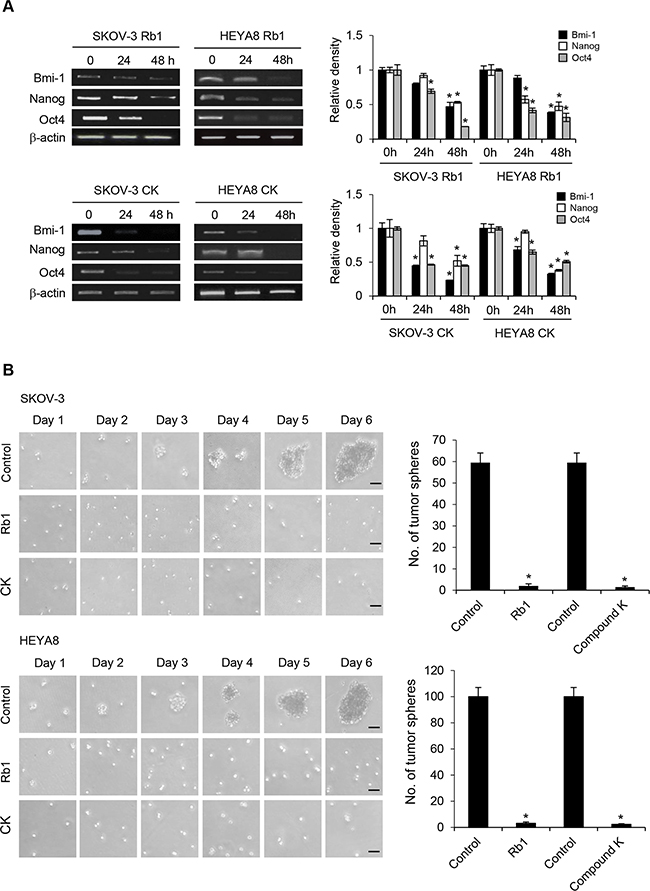
Figure 3: No regrowth/relapse in Rb1- or compound K-treated SKOV-3 and HEYA8 spheroids. A. Expression of CSC markers Bmi-1, Nanog, and Oct4 after Rb1 or CK treatment. β-actin serves as an internal control. The signal intensity was determined by densitometry. B. SKOV-3 and HEYA8 spheroids untreated or treated with Rb1 or compound K (CK) were plated as secondary spheroids. Representative views of secondary spheroids imaged until day 6 (right). The number of tumor spheres generated was photographed and counted (left). Bar, 50 μm. Experiments were repeated three times, and data are shown as mean ± SD. *, P < 0.05 vs control.
No regrowth (relapse) upon Rb1 and its metabolite compound K treatment
Because sphere forming capabilities in serial passages is an indirect marker in support of stem cell renewal, we further determined the effect of Rb1 and compound K in relapse experiment. SKOV-3 and HEYA8 primary spheroids treated with either control vehicle, 250 nM Rb1, or 125 nM compound K were dissociated and replated as secondary spheroids. By day 2, cells from the control group started growing as secondary spheroids, whereas Rb1- or compound K-treated cells did not (Figure 3B). For approximately 6 days, secondary spheroids did not form from Rb1- or compound K-treated samples, whereas control samples developed dense secondary/tertiary spheroids (Figure 3B). Furthermore, the removal of these compounds did not restore the ability of both SKOV-3 and HEYA8 to form spheroids upon serial passage (data not shown). These results suggest that Rb1 and compound K inhibit CSC self-renewal, and that this process is irreversible.
Rb1 and its metabolite compound K reduce CSC resistance to chemotherapeutic drugs
The higher drug resistance of CSCs encouraged us to assess whether Rb1 and compound K will be effective in increasing the treatment efficacy. Rb1 or compound K was used alone or in combination with cisplatin/paclitaxel, the two front-line chemotherapeutic agents currently used for treating unresectable ovarian cancer. We found cisplatin and paclitaxel at clinically relevant does of 50 μM and 100 nM had little effect of SKOV-3 and HEYA8 CSCs [11], and only a partial decrease in sphere formation was observed (Figure 4A). On the other hand, Rb1 or compound K in combination with the same doses of cisplatin or paclitaxel sensitizes these CSCs to both drugs (SKOV-3: Rb1, Rb1 + cisplatin, and Rb1+ paclitaxel, P < 0.0001; compound K, compound K + cisplatin, and compound K + paclitaxel, P < 0.0001; HEYA8: Rb1, P = 0.004, Rb1 + cisplatin, and Rb1 + paclitaxel, P < 0.0001; compound K, P = 0.004, compound K + cisplatin, P < 0.0001, and compound K + paclitaxel, P = 0.001) (Figure 4A). In addition, treatment of CSCs isolated from primary ovarian cancer samples inhibited both sphere formation and growth, demonstrating drug-induced death by Rb1 or compound K or in combination with cisplatin or paclitaxel consistently supported the in vitro observations (Rb1, CK, Rb1 + cisplatin, Rb1 + paclitaxel, compound K + cisplatin and compound K + paclitaxel, P < 0.0001) (Figure 4B).
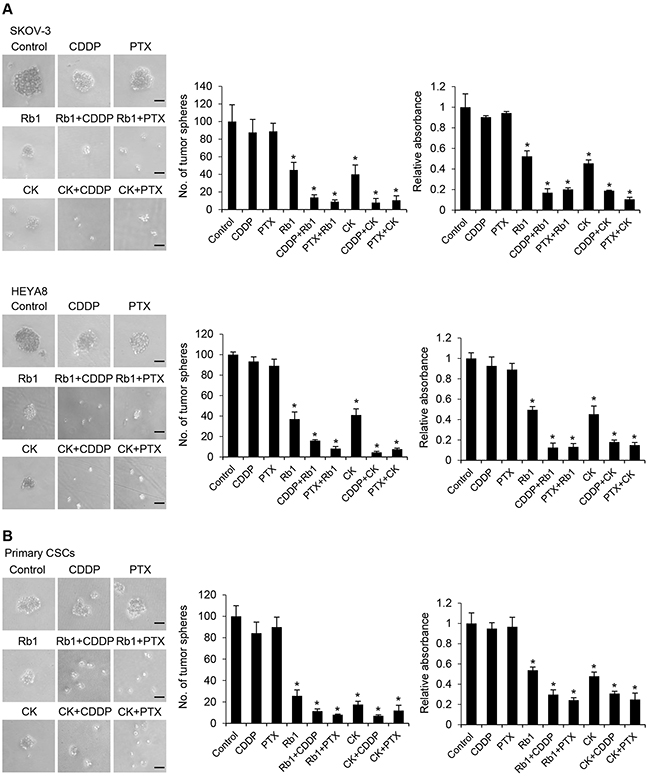
Figure 4: Rb1 and its metabolite compound K reduce chemoresistance of CSCs. A. SKOV-3 or HEYA8 CSCs or B. CSCs isolated from primary ovarian tumors were untreated or treated with cisplatin (CDDP; 50 μM) or paclitaxel (PTX; 100 nM) alone or in combination with Rb1 (250 nM) or compound K (CK) (125 nM) for 24 h. The number of tumor spheres generated were photographed (left) and counted (right). Bar, 50 μm. In parallel experiments, cell viability was determined by MTT assay. The absorbance of wells not exposed to Rb1 or CK treatment was arbitrarily set as 1, and cell growth after Rb1 or CK treatment was expressed as the fold changes compared to the control. Experiments were repeated three times, and data are shown as mean ± SD. *, P < 0.05 vs control.
Compound K inhibits EMT to exert cytotoxic effects
Rb1 is readily metabolized to compound K in the gut, and this biotransformation appears to contribute to the reported pharmacological effects of Rb1 [9]. We therefore focus on the molecular mechanisms underlying the action of compound K for the relevance. Emerging evidence suggests an intricate role of EMT in CSC self-renewal [12]. To this end, we first determined the levels of EMT transcription factors. Our results showed that treatment with compound K in the presence of cisplatin and paclitaxel was associated with a marked decrease in the expression of Snail and Slug (Figure 5B). Furthermore, in correlation with our findings from Snail/Slug, we also observed compound K substantially reverted the inhibition of E-cadherin expression, an epithelial marker, and its concomitant increase of N-cadherin expression, a mesenchymal marker (Figure 5B). Similar experiments with Rb1 revealed changes in expression indistinguishable from those in compound K (data not shown). We next determined the molecular mechanisms by which compound K inhibited Snail/Slug expression. Because studies have shown that the PI3K/Akt and ERK1/2 mitogen-activated protein kinase pathways are critical for the activation of Snail/Slug in some cell types [13, 14], we first examined the levels of phosphorylated (active) forms of Akt and ERK1/2 in SKOV-3 and HEYA8 cells treated with compound K. As shown, addition of compound K to the cells significantly decreased the phosphorylation of Akt and ERK1/2 (Figure 6A). To assess the functional significance of compound K-suppressed Akt and ERK1/2 in Snail/Slug expression, we used constitutively active constructs of Akt (E17K) and MEK1 (CA-MEK1). Both Snail and Slug expression were clearly reverted in E17K- and CA-MEK1-transfected cells upon compound K treatment (Figure 6B). These results reveal a key role of Rb1 and compound K in combination with cisplatin and paclitaxel in EMT regulation and show that these compounds also have a role in EMT-regulated CSCs.
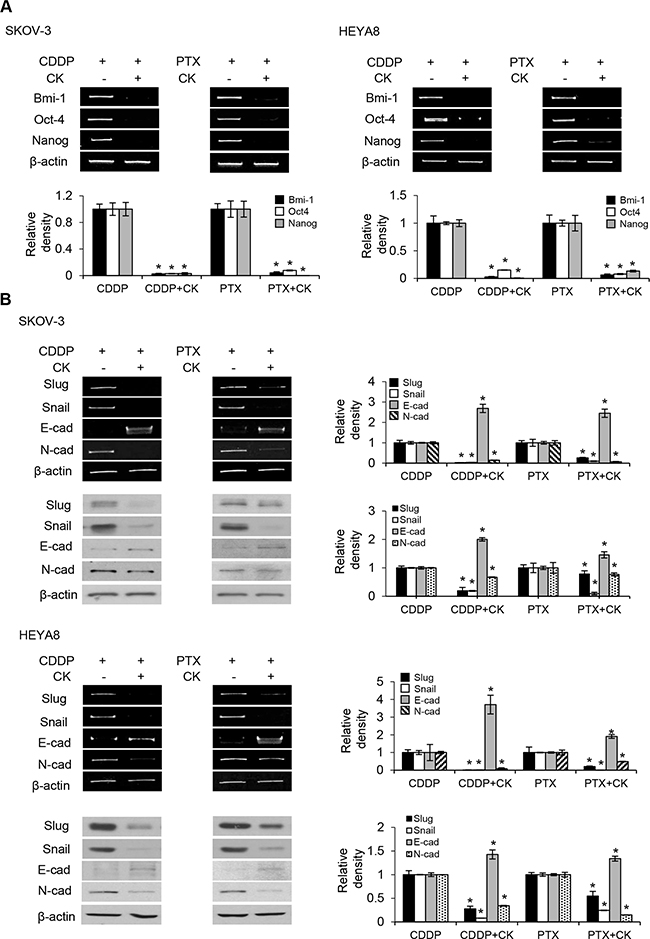
Figure 5: Compound K chemosensitizes CSCs via EMT. A. Expression of CSC markers Bmi-1, Nanog, and Oct-4 or B. EMT markers Slug, Snail, E-cadherin, and N-cadherin were determined by RT-PCR or western blot after using cisplatin (CDDP; 50 μM) or paclitaxel (PTX; 100 nM) alone or in combination with compound K (CK) for 24 h. β-actin serves as an internal control. The signal intensity was determined by densitometry. Data are shown as mean ± SD. *, P < 0.05 vs control.
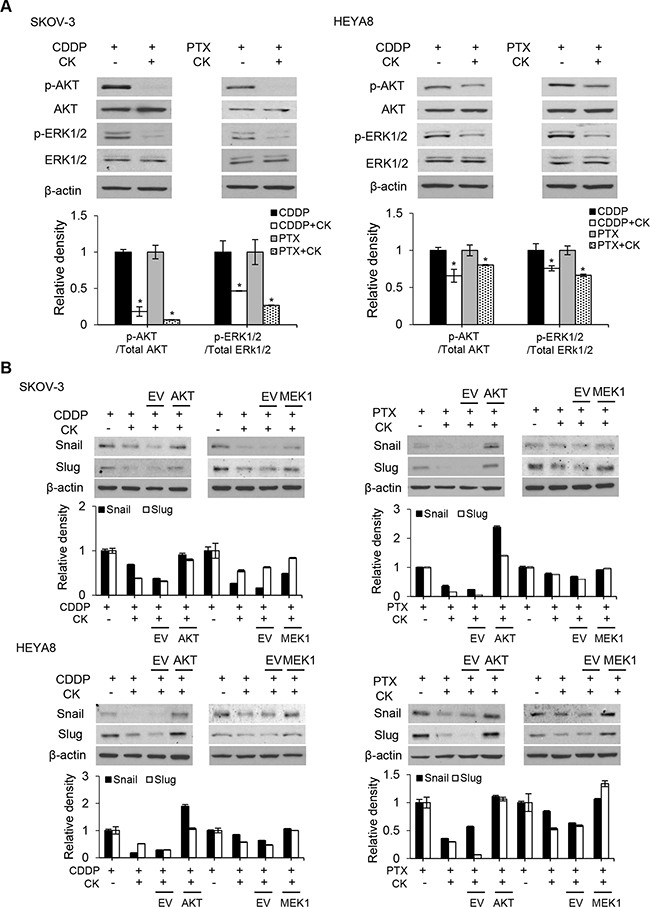
Figure 6: Compound K inhibits EMT through regulating Akt and MAPK signaling pathways. A. Phosphorylated (active) and total forms of Akt and MAPK family member ERK1/2 were detected by western blot. *, P < 0.05 vs control. B. Snail and Slug expression were observed after transfection with empty vector (EV), or constitutively active constructs of AKT or MEK1 after treated with CDDP or PTX alone, or in combination with CK. β-actin serves as an internal control. The signal intensity was determined by densitometry. Data are shown as mean ± SD.
Compound K inhibits the Wnt/β-catenin signaling pathway in CSCs
Next, we inquired the mechanism by which compound K inhibits CSC survival and its possible role in chemoresistance. β-catenin, which plays a pivotal role in self-renewal or tumorigenesis, is a particular intriguing property [15]. Since the elevated levels of β-catenin could direct cell fate [16], we examined the expression of β-catenin in compound K treated CSCs by Western blot analysis. We observed a marked decrease of β-catenin levels upon compound K treatment (Figure 7A). β-catenin is known to interact with T-cell factor (TCF)/LEF to activate the transcription of target genes [17], and evidence exists that correlate increased levels of β-catenin with increased levels of nuclear active β-catenin [18]. Hence, we studied the effect of compound K on the transcriptional activity of β-catenin/TCF using luciferase reporter plasmids encoding multimerized wildtype (TOPFLASH) or mutated (FOPFLASH) TCF binding sites, which have been widely used to characterize β-catenin/TCF-dependent gene expression [19]. In support of the observation, compound K treatment significantly inhibited TOPFLASH activation (Figure 7B) Importantly, compound K led to a sharp decrease in the expression of ABCG2 and P-glycoprotein, two major drug transporters that are implicated in the efflux chemotherapeutic drugs and are targets of β-catenin/TCF [10, 12]. The levels of ABCG2 and P-glycoprotein were reduced 12.8 and 12.5-fold in compound K treatment for SKOV-3, and 22.4 and 38.4-fold in compound K treatment for HEYA8, respectively (Figure 8A). Likewise, the chemosensitizing effect in CSCs by compound K could be reversed by expression of a stable mutant form of β-catenin (S37A) (Figure 8B). There was also a clear decrease of drug-induced cell death by the expression of constitutively active TCF (VP16-TCF) (Figure 8B). These results present the first evidence that compound K inhibit the Wnt/β-catenin signaling in CSC cells, and that these effects were mediated through its downstream CSC-driven effectors ABCG2 and P-glycoprotein.
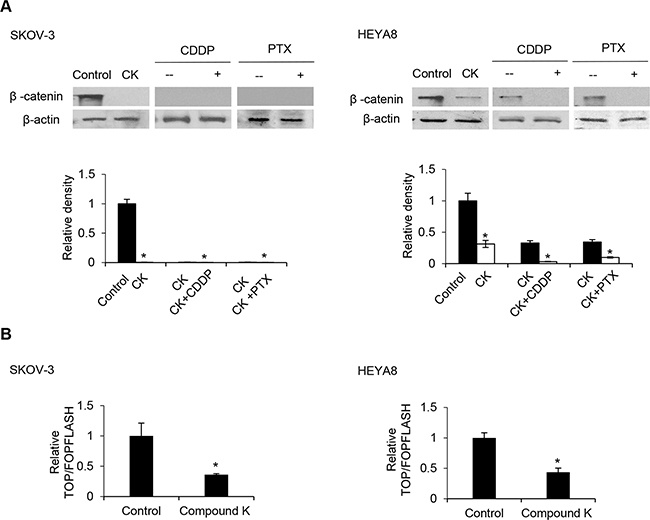
Figure 7: Compound K enhances the chemosensitizing effect through the β-catenin/TCF signaling pathway. A. Protein levels of β-catenin were analyzed by Western blot and β-actin serves as an internal control. The signal intensity was determined by densitometry. B. Cells were transfected with 0.25 μg of either the TOPFLASH or FOPFLASH luciferase reporter constructs containing wildtype and mutant TCF binding sites, respectively, together with 0.5 μg β-galactosidase for normalization of transfection efficiency. Values are normalized luciferase activity (TOPFLASH activity minus the activity devoted to FOPFLASH and normalized to β-galactosidase activity). The absorbance of wells not exposed to compound K (CK) treatment was arbitrarily set as 1. Data are shown as mean ± SD. *, P < 0.05 vs control.
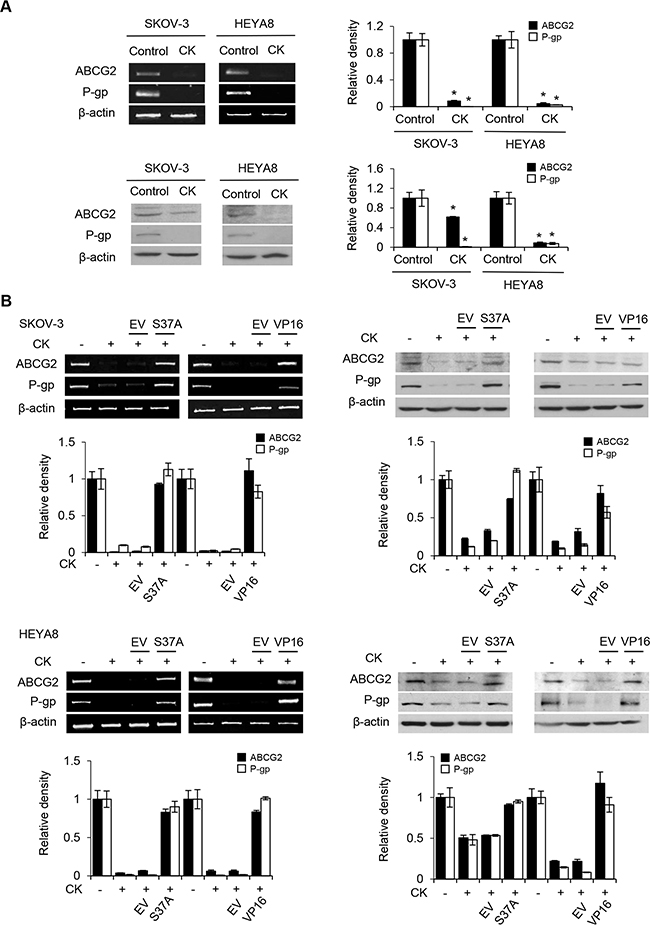
Figure 8: β-catenin activation induces ABCG2 and P-glycoprotein expression. A. CSCs were treated with compound K (CK) alone. The expression of ABCG2 and P-glycoprotein were determined by RT-PCR and Western blotting, β-actin serves as an internal control. The signal intensity was determined by densitometry. B. CSCs treated with CK were transfected with either empty vector (EV), β-catenin S37A, or VP16-TCF construct in the absence or presence of CK. The expression of ABCG2 and P-glycoprotein were determined by RT-PCR and western blot, and β-actin serves as an internal control. The absorbance of wells not exposed to CK treatment was arbitrarily set as 1. Data are shown as mean ± SD. *, P < 0.05 vs control.
Efficacy of compound K and chemotherapy in CSCs in vivo
To further validate its role in vivo, we used mouse xenograft ovarian tumors. Treatment with compound K alone was effective in inhibiting tumor growth compared with vehicle control (Figure 9A). However, treatment with compound K in combination with cisplatin or paclitaxel resulted in even greater reduction in tumor volume (compound K + cisplatin, 50.2%, P < 0.05; compound K + paclitaxel, 45.6%, P < 0.05). Furthermore, the combination therapy was statistically superior to cisplatin or paclitaxel alone (compound K + cisplatin, compound K + paclitaxel, P < 0.05) (Figure 9A). Further, compound K also induced caspase-3 expression in vivo, indicating apoptosis activation in compound K-treated HEYA8 xenografts (Figure 9C). These results demonstrate that compound K is potent in suppressing tumor growth from CSCs in vivo and that these effects are cytotoxic rather than cytostatic. The compound K regimens were well-tolerated to the tested animals. There was no abnormal behavior or weight loss noted during the entire treatment period (Figure 9B). Moreover, histopathological observations of the liver, heart, kidney, spleen, and lungs in the treated animals showed no histological alterations as compared to the controls (Figure 10A). Blood chemistry showed that compound K did not cause a significant increase in lactate dehydrogenase (LDH), aspartate aminotransferase (AST), alanine transaminase (ALT), alkaline phosphatase (ALP), creatine kinase (CK) and blood urea nitrogen (BUN) (Figure 10B), hence showing that compound K has no adverse effects to the liver, heart, and renal function.
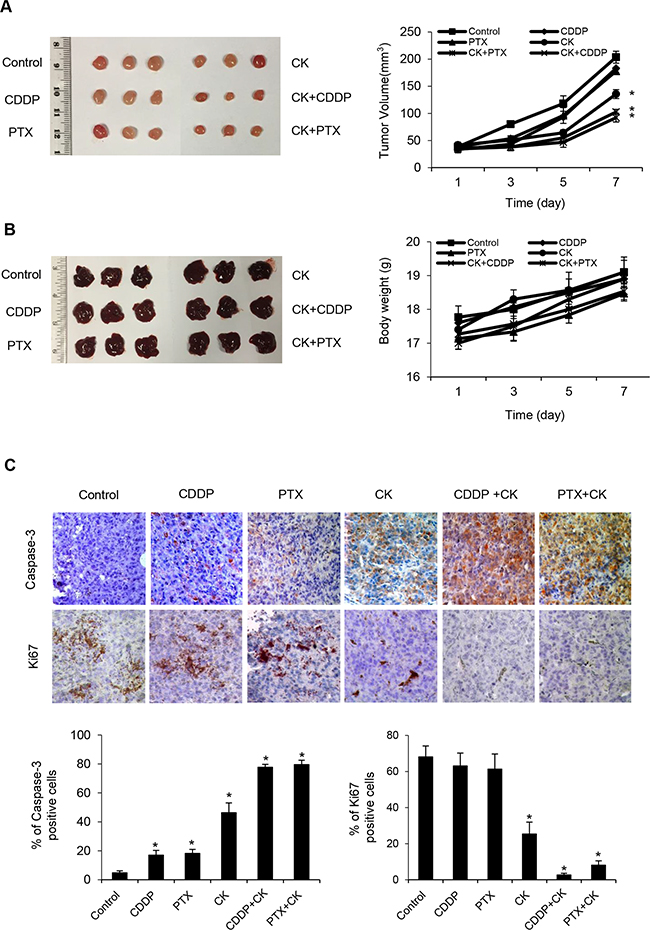
Figure 9: Therapeutic efficacy of compound K. Nude mice were injected s.c. with HEYA8 cells (106) and randomly allocated to one of the six treatment groups (n = 3, repeated three times): (i) vehicle, (ii) compound K (CK), (iii) cisplatin (CDDP), (iv) paclitaxel (PTX), (v) CK + CDDP, (vi) CK + PTX. When control animals (vehicle) began to appear moribund (28 days), all animals in an experiment were sacrificed and photographed, and A. tumor volume were measured. B. Mice weight and macroscopic aspects of characteristic livers were recorded. C. Immunohistochemical analysis of caspase-3 and Ki67 expression in tumor tissues (200x). Data are shown as mean + SEM (A, B) and mean ± SD (C). *, P < 0.05 vs control.
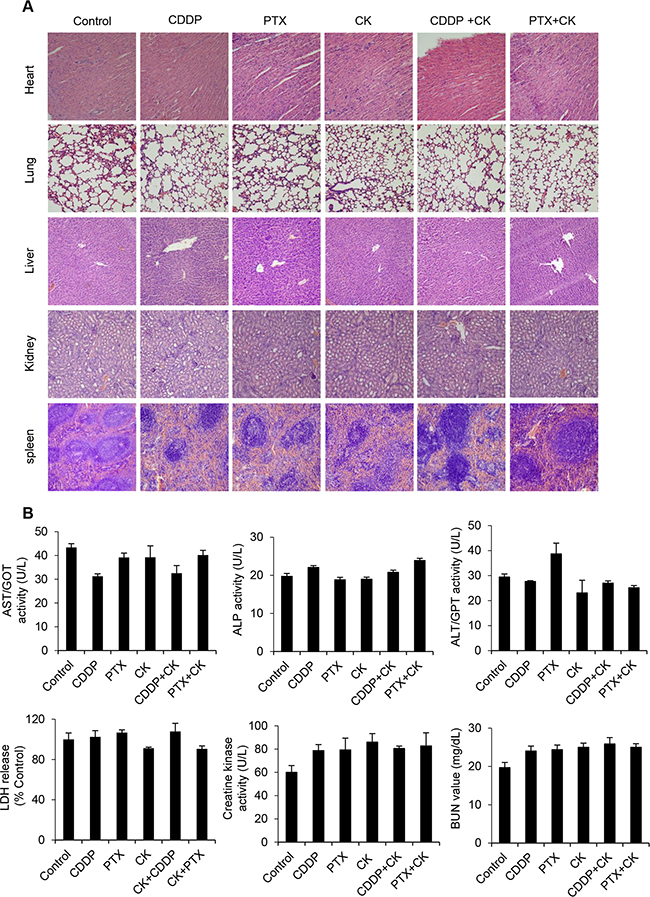
Figure 10: Compound K has no adverse effects. A. H&E staining of heart, lungs, liver, spleen and kidney tissues (200x). B. Peripheral blood was collected from retro-orbital sinus at sacrifice and plasma lactate dehydrogenase (LDH), aspartate aminotransferase (AST), alanine transaminase (ALT), alkaline phosphatase (ALP), creatine kinase and blood urea nitrogen (BUN) were measured and compared to vehicle injected mice. Data are shown as mean ± SD. *, P < 0.05 vs control.
DISCUSSION
Tumor cell resistance to chemotherapies still forms a major barrier toward effective cancer therapy. This paper presents two important findings. First, Rb1 and its metabolite compound K inhibit CSCs. Second, these compounds enhance cisplatin and paclitaxel anti-cancer effect. These effects are at least in part due to inhibition of the Wnt/β-catenin signaling pathway and EMT. To our knowledge, CSCs chemosensitization has not been documented in ginseng as well as in other traditional Chinese medicine. Our studies represent the first to report the phenomenon and elucidate the signaling mechanisms involved. The combination of these natural products and chemotherapeutic drugs could be of great therapeutic value if included in the treatment regimens.
Ginseng is listed in pharmacopeias and has been widely used in China for medicinal purposes for thousands of years due to its rich content of saponins. Ginsenoside is one of the most widely characterized saponins in the rhizome of ginseng. Ginseng also has a chemopreventive potential in the context of ovarian cancer as seen in a mouse model [5]. For example, it has been demonstrated that ginsenoside Rg3 in combination with cyclophosphamide could improve the living quality and survival time of mice with ovarian tumors [20]. Moreover, case-control studies have pointed toward preventive effects of ginseng products had a preventive effect on ovarian cancer [21]. These results suggest that ginseng might be an ideal cancer chemotherapeutic agent.
Several activities of Rb1 make it desirable both as a therapeutic and as a chemopreventive agent: (a) Unlike other saponins, which often have poor absorption and rapid excretion, Rb1 seems to exhibit a better bioavailability and a desirable duration of action [22]. (b) Rb1 disrupts many of the characteristic cancer promoting activities [23]. (c) In addition, Rb1 has shown to improve the functions of normal cells, such as cardioprotective, hepatoprotective and anti-inflammatory activities, which provide definite advantages than currently used drugs [24–27]. Rb1 has various anti-cancer properties ranging from the inhibition of cell proliferation to angiogenesis as well as in apoptosis induction [28, 29]. Structure-activity analyses suggest that ginsenosides metabolites with less sugar moieties have greater anti-cancer activities. Indeed, among all ginsenosides, compound K (one sugar residue) is found to possess the most potent tumor growth and invasion suppressing activities [30]. We observed that compound K possesses very significant anti-CSC activities as compared to Rb1. The IC50 for compound K for inhibiting CSC sphere formation and growth was 125 nM, suggesting that its anti-CSC effect is greater than that of Rb1. The difference in the number of sugar moieties may mediate their different affinities towards molecular targets.
Cisplatin and paclitaxel are two mainstays of anti-cancer drugs in the clinical treatment of ovarian cancer. In addition to the dose-limiting toxicities, their drug resistance poses another major problem for their clinical use [31]. In the present study, we demonstrate that Rb1 and compound K specifically enhances these chemotherapeutic treatments, and that these changes occur at pharmacologically attainable drug levels [11]. Moreover, non-toxic, low doses of cisplatin and paclitaxel with Rb1 and compound K can also induce an enhanced cytotoxic response in the cancer cells (data not shown), suggesting a potential avenue to improve the clinical efficacy. Our findings may also provide a probable explanation as to why Rb1 and its metabolite-containing PPD have substantial benefit in chemoresistant tumors [32]. Imatinib is used in the treatment of a number of cancers, including ovarian cancer [33–36]. We found that Rb1 and compound K had similar efficiency as imatinib in cell growth inhibition and apoptosis induction. Importantly, compound K might imatinib, as it is less sensitive to some resistance mechanisms cancer cells develop [37]. We further identified several mechanisms of action induced by compound K, such as inactivation of ABCG2 and P-gp.
The presence of activated β-catenin in a broad spectrum of human cancers makes Wnt/β-catenin signaling an attractive target for cancer treatment. Wnt/β-catenin signaling has also been implicated in ovarian cancer. While mutations in adenomatous polyposis coli or β-catenin gene are infrequent, an increased β-catenin level and/or altered Wnt/β-catenin signaling are commonly found in different ovarian cancer subtypes and that high β-catenin expression correlates with tumor grade and poor prognosis [38–40]. It is also relevant that Wnt/β-catenin is significantly overexpressed in recurrent lesions [10, 41]. Therefore, the compound K-dependent inhibition of Wnt/β-catenin may hold great value in treating patients with refractory ovarian cancer. ABC transporters are well-established efflux pumps contributing to drug resistance, which are molecular and functional properties of a distinct side population of cells. It has been demonstrated that ABCG2 and P-glycoprotein are the two most important genes for the chemoresistance of CSCs [42–44]. There is emerging evidence that inhibition of Wnt/β-catenin and drug transporters can eradicate CSCs, but to be nontoxic to normal cells/tissue stem cells [45].
There is increasing evidence that the induction of EMT in transformed ovarian cancer cells in vitro or in mouse models to generate cells with CSC characteristics, indicating that EMT may be implicated in ovarian cancer aggressiveness [46, 47]. Moreover, CSCs derived from ovarian tumors and metastatic ovarian peritoneal effusions express EMT-associated markers, and that high expression of EMT-inducing genes enhances the metastatic potential for ovarian cancer cells [48]. Our studies reveal a role for Rb1 and compound K in linking EMT and CSCs, which not only shed light on the molecular mechanisms of action but also suggest new therapeutic strategy that warrants clinical investigation.
Therapeutic selectivity is one of the most critical considerations in cancer treatment. Ginseng has been shown to be well-tolerated even at very high doses in patients but nontoxic to normal human cells [49]. We have also shown the differential effects of Rb1 and compound K in ovarian cancer and normal human ovarian surface epithelial cells (Supplementary Figure S1). Pharmacokinetics of Rb1 has not been determined in humans. However, the dosage (250 nM) utilized in this study in vitro is similar to the pharmacologically attainable levels of 145-196 nM in the plasma in consuming per gram of Rb1, based on the oral bioavailability of 4.35% of Rb1 in animal studies [50]. Treatment with Rb1 within this dosage range is highly effective in inhibiting ovarian cancer growth and does not seem to exhibit toxicity. Indeed, different phase I trials have shown that Rb1 is safe when consumed at doses as high as 10 g root and a dose of ginsenosides smaller than 300 mg/kg [51, 52].
In summary, this study shows for the first time that ginsenoside Rb1, especially its metabolite compound K, specifically target CSCs via simultaneous inhibition of Wnt/β-catenin signaling and EMT in ovarian cancer. The ability of Rb1 and its metabolite compound K to intervene different cellular pathways in CSCs suggests that Rb1 can be a wide spectrum chemopreventive agent for cancer treatment. These findings are highly clinical significant, and will provide a solid scientific background in the development of Rb1 and compound K in ginseng-based therapeutics for the treatment of relapsed cancers which currently have limited pharmaceutical options.
MATERIALS AND METHODS
Cells and cell culture
Human ovarian carcinoma cell lines SKOV-3 and HEYA8 were gifts from Dr. N. Auersperg (University of British Columbia, Vancouver, B. C., Canada) and Dr. J. Liu (MD Anderson Cancer Center, Houston, TX), respectively. Isolation and culture of CSCs from SKOV-3 and HEYA8 or primary human ovarian cancer samples were performed in serum-free stem cell-selective condition as described in Chau et al. [10]. For the relapse experiment, control/treated primary spheroids were dissociated and replated as secondary spheroids and imaged daily. Primary tumor samples were obtained from ovarian cancer patients with informed consent and approval by the Taipei Medical University Institutional Review Board. Cisplatin and paclitaxel were purchased from Calbiochem (San Diego, CA) and were used at 50 μM and 100 nM concentration, respectively.
Reverse transcription and PCR
Total RNA was extracted with Trizol and reversed transcribed, and the cDNA was synthesized using the first-stranded cDNA synthesis kit (Invitrogen). The sequences of the specific primers were as follows: Bmi-1: sense 5’-ATGTGTGTGCTTTGTGGAG-3’, antisense 5’-AGTGGTCTGGTCTTGTGAAC-3’; Nanog: sense 5’-AAGACAAGGTCCCGGTCAAG-3’, antisense 5’-CCTAGTGGTCTGCTGTATTAC-3’; Oct4: sense 5’-ATCCTGGGGGTTCTATT TGG-3’, antisense 5’-TCTCCAGGTTGCCTCTCACT-3’; ABCG2: sense 5’-CTGAGA TCCTGAGCCTTTGG-3’, antisense 5’-TGCCCATCACAACATCATCT-3’; and P-glycoprotein: sense 5’-ACCTGTGAAGAGTAGAACATGAAGA-3’, antisense 5’-AAGATCCATTCCGACCT CGC-3’. β-actin served as a control. The PCR conditions used to amplify Bmi-1, Oct4, ABCG2, and P-glycoprotein were: 33 cycles at 95°C for 30 s, 55°C for 30 s, and 72°C for 30 s. To analyze expression of Nanog, the thermocycling parameters for the PCR reactions were: 35 cycles at 95°C for 30 s, 55°C for 30 s, and 72°C for 30 s.
Western blot analysis
Equal amounts of cell lysate proteins were separated on 7.5% SDS-polyacrylamide gels and transferred to nitrocellulose membrane. Membranes were blocked for 1 h at room temperature with 5% non-fat milk, and then incubated overnight at 4°C in the primary antibody: β-catenin (1:2000) (Transduction Lab, Lexington, KY), Cleaved caspase-3 (1:1000), caspase-3(1:1000) Snail (1:1000), Slug (1:1000), phospho-Akt (Ser473) (1:1000), total Akt (1:1000), phospho-ERK1/2 (Thr202/Tyr204) (1:1000), total ERK1/2 (1:1000), ABCG2 (1:1000) and P-glycoprotein (1:1000) (Cell Signaling Technology, Beverley, MA). β-actin (1:2000) (Sigma, St. Louis, MO) was used as a loading control. The protein-antibody complexes were detected by horseradish peroxidise-conjugated secondary antibodies (Bio-Rad, Hercules, CA) followed by the enhanced chemiluminescence detection reagents (Amersham, Little Chalfont, UK).
Proliferation and chemosensitivity analysis
Rates of proliferation and sensitivity to drugs were assessed using the colorimetric MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay according to the manufacturer’s instructions (Sigma, St. Louis, MO). Briefly, 500 μl MTT solution was added to each well and the plate was incubated for 4 h at 37°C. Medium was then aspirated from each well, and 250 μl DMSO was added. Colorimetric analysis was performed at a wavelength of 570 nm using a microplate reader (Bio-Rad, Hercules, CA).
Trypan blue exclusion assay
Cells were incubated for 5 min at room temperature with an equal volume of 0.4% (w/v) trypan blue solution (Sigma, St. Louis, MO). Cells were counted using a dual-chamber hemocytometer under a light microscope. Viable and nonviable cells were recorded.
β-catenin/TCF reporter gene assay
Cells were transiently transfected with 0.5 μg of the TOPFLASH or FOPFLASH reporter plasmid (Clontech) using Lipofectamine (Invitrogen). Transfection efficiencies were determined by co-transfection of the β-galactosidase construct. Luciferase and β-galactosidase activity were assayed according to the manufacturer’s protocol using the luciferase assay kit from Promega. Luciferase activity in each well was normalized to the β-galactosidase activity. All experiments were assayed in triplicates, and the assay was performed in three independent experiments.
In vivo studies
All animal experiments were approved by the Institutional Animal Care and Use Committee at the University of Hong Kong. 106 cells were subcutaneously (s.c.) injected into the right flank of athymic nude mice. To evaluate the therapeutic effect, treatment was initiated ~21 days following tumor injection when tumors reached a detectable size 50 mm3. Mice (n = 3 per group, repeated three times) were treated by oral gavage, which was relevant to the dietary administration, with either vehicle, compound K (50 mg/kg), cisplatin (5 mg/kg), paclitaxel (2 mg/kg), compound K + cisplatin (50 mg/kg for compound K, 5 mg/kg for cisplatin), compound K + paclitaxel (50 mg/kg for compound K, 2 mg/kg for paclitaxel). Mice were monitored for adverse effects of therapy and sacrificed on day 28 or when any of the mice began to appear moribund. Mice weight and tumor volume were recorded. The length and width of a tumor were measured with a caliper and tumor volume was calculated as [(length+width)/2]3. Plasma samples were collected from retro-orbital sinus and further analyzed for hepatic, cardiac, and renal functions including LDH, AST, ALT, ALP, CK, and blood urea nitrogen. Organs including liver, heart, kidney, spleen, and lungs were fixed in formalin for paraffin embedding and stained with hematoxylin and eosin. Ki67 and caspase-3 were also tested by immunohistochemistry.
Statistical analysis
All data were presented as mean ± S.D. and repeated at least four or five times for in vitro studies. Statistical analysis was done using one-way ANOVA followed by Tukey least significant difference t test for post hoc analysis (GraphPad Software, San Diego, CA). For in vivo studies, all variables were considered nonparametric and presented as mean ± SEM or mean + SD, Kruskal Wallis test was used to evaluate the statistical significance in tumor volume and body weight data differed between control and treatment groups. P values of < 0.05 considered statistically significant.
ACKNOWLEDGMENTS
This work was supported by the Health and Medical Research Fund 11121191, HKU Strategic Research Theme on Drug, and Croucher Senior Research Fellowship to Alice S. T. Wong. We would also like to thank The Hong Kong Jockey Club Charities Trust (HKJCCT) for funding the project of “R&D Laboratory for Testing of Chinese Medicines”.
CONFLICTS OF INTEREST
The authors indicate no potential conflicts of interest.
REFERENCES
1. Dean M, Fojo T, Bates S. Tumor stem cells and drug resistance. Nat Rev Cancer. 2005; 5: 275-284.
2. Jia L, Zhao Y. Current Evaluation of the Millennium Phytomedicine- Ginseng (I): Etymology, Pharmacognosy, Phytochemistry, Market and Regulations. Curr Med Chem. 2009; 16:2475-2484.
3. Wong AS, Che CM, Leung KW. Recent advances in ginseng as cancer therapeutics: a functional and mechanistic overview. Nat Prod Rep. 2015; 32:256-272.
4. Helms S. Cancer prevention and therapeutics: Panax ginseng. Altern Med Rev. 2004; 9:259-274.
5. Yun TK. Experimental and epidemiological evidence on non-organ specific cancer preventive effect of Korean ginseng and identification of active compounds. Mutat Res. 2003; 523-524:63-74.
6. Duda RB, Zhong Y, Navas V, Li MZ, Toy BR, Alavarez JG. American ginseng and breast cancer therapeutic agents synergistically inhibit MCF-7 breast cancer cell growth. J Surg Oncol. 1999; 230-239.
7. Zhu S, Zou K, Fushimi H, Cal S, Komatsu K. Comparative study on triterpene saponins of Ginseng drugs. Planta Med. 2004; 70:666-677.
8. Wang A, Wang CZ, Wu JA, Osinski J, Yuan CS. Determination of major ginsenosides in Panax quinquefolius (American ginseng) using high-performance liquid chromatography. Phytochem Anal. 2005; 16:272-277.
9. Hasegawa H, Sung JH, Matsumiya S, Uchiyama M. Main ginseng saponin metabolites formed by intestinal bacteria. Planta Med. 1996; 62:453-457.
10. Chau WK, Ip CK, Mak AS, Wong AS. c-Kit mediates chemoresistance and tumor-initiating capacity of ovarian cancer cells through activation of Wnt/β-catenin–ATP-binding cassette G2 signaling. Oncogene. 2013; 32:2767-2781.
11. Karlsson MO, Molnar V, Freijs A, Nygren P, Bergh J, Larsson R. Pharmacokinetic models for the saturable distribution of paclitaxel. Drug Metab Dispos. 1999; 27:1220-1223.
12. Polyak K, Weinberg RA. Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer. 2009; 9:265-273.
13. Jordà M, Vinyals A, Marazuela A, Cubillo E, Olmeda D, Valero E, Cano A, Fabra A. Id-1 is induced in MDCK epithelial cells by activated Erk/MAPK pathway in response to expression of the Snail and E47 transcription factors. Exp Cell Res. 2007; 313:2389–2403.
14. Suman S, Kurisetty V, Das TP, Vadodkar A, Ramos G, Lakshmanaswamy R, Damodaran C. Activation of AKT signaling promotes epithelial-mesenchymal transition and tumor growth in colorectal cancer cells. Mol Carcinog. 2014; 53:E151-160.
15. Fodde R, Brabletz T. Wnt/beta-catenin signaling in cancer stemness and malignant behavior. Curr Opin Cell Biol. 2007; 19:150-158.
16. Chien AJ, Conrad WH, Moon RT. A Wnt survival guide: from flies to human disease. J Invest Dermatol. 2009; 129:1614-1627.
17. Molenaar M, van de Wetering M, Oosterwegel M, Peterson-Maduro J, Godsave S, Korinek V, Roose J, Destrée O, Clevers H. XTCF-3 transcription factor mediates β-catenin-induced axis formation in Xenopus embryos. Cell. 1996; 86:391-399.
18. Cheon S, Poon R, Yu C, Khoury M, Shenker R, Fish J, Alman BA. Prolonged beta-catenin stabilization and tcf-dependent transcriptional activation in hyperplastic cutaneous wounds. Lab Invest. 2005; 85:416-425.
19. Korinek V, Barker N, Morin PJ, van Wichen D, de Weger R, Kinzler KW, Vogelstein B, Clevers H. Constitutive transcriptional activation by a beta-catenin-Tcf complex in APC-/- colon carcinoma. Science. 1997; 275:1784-1787.
20. Xu TM, Xin Y, Cui MH, Jiang X, Gu LP. Inhibitory effect of ginsenoside Rg3 combined with cyclophosphamide on growth and angiogenesis of ovarian cancer. Chin Med J (Engl). 2007; 120:584-588.
21. Yun TK, Choi SY. Preventive effect of ginseng intake against various human cancers: a case-control study on 1987 pairs. Cancer Epidemiol Biomarkers Prev. 1995; 4:401-408.
22. Kim EH, Lim S, Kim SO, Ahn SH, Choi YJ. Optimization of enzymatic treatment for compound K production from white ginseng extract by response surface methodology. Biosch Biotechnol Biochem. 2013; 77:1138-1140.
23. Na JY, Kim S, Song K, Lim KH, Shin GW, Kim JH, Kim B, Kwon YB, Kwon J. Anti-apoptotic Activity of Ginsenoside Rb1 in hydrogen peroxide-treated chondrocytes: stabilization of mitochondria and the inhibition of caspase-3. J Ginseng Res. 2012; 36:242-247.
24. Li J, Shao ZH, Xie JT, Wang CZ, Ramachandran S, Yin JJ, Aung H, Li CQ, Qin G, Vanden Hoek T, Yuan CS. The effects of ginsenoside Rb1 on JNK in oxidative injury in cardiomyocytes. Arch Pharm Res. 2012; 35:1259-1267.
25. Hou YL, Tsai YH, Lin YH, Chao JC. Ginseng extract and ginsenoside Rb1 attenuate carbon tetrachloride-induced liver fibrosis in rats. BMC Complement. Altern Med. 2014; 14:415.
26. Wang J, Qiao L, Li Y, Yang G. Ginsenoside Rb1 attenuates intestinal ischemia-reperfusion-induced liver injury by inhibiting NF-κB activation. Exp Mol Med. 2008; 40:686-698.
27. Cheng W, Wu D, Zuo Q, Wang Z, Fan W. Ginsenoside Rb1 prevents interleukin-1 beta induced inflammation and apoptosis in human articular chondrocytes. Int Orthop. 2013; 37: 2065-2070.
28. Lee DG, Jang SI, Kim YR, Yang KE, Yoon SJ, Lee ZW, An HJ, Jang IS, Choi JS, Yoo HS. Anti-proliferative effects of ginsenosides extracted from mountain ginseng on lung cancer. Chin J Integr Med. 2014 [Epub ahead of print].
29. Zheng Y, Nan H, Hao M, Song C, Zhou Y, Gao Y. Antiproliferative effects of protopanaxadiol ginsenosides on human colorectal cancer cells. Biomed Rep. 2013; 1:555-558.
30. Nag SA, Qin JJ, Wang W, Wang MH, Wang H, Zhang R. Ginsenosides as anticancer agents: In vitro and in vivo activities, structure-activity relationships, and molecular mechanisms of action. Front Pharmacol. 2012; 3:25.
31. Agarwal R, Kaye SB. Ovarian cancer: strategies for overcoming resistance to chemotherapy. Nat Rev Cancer. 2003; 3:502-516.
32. Lee SJ, Sung JH, Lee SJ, Moon CK, Lee BH. Antitumor activity of a novel ginseng saponin metabolite in human pulmonary adenocarcinoma cells resistant to cisplatin. Cancer Lett. 1999; 144:39-43.
33. Alberts DS, Liu PY, Wilczynski SP, Jang A, Moon J, Ward JH, Beck JT, Clouser M, Markman M. Phase II trial of imatinib mesylate in recurrent, biomarker positive, ovarian cancer (Southwest Oncology Group Protocol S0211). Int J Gynecol Oncol 2007; 17: 784–788.
34. Posadas EM, Kwitkowski V, Kotz HL, Espina V, Minasian L, Tchabo N, Premkumar A, Hussain MM, Chang R, Steinberg SM, Kohn EC. A prospective analysis of imatinib-induced c-KIT modulation in ovarian cancer: a phase II clinical study with proteomic profiling. Cancer. 2007; 110:309-317.
35. Schilder RJ, Sill RW, Lee RB, Shaw TJ, Senterman MK, Klein-Szanto AJ, Miner Z, Vanderhyden BC. Phase II evaluation of imatinib mesylate in the treatment of recurrent or persistent epithelial ovarian or primary peritoneal carcinoma: a Gynecologic Oncology Group Study. J Clin Oncol. 2008; 26:3418-3425.
36. Matei D, Emerson RE, Shcilder J, Menning N, Baldridge LA, Johnson CS, Breen T, McClean J, Stephens D, Whalen C, Sutton G. Imatinib mesylate in combination with docetaxel for the treatment of patients with advanced, platinum-resistant ovarian cancer and primary peritoneal carcinomatosis: a Hoosier Oncology Group trial. Cancer. 2008; 113:723-732.
37. Milojkovic D, Apperley J. Mechanisms of resistance to imatinib and second-generation tyrosine inhibitors in chronic myeloid leukemia. Clin Cancer Res. 2009; 15:7519-7527.
38. Gamallo C, Palacios J, Moreno G, Calvo de Mora J, Suárez A, Armas A. Beta-catenin expression pattern in stage I and II ovarian carcinomas: relationship with beta-catenin gene mutations, clinicopathological features, and clinical outcome. Am J Pathol. 1999; 155:527-536.
39. Lee E, Salic A, Krüger R, Heinrich R, Kirschner MW. The roles of APC and Axin derived from experimental and theoretical analysis of the Wnt pathway. PLoS Biol. 2003; 1:E10.
40. Kildal W, Risberg B, Abeler VM, Kristensen GB, Sudbø J, Nesland JM, Danielsen HE. Beta-catenin expression, DNA ploidy and clinicopathological features in ovarian cancer: a study in 253 patients. Eur J Cancer. 2005; 41:1127-1134.
41. Steg AD, Bevis KS, Katre AA, Ziebarth A, Dobbin ZC, Alvarez RD, Zhang K, Conner M, Landen CN. Stem cell pathways contribute to clinical chemoresistance in ovarian cancer. Clin. Cancer Res. 2012; 18:869-881.
42. Zhou S, Schuetz JD, Bunting KD, Colapietro AM, Sampath J, Morris JJ, Lagutina I, Grosveld GC, Osawa M, Nakauchi H, Sorrentino BP. The ABC transporter Bcrp1/ABCG2 is expressed in a wide variety of stem cells and is a molecular determinant of the side-population phenotype. Nat Med. 2001; 7:1028-1034.
43. An Y, Ongkeko WM. ABCG2: the key to chemoresistance in cancer stem cells? Expert Opin Drug Metab Toxicol. 2009; 5:1529-1542.
44. Hirschmann-Jax C, Foster AE, Wulf GG, Nuchtern JG, Jax TW, Gobel U, Goodell MA, Brenner MK. A distinct “side population” of cells with high drug efflux capacity in human tumor cells. Proc Natl Acad Sci U S A. 2004; 101:14228-14233.
45. Takebe N, Warren RQ, Ivy SP. Breast cancer growth and metastasis: interplay between cancer stem cells, embryonic signaling pathways and epithelial-to-mesenchymal transition. Breast Cancer Res. 2011; 13:211.
46. Kang KS, Choi YP, Gao MQ, Kang S, Kim BG, Lee JH, Kwon MJ, Shin YK, Cho NH. CD24+ ovary cancer cells exhibit an invasive mesenchymal phenotype. Biochem Biophys Res Commun. 2013; 432:333-338.
47. Latifi A, Abubaker K, Castrechini N, Ward AC, Liongue C, Dobill F, Kumar J, Thompson EW, Quinn MA, Findlay JK, Ahmed N. Cisplatin treatment of primary and metastatic epithelial ovarian carcinomas generates residual cells with mesenchymal stem cell-like profile. J Cell Biochem. 2011; 112:2850-2864.
48. Davidowitz RA, Selfors LM, Iwanicki MP, Elias KM, Karst A, Piao H, Ince TA, Drage MG, Dering J, Konecny GE, Matulonis U, Mills GB, Slamon DJ, et al. Mesenchymal gene program-expressing ovarian cancer spheroids exhibit enhanced mesothelial clearance. Clin Invest. 2014; 124:2611-2615.
49. Coon JT, Ernst E. Panax ginseng: a systematic review of adverse effects and drug interactions. Drug Saf. 2002; 25:323-344.
50. Xu QF, Fang XL, Chen DF. Pharmacokinetics and bioavailability of ginsenoside Rb1 and Rg1 from Panax notoginseng in rats. J Ethnopharmacol. 2003; 84:187-192.
51. Wang CZ, Kim KE, Du GJ, Qi LW, Wen XD, Li P, Bauer BA, Bissonnette MB, Musch MW, Chang EB, Yuan CS. Ultra-performance liquid chromatography and time-of-flight mass spectrometry analysis of gensenoside metabolites in human plasma. Am J Chin Med. 2011; 39:1161-1171.
52. Kim HK. Pharmacokinetics of ginsenoside Rb1 and its metabolite compound K after oral administration of Korean Red Ginseng extract. J Ginseng Res. 2013; 37:451-456.