
INTRODUCTION
Proteases comprise a diverse group of enzymes with the ability to cleave peptide bonds. Their importance in human physiology is illustrated by their participation in numerous biological processes, including embryonic development and differentiation, cell proliferation and apoptosis, tissue remodelling and wound healing or cell migration and angiogenesis [1-2]. The analysis of different mammalian genomes has shown that protease-coding genes constitute about 2% of all protein-coding genes, with at least 570 proteases in human and other primates, and more than 630 in rodents [3-5]. This large number of genes, and the fact that many of them work together in specific networks, has led to the introduction of novel concepts for the global analysis of proteases. In this regard, the term degradome has been coined to define the complete set of protease genes present in a genome, and in the case of cancer, the tumor degradome constitutes the set of genes expressed by a tumor during disease progression [6-7].
Tumor proteases have been widely studied during the last decades due to their ability to cleave the extracellular matrix, facilitating tumor invasion and metastasis, or to activate specific cytokines and growth factors necessary for tumor growth and angiogenesis [8-10]. The relevance of proteases in cancer is underscored by the finding that a growing number of protease genes are mutated in different malignancies [11-17]. Furthermore, changes in the expression profile of proteolytic genes have been widely associated with tumor development. Thus, overexpression of MMP1 or MMP2 has been shown to be necessary to form lung metastasis by breast tumor cells [18] and expression of specific proteases is a hallmark of many tumor types [19-21]. Due to the initial discovery of proteases with tumor promoting activities, most expression profiling studies have focused their attention on proteases overexpressed by tumor cells, while little attention has been paid to proteases whose expression was repressed during malignant transformation. However, a growing body of evidence is showing that certain proteases can have tumor-defying functions, with some of them constituting bona fide tumor suppressors. This is the case of CYLD1, whose mutations cause cylindromatosis; A20, in which chromosomal deletions and inactivating mutations have been found in several lymphoma subtypes; BAP1, with point mutations and deletions described in breast and lung cancer and melanoma; CASP8, mutated in lymphoproliferative syndromes and different carcinomas, or USP7, implicated in p53 deubiquitylation [22-23]. Remarkably, some proteases hamper tumor growth or progression when either produced by tumor cells or by the tumor stroma [24-26]. In addition, the recent sequencing of cancer genomes is identifying novel somatic mutations affecting protease-coding genes [27-30], reinforcing the hypothesis that inactivation of certain proteases, by either somatic mutation or gene repression, might contribute to cancer development.
In this work, we have designed a new quantitative qPCR-based device for profiling the entire degradome in human. The use of a TaqMan-based approach allows a better quantification of differences in expression between biological samples, as well as provides an unmatched sensitivity to detect transcriptional changes affecting genes with low expression levels, which are usually difficult to determine when using traditional hybridization-based detection methods. We have used this new platform to assess and compare protease expression levels in normal mucosa and colorectal tumor samples. Thus, we have centered our attention on proteases whose expression was repressed in colorectal carcinoma providing the utility of degradome profiling as a good instrument to identify novel proteases with antitumor properties.
RESULTS
Expression of extracellular proteases is largely altered in colorectal carcinomas
To identify proteases differentially expressed in colon cancer, we obtained RNA from colon and rectal carcinomas as well as matched normal mucosa from 14 different patients diagnosed with colon cancer at different stages of progression, and subjected to surgery (Supplementary Table S1). Quantitative expression of human proteases and protease inhibitor genes was analyzed using two custom-designed TLDAs, with specific probes for 545 different human proteases, and 65 protease inhibitor genes. A comparison between tumor and normal samples resulted in the identification of genes with changes in expression of more than 4 RQs between tumor and normal samples. These included 21 protease genes overexpressed in tumor tissue, and 35 protease genes which were downregulated (Figure 1 and Table 1). Interestingly, we found a significant difference in the subcellular localization of proteases with altered expression in the tumor. This effect was evident for proteases overexpressed in tumor samples, as more than 90% of them have an extracellular localization (19 extracellular vs. 2 intracellular, p<0.001). This difference in the subcellular localization cannot be attributed to differences in the content of the arrays (266 vs. 264) nor to the normal expression of proteases in colon tissue, as intracellular and extracellular protease-coding genes are similarly expressed in these samples (219 extracellular vs. 255 intracellular). A similar trend was observed for protease genes downregulated in the tumor (24 extracellular vs. 11 intracellular, p=0.03). Additionally, analysis of protease inhibitors allowed us to identify three extracellular protease inhibitors overexpressed in tumor samples, while only one intracellular inhibitor was repressed (Table 1), suggesting that inhibitors follow a similar trend as proteases. Together, these data suggest that changes in the regulatory pathways in colon carcinoma cells predominantly affect proteases exerting their activity in the extracellular matrix and on the cell surface compartments.
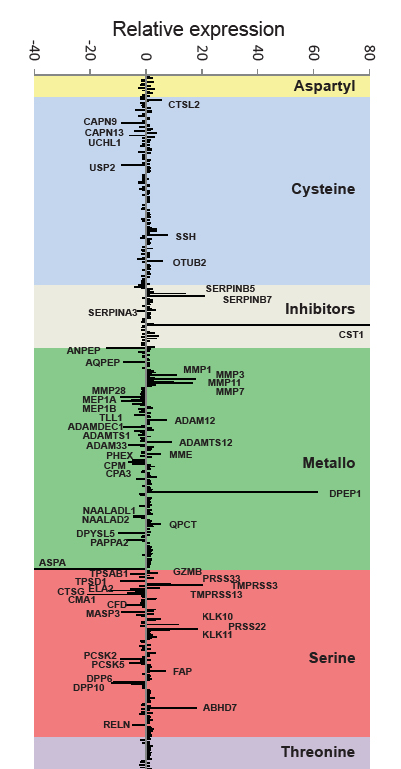
Figure 1: Degradome expression profiling of colorectal carcinoma. Proteolytic genes are classified according to their catalytic class, and changes in the expression between colorectal carcinoma and normal mucosa are represented as median RQ (RQ=2-ΔΔCt) for genes upregulated in cancer samples and as -1/RQ for genes with higher expression in the normal tissue. Gene symbols are shown for those genes with RQ>4 or -1/RQ<-4.
Table 1: Proteases and protease inhibitors differentially expressed in colorectal cancer
Overexpressed in tumor tissue
Protease |
Gene |
RQ |
Localization |
Process |
Activity |
Dipeptidase 1 (renal) |
DPEP1 |
61.2 |
EC-TM |
Renal metabolism |
Protease |
Transmembrane protease, serine 3 |
TMPRSS3 |
20.0 |
EC-TM |
Development of the inner ear |
Protease |
Brain serine proteinase 2 |
PRSS22 |
18.3 |
EC-S |
Inmune response |
Protease |
Epoxide hydrolase 4 |
ABHD7 |
18.0 |
EC-TM |
Unknown |
NPH |
Stromelysin 1 |
MMP3 |
17.8 |
EC-S |
EC matrix degradation |
Protease |
Matrix metallopeptidase 7 |
MMP7 |
16.6 |
EC-S |
EC matrix degradation |
Protease |
Kallikrein-related peptidase 10 |
KLK10 |
11.5 |
EC-S |
EC matrix degradation |
Protease |
Matrix metallopeptidase 1 |
MMP1 |
10.7 |
EC-S |
EC matrix degradation |
Protease |
Matrix metallopeptidase 11 |
MMP11 |
9.9 |
EC-S |
EC matrix degradation |
Protease |
ADAMTS12 |
ADAMTS12 |
8.9 |
EC-S |
EC matrix degradation |
Protease |
Protease, serine 33 |
PRSS33 |
8.9 |
EC-S |
Unknown |
Protease |
Kallikrein-related peptidase 11 |
KLK11 |
8.3 |
EC-S |
EC matrix degradation |
Protease |
Sonic hedgehog homolog (Drosophila) |
SHH |
7.8 |
EC-TM |
Embryo patterning |
Protease |
ADAM metallopeptidase domain 12 |
ADAM12 |
7.3 |
EC-TM |
Adipogenesis and myogenesis |
Protease |
Seprase |
FAP |
7.1 |
EC-TM |
Inmune response |
Protease |
Otubain 2 |
OTUB2 |
5.7 |
IC |
Unknown |
Protease |
Cathepsin L2 |
CTSL2 |
5.3 |
IC |
Corneal physiology |
Protease |
Neprilysin |
MME |
5.3 |
EC-TM |
Renal metabolism |
Protease |
Glutaminyl-peptide cyclotransferase |
QPCT |
5.3 |
EC-S |
Peptides cyclization |
NPH |
Transmembrane protease, serine 13 |
TMPRSS13 |
4.8 |
EC-TM |
Growth factor processing |
Protease |
Granzyme B |
GZMB |
4.2 |
EC-S |
Inmune response |
Protease |
Cystatin SN |
CST1 |
80.0 |
EC-S |
Cysteine protease inhibitor |
Inhibitor |
serpin peptidase inhibitor, clade B, member 7 |
SERPINB7 |
20.7 |
EC-S |
Serine protease inhibitor |
Inhibitor |
serpin peptidase inhibitor, clade B, member 5 |
SERPINB5 |
14.0 |
EC-S |
Serine protease inhibitor |
Inhibitor |
Downregulated in tumor tissue
Protease |
Gene |
RQ |
Localization |
Process |
Activity |
Aspartoacylase |
ASPA |
40.1 |
IC |
NAA to aspartate and acetate conversion |
NPH |
Chymase 1, mast cell |
CMA1 |
21.0 |
EC-S |
EC matrix degradation |
Protease |
Alanyl (membrane) aminopeptidase |
ANPEP |
13.9 |
EC-TM |
Digestion |
Protease |
Cathepsin G |
CTSG |
13.2 |
EC-S |
Inmune response |
Protease |
Dipeptidyl-peptidase 10 |
DPP10 |
12.1 |
EC-TM |
Voltage-gated potassium channels binding |
NPH |
Dipeptidyl-peptidase 6 |
DPP6 |
11.6 |
EC-TM |
Voltage-gated potassium channels binding |
NPH |
Dihydropyrimidinase-related protein 5 |
DPYSL5 |
9.9 |
IC |
Development. Differentiation |
NPH |
Matrix metallopeptidase 28 |
MMP28 |
9.2 |
EC-S |
Tissue homeostasis. Wound repair |
Protease |
Tryptase delta 1 |
TPSD1 |
9.0 |
EC-S |
Unknown |
Protease |
Proprotein convertase subtilisin/kexin type 2 |
PCSK2 |
8.9 |
IC |
Prohormones processing |
Protease |
Meprin beta subunit |
MEP1B |
8.7 |
EC-TM |
Unknown |
Protease |
Mannan-binding lectin serine peptidase 3 |
MASP3 |
8.7 |
EC-S |
Lectin pathway of complement activation |
Protease |
Calpain 9 |
CAPN9 |
8.7 |
IC |
Digestion |
Protease |
Ubiquitin specific peptidase 2 |
USP2 |
8.6 |
IC |
Ubiquitin-dependent catabolic process |
Protease |
Aminopeptidase Q |
AQPEP |
7.9 |
EC-TM |
Trophoblast implantation |
Protease |
ADAM-like, decysin 1 |
ADAMDEC1 |
7.9 |
EC-S |
Dendritic cell function |
Protease |
Complement factor D (adipsin) |
CFD |
7.3 |
EC-S |
Alternative complement pathway |
Protease |
Pappalysin 2 |
PAPPA2 |
6.8 |
EC-S |
IGF processing |
Protease |
Granzyme M |
GZMM |
6.5 |
IC |
Immune response |
Protease |
Carboxypeptidase M |
CPM |
6.4 |
EC-TM |
Monocyte to macrophage differentiation |
Protease |
ADAM metallopeptidase domain 33 |
ADAM33 |
6.3 |
EC-TM |
Asthma & bronchial hyperresponsiveness |
Protease |
Ubiquitin carboxyl-terminal esterase L1 |
UCHL1 |
5.9 |
IC |
Ubiquitin-dependent catabolic process |
Protease |
Proprotein convertase subtilisin/kexin type 5 |
PCSK5 |
5.9 |
IC |
Integrin processing |
Protease |
Elastase, neutrophil expressed |
ELA2 |
5.6 |
EC-S |
Immune response |
Protease |
Tryptase alpha/beta 1, tryptase beta 2 |
TPSAB1 |
5.4 |
EC-S |
Immune response |
Protease |
Meprin alpha subunit |
MEP1A |
5.2 |
EC-TM |
Unknown |
Protease |
Abhydrolase domain containing 12B |
ABHD12B |
5.1 |
IC |
Esterase |
NPH |
Reelin |
RELN |
4.9 |
EC-S |
Neural development |
Protease |
PHEX endopeptidase |
PHEX |
4.9 |
IC |
Bone mineralization |
Protease |
Tolloid-like 1 |
TLL1 |
4.9 |
EC-S |
Development. Differentiation |
Protease |
Carboxypeptidase A3 |
CPA3 |
4.6 |
EC-S |
Secretory granule peptidase |
Protease |
N-acetylated α-linked acidic dipeptidase-like 1 |
NAALADL1 |
4.4 |
EC-TM |
Neuropeptide alpha-NAAG hydrolysis |
Protease |
N-acetylated α-linked acidic dipeptidase 2 |
NAALAD2 |
4.4 |
EC-TM |
Neuropeptide alpha-NAAG hydrolysis |
Protease |
Calpain 13 |
CAPN13 |
4.1 |
IC |
Unknown |
Protease |
ADAM with thrombospondin type 1 motif, 1 |
ADAMTS1 |
4.1 |
EC-S |
EC matrix degradation |
Protease |
Serpin peptidase inhibitor, clade A, member 3 |
SERPINA3 |
4.2 |
EC-S |
Immune response |
Inhibitor |
IC, intracellular; EC, extracellular; S, secreted; TM, transmembrane; NPH, non-protease homologue
Identification of novel proteases differentially expressed in colorectal cancer
The degradome expression profiling of colorectal cancer using TLDAs allowed us to identify several proteases with consistent changes in the expression pattern between tumor and normal tissue in different patients (Figure 1 and Table 1). As expected, among the overexpressed genes in tumor samples we found several genes previously identified as upregulated in this pathology, including several members of the matrix metalloproteinase family (MMP1, MMP3, MMP7, MMP10, MMP11), members of the kallikrein family of serine-peptidases (KLK6, KLK8, KLK10), as well as DPEP1 or PRSS22 among others [31-34]. The expression of some of these genes was up to 800 times higher in the tumor than in normal tissue, and their implication in colon cancer progression and invasion has already been demonstrated for some of them [35], validating the utility of degradome TLDAs to identify genes causally implicated in this pathology. Additionally, we were able to identify other genes highly upregulated in tumor samples, including ABHD7, TMPRSS3 or OTUB2, which had not been previously associated with this pathology, and might constitute novel candidate genes for colon cancer. Similarly, a detailed analysis of genes downregulated in tumor samples resulted in the identification of protease genes including MMP28, ANPEP, ADAMTS1 and ADAMTS15, previously known to be repressed in colon carcinoma or in other tumor types [36-38]. However, most of the genes with reduced expression in tumor samples had not been previously linked to this disease.
Although proteolytic enzymes have been widely characterized due to their pro-tumoral activities, recent reports have shown that some of these enzymes are tumor suppressors or have anti-tumoral effects [22, 24], suggesting that downregulation of their expression in tumor cells might contribute to tumor growth. In order to test this hypothesis, and to reduce the number of candidate genes, we first compared our list of the top fifty protease genes with altered expression in colon cancer to a list of genes somatically mutated in colon and breast cancers [27, 39]. This resulted in the identification of two upregulated genes (DPEP1 and MMP11) as mutated in colon cancer and another three protease genes mutated in breast cancer (TMPRSS3, ADAM12 and MMP10). In parallel studies for downregulated genes, we were able to identify seven genes with mutations in colon cancer (DPP10, PCSK2, ADAM33, RELN, CAPN13, ADAMTS1 and ADAMTS15), and five genes with mutations in breast carcinomas (MASP3, ABHD12B, TLL1, CPA3 and DPP6). The proportion of downregulated protease genes with somatic mutations in colon cancer was significantly higher when compared to upregulated ones (14% vs. 4%, p<0.05). This difference could not be attributed to changes in the transcript length between both groups (3396 bp vs. 4251 bp), suggesting that it might constitute a functional difference.
To try to confirm the repression of these genes in colorectal cancer, we performed qPCR analysis of this subset of genes in a validation series of 28 additional patients, consisting of pairs of tumor and matched normal mucosa (Supplementary Table S2). This analysis allowed us to corroborate that several of these protease-coding genes were expressed in normal colon mucosa, but consistently downregulated in colorectal carcinoma samples (Figure 2a). In fact, in some samples the expression of three of these genes, encoding the non-protease homologue DPP10, the proprotein convertase PCSK2 and the mannan-binding lectin serine peptidase 3 MASP3, are reduced up to a thousand fold in tumor tissue when compared to normal mucosa. No significant correlation between clinical stage and the expression of these genes could be established. Among the genes downregulated in colorectal carcinomas, we decided to focus on MASP3, as this gene encodes a bona fide protease whose participation in tumor processes has not been studied before, and its role in immunological processes could contribute to the tumorigenic process. To further confirm these results, we performed an immunohistochemical analysis of normal and tumor colorectal sections to investigate the expression of MASP3. Thus, in agreement with the qPCR results, MASP3 was absent in the tumor tissue while its expression could be detected in the corresponding normal colon mucosa (Figure 2b). Together, these results suggest that diminished activity of this protease, either through changes in gene expression or through the acquisition of somatic mutations, are common to colon tumors, and this gene could be causally implicated in the development of colorectal cancer.
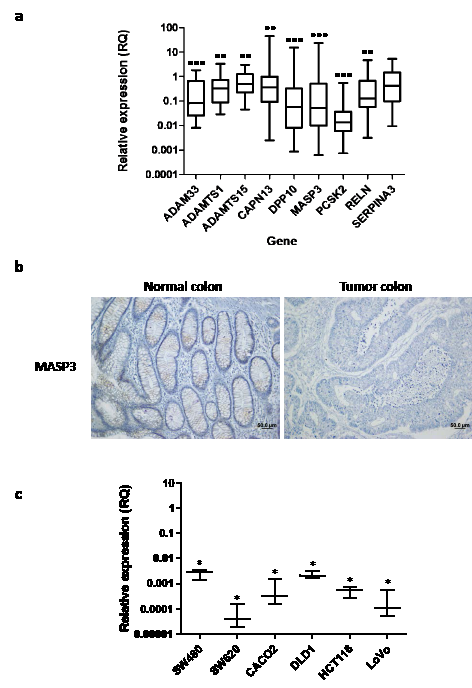
Figure 2: Identification of protease genes downregulated in colorectal cancer. (a) Gene expression was quantified for nine different genes in a validation cohort of 28 colorectal carcinoma-normal mucosa pairs using qPCR. This led to the identification of proteolytic genes consistently downregulated in colorectal carcinomas. Fold change between tumor and normal tissue is expressed as RQ values and significant differences were assessed by a non-parametric Mann Whitney-Wilcoxon test (**, p<0.01; ***, p<0.001). (b) Immunohistochemical analysis of MASP3 expression in tumor and normal colorectal samples. MASP3 staining could be detected in normal colon mucosa (left panel), while no staining is detected in tumor tissue (right panel). (c) Expression of MASP3 was quantified in different colorectal cancer cell lines. Colon fibroblasts were used as control for comparison. Data are expressed as fold change using RQ values. Significant differences were assessed by a non-parametric Mann Whitney-Wilcoxon test (*, p<0.05).
Antitumoral effects of MASP3 in colorectal carcinomas
To further characterize this hypothesis as well as to perform functional analysis of this gene in vitro and in vivo, we studied the expression of MASP3 in different colorectal cancer cell lines (CaCo2, DLD-1, HCT116, LoVo, SW480, SW620), as well as in colon fibroblasts. This analysis revealed that MASP3 is expressed in normal colon fibroblasts, while its expression is significantly reduced in all six cancer cell lines examined, reaching changes greater than 1,000-fold (Figure 2c). These results suggest that these cell lines represent an adequate model to investigate whether expression of this protease might repress tumor growth in vitro or in vivo. To do that, we transfected the colon carcinoma cell lines HCT116 Luc2 and DLD-1 with an expression vector encoding MASP3, as well as with the empty vector. The overexpression of this gene was confirmed by Western Blot analysis (Figure 3a).
Overexpression of MASP3 significantly reduced cell proliferation in vitro when compared to control cells (Figure 3b). In order to investigate if the observed reduction in proliferative potential was actually due to possible pro-apoptotic effects of this protein, we decided to compare the levels of apoptosis between HCT116 cells overexpressing MASP3 and control cells. The analysis of PARP1 caspase cleavage products revealed no differences between both types of cells (Supplementary Figure S1), indicating that the antiproliferative action of MASP3 is not derived from pro-apoptotic effects. Next, to investigate the in vivo significance of these results, we assayed the impact exerted by the ectopic expression of this protease on the tumorigenic potential of colon cancer cells, using a mouse xenograft model. Thus, HCT116 Luc2 cells transfected with the empty vector readily yielded fast-growing tumors when injected subcutaneously in nude mice. By contrast, overexpression of MASP3 in this cell line abrogated its ability to produce detectable tumors in the same experiments (Figure 3c). MASP3-overexpressing DLD-1 cells produced detectable tumors, but their growth was significantly reduced when compared to control cells (Figure 3c).
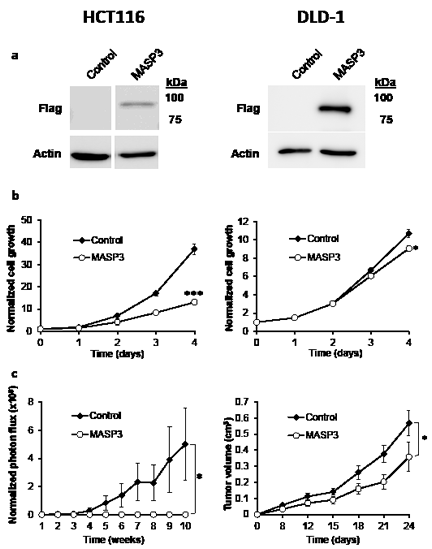
Figure 3. MASP3 expression reduces the proliferation of colorectal cancer cells and restrain tumor growth in a xenograft mouse model. HCT116 Luc2 (left side) and DLD- 1 (right side) colorectal cancer cells were transfected with a vector expressing MASP3 cDNA, or with the empty vector as control. (a) Western-blot analysis showing the overexpression of MASP3 in transfected cells. (b) Cell proliferation was quantified using an MTT-based assay. Each point was normalized with respect to 0 h and the mean ± SEM was represented (*, p<0.05; ***, p<0.001). Expression of MASP3 resulted in a reduction in cell proliferation when compared to control cells. (c) Tumor xenograft experiments were carried out with cells overexpressing MASP3 or transfected with the empty vector. Normalized photon flux ± SEM (HCT116 Luc2) or tumor volume ± SEM (DLD-1) was calculated for each group at the indicated times after injection and significant differences were assessed by a linear mixed-effects model (*, p<0.05). Ectopic expression of MASP3 blocked tumor formation compared to cells transfected with empty vector.
As a complementary approach and due to the identification of MASP3 as a gene consistently downregulated in colon carcinomas, we investigated whether depletion of this gene could increase the tumorigenic potential of HCT116 and DLD-1 cancer cells. Silencing of MASP3 by transduction with lentiviral vectors encoding specific shRNAs resulted in a significant increase in tumor growth rate when compared to control cells (Figure 4). Together, these data suggest that MASP3 downregulation in colon carcinomas might be a required step in the development of the malignant phenotype by colon cancer cells, facilitating cell proliferation and tumor growth.
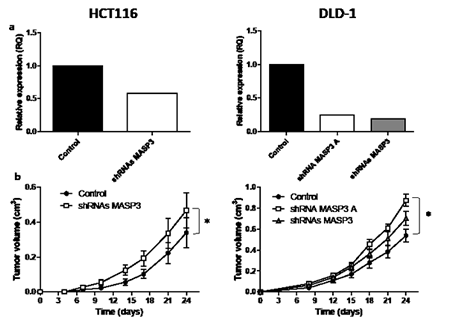
Figure 4. MASP3 downregulation increases the tumorigenic potential of colorectal cancer cells. HCT116 (left side) and DLD-1 (right side) colorectal cancer cells were transduced with MASP3 shRNA vectors or with the empty vector as control. (a) The relative expression levels of MASP3 were assayed by quantitative RT-PCR. (b) Tumor xenograft experiments were carried out with MASP3-silenced and control cells. Tumor volume was calculated for each group at the indicated times after injection and significant differences were assessed by a linear mixed-effects model (*, p<0.05). shRNAs MASP3 denotes the pool of 4 available silencing vectors, shRNA MASP3 A denotes the most efficient individual vector.
DISCUSSION
Proteases play critical roles in cancer biology, and all steps of cancer progression, from cell transformation to tumor invasion and metastasis, depend on proteolytic activities [8-10]. In consequence, alterations in sequence or expression of protease-encoding genes are probably a nearly universal feature of cancer, prompting us to hypothesize that most, if not all, human cancers present “protease addiction”, i.e. an exacerbated dependence on the activity of specific proteolytic enzymes, which potentially constitute valuable targets for anti-cancer therapies [40]. Importantly, not all proteases relevant to cancer biology play cancer-promoting roles, but a growing number of them present anti-tumor properties [22-26]. Taken together, these facts support the interest of implementing reliable procedures for profiling protease expression in human cancers.
Different approaches are currently available that allow global transcriptional profiling of biological samples. Most studies performed in this direction have been based on the use of microarray hybridization platforms, which provide a nearly complete coverage of the human genome, including the degradome. Over the last 15 years, such studies have represented the most powerful tool for global analysis of gene expression and they have generated expression data from tens of thousands of tumor samples [41]. The recent development of RNA-Seq has provided an alternative approach for global transcriptional profiling, with the clear advantage of allowing to detect the expression of previously uncharacterized loci [42]. In this work we have developed a quantitative PCR-based approach for profiling the entire degradome, with the advantages of simplicity, sensitivity and reduced cost compared to the mentioned global approaches.
TLDA-based degradome profiling of colorectal carcinomas uncovered significant transcriptional changes, which preferentially affected to extracellular proteases and inhibitors. This enrichment of extracellular molecules among both overexpressed and downregulated transcripts points to the relevance of the extracellular medium in cancer biology and highlights the relevance of the degradome in its regulation. Remarkably, a significant proportion of protease genes found in this work to be downregulated in colorectal cancer had been reported to be somatically mutated in colorectal or breast cancer. Among these genes, we chose to focus on MASP3, which encodes a bona fide protease, whose immunological role could be involved in tumor progression. Overexpression of this enzyme in two different colorectal cancer cell lines led to a marked drop of their proliferation in cell culture and to the reduction or abrogation of their tumorigenicity when xenografted in nude mice. Remarkably, shRNA-mediated silencing of MASP3 increased the tumorigenicity of the assayed colorectal cancer cell lines, despite the already very low basal expression level in these cells. These results, along with the consistently reduced expression of this protease in colorectal carcinomas and cancer cell lines, demonstrate its anticancer effects. It is noteworthy that the experimental system used in this work allowed us to investigate the biological relevance of MASP3 produced exclusively by the cancer cells, regardless of the levels of this protease synthesized by stromal or inflammatory components of the tumor. The above results indicate that the antitumorigenic effect of MASP3 does not require an alteration of its levels in stromal or inflammatory cells, since these components are expected to be the same in animals injected with control cells or with protease-overexpressing cells.
To date, very little is known about the role of MASP3 in tumor development. MASP3 is a mannan-binding lectin-associated serine protease with an immunological role through activation of the complement system [43]. Three different human MASP proteins have been described. MASP1 and 3 are encoded by the same gene, generated by alternative splicing [43]. They share the same heavy chain, but have a different light chain. Even though the expression of both isoforms has been quantified in our TLDA-based study, only MASP3 showed significant changes in tumor samples compared to normal mucosa. Based on the role of MASP proteins in complement activation, we hypothesized that complement could be involved in MASP3 antitumoral effects. As a first approach to investigate this possibility, we tested in parallel the effects of MASP3 overexpression and silencing on colon cancer proliferation using heat inactivated and non-inactivated serum in the culture medium. Since we did not observe any significant difference in these experiments, we cannot conclude that complement is involved in this effect. Natural substrates have not been identified for MASP3, but it has been reported to cleave recombinant insulin-like growth factor binding protein 5 (IGFBP-5) in vitro [44]. IGFBP-5, through its binding to IGFs and the regulation of their interaction with IGF receptors, is an essential regulator of several physiological processes [45-46]. Thus, MASP3-mediated cleavage of IGFBP-5 might hinder its ability to bind IGF-I suggesting that this linkage between MASP3 and IGFs could constitute an important mechanism for colon cancer progression. We have explored this possibility by studying the maturation of IGFBP-5 in cells overexpressing MASP3, finding that the IGFBP-5 processed products and their relative levels were indistinguishable in control and MASP3-overexpressing cells (data not shown), indicating that processing of this IGF binding protein does not account for the antitumorigenic effects of MASP3 in colorectal cells. The lack of information on the biochemical properties and function of this protease prevents the design of hypothesis-driven experiments to explore the mechanisms underlying its antitumoral effects in colorectal cells and further studies will be required to identify the MASP-3 substrates involved in its anticancer effects.
In summary, in this work we have designed a new qPCR-based tool for profiling the human degradome in normal and pathological conditions, including cancer. We have used these TLDAs to evaluate protease expression levels in colorectal carcinomas with the finding that most proteases with altered expression in these tumors exert their function in the extracellular compartment. In addition, we have found that among repressed proteases there was a higher proportion of genes with somatic mutations in colorectal cancer when compared to upregulated proteases. Further studies with some of these genes have demonstrated that they are consistently repressed in colorectal carcinomas and cancer cell lines when compared to normal colonic mucosa. Additionally, functional analysis have confirmed that MASP3 is a bona fide candidate antitumor protease, as it is either mutated or downregulated in tumor samples, and it plays a role in the regulation of cell proliferation and subcutaneous tumor formation. Taken collectively, these findings strongly support the utility of degradome profiling to identify novel proteases with antitumor functions and provide new insights into the functional diversity of proteases associated with human malignancies.
METHODS
Low-density array design
The human degradome database [47] was used to design two TaqMan Low Density Arrays (TLDA) containing specific primers and probes for all human proteases and a subset of protease inhibitors (Applied Biosystems). An EP-array was designed for the expression profiling of extracellular proteases and inhibitors, and membrane-bound proteases. Additionally, we designed an IP-array for the analysis of intracellular proteases and inhibitors. As a quality control to measure the number of probes able to detect the expression of a human protease, we performed qPCR with these arrays and using as starting material cDNAs derived from 10 different human tissues (brain, colon, liver, lung, muscle, pancreas, peripheral blood leukocytes, prostate, stomach and testis). Using this approach, more than 96.5% (608/630) of the probes were able to detect the expression of a protease gene in at least some of these tissues.
RNA samples and qPCR amplification
Colon and rectal carcinoma samples, as well as matched normal mucosa from the same patients were obtained from the Tumor Bank of the Hospital Universitario Central de Asturias (Oviedo, Spain) and from Hospital Clinic (Barcelona, Spain). Surgical specimens were collected in the operating theatre immediately after their removal, and they were quickly transported in an unfixed state to the pathology department. Samples in which time to freezing exceeded 30 min were discarded. Areas with massive ischaemic and/or necrotic phenomena were avoided. Samples were embedded in OCT medium (Bayer), and snap-frozen on liquid nitrogen-freezing isopentane. RNA was obtained from 10 mg of frozen tissue using an RNA purification kit from Qiagen, and the purity and integrity of the RNA was determined using a BioAnalyzer (Agilent Technologies). Only RNA samples with a RIN>=7 were used for degradome expression profiling. One µg of total RNA was used for cDNA synthesis, and qPCR amplification was performed following manufacturer’s instructions (Applied Biosystems), using an Applied Biosystems 7900HT Fast Real-Time PCR system. Data analysis was performed with SDS 2.1 (Applied Biosystems).
Immunostaining
Normal and tumor colorectal samples (n=40) were obtained from the Tumor Bank of the Hospital Universitario Central de Asturias (Oviedo, Spain) and used to evaluate the expression of the most downregulated genes (MASP3, DPP10 and PCSK2). Pilot experiments revealed that MASP3 protein expression was reproducibly detected in normal mucosa samples, while antibodies against DPP10 and PCSK2 failed to yield a reliable signal in these samples. Therefore, we decided to focus our analysis on MASP3. The automated system DISCOVERY® (Ventana Medical System, Tucson, AZ) was used to carry out the immunohistochemical protein detection. Sections were deparaffinized and rehydrated in EZ Prep® (Ventana Medical System) for 20 minutes. Antigen retrieval was done by heating (CC1 HCl-Tris buffer solution, pH 9.0) (Ventana Medical System). Endogenous peroxidase activity was blocked with H2O2 solution (Inhibitor®, Ventana Medical System) for 4 minutes. Samples were incubated with primary antibody at 37 ºC with polyclonal anti-MASP3 (Atlas Antibodies). Slides were incubated with the secondary antibody (OmniMap® Ventana medical System) for 30 minutes at room temperature. Then, samples were visualized with DAB (3-3´-Diaminobenzidine) (Ventana Medical System). Finally, samples were counterstained with hematoxyline (Ventana Medical System), dehydrated and mounted in Entellan® (Merck, Germany). Sections were studied and photographed (20X) under a light microscope (Nikon-Eclipse 80i).
DNA constructs
Vector containing human cDNA from MASP3 was purchased from GeneService. The cDNA was tagged with two consecutive FLAG epitopes in the carboxy terminus and subcloned into the pcDNA3.1 and pCEP-Pu expression vectors. The constructs were verified by capillary sequencing. For RNA interference experiments, four shRNA vectors were purchased for MASP3 (Open Biosystems), and their ability to repress the expression of the protease gene was evaluated by quantitative RT-PCR on RNA samples from cells transduced with the respective vectors, individually and combined. Due to the very low basal expression of this protease, only the complete set combined or the vector designed as shRNA MASP3 A produced a significant silencing of this protease in colon cancer cells.
Cell culture
Tumor cell lines 293T, HCT116, SW480, SW620, LoVo, DLD-1, and Caco-2 were purchased from the American Type Culture Collection. The luciferase-expressing cell line HCT116 Luc2 was purchased from Caliper Life Sciences. Cells were routinely maintained in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% heat-inactivated fetal bovine serum (FBS), 100 U/ml penicillin and 50 µg/ml streptomycin.
Transfection
HCT116 Luc2 and DLD-1 cells were transfected with a MASP3 cDNA cloned into pcDNA3.1 and pCEP-Pu, respectively, using Lipofectamine reagent (Invitrogen). Cells were grown at 37 ºC for 2 days in culture medium before changing to selection medium, 1 mg/ml G418 (Invitrogen) for HCT116 Luc2 and 1 µg/ml Puromycin (Sigma) for DLD-1 cells, respectively. After selection, cell populations expressing the desired gene were obtained along with control cell lines transfected with the empty vectors.
Viral package and cell infection
Lentiviruses were packaged in HEK-293T cells using a VSVG-based package system kindly provided by Dr JM Silva (Columbia University, New York, USA). Cells were transfected using TransITs-LT1 Transfection Reagent (Mirus) and a mixture of 2 μg of the desired plasmid and 1 μg of each lentiviral helper, following the manufacturer’s instructions. Transfection medium was removed 24 h after transfection and fresh medium was added to the plate. Cell supernatants were collected at 24 and 48 h and filtered through a 0.45-μm sterile filter. HCT116 and DLD-1 cells were seeded in 6-well plates at 20–30% confluence 24 h before infection. The following day, 1 ml of viral supernatant supplemented with 5 mg/ml of polybrene (Millipore) was added to growing cells. This step was repeated twice and cells were left recovering for 24 h in growing media before puromycin selection (1 µg/ml).
Western Blot analysis
Forty eight hours after transfection, cells were washed twice with 1X PBS and lysed in 50 mM Tris buffer, pH 7.4, containing 150 mM NaCl, 1% Triton X-100, 10 mM EDTA, and complete protease inhibitor cocktail (Roche Applied Science). The protein concentration was evaluated by the bicinchoninic acid technique (BCA protein assay kit; Pierce Biotechnology Inc.). A protein sample (10 μg) was loaded on SDS-polyacrylamide gels. After electrophoresis, gels were electrotransferred onto PVDF filters, and the filters were blocked with 5% nonfat dried milk in TBS-T (TBS with 0.05% Tween-20) and incubated with an anti-FLAG antibody following the recommendations of the supplier (Cell Signalling). After 3 washes with TBS-T, filters were incubated with the corresponding secondary antibody in 1.5% nonfat dry milk in TBS-T, and developed with Immobilon Western Chemiluminescent HRP substrate (Millipore) in a LAS-3000 Imaging System (Fujifilm).
Proliferation assay
To quantify cell proliferation, a Cell Titer 96 Non Radioactive cell proliferation kit was used following manufacturer’s instructions (Promega Corp.). Briefly, HCT116 Luc2 and DLD-1 transfected cells were seeded into 96-well plates at a density of 5x103 cells per well (100 µl) and incubated at 37 ºC, 5% CO2 for 4 days. Cell proliferation was quantified by measuring the conversion of a tetrazolium salt into formazan in living cells. At the desired time points (0 h, 24 h, 48 h and 72 h), 15 µl of Dye solution was added into each well (n=4) and cells were incubated at 37 ºC for 2 h. Then, 100 µl of solubilization/stop mixture was added into each well. After 1 h of incubation at 37 ºC the absorbance was measured at 570 nm with a Power Wave XS Microplate reader (Biotek). Then, each point was normalized with time 0 h and mean ± SEM was calculated and represented. Statistical significance was assessed using a non-parametric Mann Whitney-Wilcoxon test (*, p<0.05; ***, p<0.001).
Mouse xenograft model
Six-week-old athymic Nude-Foxn1nu/nu mice (Charles River) were used for tumor xenograft experiments with HCT116 Luc2 and DLD-1 cell lines, transfected with pcDNA3 or pCEP-Pu constructs or HCT116 and DLD-1 cell lines transduced with shRNA vectors. Eight mice were used for each experiment. Both flanks of each animal were injected subcutaneously with 2 × 106 (HCT116 Luc2) or 3 × 106 (DLD-1) cells in 100 μl PBS resulting in eight flanks per construction. To monitor growth of tumors derived from HCT116 Luc2 cells (transfected with the MASP3 expression vector or with empty pcDNA3.1), tumor size was measured every week by chemiluminescence detection using a Xenogen IVIS system (Caliper Life Sciences). Normalized photon flux ± SEM was calculated for each group. For xenografts of colon cancer cells without a luciferase reporter, tumor size was measured twice a week with a caliper and tumor volume was determined using the formula: V=0.4 x A x B2, where A is the largest and B is the smallest dimension of the tumor. For both types of measurements, significant differences were assessed by a linear mixed-effects model (*, p<0.05).
ACKNOWLEDGMENTS
We thank S. Álvarez, M. Fernández and D. Álvarez for excellent technical assistance, X. Cristina (Applied Biosystems) for help in the design of the low density arrays and R. González for helpful comments. This work has been supported by grants from Ministerio de Ciencia e Innovación-Spain, Fundación “M. Botín”, and European Union (FP7 MicroEnviMet). The Instituto Universitario de Oncología is supported by Obra Social Cajastur and Acción Transversal del Cáncer-RTICC.
References
1. Lopez-Otin C and Bond JS. Proteases: multifunctional enzymes in life and disease. J Biol Chem. 2008; 283(45):30433-30437.
2. Turk B, Turk DS and Turk V. Protease signalling: the cutting edge. EMBO J. 2012.
3. Puente XS and Lopez-Otin C. A genomic analysis of rat proteases and protease inhibitors. Genome Res. 2004; 14(4):609-622.
4. Puente XS, Gutierrez-Fernandez A, Ordonez GR, Hillier LW and Lopez-Otin C. Comparative genomic analysis of human and chimpanzee proteases. Genomics. 2005; 86(6):638-647.
5. Quesada V, Velasco G, Puente XS, Warren WC and Lopez-Otin C. Comparative genomic analysis of the zebra finch degradome provides new insights into evolution of proteases in birds and mammals. BMC Genomics. 2010; 11(1):220.
6. Lopez-Otin C and Overall CM. Protease degradomics: a new challenge for proteomics. Nat Rev Mol Cell Biol. 2002; 3(7):509-519.
7. Overall CM and Lopez-Otin C. Strategies for MMP inhibition in cancer: innovations for the post-trial era. Nat Rev Cancer. 2002; 2(9):657-672.
8. Kessenbrock K, Plaks V and Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell. 2010; 141(1):52-67.
9. Lopez-Otin C and Hunter T. The regulatory crosstalk between kinases and proteases in cancer. Nat Rev Cancer. 2010; 10(4):278-292.
10. Mason SD and Joyce JA. Proteolytic networks in cancer. Trends Cell Biol. 2011; 21(4):228-237.
11. Wei X, Prickett TD, Viloria CG, Molinolo A, Lin JC, Cardenas-Navia I, Cruz P, Rosenberg SA, Davies MA, Gershenwald JE, Lopez-Otin C and Samuels Y. Mutational and functional analysis reveals ADAMTS18 metalloproteinase as a novel driver in melanoma. Mol Cancer Res. 2010; 8(11):1513-1525.
12. Wei X, Moncada-Pazos A, Cal S, Soria-Valles C, Gartner J, Rudloff U, Lin JC, Rosenberg SA, Lopez-Otin C and Samuels Y. Analysis of the disintegrin-metalloproteinases family reveals ADAM29 and ADAM7 are often mutated in melanoma. Hum Mutat. 2011; 32(6):E2148-2175.
13. Saggar S, Chernoff KA, Lodha S, Horev L, Kohl S, Honjo RS, Brandt HR, Hartmann K and Celebi JT. CYLD mutations in familial skin appendage tumours. J Med Genet. 2008; 45(5):298-302.
14. Harbour JW, Onken MD, Roberson ED, Duan S, Cao L, Worley LA, Council ML, Matatall KA, Helms C and Bowcock AM. Frequent mutation of BAP1 in metastasizing uveal melanomas. Science. 2010; 330(6009):1410-1413.
15. Honma K, Tsuzuki S, Nakagawa M, Tagawa H, Nakamura S, Morishima Y and Seto M. TNFAIP3/A20 functions as a novel tumor suppressor gene in several subtypes of non-Hodgkin lymphomas. Blood. 2009; 114(12):2467-2475.
16. Soung YH, Lee JW, Kim SY, Jang J, Park YG, Park WS, Nam SW, Lee JY, Yoo NJ and Lee SH. CASPASE-8 gene is inactivated by somatic mutations in gastric carcinomas. Cancer Res. 2005; 65(3):815-821.
17. Harada K, Toyooka S, Shivapurkar N, Maitra A, Reddy JL, Matta H, Miyajima K, Timmons CF, Tomlinson GE, Mastrangelo D, Hay RJ, Chaudhary PM and Gazdar AF. Deregulation of caspase 8 and 10 expression in pediatric tumors and cell lines. Cancer Res. 2002; 62(20):5897-5901.
18. Gupta GP, Nguyen DX, Chiang AC, Bos PD, Kim JY, Nadal C, Gomis RR, Manova-Todorova K and Massague J. Mediators of vascular remodelling co-opted for sequential steps in lung metastasis. Nature. 2007; 446(7137):765-770.
19. Joyce JA, Baruch A, Chehade K, Meyer-Morse N, Giraudo E, Tsai FY, Greenbaum DC, Hager JH, Bogyo M and Hanahan D. Cathepsin cysteine proteases are effectors of invasive growth and angiogenesis during multistage tumorigenesis. Cancer Cell. 2004; 5(5):443-453.
20. Urban P, Vuaroqueaux V, Labuhn M, Delorenzi M, Wirapati P, Wight E, Senn HJ, Benz C, Eppenberger U and Eppenberger-Castori S. Increased expression of urokinase-type plasminogen activator mRNA determines adverse prognosis in ErbB2-positive primary breast cancer. J Clin Oncol. 2006; 24(26):4245-4253.
21. Luise C, Capra M, Donzelli M, Mazzarol G, Jodice MG, Nuciforo P, Viale G, Di Fiore PP and Confalonieri S. An atlas of altered expression of deubiquitinating enzymes in human cancer. PLoS One. 2011; 6(1):e15891.
22. Lopez-Otin C and Matrisian LM. Emerging roles of proteases in tumour suppression. Nat Rev Cancer. 2007; 7(10):800-808.
23. Fraile JM, Quesada V, Rodriguez D, Freije JM and Lopez-Otin C. Deubiquitinases in cancer: new functions and therapeutic options. Oncogene. 2012; 31(19):2373-2388.
24. Balbin M, Fueyo A, Tester AM, Pendas AM, Pitiot AS, Astudillo A, Overall CM, Shapiro SD and Lopez-Otin C. Loss of collagenase-2 confers increased skin tumor susceptibility to male mice. Nat Genet. 2003; 35(3):252-257.
25. Acuff HB, Sinnamon M, Fingleton B, Boone B, Levy SE, Chen X, Pozzi A, Carbone DP, Schwartz DR, Moin K, Sloane BF and Matrisian LM. Analysis of host- and tumor-derived proteinases using a custom dual species microarray reveals a protective role for stromal matrix metalloproteinase-12 in non-small cell lung cancer. Cancer Res. 2006; 66(16):7968-7975.
26. Houghton AM, Grisolano JL, Baumann ML, Kobayashi DK, Hautamaki RD, Nehring LC, Cornelius LA and Shapiro SD. Macrophage elastase (matrix metalloproteinase-12) suppresses growth of lung metastases. Cancer Res. 2006; 66(12):6149-6155.
27. Sjoblom T, Jones S, Wood LD, Parsons DW, Lin J, Barber TD, Mandelker D, Leary RJ, Ptak J, Silliman N, Szabo S, Buckhaults P, Farrell C, Meeh P, Markowitz SD, Willis J, et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006; 314(5797):268-274.
28. Puente XS, Pinyol M, Quesada V, Conde L, Ordonez GR, Villamor N, Escaramis G, Jares P, Bea S, Gonzalez-Diaz M, Bassaganyas L, Baumann T, Juan M, Lopez-Guerra M, Colomer D, Tubio JM, et al. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature. 2011; 475(7354):101-105.
29. Quesada V, Conde L, Villamor N, Ordonez GR, Jares P, Bassaganyas L, Ramsay AJ, Bea S, Pinyol M, Martinez-Trillos A, Lopez-Guerra M, Colomer D, Navarro A, Baumann T, Aymerich M, Rozman M, et al. Exome sequencing identifies recurrent mutations of the splicing factor SF3B1 gene in chronic lymphocytic leukemia. Nat Genet. 2012; 44(1):47-52.
30. Stratton MR. Exploring the genomes of cancer cells: progress and promise. Science. 2011; 331(6024):1553-1558.
31. Goerge T, Barg A, Schnaeker EM, Poppelmann B, Shpacovitch V, Rattenholl A, Maaser C, Luger TA, Steinhoff M and Schneider SW. Tumor-derived matrix metalloproteinase-1 targets endothelial proteinase-activated receptor 1 promoting endothelial cell activation. Cancer Res. 2006; 66(15):7766-7774.
32. Lynch CC, Crawford HC, Matrisian LM and McDonnell S. Epidermal growth factor upregulates matrix metalloproteinase-7 expression through activation of PEA3 transcription factors. Int J Oncol. 2004; 24(6):1565-1572.
33. Henkhaus RS, Gerner EW and Ignatenko NA. Kallikrein 6 is a mediator of K-RAS-dependent migration of colon carcinoma cells. Biol Chem. 2008; 389(6):757-764.
34. McIver CM, Lloyd JM, Hewett PJ and Hardingham JE. Dipeptidase 1: a candidate tumor-specific molecular marker in colorectal carcinoma. Cancer Lett. 2004; 209(1):67-74.
35. Wilson CL, Heppner KJ, Labosky PA, Hogan BL and Matrisian LM. Intestinal tumorigenesis is suppressed in mice lacking the metalloproteinase matrilysin. Proc Natl Acad Sci U S A. 1997; 94(4):1402-1407.
36. Lind GE, Kleivi K, Meling GI, Teixeira MR, Thiis-Evensen E, Rognum TO and Lothe RA. ADAMTS1, CRABP1, and NR3C1 identified as epigenetically deregulated genes in colorectal tumorigenesis. Cell Oncol. 2006; 28(5-6):259-272.
37. Bister VO, Salmela MT, Karjalainen-Lindsberg ML, Uria J, Lohi J, Puolakkainen P, Lopez-Otin C and Saarialho-Kere U. Differential expression of three matrix metalloproteinases, MMP-19, MMP-26, and MMP-28, in normal and inflamed intestine and colon cancer. Dig Dis Sci. 2004; 49(4):653-661.
38. Viloria CG, Obaya AJ, Moncada-Pazos A, Llamazares M, Astudillo A, Capella G, Cal S and Lopez-Otin C. Genetic inactivation of ADAMTS15 metalloprotease in human colorectal cancer. Cancer Res. 2009; 69(11):4926-4934.
39. Wood LD, Parsons DW, Jones S, Lin J, Sjoblom T, Leary RJ, Shen D, Boca SM, Barber T, Ptak J, Silliman N, Szabo S, Dezso Z, Ustyanksky V, Nikolskaya T, Nikolsky Y, et al. The genomic landscapes of human breast and colorectal cancers. Science. 2007; 318(5853):1108-1113.
40. Freije JM, Fraile JM and Lopez-Otin C. Protease addiction and synthetic lethality in cancer. Front Oncol. 2011; 1:25.
41. Quackenbush J. Microarray analysis and tumor classification. N Engl J Med. 2006; 354(23):2463-2472.
42. Bradford JR, Hey Y, Yates T, Li Y, Pepper SD and Miller CJ. A comparison of massively parallel nucleotide sequencing with oligonucleotide microarrays for global transcription profiling. BMC Genomics. 2010; 11:282.
43. Schwaeble W, Dahl MR, Thiel S, Stover C and Jensenius JC. The mannan-binding lectin-associated serine proteases (MASPs) and MAp19: four components of the lectin pathway activation complex encoded by two genes. Immunobiology. 2002; 205(4-5):455-466.
44. Cortesio CL and Jiang W. Mannan-binding lectin-associated serine protease 3 cleaves synthetic peptides and insulin-like growth factor-binding protein 5. Arch Biochem Biophys. 2006; 449(1-2):164-170.
45. Tonner E, Allan G, Shkreta L, Webster J, Whitelaw CB and Flint DJ. Insulin-like growth factor binding protein-5 (IGFBP-5) potentially regulates programmed cell death and plasminogen activation in the mammary gland. Adv Exp Med Biol. 2000; 480:45-53.
46. Akkiprik M, Feng Y, Wang H, Chen K, Hu L, Sahin A, Krishnamurthy S, Ozer A, Hao X and Zhang W. Multifunctional roles of insulin-like growth factor binding protein 5 in breast cancer. Breast Cancer Res. 2008; 10(4):212.
47. Quesada V, Ordonez GR, Sanchez LM, Puente XS and Lopez-Otin C. The Degradome database: mammalian proteases and diseases of proteolysis. Nucleic Acids Res. 2009; 37(Database issue):D239-243.