
INTRODUCTION
The members of the PYGOPUS and BCL9 protein families have been discovered as novel co-factors of canonical Wnt/ß-catenin signaling [1–3]. These co-activators form a complex with ß-catenin-LEF/TCF to activate transcription of Wnt target genes and to enhance the signaling output [4–8]. However, the knockout of Pygopus and Bcl9’s in the mouse has resulted in only limited embryonic defects compared to other components of the Wnt/ß-catenin pathway [9, 10]. Therefore, PYGOPUS and BCL9 proteins may not be absolutely essential for Wnt/ß-catenin signaling during embryonic development in vertebrates. On the other hand, several studies revealed a specific role in particular for Pygo2 and Bcl9-2 in diseases such as colon cancer to hyperactivate canonical Wnt signaling [3, 8]. Deregulation of the Wnt/ß-catenin signaling pathway was shown to be the primary driver of colon cancer development: the vast majority of sporadic human colon cancers harbor loss-of-function (LOF) mutations of the adenomatous polyposis coli gene (Apc) and less frequently gain of function (GOF) mutations of the ß-catenin encoding proto-oncogene Ctnnb1 [11]. These mutations constitutively activate Wnt/ß-catenin signaling by preventing ß-catenin degradation, which results in accumulation and nuclear translocation of stabilized ß-catenin. In the nucleus, ß-catenin interacts with TCF/LEF transcription factors to activate target gene transcription [12, 13].
Overactivated Wnt/ß-catenin signaling in colon cancer may be further increased by deregulated expression of different members of the Pygopus and Bcl9 coactivator family. Indeed, our previous studies demonstrated significant overexpression of PYGO2 in tumors of APCMin/+ mice, colon cancer cells and in human colon cancer, indicating a possible role in tumor development. In addition, Pygo2 knockdown in colon cancer cells is able to suppress Wnt target gene transcription [8]. Our and other previous in vitro and in vivo studies suggested that Bcl9-2 also acts as an oncogene that enhances Wnt signaling activity in cancer [8, 9, 14–16]. Besides colon cancer, Pygo2 was also suggested to have a potential role in various other malignancies including breast, ovarian, lung, glioblastomas and liver cancers [17–21].
However, the functional relevance of the Pygopus homologues during different stages of colon cancer has not yet been studied in vivo. From our previous studies, we found that BCL9, BCL9-2 and PYGO2, but not PYGO1, were expressed in the epithelial cells of the normal intestinal mucosa [8]. Despite PYGO2 overexpression in colon cancer, our preliminary studies revealed that the knockout of Pygo2 in the intestine had no influence on embryonic intestinal development and for adult intestinal homeostasis (Schelp and Brembeck, unpublished data). Therefore, the present study was designed to investigate the role of Pygo2 during colon cancer initiation and progression in vivo.
Mouse models are valuable tools to study the crucial events during initiation and progression of colon cancer. Several mouse models of Apc LOF and Ctnnb1 GOF were engineered that mimic the different stages of human colon cancer development [22]. Intercrossing of these mouse models with other mutants revealed the importance of several additional genes for colon carcinogenesis [23]. In our study, we first analyzed a chemically induced colon cancer mouse model with and without constitutive knockout of Pygo2. In addition, we have generated inducible compound mutant mice to analyze the role of Pygo2 during different stages of tumorigenesis. For this, we have intercrossed mice harboring either hetero- or homozygous intestine specific deletion of Pygo2 with inducible mutants of stabilized ß-catenin (Ctnnb1) [24] or homo- and heterozygous truncated Apc [25]. Our results presented here indicate that Pygo2 synergizes intestinal tumor formation in vivo that is primarily driven by aberrant Wnt signaling. We describe that Pygo2 knockout reduced tumor formation in chemically induced colon tumors. Moreover, we show that Pygo2 is essential for early stages of intestinal tumorigenesis induced by Ctnnb1 mutation, but not in the context of Apc mutations. Thus, targeting Pygo2 may represent an attractive therapeutic option to suppress or arrest tumor growth in human colon cancer in a context dependent manner. This is of particular interest, since Pygo2 appears to be dispensable for normal intestinal homeostasis.
RESULTS
Pygo2 knockout delays the progression of chemically induced colon tumors.
Our previous data indicated that Pygo2 might be important for intestinal tumorigenesis [1, 8, 26]. We have previously analyzed in detail the phenotype of mutant mice with constitutive deletion of both Pygo2 alleles in the intestine (Schelp and Brembeck, unpublished data) and compared them with control littermates (see Methods, Schäffer and Birchmeier). Analysis of mutant mice revealed that loss of Pygo2 neither disturbed normal embryonic development of the intestine nor impaired intestinal homeostasis or lineage commitment in adults (Schelp and Brembeck, unpublished data). Of note, we also did not observe any effect of intestinal deletion of both Pygo1 and Pygo2 genes, indicating that both genes are completely dispensable for normal intestinal function (Schelp and Brembeck, unpublished data).
To test the potential pro-oncogenic role of Pygo2 in intestinal tumors, we first challenged constitutive, intestine specific Pygo2 deficient mice (“Pygo2−/−”; corresponding to the genotype VilCre; Pygo2 Δ/Δ) and control littermates (Pygo2lox(ex3)/lox(ex3)). We treated the mice with an adapted scheme for chemically induced tumor formation in the colon [27]. 6- to 8-week old mice were injected intraperitoneally with one single dose of azoxymethane (AOM). One week later, mice were treated with dextran sodium sulfate (DSS) in the drinking water for five consecutive days. Additionally, control and Pygo2 knockout mice were treated with DSS alone, to analyze a role of Pygo2 during intestinal epithelial regeneration following inflammation. However, independent of Pygo2 loss, all mice recovered from the initial phase of DSS induced inflammation as monitored by changes in body weight and survival (Supplementary Figure S1A and data not shown). Our histopathological analysis at 14 and 28 days after beginning the treatment did not reveal any disturbance of intestinal regeneration by Pygo2 ablation (Supplementary Figure S1B, S1C). These observations corroborate our results that Pygo2 is apparently dispensable for normal intestinal homeostasis in the adult.
We monitored survival and tumor development in chemically treated mice for six months. During this time, most animals developed massive signs of anal bleeding and weight loss. In total, 86 percent of control mice (25 of 29 animals) and 83 percent of Pygo2 knockout animals (30 of 36 mice) were available for analysis at this endpoint. Anal prolapse was seen in 18 of 25 control mice (72%), but only in 5 of 30 Pygo2 deficient animals (17%). We carefully examined the entire intestine and measured the size of tumors, which were all located within the colon (Figure 1A). The total number of colon tumors was highly significantly reduced in Pygo2 mutant animals (mean number of tumors 2.5 in Pygo2 knockouts versus 4.2 in controls, P = 0.0023; Figure 1A). Moreover, the number of larger tumors (3 mm diameter or more) was also strongly reduced compared to controls (0.5 versus 2.2, P = 0.0002; Figure 1A).
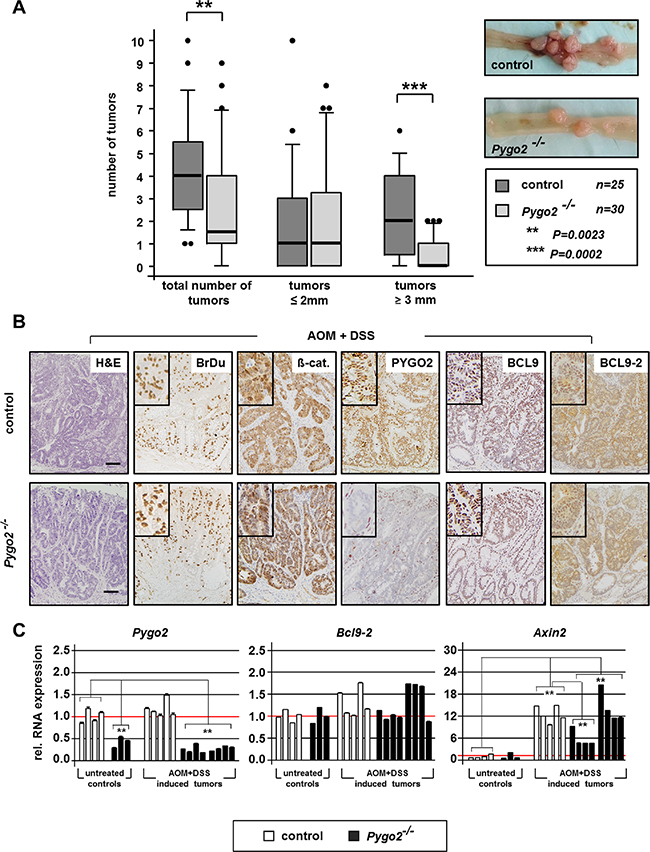
Figure 1: Knockout of Pygo2 delays the progression of chemically induced intestinal tumors. (A) Boxplot analysis illustrates number and size of colon tumors in control (Pygo2lox(ex3)/lox(ex3); n = 30) and constitutive Pygo2 deficient mice (marked with Pygo2−/− corresponding to VilCre; Pygo2Δ/Δ; n = 35). All mice were sacrificed six months after beginning with AOM and DSS treatment. Pygo2 deficient animals were compared to age-matched littermate controls. Significant difference is indicated with ** for P < 0.01 and *** for P < 0.001. Microscopic views of control and Pygo2 deficient colon tumors show tumor burden. (B) Representative H&E stains and immunostains with indicated antibodies on tumor sections from control and Pygo2 knockout animals. Scale bars in the pictures represent 200 μm for all IHC. Inserts show the staining at higher magnification. (C) qRT-PCR analyses of RNA extracted from colon tumors of control and Pygo2 deficient animals and colon tissues obtained from untreated mice with the same genotype. Each bar represents the relative RNA expression of the indicated gene for single animal; each result represents at least three independent experiments. Significances were calculated for the mean expression level compared to untreated controls and relative to the expression levels of control tumors, respectively. All significant differences are marked with * for P < 0.05 and ** for P < 0.01.
We further analyzed the tumors by immunohistochemistry and performed qRT-PCR using total RNA extracted from tumor tissues and whole tissues derived from the colon of untreated controls (Figure 1B, 1C). Colon tumors from controls and Pygo2 knockout animals showed partially invasiveness into the submucosa and high proliferation as detected by BrdU stains (Figure 1B). Most tumors also demonstrated nuclear ß-catenin, which suggests that chemically induced tumors are linked to hyperactivated Wnt/ß-catenin signaling. PYGO2 was strongly expressed in the nuclei of the tumor cells in controls, but not detectable in tumors of Pygo2 deficient mice. On the RNA level, Pygo2 was not transcriptionally induced in the tumors, and significantly downregulated in Pygo2 knockout tumors. We also analyzed the expression of the co-factors Bcl9 and Bcl9- 2. The expression of BCL9 was NOT increased as we have previously reported in human colon cancers [8]. BCL9-2 was moderately upregulated in chemically induced colon tumors (Figure 1B, 1C). This is in contrast to our previous observation that BCL9-2 expression is strongly induced in adenoma of APCMin/+ mice and human colon cancer [8].
Moreover, Axin2, representing a reliable marker of activated Wnt/ß-catenin signaling [28], was strongly transcriptionally induced in the tumors of control and Pygo2 deficient animals. Of note, a subgroup of colon tumors derived from Pygo2 deficient animals showed significant repression of Axin2, suggesting that Wnt/ß-catenin signaling is partially inhibited in these tumors of Pygo2 deficient mice.
Taken together, ablation of Pygo2 can partially rescue the tumor formation of chemically induced colon tumors that may be linked to hyperactivated Wnt/ß-catenin signaling. Thus, loss of Pygo2 cannot prevent colon tumor development in the chemically induced carcinogenesis model, but may delay tumor progression in terms of tumor number and size.
Deletion of Pygo2 rescues the initial phase of intestinal hyperproliferation induced by ß-catenin (Ctnnb1) gain-of-function, but not hyperproliferation and adenoma formation induced by Apc loss-of-function
Our preliminary results indicated that deletion of Pygo2 exerts different effects in early phases of intestinal tumor development in contrast to APCMin/+ mice, in which the formation of adenoma was not prevented by constitutive ablation of Pygo2 (Schelp and Brembeck, unpublished data). In our study reported here, we used inducible intestine-specific ablation of either one or both alleles of Pygo2 (hereafter labeled with Pygo2+/− and Pygo2-/-; corresponding to the genotypes VilCre-ERT; Pygo2 Δ/+ and VilCre-ERT; Pygo2 Δ/Δ respectively). Pygo2 deficient mice were crossed with genetic mutants harboring either hetero- or homozygous truncated Apc (Apc LOF) [25] or heterozygous stabilized ß-catenin (Ctnnb1 GOF) to generate compound animals [24]. Details on mutant mice are provided in Methods. We induced genetic recombination in the intestine by intraperitoneal injection of tamoxifen for five consecutive days [29]. With this, we achieved almost complete deletion of Pygo2 expression throughout the small and large intestine (data not shown) and introduced Apc or Ctnnb1 mutations in the same targeted epithelial cells. In all studies, Pygo2 deficient animals were compared to appropriate control littermates with the corresponding genetic background.
First we compared the effects of Pygo2 loss during the acute phase of intestinal tumor initiation induced by homozygous truncated Apc (Apc LOF; Pygo2+/+, +/− and −/−) and by heterozygous stabilized ß-catenin (Ctnnb1 GOF; Pygo2+/+, +/− and −/− ; Figures 2, 3). Loss of function of Apc led to a severe intestinal phenotype within one week post induction and all mutant mice died within seven to eight days. The survival of Apc mutant animals was not increased by the loss of one or both alleles of Pygo2 Figure 2A). Analyses of intestinal tissues of mice sacrificed on day six after start of induction demonstrated massive intestinal hyperproliferation with elongated crypts, strong proliferation as detected by BrdU stain and upregulation of cytoplasmatic and nuclear ß-catenin (Figure 2B). However, knockout of Pygo2 did not rescue the intestinal hyperproliferation induced by Apc LOF.
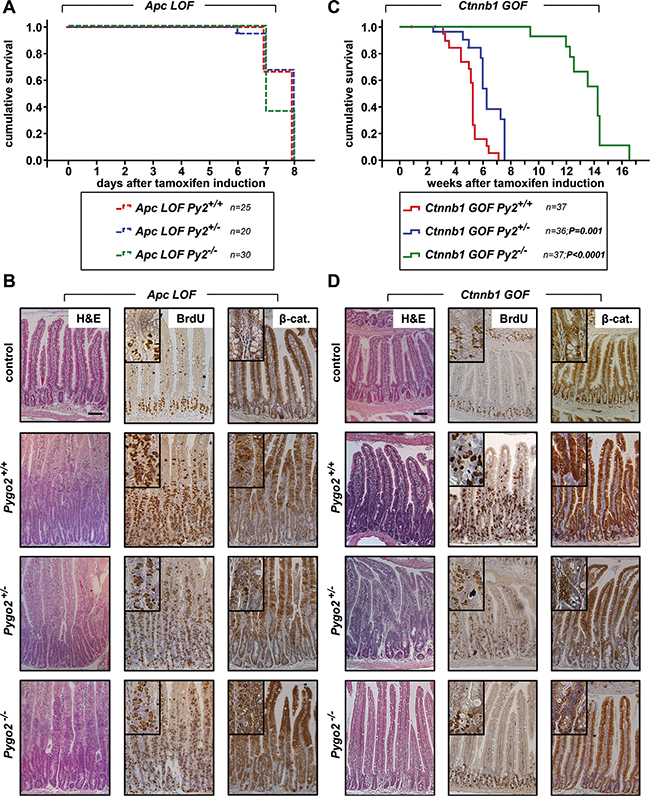
Figure 2: Knockout of Pygo2 completely rescues the initial phase of intestinal hyperproliferation induced by ß-catenin (Ctnnb1) gain-of-function, but not of Apc loss-of-function. (A) Kaplan-Meier survival analysis of compound homozygous APC loss-of-function animals (LOF) with wild type Pygo2 expression (labelled with Apc LOF Py2+/+ corresponding to the genotype VilCre-ERT; ApcΔex15/Δex15; Pygo2+/+, n = 25) compared to heterozygous Pygo2 deficient mice (marked with Apc LOF Py2+/− corresponding to VilCre-ERT; ApcΔex15/Δex15; Pygo2 Δ/+; n = 20) and homozygous Pygo2 knockout mice (labelled with Apc LOF Py2−/− corresponding to VilCre-ERT; ApcΔex15/Δex15; Pygo2 Δ/Δ; n = 30). All mice were induced by tamoxifen injection for five consecutive days. (B) Representative immunostains on intestinal sections from compound homozygous Apc LOF mutant animals with wild type Pygo2 expression (Pygo2+/+) compared to mice with hetero- and homozygous ablation of Pygo2 (Pygo2+/− and Pygo2−/−). The upper panel shows intestinal tissues from uninduced control mice (with the genotype Apclox(ex15)/lox(ex15); Pygo2lox(ex3)/lox(ex3)). Animals were analyzed on day six after start of induction with tamoxifen. Tissue sections were stained by H&E and with indicated antibodies. (C) Kaplan-Meier survival analysis of compound heterozygous ß-catenin gain-of-function animals (GOF) with wild type Pygo2 expression (labelled with Ctnnb1 GOF Py2+/+ corresponding to the genotype VilCre-ERT; Ctnnb1Δex3/+; Pygo2+/+; n = 37) compared to heterozygous Pygo2 deficient mice (marked with Ctnnb1 GOF Py2+/− corresponding to VilCre-ERT; Ctnnb1Δex3/+; Pygo2 Δ/+; n = 36) and homozygous Pygo2 knockout mice (labelled with Ctnnb1 GOF Py2−/− corresponding to VilCre-ERT; Ctnnb1Δex3/+; Pygo2 Δ/Δ; n = 37). Recombination was induced by tamoxifen injections on five consecutive days. Significance was calculated using the P Log Rank Test with p = 0.001 for heterozygous and p < 0.0001 for homozygous Pygo2 deficient mice compared to wild type Pygo2 expressing animals. (D) Immunostains with the indicated antibodies on intestinal tissue sections from Ctnnb1 GOF animals with different Pygo2 background. The upper panel shows stainings from uninduced control mice (Ctnnb1lox(ex3)/+; Pygo2lox(ex3)/lox(ex3)). Mice were analyzed on day 18 after beginning of tamoxifen treatment. Scale bars in the pictures represent 200 μm for all IHC. Inserts show the staining at higher magnification.
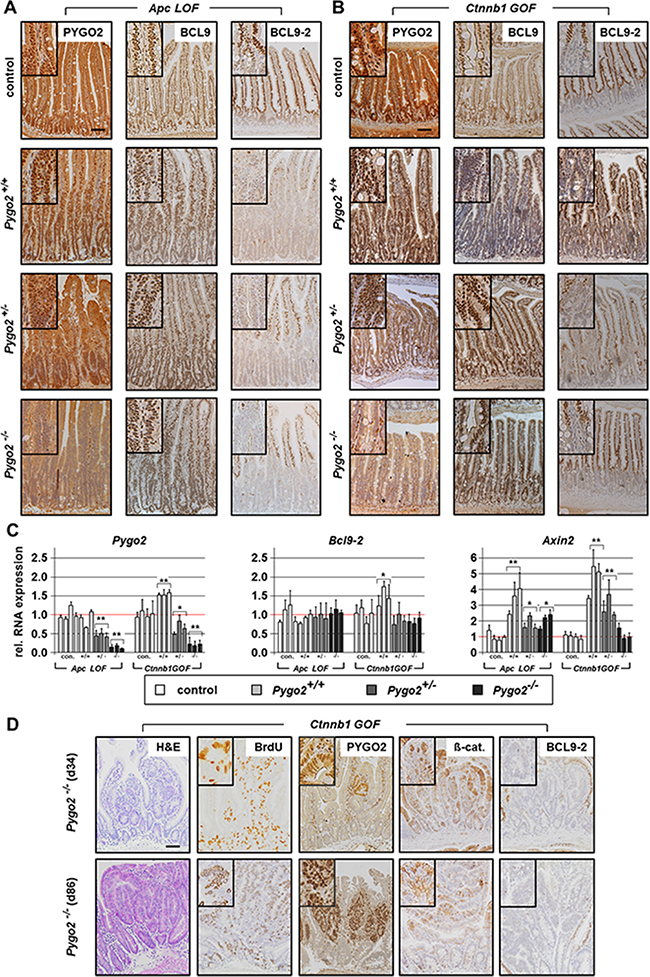
Figure 3: Analysis of Pygo2, Bcl9 co-factors, and Axin2 in Apc LOF and Ctnnb1 GOF mutants; failed Pygo2 deletion by recombination induced microadenoma formation in Ctnnb1 GOF-Pygo2 deficient animals. (A, B) Immunostainings with indicated antibodies on intestinal tissue sections from compound Apc homozygous LOF mice (A) and heterozygous Ctnnb1 GOF animals (B) with wild type Pygo2 expression and hetero- and homozygous Pygo2 ablation. The upper panel shows the same stains from controls. (C) qRT-PCR analyses show RNA expression of Pygo2, Bcl9-2 and Axin2. RNA is extracted from intestinal epithelial cells of Apc LOF mutants on day 6 and Ctnnb1 GOF mutants on day 18 post induction. Each bar represents one animal. Significances were calculated for the mean expression level compared to the respective control group. The significant differences are marked with * for P < 0.05, and ** for P < 0.01. (D) H&E stains and immunohistochemical analyses of intestinal tissues from compound Pygo2 deficient Ctnnb1 GOF mice, which were sacrificed on day 34 and day 86. Note that the intestine shows small areas with expression of PYGO2 (upper panel). These animals develop at later stages multiple microadenoma (lower panel) that express PYGO2, but not BCL9-2, with increased BrdU staining and concomitant nuclear ß-catenin. Scale bars in the pictures represent 200 μm for all IHC. Inserts show the staining at higher magnification.
In contrast, introduction of one mutant Ctnnb1 allele led to a less severe phenotype and all mice died between two and to seven weeks post induction (Figure 2C). Surprisingly, ablation of one allele of Pygo2 prolonged the survival of Ctnnb1 GOF animals significantly (n = 36, p < 0.001; Figure 2C). Complete knockout of Pygo2 prolonged the survival of Ctnnb1 mutants in the initial phase for up to ten weeks (n = 37; p < 0.0001). We also analyzed the intestines of these compound mutant mice by immunohistochemistry. Intestinal hyperproliferation induced by Ctnnb1 GOF was mild starting on day six after induction and was most prominent on day 18, when compound Ctnnb1 GOF, Pygo2 wild type animals started to die (Figure 2D and data not shown). The intestines of Ctnnb1 GOF mutant animals with PYGO2 wild type expression showed strong enlargement of crypts, increased proliferation and high levels of cytoplasmatic und nuclear ß-catenin. Remarkably, hyperproliferation was reduced in animals after ablation of one allele of Pygo2. Most notably, ablation of both Pygo2 alleles completely rescued the phenotype of Ctnnb1 GOF animals: The intestinal architecture was similar to controls with normal crypt-villus axes, and proliferative cells were restricted to the stem cell zone (Figure 2D).
We next analyzed the expression of the Pygo and Bcl9 cofactors by immunohistochemistry. Moreover, we enriched intestinal tissues for epithelial cells (see Material and Methods) and performed qRT-PCRs from the extracted epithelial RNA (Figure 3A–3C). PYGO2 was strongly expressed within the hyperproliferative cells of the intestine in both Apc LOF and Ctnnb1 GOF mutant animals with Pygo2 wild type expression. On the RNA level, Pygo2 was slightly increased only in Ctnnb1 GOF mice. Knockout of one Pygo2 allele reduced RNA levels to approximately 50 percent. The complete knockout of both Pygo2 alleles almost completely reduced Pygo2 RNA levels, and PYGO2 protein was not detectable in the epithelial cells of the intestine, both in Apc LOF and Ctnnb1 GOF mutant mice. As expected, BCL9 expression remained unchanged in both mouse models. Bcl9-2 transcription was not induced and the protein was completely absent in the hyperproliferative regions in Apc LOF mutants (Figure 3A, 3C). In Ctnnb1 GOF-Pygo2 wild type mice, Bcl9-2 was only slightly increased on the RNA level, but was not detectable on the protein level in the hyperproliferative cells (Figure 3B, 3C). These results provide in vivo evidence that Bcl9-2 is not a target of deregulated Wnt/ß-catenin signaling during tumor initiation. However, transcription of Axin2 as marker of activated Wnt signaling was strongly induced in both Apc LOF and Ctnnb1 GOF mutant animals (Figure 3C). Remarkably, Axin2 was reduced in compound heterozygous and homozygous Pygo2 deficient mice in both models. However, knockout of Pygo2 completely reduced Axin2 expression only in Ctnnb1 GOF mice to the control level, but not in Apc LOF animals. These findings further corroborate that Pygo2 knockout completely rescued the initial phase of intestinal hyperproliferation induced by stabilized ß-catenin (Ctnnb1), but not of truncated Apc.
During the survival analyses of Ctnnb1 mutant mice, we observed that homozygous Pygo2 knockout animals survived the initial phase of tumor initiation for up to ten weeks after induction. However, these animals also died within four months (Figure 2C). We therefore analyzed the intestines of mutant animals by immunohistochemistry at later points of time. Approximately one month post induction, when all Pygo2 wild type animals already died, we found small areas with increased proliferation and most importantly with re-expression of PYGO2 in single cells (Figure 3D upper panel). At later time points (approximately after three months; Figure 3D lower panel), the intestines of these mice were covered with multiple small adenoma that showed high proliferation and increased ß-catenin. PYGO2 was strongly expressed in these microadenoma, while the surrounding intestinal epithelial cells did not express PYGO2. Of note, these small adenoma did not express BCL9-2. These observations led us to hypothesize that these tumors may arise from single intestinal cells that may have escaped the initial recombination for the loss of Pygo2. Thus, re-expression of PYGO2 appears to promote microadenoma formation in compound Ctnnb1 GOF-Pygo2 knockout animals at later stages.
We also analyzed in detail adenoma formation by the inducible loss of only one functional allele of Apc, which results in an ApcMin/+ -like phenotype [25]. We compared animals that are wild type for Pygo2 with hetero- and homozygous Pygo2 deficient animals (Supplementary Figure S2). Survival and tumor development of animals was monitored and adenoma were analyzed by immunohistochemistry. Deletion of one or two alleles of Pygo2 did not significantly change the overall survival of Apc mutant animals, which died between six and seven months post induction (Supplementary Figure S2A). Analyses of the intestines of mutant animals revealed that the tissues exhibited multiple small adenoma without any gross difference between Pygo2 wild type and knockout (not shown). Loss of PYGO2 expression in the adenoma was confirmed by immunohistochemistry in Pygo2 deficient animals. Moreover, all adenoma of the analyzed genotypes were highly proliferative, showed expression of nuclear ß-catenin and upregulation of BCL9-2 (Supplementary Figure S2B). Thus, Pygo2 ablation apparently cannot prevent adenoma formation that is genetically induced by loss of one functional Apc allele in vivo, which also exhibits loss of the second allele due to loss-of-heterozygosity (LOH) [30, 31].
In summary, we provide in vivo evidence that Pygo2 loss cannot compensate the intestinal hyperproliferation and adenoma formation resulting from Apc truncation. In contrast, Pygo2 ablation rescues the initial phase of intestinal hyperproliferation that is induced by stabilized ß-catenin.
Pygo2 differentially activates key target genes during the initial phase of intestinal tumorigenesis in Apc versus Ctnnb1 mutant mice and in chemically induced tumors
Next, we characterized the in vivo role of Pygo2 for the transcriptional activation of Wnt/ß-catenin target genes and of genes that are implicated in intestinal tumor initiation and progression. We and others have previously shown that the nuclear co-factors of the Bcl9 and Pygopus gene families are not general co-activators of canonical Wnt signaling, but that they activate a specific gene signature in normal and transformed cells [1, 8, 32–34]. Therefore, we hypothesized that Pygo2 might also differentially activate such target genes in vivo during intestinal tumorigenesis.
First, we compared the initial phase of intestinal hyperproliferation induced by Apc LOF and Ctnnb1 GOF on day six and 18, respectively. We assessed the expression of the Lef/Tcf transcription factors, which are activated by Wnt/ß-catenin-signaling [35] (Figure 4A–4C and Supplementary Figure S3A–S3B). In the normal intestine, TCF4 is expressed in the epithelial cells of the crypt compartment and along the crypt-villus axis with highest expression in the villi [36], which we also found in controls. During the initial phase of hyperproliferation, we established that Tcf4 was not upregulated at the protein or RNA level in Apc LOF mice. Tcf4 was moderately increased in Ctnnb1 GOF animals, both on protein and RNA expression levels. Remarkably, loss of one or two alleles of Pygo2 downregulated Tcf4 RNA in both mouse models, even below the expression level of controls. In contrast to Tcf4, we found that LEF1 protein was barely detectable in the normal intestine. Lef1 protein and RNA levels were strongly upregulated, both in Apc LOF and Ctnnb1 GOF mice. Remarkably, elevated Lef1 was strongly suppressed in Pygo2 heterozygous and even stronger in homozygous knockout animals in both mouse models. However, Lef1 was still higher in Pygo2 deficient intestines of Apc and Ctnnb1 mutant mice compared to controls. Full-length Tcf1, previously described as target of Tcf4 [37], was also increased in Apc and Ctnnb1 mutant tissues. However, ablation of Pygo2 only moderately decreased overexpressed Tcf1 levels. Taken together, Pygo2 ablation reduces the overexpression of Lef1 and Tcf1, and downregulates Tcf4 in Apc mutant animals, but does not rescue intestinal hyperproliferation. In Ctnnb1 mutants, however, the same repression of Tcf1, Tcf4 and Lef1 is seen after loss of Pygo2, but the intestinal hyperproliferation is completely rescued. Thus, the effect of Pygo2 ablation on Lef/Tcf expression during the initial phase of tumorigenesis is apparently not linked to the outcome of the Apc LOF or Ctnnb1 GOF phenotype.
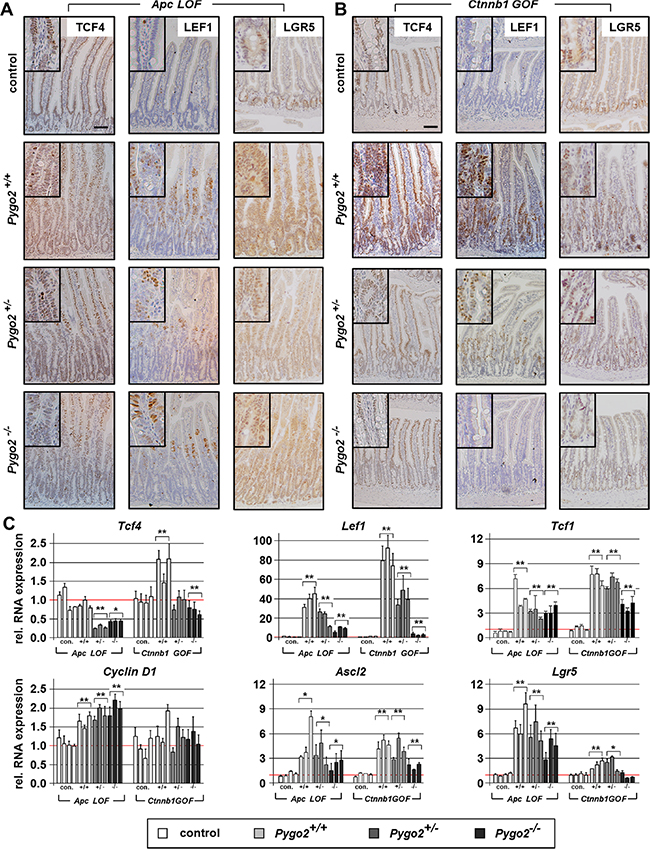
Figure 4: Pygo2 ablation downregulates different Wnt/ß-catenin target genes overexpressed during intestinal hyperproliferation induced by Apc LOF and Ctnnb1 GOF. (A, B) Representative Immunostains with the indicated antibodies on intestinal tissue sections from compound homozygous Apc LOF mice (A) and heterozygous Ctnnb1 GOF animals (B). The upper panel shows the stainings from controls. Pictures from immunostains of compound mutant animals with wild type Pygo2 expression and hetero- and homozygous Pygo2 ablation are provided below. Scale bars in the pictures represent 200 μm for all IHC. Inserts show the staining at higher magnification. (C) Relative RNA expression of Wnt/ß-catenin target genes in Apc LOF and Ctnnb1 GOF mutant animals. qRT-PCR was performed with RNA extracted from intestinal epithelial cells of compound mutant animals as indicated below the graphs. Significances are marked with *P < 0.05, and **P < 0.01 for the mean expression level compared to the respective control group.
We further assessed the regulation of other known Wnt/ß-catenin target genes such as Cyclin D1 (Figure 4C and Supplementary Figure S3A–S3B). Cyclin D1 transcription and protein expression was also increased in Apc mutant animals and remained unchanged by ablation of Pygo2. In contrast, Cyclin D1 RNA was not increased but the protein was clearly expressed in the hyperproliferative cells in Ctnnb1 GOF animals. Again, CyclinD1 protein expression was completely normal and restricted to the stem cell compartment in Ctnnb1 GOF animals with ablation of Pygo2, similar to controls. Moreover, we characterized the expression of the intestinal stem cell markers Ascl2 and Lgr5, which are also Wnt/ß-catenin target genes in the cycling columnar cells at the crypt base [38–41] (Figure 4A–4C). As expected, the expression of both stem cell markers was increased in Apc and Ctnnb1 mutant animals. Lgr5 and Ascl2 were highly increased in Apc mutant animals, and the overexpression was only slightly reduced by deletion of Pygo2. In contrast, ablation of Pygo2 reduced upregulated Ascl2 and completely suppressed the upregulation of Lgr5 in Ctnnb1 GOF intestines to control levels. Of note, Lgr5 was only moderately overexpressed in Ctnnb1 mice compared to Ascl2. This observation may indicate that deletion of Pygo2 can reduce deregulated Wnt signaling to a certain threshold.
In summary, the overexpression of Wnt/ß-catenin target genes such as Lef/Tcf’s, Axin2, Cyclin D1, Ascl2 and Lgr5 induced by truncated Apc cannot be rescued by loss of Pygo2. In contrast, ablation of Pygo2 in the context of stabilized ß-catenin (Ctnnb1) counteracts the overexpression of these target genes. Moreover, these data suggest that the less severe phenotype of Ctnnb1 mutant intestines is also linked to lower levels of increased Wnt/ß-catenin target gene expression. Thus, knockout of Pygo2 is apparently sufficient to reduce target gene expression to normal levels in this model. In summary, Pygo2 loss apparently reduces deregulated Wnt signaling gene output to a certain threshold.
Next, we characterized the importance of Pygo2 for the expression of differentiation markers in Apc and ß-catenin (Ctnnb1) mutant intestines (Figure 5A– 5C, Supplementary Figure S3C–S3E). Paneth cells located in the crypt bottom are characterized by the expression of Lysozyme and of the Wnt/ß-catenin target gene Sox9 [42, 43]. Following Apc induced intestinal hyperproliferation we found Paneth-like cells throughout the hyperproliferative epithelium as indicated by Lysozyme stains. Moreover, Sox9 RNA was strongly induced and the protein was almost completely expressed in all cells along the crypt-villus axis. These Apc induced changes of Sox9 expression and cell differentiation were unchanged by ablation of Pygo2. Overexpression of Sox9 and mislocalization of Paneth-like cells were also present in Ctnnb1 GOF intestine, although less pronounced. Knockout of Pygo2 only partially rescued these changes. In particular, SOX9 was still moderately overexpressed (Figure 5A–5C, Supplementary Figure S3C–S3D). Thus, loss of Pygo2 has no or little effect on the generation of Paneth-like cells in the hyperproliferative intestine of Apc or Ctnnb1 mutants.
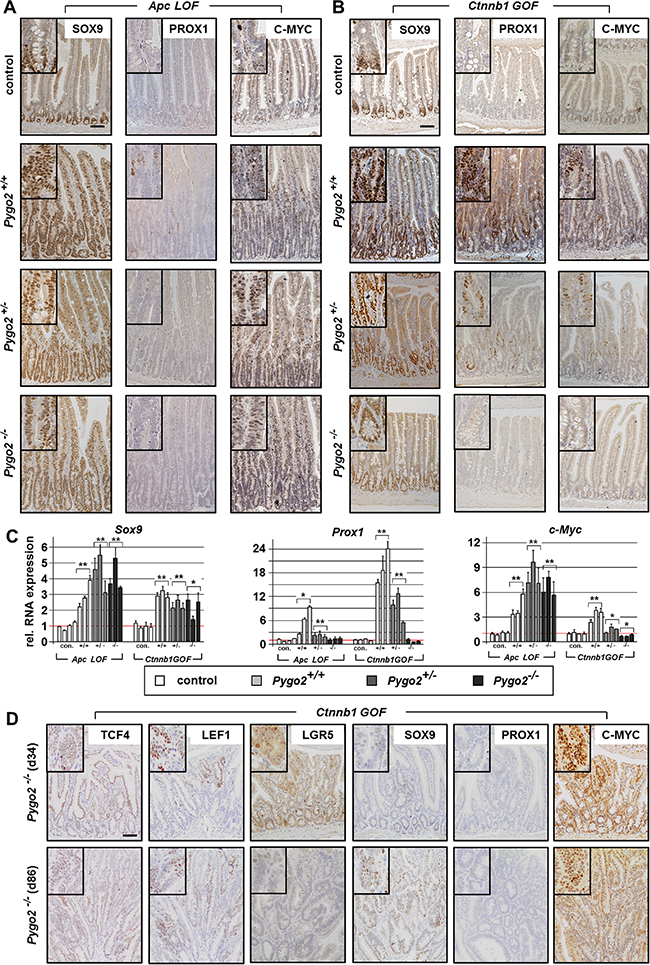
Figure 5: Pygo2 ablation completely suppresses the upregulation of Prox1, but fails to decrease the overexpression of c-Myc during early intestinal tumorigenesis in Apc LOF mice. (A, B) Immunostains for detecting tumor progression markers SOX9, PROX1 and C-MYC on intestinal tissue sections of compound Apc LOF (A) and Ctnnb1 GOF animals (B). (C) qRT-PCR analyses for the relative RNA expression of Sox9, Prox1 and C-myc. Each bar represents one animal. Each graph represents the mean of at least three independent experiments from RNA of mice with the indicated genotype. Significant difference for the mean expression level relative to the corresponding control group is marked * for P < 0.05, and ** for P < 0.01. (D) Immunohistochemical analyses of intestinal tissues from compound Pygo2 deficient Ctnnb1 GOF mice on day 34 and day 86 post induction. Sections were stained with indicated antibodies for detecting Wnt/ß-catenin targets and tumor progression markers. Note that microadenoma also do not express PROX1, but show overexpression of C-MYC in the adenomatous cells. Scale bars in the pictures represent 200 μm for all IHC. Inserts show the staining at higher magnification.
We also studied the expression of Dclk1 that marks Tuft cells in the normal intestine and was recently suggested to be a tumor stem cell marker in the intestine [44] (Supplementary Figure S3C–S3E). Dclk1 expression was unchanged in Ctnnb1 GOF mutants and not affected by Pygo2 loss. Remarkably, Dclk1 was completely lost in Apc LOF mutants, both on Pygo2 wild type and knockout background. This might reflect the differentiation defects due to relatively severe hyperproliferation in Apc mutants.
Finally, we characterized transcription factors, which are important for tumor progression during intestinal carcinogenesis. We analyzed Prox1, a stem cell regulator in intestinal tumors that promotes the transition from benign to a malignant phenotype by oncogenic Wnt/ß-catenin signaling (Figure 5A–5C) [45, 46]. In fact, we found strong upregulation of Prox1 in Apc LOF and even more prominent in Ctnnb1 GOF mice. Most interestingly, Pygo2 knockout completely downregulated Prox1 levels to control levels in both animal models. However, despite the downregulation of Prox1 in Pygo2 deficient animals, the hyperproliferation induced by mutant Apc was not rescued. These data suggest that Pygo2 function on Prox1 expression is not essential for intestinal tumorigenesis during early stages in our models.
Last, we analyzed c-Myc, a key transcription factor for early stages of intestinal tumorigenesis that is activated by Wnt/ß-catenin signaling [47] (Figure 5A–5C). Of note, genetic deletion of c-Myc was shown to completely rescue the phenotype of Apc induced early tumorigenesis [48]. In fact, we found strong upregulation of c-Myc RNA levels and protein expression in Apc LOF mice. However, knockout of Pygo2 failed to downregulate c-Myc overexpression in Apc mutant animals. Similarly, c-Myc was also upregulated in Ctnnb1 GOF intestines however, this was less pronounced compared to Apc mutants. Importantly, deletion of one Pygo2 allele downregulated c-Myc and knockout of both Pygo2 alleles completely suppressed c-Myc expression even below control levels. Thus, the function of Pygo2 for hyperactivated Wnt/ß-catenin signaling appears to increase the signaling output to a certain threshold. Overexpressed c-Myc may represent a key target, which can be downregulated to a certain threshold by Pygo2 deletion during tumorigenesis.
We also analyzed the target genes in microadenoma of Pygo2 deficient-Ctnnb1 GOF mice within three months after induction (Figure 5D and data not shown). These tumors emerged from Pygo2 escapers (see Figure 3D) and demonstrated also overexpression of TCF4, LEF1 and SOX9, but still lacked PROX1. Moreover, LGR5 was only slightly increased within the adenomatous cells. Most importantly, the microadenoma showed also strong expression of C-MYC, which further supports that C-MYC expression was apparently activated by Pygo2 in Ctnnb1 GOF mutants.
Of particular note, intestinal epithelial specific conditional loss of Pygo2 in mice did not influence the expression of most of the Wnt/ß-catenin target genes analyzed in our study (Supplementary Figure S4). Only the relative RNA expression of Tcf4 and Lef1 was slightly, but significantly, downregulated compared to controls. However, these minor changes in gene expression are apparently not sufficient to result in any phenotypic changes. These data further suggest that Pygo2 is apparently dispensable for the expression of target genes in the normal intestine.
Recently, Pygo2 was also implicated in Wnt/ß-catenin-independent functions, for instance to increase Notch signaling in mammary gland cells [49]. We therefore asked if there might be a similar effect in Pygo2 deficient intestines in our animal models (Supplementary Figures S3E, S4 and S5B). We analyzed the RNA levels of Hes1 as the most abundantly expressed Notch target gene in the intestine [55]. Hes1 is expressed in proliferating crypt cells that differentiate to absorptive enterocytes [50]. However, Hes1 RNA was not upregulated in the hyperproliferative intestine of both Apc LOF and Ctnnb1 GOF mice and in chemically induced tumors. Genetic ablation of Pygo2 did only slightly downregulate Hes1 transcription in Ctnnb1 GOF animals, but not in Apc LOF mice and in chemically induced tumors. Moreover, Hes1 was also not suppressed in the normal intestine by loss of Pygo2. This may suggest that Notch signaling is not a major target of Pygo2 in the intestine.
Finally, we analyzed if these target genes were also affected in chemically induced intestinal tumors following Pygo2 knockout. We analyzed the expression by immunohistochemistry and qRT-PCR on RNA extracted from the tumors (Figure 6A, 6B and Supplementary Figure S5). In fact, all target genes with the exception of Tcf4 were also strongly overexpressed in the tumors of control and Pygo2 deficient animals, both on the protein and RNA level. However, a subgroup of chemically induced tumors derived from Pygo2 deficient animals showed also significant reduction of the overexpression of Lef1, Ascl2, Lgr5, Sox9 and Prox1 RNA levels compared to control tumors (Figure 6B). In contrast and most importantly, the transcription of c-Myc was not downregulated by Pygo2 ablation. Thus, also chemically induced intestinal tumors show a similar pattern of gene regulation in the context of Pygo2 knockout as seen in early stages of tumorigenesis induced by Apc LOF or Ctnnb1 GOF. Moreover, deletion of Pygo2 cannot rescue the overexpression of c-Myc in this tumor model.
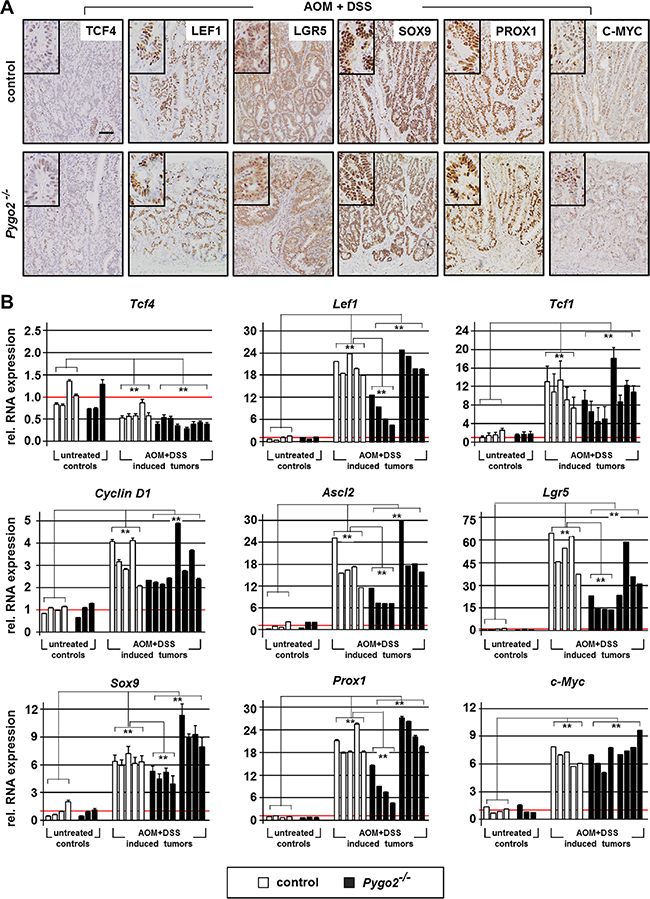
Figure 6: Knockout of Pygo2 downregulates overexpressed Wnt/ß-catenin targets and tumor progression genes in a subgroup of Pygo2 deficient chemically induced intestinal tumors. (A) Representative immunohistochemical stainings of chemically induced intestinal tumors for the indicated markers. Tissues from controls (Pygo2lox(ex3)/lox(ex3)) and Pygo2 knockout animals (VilCre; Pygo2Δ/Δ) were collected six months after treatment with AOM and DSS. Scale bars in the pictures represent 200 μm for all IHC. Inserts show the staining at higher magnification. (B) qRT-PCR analyses of RNA extracted from tumors and from colon tissues of untreated mice, both of controls and Pygo2 deficient animals. Each graph shows the relative RNA expression of the indicated gene for one single animal. Significances were calculated for the mean expression level compared to untreated controls and relative to the expression levels of control tumors, respectively. ** marks significant differences between the respective groups with P < 0.01.
Taken together, we have identified molecular targets that are differentially activated by Pygo2 during intestinal tumorigenesis in our mouse models. Pygo2 ablation reduces the overexpression of several key Wnt/ß-catenin target genes and apparently reduces the signaling threshold. In particular, the overexpression of c-Myc is not rescued by Pygo2 knockout in Apc LOF tissues and in chemically induced tumors. This might explain why Pygo2 knockout cannot prevent tumorigenesis, but reduces tumor formation in vivo.
DISCUSSION
Pygopus was discovered as Wnt signaling co-activator important for embryonic development in Drosophila [51]. Since this discovery, the role of the vertebrate homologues Pygo1 and Pygo2 has been studied and it was shown that they exert a limited role during Wnt/ß-catenin dependent embryonic development [52, 53]. Recent studies focused on their oncogenic role in the context of deregulated Wnt signaling during cancer development [54, 55]. We have previously shown that Pygo2, but not Pygo1, is overexpressed in different stages of human colon cancer and in adenoma of APCMin/+ mice [8]. In the present study, we have analyzed the in vivo role of Pygo2 using chemically induced colon cancer and conditional Apc LOF and Ctnnb1 GOF mouse models. Intriguingly, we found that Pygo2 deletion delayed chemically induced colon tumor formation and totally suppressed intestinal hyperproliferation induced by stabilized ß-catenin. In contrast, Pygo2 knockout failed to suppress intestinal hyperproliferation and adenoma formation in Apc loss of function driven intestinal tumorigenesis. We hypothesized that this different effect may critically be linked to varying dosages of Wnt signaling output that are activated by Pygo2. Based on our results, we indeed found that several genes were differentially suppressed by Pygo2 knockout in our tumor models.
We confirmed in our chemically induced colon tumor model that Wnt signaling is in fact overactivated. This was indicated by elevated nuclear ß-catenin and overexpression of Axin2 in the tumors. These results are in line with previous reports that described hyperactive Wnt signaling and mutations of the Apc or Ctnnb1 genes in this tumor model [56, 57]. Most importantly, Pygo2 deletion significantly reduced tumor number and size. Although we did not see any apparent difference in tumor invasiveness or angiogenesis between controls and knockout animals, we suggest that Pygo2 is involved in tumor progression with respect to growth of the primary tumor. Similar to our study, delayed tumor development following chemical induction was also observed in Bcl9/Bcl9-2 double mutants [9]. Of note, these co-activators are also dispensable for normal intestinal homeostasis similar to Pygo2 [58]. Besides deregulated Wnt-signaling, chemically induced tumors have been shown to harbor additional mutations in other pathways such as TGF-ß and K-ras signaling [27, 59]. It is therefore possible that Pygo2 loss and the effects on Wnt signaling are not sufficient to completely suppress colon tumor formation in this animal model. However, our data indicate that Pygo2 deletion delays tumor growth by inhibition of Wnt signaling in chemically induced colon tumors. Together, these data suggest that the Wnt signaling co-activators of the Bcl9 and Pygopus family are apparently dispensable for normal intestinal homeostasis. In contrast, Pygo2, similar to Bcl9/Bcl9-2, may drive intestinal tumor formation by increasing aberrant Wnt signaling in vivo.
Our findings regarding the phenotype of homozygous Apc LOF and heterozygous Ctnnb1 GOF mutants with Pygo2 wildtype background are in accordance with previous reports [24, 25, 48]. Mice homozygous for Apc LOF showed enlarged crypt-villus axes within few days, indicating massive intestinal hyperproliferation, and survived only seven to eight days after induction. The phenotype was less severe in heterozygous Ctnnb1 mutants with intestinal hyperproliferation starting after two weeks and the overall survival of Pygo2 wildtype animals was less than three weeks. Most importantly, Pygo2 ablation completely rescued the intestinal phenotype in heterozygous Ctnnb1 GOF mutants, but not in Apc LOF mutants. Although Pygo2 knockout in Ctnnb1 GOF mice completely rescued the initial intestinal phenotype, these mice however also died several weeks later than Pygo2 wild type animals. Remarkably, we found PYGO2 re-expression in single intestinal cells of knockout animals. This is most likely due to failed recombination of Pygo2 deletion as reported for other knockout models [60]. These Pygo2 “escaper” cells induced apparently microadenoma, which led to a lethal phenotype of Pygo2 knockout-Ctnnb1 GOF mice at much later points of time. This is a further in vivo evidence for the crucial role of Pygo2 during intestinal tumorigenesis induced by Ctnnb1 GOF. Thus, complete loss of Pygo2 may in fact extend the life span of Ctnnb1 GOF mice that is comparable to normal mice.
We also studied the phenotype of heterozygous Apc LOF mice. These mice exhibited an ApcMin/+-like phenotype with development of multiple small intestinal adenoma [61]. In this tumor model, the second functional Apc allele is lost over time in single epithelial cells (loss of heterozygosity, LOH), which induces adenoma formation[31]. In fact, we found upregulation of nuclear ß-catenin and overexpression of Pygo2 in the adenoma. However, deletion of Pygo2 did also not rescue adenoma formation in this Apc LOF model. Thus, Pygo2 deletion is apparently not sufficient to re-establish normal levels of Wnt signaling and to prevent tumorigenesis in either of the Apc LOF mutants studied here, in contrast to Ctnnb1 GOF mutants.
The overall persistence of the phenotype in Apc LOF animals despite Pygo2 knockout may be linked to additional Wnt-independent functions of Apc. Among these, Apc is crucial for chromosome segregation, migration, apoptosis and differentiation [62, 63]. In addition, it was shown that loss of Apc drives chromosomal instability in adenoma of Apc mutants and in chemically induced colon tumors of mice [64, 65]. Thus, Pygo2 loss might not have compensated these Wnt-independent functions of Apc. Indeed, a recent study revealed that restoration of APC wild type protein rescued intestinal tumorigenesis in mice even in the presence of K-ras and TP53 mutations [66].
We have analyzed in our tumor models the expression of all four members of the Pyogous and Bcl9 co-factor families (summarized in Table 1). We found in all animal models induction of PYGO2 expression, but not of PYGO1. PYGO2 was overexpressed in the hyperproliferative regions during early tumorigenesis, but also in adenoma and colon tumors in mice with Pygo2 wild type background. These results are in accordance with our previous studies in APCMin/+ mice, human colon cancers and colon cancer cell lines [8]. In Pygo2 knock out mice, PYGO2 was clearly downregulated to undetectable levels in all tumor models. Of note, we did not see compensatory upregulation of PYGO1 in Pygo2 knockout tissues. In fact, we have previously not detected PYGO1 expression in the normal intestine and during different stages of tumorigenesis in ApcMin/+ tumors and human colon cancer cells (this study and [8]). Therefore, it appears unlikely that Pygo1 may have compensated the intestinal phenotype of Pygo2 knockout animals. BCL9 expression was also not induced in our tumor models, similar to our previous studies [8]. In contrast, BCL9-2 was expressed in chemically induced tumors, and strongly overexpressed in adenoma of heterozygous Apc LOF mice. In contrast, BCL9-2 was not induced in the hyperproliferative regions of homozygous Apc LOF and only slightly increased in heterozygous Ctnnb1 GOF mice. Thus, BCL9-2 overexpression appears to be linked to more advanced stages of tumorigenesis, as we found in APCMin/+ mice and human colon cancers [8]. Taken together, current results support our previous results, that BCL9 and BCL9-2 are not targets of active Wnt/ß-catenin signaling, in contrast to other studies [15]. Thus, BCL9-2 overexpression in advanced stages of tumorigenesis might be linked to additional genetic events such as genomic instability leading to overexpression of the gene as previously found for BCL9 [67]. Moreover, upregulation of BCL9-2 in tumors and adenoma was unchanged by Pygo2 ablation, indicating that Bcl9-2 expression is not activated by Pygo2. In fact, BCL9-2 expression is apparently suppressed by active Wnt signaling as we found loss of BCL9-2 expression in the hyperproliferative regions in both Apc and Ctnnb1 mutant models. This is in line with our finding, that BCL9-2 is absent in the crypts of the normal epithelium where Wnt signaling is active [8]. Moreover, ß-catenin negatively regulated the expression of Bcl9-2 in human colon cancer cells [8]. Thus, we speculate that the absence of BCL9-2 in the hyperproliferative regions might be a direct or indirect effect of active Wnt/ß-catenin signaling. Overall, Bcl9-2 upregulation in adenoma of heterozygous Apc mice and chemically induced tumors may further drive tumor progression, which cannot be compensated by loss of Pygo2 [8, 14, 16]. However, the exact molecular mechanism for the regulation of the Pygo and Bcl9 co-factors and their role in Apc- or Ctnnb1-associated colorectal cancer requires further analyses.
Table 1: Summary for the relative regulation of target gene expression in the tumor models with and without knockout of Pygo2
tumor model |
AOM+DSS tumors |
Apc LOF |
Ctnnb1 GOF |
|||
|---|---|---|---|---|---|---|
genotype |
Pygo2 WT |
Pygo2 KO |
Pygo2 WT |
Pygo2 KO |
Pygo2 WT |
Pygo2 KO |
Wnt/ß-catenin signaling co-factors |
||||||
Pygo2 |
↑ |
↓↓↓ |
↑ |
↓↓↓ |
↑↑ |
↓↓↓ |
Pygo1 |
n.e. |
n.e. |
n.e. |
n.e. |
n.e. |
n.e. |
Bcl9 |
≈ |
≈ |
≈ |
≈ |
≈ |
≈ |
Bcl9-2 |
↑↑ |
↑↑ |
≈ |
≈ |
(↑) |
≈ |
Classical Wnt target genes |
||||||
Axin2 |
↑↑↑ |
↑ |
↑↑ |
↑ |
↑↑ |
≈ |
CyclinD1 |
↑↑ |
↑↑ |
↑↑ |
↑↑ |
↑ |
≈ |
Tcf1 |
↑↑↑ |
↑↑ |
↑↑↑ |
↑↑ |
↑↑↑ |
↑↑ |
Tcf4 |
≈ |
↓ |
≈ |
↓ |
↑ |
↓ |
Lef1 |
↑↑↑ |
↑ |
↑↑↑ |
↑ |
↑↑↑ |
↑ |
Stem cell marker |
||||||
Ascl2 |
↑↑↑ |
↑↑ |
↑↑↑ |
↑↑ |
↑↑ |
↑ |
Lgr5 |
↑↑↑ |
↑↑ |
↑↑↑ |
↑↑ |
↑ |
≈ |
Dclk1 |
↓ |
↓ |
↓ |
↓ |
≈ |
≈ |
Tumor progression marker |
||||||
Sox9 |
↑↑↑ |
↑↑ |
↑↑↑ |
↑↑↑ |
↑↑ |
↑ |
Prox1 |
↑↑↑ |
↑↑ |
↑↑ |
≈ |
↑↑↑ |
≈ |
c-myc |
↑↑ |
↑↑ |
↑↑↑ |
↑↑↑ |
↑ / ↑↑ |
≈ |
Relative expression level of target genes analyzed in this study. The summary is based on RNA expression as determined by qRT-PCR analyses and protein expression as detected by IHC. The increase ↑ or decrease ↓ is indicated relative to the respective uninduced controls in normal intestinal tissues. ≈ marks unchanged expression compared to control. Shown are the results from chemically induced tumors (AOM+DSS tumors) on Pygo2 wild type background and the subgroup of tumors with reduced expression following Pygo2 ablation (left column). The middle and right columns summarize the relative expression in homozygous Apc LOF and heterozygous Ctnnb1 GOF mutants with Pygo2 wild type expression compared to Pygo2 knockout animals.
Our data suggested that Pygo2 knockout might have rescued the intestinal phenotype in Ctnnb1 GOF mutants by suppressing specific Wnt target genes that are overexpressed during tumorigenesis. We speculate that such genes might be strongly deregulated in the early phase of Apc LOF animals and chemically induced tumors, which cannot be compensated by Pygo2 loss to rescue the phenotype. This hypothesis was first confirmed in Ctnnb1 GOF mice by the complete downregulation of increased Axin2, the best marker of active Wnt signaling [68]. In contrast, Axin2 was strongly overexpressed in APC LOF mutants and in chemically induced tumors, and only partially suppressed by Pygo2 knockout. We further analyzed several classical Wnt target genes and genes, which have been shown to be important for intestinal tumorigenesis. Our results provide several remarkable observations in the tumor models studied here with respect to their described role during tumorigenesis. We have summarized our results based on RNA and protein expression studied by qRT-PCR and IHC respectively in Table 1.
The overexpression of CyclinD1, another classical Wnt target gene that is deregulated during tumorigenesis [69], was suppressed by Pygo2 knockout, similar to Axin2. CyclinD1 was strongly upregulated in chemically induced tumors and Apc LOF mutants as previously described [70, 71]. Overexpressed CyclinD1 however was only partially downregulated by Pygo2 loss in these two animal models. In contrast, CyclinD1 was only moderately increased in Ctnnb1 mutants and completely reduced to control levels in Pygo2 deficient animals.
The different members of the Lef/Tcf transcription factor family have been implicated in tumorigenesis[72], and we found strong upregulation of Tcf1 and Lef1 in the tumor models, which were only in part downregulated by Pygo2 loss. Tcf4 was only induced in Ctnnb1 mutants, and the expression was downregulated by Pygo2 knockout. Remarkably, Tcf4 was not increased in chemically induced tumors and Apc LOF mutants, and was suppressed even below control levels in Pygo2 deficient animals. In fact, Tcf4 might represent a tumor suppressor as previously suggested by studies with Tcf4 knockout animals that developed hyperproliferation in the colon [73]. Another study suggested that Tcf4 appears to be important for intestinal homeostasis [74]. Of note, we found that the expression of Tcf4 was suppressed by Pygo2 loss in normal intestinal cells, indicating that Tcf4 is a Pygo2 dependent Wnt-target gene. However, since the Pygo2 knockout in the intestine has no influence on intestinal homeostasis (Schelp and Brembeck, unpublished data), it is most likely that Pygo2 is not essential for Tcf4 expression due to the lack of an apparent phenotype.
We characterized intestinal stem cell markers that are activated by Wnt/ß-catenin signaling. Lgr5 and Ascl2 are crypt stem cell markers expressed in the cycling stem cells of the crypt[39, 41]. In line with human intestinal cancers [38, 75], Lgr5 and Ascl2 were overexpressed in all three tumor models indicating an increased number of stem-like cells as a result of aberrant Wnt signaling [76, 77]. Ascl2 was strongly induced in all animal models and partially downregulated by Pygo2 knockout. Lgr5 was moderately induced in Ctnnb1 GOF mice and ablation of Pygo2 resulted in complete downregulation of Lgr5. In contrast, the strong overexpression of Lgr5 in chemically induced tumors and in Apc LOF mice was only partially downregulated by Pygo2 ablation. This may indicate that the tumor promoting function of Pygo2 in the Ctnnb1 GOF model may be partly regulated by the induction of Lgr5-positive stem-like cells that are known to be the origin of intestinal cancer [78].
Recently, Dclk1 was suggested as putative cancer stem cell marker in the intestine since Dclk1 ablation induced the regression of adenoma in APCMin/+ mice [44]. Dclk1 is normally expressed in tuft cells of the intestine [44]. However, Dclk1 was completely lost in chemically induced tumors and in Apc LOF mice and unchanged in Ctnnb1 GOF mutants. Moreover, Pygo2 ablation did not change Dclk1 expression. The loss of Dclk1 in our tumor models might reflect the disturbed differentiation in the colon tumors and the relatively high Wnt signaling level in Apc LOF mice [44]. In fact, a recent study showed that Dclk1 is silenced in human colon cancer by promoter methylation [79]. Therefore, further studies are required to assess the reliability of Dclk1 as a cancer stem cell marker.
Finally, we addressed the expression of known tumor progression markers, which have been implicated in intestinal tumorigenesis. Sox9, a target of Wnt signaling expressed in Paneth cells, was shown to be overexpressed in several human malignancies including colon cancer [80, 81]. We also found Sox9 overexpression in chemically induced colon tumors and in Apc and Ctnnb1 mutants. Pygo2 failed to suppress Sox9 overexpression in Apc LOF, and only partially reduced elevated Sox9 in colon tumors and Ctnnb1 GOF mice. The transcription factor Prox1, another Wnt target gene, was characterized as an oncogene: Prox1 ablation inhibited, and overexpression promoted tumor formation in ApcMin/+ mice, respectively [45, 46]. Remarkably, overexpressed Prox1 in the early stage of intestinal hyperproliferation in Apc LOF and Ctnnb1 GOF mutants was completely downregulated to control levels in Pygo2 deficient animals. Thus, despite the complete downregulation of Prox1, Pygo2 ablation did not rescue the phenotype of Apc mutants, indicating that Prox1 overexpression is not important for intestinal hyperproliferation. Moreover, Prox1 was strongly overexpressed in chemically induced tumors, and partially reduced by Pygo2 ablation. Therefore, Prox1 is apparently a Wnt target gene that requires the co-activator function of Pygo2, but is not essential for early intestinal tumorigenesis in our models. The most important finding for the role of Pygo2 in our study was the regulation of c-Myc as an essential transcription factor for colon cancer [82]. The strong c-Myc overexpression in chemically induced tumors and in Apc LOF animals was unchanged by Pygo2 ablation. In contrast, the moderate overexpression of c-Myc in Ctnnb1 GOF animals was completely reduced to control levels. Of note, previous studies have shown that knockout of c-Myc rescued tumorigenesis in Apc LOF mice [48, 83]. Thus, Pygo2 loss may have rescued the intestinal hyperproliferation of Ctnnb1 GOF mice predominantly by downregulating c-Myc representing a global gene amplifier [84]. Therefore, we propose that the role of Pygo2 in intestinal tumorigenesis is dependent on the Wnt signaling dosage and the deregulation of specific target genes. Different dosages of Wnt signaling output have been studied in the context of embryonic stem cell differentiation and also for intestinal tumorigenesis [85–87]. In particular, specific Apc mutations induce different Wnt signaling dosages and thus determine the susceptibility for intestinal tumorigenesis [85]. Moreover, similar to the effects in our study in Ctnnb1 GOF, it was recently shown that Pygo2 loss suppressed skin hyperplasia induced by stabilized ß-catenin in vivo [88]. This further underscores the importance of Pygo2 for the deregulated Wnt signaling output induced by mutant ß-catenin. Further studies are required to elucidate the underlying molecular mechanism.
Altogether, our study highlights that deregulated Wnt signaling induced by stabilized ß-catenin can be repressed by Pygo2 loss. This function may require a certain level of hyperactive Wnt signaling. Thus, targeting Pygo2 in malignant tumors that harbor Ctnnb1 mutations may result in suppression of tumor formation and growth. Interestingly, several human cancers have been characterized with ß-catenin mutations at higher frequencies compared to colon cancer. Among these tumors are hepatocellular carcinomas (HCC), solid-pseudopapillary tumors of the pancreas and almost all ovarian solid pseudopapillary tumors [89–91]. Therefore, small molecules that inhibit the interaction of PYGO2 with ß-catenin via BCL9/BCL9-2 may be of therapeutic value. In fact, Pyrvinium pamoate, an anti-helminth drug, was shown to decrease PYGO2 protein levels in colon cancer cells lines with Apc and Ctnnb1 mutations [92], and in ApcMin/+ mice [93]. However, since this drug apparently has also toxic effects, further compounds have to be developed and tested in vivo to target Pygo2 for the treatment of cancers with mutant ß-catenin. This is an interesting therapeutic strategy, since targeting Pygo2 in normal cells apparently will not disturb tissue homeostasis.
MATERIALS AND METHODS
Animal maintenance
All animal experiments were performed in accordance with German guidelines (TierSchG) and approved by governmental authorities. Animals were maintained on a pure C57BL/6 background. In all experiments, age-matched, non-transgenic littermates were used as controls. All mouse strains were grown and bred in European Neuroscience Institute, and Animal facility in University Medical Center, Göttingen.
Animal models
The generation of ß-catenin conditional mutants was previously described [24]. In brief, Ctnnb1 GOF animals used in our study harbor one mutant beta-catenin allele with loxP sites flanking exon 3 (Ctnnb1lox(ex3)/+). This results after conditional recombination in deletion of 76 amino acids encoding the ß-catenin phosphorylation sites (Ctnnb1Δex3/+). Conditional Ctnnb1 GOF mice express a truncated mutant ß-catenin, which cannot be degraded, and therefore results in aberrant Wnt signaling activation [24].
Apc LOF animals were conditionally generated with mice harboring Apc mutant alleles with loxP sites flanking exon 15 (Apclox(ex15)/lox(ex15); [25]. After recombination, homozygous mutant mice express truncated APC protein lacking almost all functional APC domains that results in hyperactivated Wnt signaling (ApcΔex15/Δex15; [25]. In addition, heterozygous Apc mutant mice were generated (ApcΔex15/+), which develop multiple intestinal adenoma after loss of the second allele as previously described [25].
The conditional Pygo2 knockout was generated by insertion of the loxP-flanked deletion cassette in exon 3 that encodes the essential NHD and PHD domains of the PYGO2 protein (Pygo2lox(ex3)/lox(ex3); H.J. Schäffer and W. Birchmeier, unpublished) and results in a null-phenotype after recombination. Details on generation of mice and phenotype will be described elsewhere. In our study, Pygo2 mutant mice were generated by mating hetero- or homozygous floxed mice with intestine specific transgenic Cre-mice (Pygo2Δ/+ and Pygo2 Δ/Δ). For induction of recombination, we used constitutive Vil-Cre and inducible Vil-CreERT transgenic mice that were previously described ( VilCre; VilCre-ERT[29].To induce conditional intestine specific recombination, six- to eight-weeks old mice were injected intraperitoneally with 1 mg tamoxifen per mouse for five continuous days. All Apc LOF homozygous mice were sacrificed on day six. Apc LOF heterozygous mice were sacrificed on day 70. ß-catenin (Ctnnb1 GOF) heterozygous mice were sacrificed on day 6 and 18. Compound Pygo2 knockout Ctnnb1 GOF animals were sacrificed in addition on day 30, 34 and 86, respectively. During the survival study, all mice were monitored daily to record the date of death. In all experiments, uninduced Cre-negative littermates with the respective genotype were used as controls.
AOM and DSS treatment
For the regeneration studies dextran sodium sulphate (DSS, MP Bio medicals, 216011050) was orally administered. Six- to eight-weeks old mice (control and Pygo2−/−) were fed with 1.5% (w/v) DSS in drinking water for five consecutive days. Mice were monitored regularly for the health status and sacrificed on day 14 and day 28 for the histological examinations. For chemically induced colon tumorigenesis, a single dose of 10 μg of azoxymethane (AOM, Sigma, A5486) per gram body weight was injected intraperitoneally in control and constitutive Pygo2 knockout mice. Seven days after AOM treatment, DSS 1.5% (w/v) was supplied in drinking water for five continuous days. Mice were sacrificed six months after the onset of AOM treatment. The intestine was cut open longitudinally and tumor numbers and sizes were recorded.
Epithelial cell extraction from small intestine and colon
Isolation of intestinal epithelial cells from mice was performed as previously described [94, 95] with some modifications. Immediately after sacrificing, the intestines were cut longitudinally and rinsed in PBS. Further, they were incubated in a HBSS/EDTA (pH 7.4) solution at 37°C for 15 minutes to destabilize the outer epithelial layer of the intestine. To extract the epithelial cells into the HBSS solution, tissues were vortexed for 2 min and cells were collected by centrifugation at 1600 rpm for 10 min. Finally, the pellet containing predominantly intestinal epithelial cells were stored in 1ml of TRIZOL at -80°C until use.
Immunohistochemistry (IHC)
All tissues were fixed in 4% PFA and 3-micron tissue sections were used. In all experiments, standardized protocols were used. For BrdU for in vivo DNA labeling, mice were injected intraperitoneally 2 hours prior to sacrificing with 100μg BrdU (Roche, 10280879001) per gram body weight. For immunohistochemistry, antigen retrieval of tissue sections was performed by heating in 10 mM Tris, 1 mM EDTA (pH 9.0). After blocking, sections were incubated overnight with respective primary antibodies (see Supplementary Table S2). HRP conjugated secondary antibodies were applied and the staining visualized by DAB according to manufacturer`s protocol.
cDNA synthesis and qRT-PCR
RNA was isolated using Tri-Reagent (Ambion-Invitrogen) and reverse-transcribed using MMLV-RT (Thermo Fisher Scientific). qRT-PCR was performed with SYBR green (Sigma-Aldrich) on the ABI Prism 7900HT (Applied Bio-systems). Primers used in the study are listed in Supplementary Table S1. Relative gene expression was calculated using HPRT as a housekeeper gene.
Microscopy
All bright field images were acquired by an OLYMPUS SC30 integrated to an OLYMPUS bx43 microscope. CellSens Dimension 1.6 software was utilized for image acquisition.
Statistical analysis
Data of qRT-PCRs were analyzed using Microsoft excel. For statistical significance, the two-sided student t-test was used. Graph Pad Prism6 assisted Kaplan-Meier survival curve was used to compare the survival time of mice. Box plot analysis was performed for the comparison of tumor size and number of chemically induced tumors.
ACKNOWLEDGMENTS
We thank Nadine Schelp for providing material from mutant mice, Maria Wiese for providing material and critical discussion, and Jens Bunzendahl for technical assistance (Tumor Biology and Signal Transduction, Dept. of Hematology and Medical Oncology, Georg-August-University Göttingen/Germany).
Mutant mice were kindly provided by Sylvie Robine (Equipe de Morphogenèse et Signalisation cellulaires, Institute Curie/France), Makoto Mark Taketo (Dept. of Pharmacology, Graduate School of Medicine, Kyoto University Yoshida-Konoé-cho, Sakyo, Kyoto/Japan), Ron Smits (Netherlands Cancer Institute, Amsterdam/The Netherlands) and Walter Birchmeier (Max-Delbrueck-Center for Molecular Medicine, Berlin/Germany).
We thank Heidi Hahn and Holger Bastians (Georg-August-University Göttingen/Germany) for discussion and critical reading of the manuscript.
CONFLICTS OF INTEREST
All authors declare no conflicts of interest.
GRANT SUPPORT
This work was supported by grant of the Wilhelm Sander Stiftung to F.H.B (2011.093.1).
FINANCIAL DISCLOSURE
All authors have no financial conflicts.
Authors’ contributions
Study concept, design and supervision: F.H.B.; acquisition of data: S.B.T.; analysis and interpretation of data: S.B.T., F.H.B.; drafting of the manuscript: S.B.T., F.H.B.; obtained funding: F.H.B.
REFERENCES
1. Brembeck FH, Schwarz-Romond T, Bakkers J, Wilhelm S, Hammerschmidt M, Birchmeier W. Essential role of BCL9-2 in the switch between beta-catenin's adhesive and transcriptional functions. Genes Dev. 2004; 18:2225–2230.
2. Kramps T, Peter O, Brunner E, Nellen D, Froesch B, Chatterjee S, Murone M, Zullig S, Basler K. Wnt/wingless signaling requires BCL9/legless-mediated recruitment of pygopus to the nuclear beta-catenin-TCF complex. Cell. 2002; 109:47–60.
3. Thompson B, Townsley F, Rosin-Arbesfeld R, Musisi H, Bienz M. A new nuclear component of the Wnt signalling pathway. Nat. Cell Biol. 2002; 4:367–373.
4. Townsley FM, Thompson B, Bienz M. Pygopus residues required for its binding to Legless are critical for transcription and development. J. Biol. Chem. 2004; 279:5177–5183.
5. Krieghoff E, Behrens J, Mayr B. Nucleo-cytoplasmic distribution of beta-catenin is regulated by retention. J. Cell. Sci. 2006; 119:1453–1463.
6. Andrews PG, He Z, Popadiuk C, Kao KR. The transcriptional activity of Pygopus is enhanced by its interaction with cAMP-response-element-binding protein (CREB)-binding protein. Biochem. J. 2009; 422:493–501.
7. Chen J, Luo Q, Yuan Y, Huang X, Cai W, Li C, Wei T, Zhang L, Yang M, Liu Q, Ye G, Dai X, Li B. Pygo2 associates with MLL2 histone methyltransferase and GCN5 histone acetyltransferase complexes to augment Wnt target gene expression and breast cancer stem-like cell expansion. Mol. Cell. Biol. 2010; 30:5621–5635.
8. Brembeck FH, Wiese M, Zatula N, Grigoryan T, Dai Y, Fritzmann J, Birchmeier W. BCL9-2 promotes early stages of intestinal tumor progression. Gastroenterology. 2011; 141:1359–1370, 1370 e1351–1353.
9. Deka J, Wiedemann N, Anderle P, Murphy-Seiler F, Bultinck J, Eyckerman S, Stehle JC, Andre S, Vilain N, Zilian O, Robine S, Delorenzi M, Basler K, et al. Bcl9/Bcl9l are critical for Wnt-mediated regulation of stem cell traits in colon epithelium and adenocarcinomas. Cancer Res. 2010; 70:6619–6628.
10. Li B, Rheaume C, Teng A, Bilanchone V, Munguia JE, Hu M, Jessen S, Piccolo S, Waterman ML, Dai X. Developmental phenotypes and reduced Wnt signaling in mice deficient for pygopus 2. Genesis. 2007; 45:318–325.
11. Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, Kinzler KW. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science. 1997; 275:1787–1790.
12. Daniels DL, Weis WI. Beta-catenin directly displaces Groucho/TLE repressors from Tcf/Lef in Wnt-mediated transcription activation. Nat. Struct. Mol. Biol. 2005; 12:364–371.
13. Molenaar M, van de Wetering M, Oosterwegel M, Peterson-Maduro J, Godsave S, Korinek V, Roose J, Destree O, Clevers H. XTcf-3 transcription factor mediates beta-catenin-induced axis formation in Xenopus embryos. Cell. 1996; 86:391–399.
14. Adachi S, Jigami T, Yasui T, Nakano T, Ohwada S, Omori Y, Sugano S, Ohkawara B, Shibuya H, Nakamura T, Akiyama T. Role of a BCL9-related beta-catenin-binding protein, B9L, in tumorigenesis induced by aberrant activation of Wnt signaling. Cancer Res. 2004; 64:8496–8501.
15. de la Roche M, Worm J, Bienz M. The function of BCL9 in Wnt/beta-catenin signaling and colorectal cancer cells. BMC cancer. 2008; 8:199.
16. Sakamoto I, Ohwada S, Toya H, Togo N, Kashiwabara K, Oyama T, Nakajima T, Ito H, Adachi S, Jigami T, Akiyama T. Up-regulation of a BCL9-related beta-catenin-binding protein, B9L, in different stages of sporadic colorectal adenoma. Cancer Sci. 2007; 98:83–87.
17. Andrews PG, Lake BB, Popadiuk C, Kao KR. Requirement of Pygopus 2 in breast cancer. Int J Oncol. 2007; 30:357–363.
18. Popadiuk CM, Xiong J, Wells MG, Andrews PG, Dankwa K, Hirasawa K, Lake BB, Kao KR. Antisense suppression of pygopus2 results in growth arrest of epithelial ovarian cancer. Clin. Cancer Res. 2006; 12:2216–2223.
19. Liu Y, Dong QZ, Wang S, Fang CQ, Miao Y, Wang L, Li MZ, Wang EH. Abnormal expression of Pygopus 2 correlates with a malignant phenotype in human lung cancer. BMC cancer. 2013; 13:346.
20. Wang ZX, Chen YY, Li BA, Tan GW, Liu XY, Shen SH, Zhu HW, Wang HD. Decreased pygopus 2 expression suppresses glioblastoma U251 cell growth. J Neurooncol. 2010; 100:31–41.
21. Zhang S, Li J, Liu P, Xu J, Zhao W, Xie C, Yin Z, Wang X. Pygopus-2 promotes invasion and metastasis of hepatic carcinoma cell by decreasing E-cadherin expression. Oncotarget. 2015; 6:11074–11086. doi: 10.18632/oncotarget.3570.
22. Johnson RL, Fleet JC. Animal models of colorectal cancer. Cancer Metastasis Rev. 2013; 32:39–61.
23. Janssen KP, Alberici P, Fsihi H, Gaspar C, Breukel C, Franken P, Rosty C, Abal M, El Marjou F, Smits R, Louvard D, Fodde R, Robine S. APC and oncogenic KRAS are synergistic in enhancing Wnt signaling in intestinal tumor formation and progression. Gastroenterology. 2006; 131:1096–1109.
24. Harada N, Tamai Y, Ishikawa T, Sauer B, Takaku K, Oshima M, Taketo MM. Intestinal polyposis in mice with a dominant stable mutation of the beta-catenin gene. EMBO J. 1999; 18:5931–5942.
25. Robanus-Maandag EC, Koelink PJ, Breukel C, Salvatori DC, Jagmohan-Changur SC, Bosch CA, Verspaget HW, Devilee P, Fodde R, Smits R. A new conditional Apc-mutant mouse model for colorectal cancer. Carcinogenesis. 2010; 31:946–952.
26. Brembeck FH, Rosario M, Birchmeier W. Balancing cell adhesion and Wnt signaling, the key role of beta-catenin. Curr. Opin. Genet. Dev. 2006; 16:51–59.
27. De Robertis M, Massi E, Poeta ML, Carotti S, Morini S, Cecchetelli L, Signori E, Fazio VM. The AOM/DSS murine model for the study of colon carcinogenesis: From pathways to diagnosis and therapy studies. J. Carcinog. 2011; 10:9.
28. Lustig B, Jerchow B, Sachs M, Weiler S, Pietsch T, Karsten U, van de Wetering M, Clevers H, Schlag PM, Birchmeier W, Behrens J. Negative feedback loop of Wnt signaling through upregulation of conductin/axin2 in colorectal and liver tumors. Mol. Cell. Biol. 2002; 22:1184–1193.
29. el Marjou F, Janssen KP, Chang BH, Li M, Hindie V, Chan L, Louvard D, Chambon P, Metzger D, Robine S. Tissue-specific and inducible Cre-mediated recombination in the gut epithelium. Genesis. 2004; 39:186–193.
30. Ichii S, Horii A, Nakatsuru S, Furuyama J, Utsunomiya J, Nakamura Y. Inactivation of both APC alleles in an early stage of colon adenomas in a patient with familial adenomatous polyposis (FAP). Hum. Mol. Gen. 1992; 1:387–390.
31. Luongo C, Moser AR, Gledhill S, Dove WF. Loss of Apc+ in intestinal adenomas from Min mice. Cancer Res. 1994; 54:5947–5952.
32. Cantu C, Zimmerli D, Hausmann G, Valenta T, Moor A, Aguet M, Basler K. Pax6-dependent, but beta-catenin-independent, function of Bcl9 proteins in mouse lens development. Genes Dev. 2014; 28:1879–1884.
33. Song N, Schwab KR, Patterson LT, Yamaguchi T, Lin X, Potter SS, Lang RA. pygopus 2 has a crucial, Wnt pathway-independent function in lens induction. Development. 2007; 134:1873–1885.
34. Zatula N, Wiese M, Bunzendahl J, Birchmeier W, Perske C, Bleckmann A, Brembeck FH. The BCL9-2 proto-oncogene governs estrogen receptor alpha expression in breast tumorigenesis. Oncotarget. 2014; 5:6770–6787. doi: 10.18632/oncotarget.2252.
35. Clevers H, Nusse R. Wnt/beta-catenin signaling and disease. Cell. 2012; 149:1192–1205.
36. Angus-Hill ML, Elbert KM, Hidalgo J, Capecchi MR. T-cell factor 4 functions as a tumor suppressor whose disruption modulates colon cell proliferation and tumorigenesis. Proc. Natl. Acad. Sci. USA. 2011; 108:4914–4919.
37. Roose J, Huls G, van Beest M, Moerer P, van der Horn K, Goldschmeding R, Logtenberg T, Clevers H. Synergy between tumor suppressor APC and the beta-catenin-Tcf4 target Tcf1. Science. 1999; 285:1923–1926.
38. Jubb AM, Chalasani S, Frantz GD, Smits R, Grabsch HI, Kavi V, Maughan NJ, Hillan KJ, Quirke P, Koeppen H. Achaete-scute like 2 (ascl2) is a target of Wnt signalling and is upregulated in intestinal neoplasia. Oncogene. 2006; 25:3445–3457.
39. Barker N, van Es JH, Kuipers J, Kujala P, van den Born M, Cozijnsen M, Haegebarth A, Korving J, Begthel H, Peters PJ, Clevers H. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature. 2007; 449:1003–1007.
40. Barker N, Ridgway RA, van Es JH, van de Wetering M, Begthel H, van den Born M, Danenberg E, Clarke AR, Sansom OJ, Clevers H. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature. 2009; 457:608–611.
41. van der Flier LG, van Gijn ME, Hatzis P, Kujala P, Haegebarth A, Stange DE, Begthel H, van den Born M, Guryev V, Oving I, van Es JH, Barker N, Peters PJ, et al. Transcription factor achaete scute-like 2 controls intestinal stem cell fate. Cell. 2009; 136:903–912.
42. Bastide P, Darido C, Pannequin J, Kist R, Robine S, Marty-Double C, Bibeau F, Scherer G, Joubert D, Hollande F, Blache P, Jay P. Sox9 regulates cell proliferation and is required for Paneth cell differentiation in the intestinal epithelium. J. Cell Biol.2007; 178:635–648.
43. Mori-Akiyama Y, van den Born M, van Es JH, Hamilton SR, Adams HP, Zhang J, Clevers H and de Crombrugghe B. SOX9 is required for the differentiation of paneth cells in the intestinal epithelium. Gastroenterology. 2007; 133:539–546.
44. Nakanishi Y, Seno H, Fukuoka A, Ueo T, Yamaga Y, Maruno T, Nakanishi N, Kanda K, Komekado H, Kawada M, Isomura A, Kawada K, Sakai Y, et al. Dclk1 distinguishes between tumor and normal stem cells in the intestine. Nat. Genet. 2013; 45:98–103.
45. Petrova TV, Nykanen A, Norrmen C, Ivanov KI, Andersson LC, Haglund C, Puolakkainen P, Wempe F, von Melchner H, Gradwohl G, Vanharanta S, Aaltonen LA, Saharinen J, et al. Transcription factor PROX1 induces colon cancer progression by promoting the transition from benign to highly dysplastic phenotype. Cancer cell. 2008; 13:407–419.
46. Ragusa S, Cheng J, Ivanov KI, Zangger N, Ceteci F, Bernier-Latmani J, Milatos S, Joseph JM, Tercier S, Bouzourene H, Bosman FT, Letovanec I, Marra G, et al. PROX1 promotes metabolic adaptation and fuels outgrowth of Wnt(high) metastatic colon cancer cells. Cell Rep. 2014; 8:1957–1973.
47. van de Wetering M, Sancho E, Verweij C, de Lau W, Oving I, Hurlstone A, van der Horn K, Batlle E, Coudreuse D, Haramis AP, Tjon-Pon-Fong M, Moerer P, van den Born M, et al. The beta-catenin/TCF-4 complex imposes a crypt progenitor phenotype on colorectal cancer cells. Cell. 2002; 111:241–250.
48. Sansom OJ, Meniel VS, Muncan V, Phesse TJ, Wilkins JA, Reed KR, Vass JK, Athineos D, Clevers H, Clarke AR. Myc deletion rescues Apc deficiency in the small intestine. Nature. 2007; 446:676–679.
49. Gu B, Watanabe K, Sun P, Fallahi M, Dai X. Chromatin effector Pygo2 mediates Wnt-notch crosstalk to suppress luminal/alveolar potential of mammary stem and basal cells. Cell stem cell. 2013; 13:48–61.
50. Fre S, Huyghe M, Mourikis P, Robine S, Louvard D, Artavanis-Tsakonas S. Notch signals control the fate of immature progenitor cells in the intestine. Nature. 2005; 435:964–968.
51. Kramps T, Peter O, Brunner E, Nellen D, Froesch B, Chatterjee S, Murone M, Zullig S, Basler K. Wnt/wingless signaling requires BCL9/legless-mediated recruitment of pygopus to the nuclear beta-catenin-TCF complex. Cell. 2002; 109:47–60.
52. Li B, Rheaume C, Teng A, Bilanchone V, Munguia JE, Hu M, Jessen S, Piccolo S, Waterman ML, Dai X. Developmental phenotypes and reduced Wnt signaling in mice deficient for pygopus 2. Genesis. 2007; 45:318–325.
53. Schwab KR, Patterson LT, Hartman HA, Song N, Lang RA, Lin X, Potter SS. Pygo1 and Pygo2 roles in Wnt signaling in mammalian kidney development. BMC biology. 2007; 5:15.
54. Andrews PG, Lake BB, Popadiuk C, Kao KR. Requirement of Pygopus 2 in breast cancer. Int. J. Oncol. 2007; 30:357–363.
55. Popadiuk CM, Xiong J, Wells MG, Andrews PG, Dankwa K, Hirasawa K, Lake BB, Kao KR. Antisense suppression of pygopus2 results in growth arrest of epithelial ovarian cancer. Clin. Cancer Res. 2006; 12:2216–2223.
56. Koesters R, Hans MA, Benner A, Prosst R, Boehm J, Gahlen J, Doeberitz MK. Predominant mutation of codon 41 of the beta-catenin proto-oncogene in rat colon tumors induced by 1,2-dimethylhydrazine using a complete carcinogenic protocol. Carcinogenesis. 2001; 22:1885–1890.
57. Maltzman T, Whittington J, Driggers L, Stephens J, Ahnen D. AOM-induced mouse colon tumors do not express full-length APC protein. Carcinogenesis. 1997; 18:2435–2439.
58. Deka J, Wiedemann N, Anderle P, Murphy-Seiler F, Bultinck J, Eyckerman S, Stehle JC, Andre S, Vilain N, Zilian O, Robine S, Delorenzi M, Basler K, et al. Bcl9/Bcl9l are critical for Wnt-mediated regulation of stem cell traits in colon epithelium and adenocarcinomas. Cancer Res. 2010; 70:6619–6628.
59. Suzuki R, Miyamoto S, Yasui Y, Sugie S, Tanaka T. Global gene expression analysis of the mouse colonic mucosa treated with azoxymethane and dextran sodium sulfate. BMC cancer. 2007; 7:84.
60. Chmielowiec J, Borowiak M, Morkel M, Stradal T, Munz B, Werner S, Wehland J, Birchmeier C, Birchmeier W. c-Met is essential for wound healing in the skin. J. Cell Biol. 2007; 177:151–162.
61. Robanus-Maandag EC, Koelink PJ, Breukel C, Salvatori DC, Jagmohan-Changur SC, Bosch CA, Verspaget HW, Devilee P, Fodde R, Smits R. A new conditional Apc-mutant mouse model for colorectal cancer. Carcinogenesis. 2010; 31:946–952.
62. Kaplan KB, Burds AA, Swedlow JR, Bekir SS, Sorger PK, Nathke IS. A role for the Adenomatous Polyposis Coli protein in chromosome segregation. Nature Cell Biol. 2001; 3:429–432.
63. Nathke IS. The adenomatous polyposis coli protein: the Achilles heel of the gut epithelium. Annu. Rev. Cell Dev. Biol. 2004; 20:337–366.
64. Fodde R, Kuipers J, Rosenberg C, Smits R, Kielman M, Gaspar C, van Es JH, Breukel C, Wiegant J, Giles RH, Clevers H. Mutations in the APC tumour suppressor gene cause chromosomal instability. Nature Cell Biol. 2001; 3:433–438.
65. Gerling M, Glauben R, Habermann JK, Kuhl AA, Loddenkemper C, Lehr HA, Zeitz M, Siegmund B. Characterization of chromosomal instability in murine colitis-associated colorectal cancer. PLOS ONE. 2011; 6:e22114.
66. Dow LE, O'Rourke KP, Simon J, Tschaharganeh DF, van Es JH, Clevers H, Lowe SW. Apc Restoration Promotes Cellular Differentiation and Reestablishes Crypt Homeostasis in Colorectal Cancer. Cell. 2015; 161:1539–1552.
67. Willis TG, Zalcberg IR, Coignet LJ, Wlodarska I, Stul M, Jadayel DM, Bastard C, Treleaven JG, Catovsky D, Silva ML, Dyer MJ. Molecular cloning of translocation t(1;14)(q21;q32) defines a novel gene (BCL9) at chromosome 1q21. Blood. 1998; 91:1873–1881.
68. Lustig B, Jerchow B, Sachs M, Weiler S, Pietsch T, Karsten U, van de Wetering M, Clevers H, Schlag PM, Birchmeier W, Behrens J. Negative feedback loop of Wnt signaling through upregulation of conductin/axin2 in colorectal and liver tumors. Mol. Cell. Biol. 2002; 22:1184–1193.
69. Bahnassy AA, Zekri AR, El-Houssini S, El-Shehaby AM, Mahmoud MR, Abdallah S, El-Serafi M. Cyclin A and cyclin D1 as significant prognostic markers in colorectal cancer patients. BMC gastroenterology. 2004; 4:22.
70. Shinozaki H, Yang K, Fan K, Edelmann W, Kucherlapati R, Weinstein IB, Lipkin M. Cyclin D1 expression in the intestinal mucosa and tumors of Apc1638N mice. Anticancer Res.2003; 23:2217–2226.
71. Zhang T, Nanney LB, Luongo C, Lamps L, Heppner KJ, DuBois RN, Beauchamp RD. Concurrent overexpression of cyclin D1 and cyclin-dependent kinase 4 (Cdk4) in intestinal adenomas from multiple intestinal neoplasia (Min) mice and human familial adenomatous polyposis patients. Cancer Res. 1997; 57:169–175.
72. Kriegl L, Horst D, Reiche JA, Engel J, Kirchner T, Jung A. LEF-1 and TCF4 expression correlate inversely with survival in colorectal cancer. J. Transl. Med. 2010; 8:123.
73. Angus-Hill ML, Elbert KM, Hidalgo J, Capecchi MR. T-cell factor 4 functions as a tumor suppressor whose disruption modulates colon cell proliferation and tumorigenesis. Proc. Natl. Acad. Sci. USA. 2011; 108:4914–4919.
74. van Es JH, Haegebarth A, Kujala P, Itzkovitz S, Koo BK, Boj SF, Korving J, van den Born M, van Oudenaarden A, Robine S, Clevers H. A critical role for the Wnt effector Tcf4 in adult intestinal homeostatic self-renewal. Mol. Cell. Biol. 2012; 32:1918–1927.
75. Uchida H, Yamazaki K, Fukuma M, Yamada T, Hayashida T, Hasegawa H, Kitajima M, Kitagawa Y, Sakamoto M. Overexpression of leucine-rich repeat-containing G protein-coupled receptor 5 in colorectal cancer. Cancer Sci. 2010; 101:1731–1737.
76. Chen X, Wei B, Han X, Zheng Z, Huang J, Liu J, Huang Y, Wei H. LGR5 is required for the maintenance of spheroid-derived colon cancer stem cells. Int. J. Mol. Med. 2014; 34:35–42.
77. Jubb AM, Chalasani S, Frantz GD, Smits R, Grabsch HI, Kavi V, Maughan NJ, Hillan KJ, Quirke P, Koeppen H. Achaete-scute like 2 (ascl2) is a target of Wnt signalling and is upregulated in intestinal neoplasia. Oncogene. 2006; 25:3445–3457.
78. Barker N, Ridgway RA, van Es JH, van de Wetering M, Begthel H, van den Born M, Danenberg E, Clarke AR, Sansom OJ, Clevers H. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature. 2009; 457:608–611.
79. Vedeld HM, Skotheim RI, Lothe RA, Lind GE. The recently suggested intestinal cancer stem cell marker DCLK1 is an epigenetic biomarker for colorectal cancer. Epigenetics. 2014; 9:346–350.
80. Lu B, Fang Y, Xu J, Wang L, Xu F, Xu E, Huang Q, Lai M. Analysis of SOX9 expression in colorectal cancer. Am. J. Clin. Pathol. 2008; 130:897–904.
81. Matheu A, Collado M, Wise C, Manterola L, Cekaite L, Tye AJ, Canamero M, Bujanda L, Schedl A, Cheah KS, Skotheim RI, Lothe RA, Lopez de Munain A, et al. Oncogenicity of the developmental transcription factor Sox9. Cancer Res. 2012; 72:1301–1315.
82. He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, Morin PJ, Vogelstein B, Kinzler KW. Identification of c-MYC as a target of the APC pathway. Science. 1998; 281:1509–1512.
83. Ignatenko NA, Holubec H, Besselsen DG, Blohm-Mangone KA, Padilla-Torres JL, Nagle RB, de Alboranc IM, Guillen RJ, Gerner EW. Role of c-Myc in intestinal tumorigenesis of the ApcMin/+ mouse. Cancer Biol. Ther. 2006; 5:1658–1664.
84. Miller DM, Thomas SD, Islam A, Muench D, Sedoris K. c-Myc and cancer metabolism. Clin. Cancer Res. 2012; 18:5546–5553.
85. Gaspar C, Fodde R. APC dosage effects in tumorigenesis and stem cell differentiation. Int. J. Dev. Biol. 2004; 48:377–386.
86. Kielman MF, Rindapaa M, Gaspar C, van Poppel N, Breukel C, van Leeuwen S, Taketo MM, Roberts S, Smits R, Fodde R. Apc modulates embryonic stem-cell differentiation by controlling the dosage of beta-catenin signaling. Nat. Genet. 2002; 32:594–605.
87. Luis TC, Naber BA, Roozen PP, Brugman MH, de Haas EF, Ghazvini M, Fibbe WE, van Dongen JJ, Fodde R, Staal FJ. Canonical wnt signaling regulates hematopoiesis in a dosage-dependent fashion. Cell stem cell. 2011; 9:345–356.
88. Sun P, Watanabe K, Fallahi M, Lee B, Afetian ME, Rheaume C, Wu D, Horsley V, Dai X. Pygo2 regulates beta-catenin-induced activation of hair follicle stem/progenitor cells and skin hyperplasia. Proc. Natl. Acad. Sci. USA. 2014; 111:10215–10220.
89. Abraham SC, Klimstra DS, Wilentz RE, Yeo CJ, Conlon K, Brennan M, Cameron JL, Wu TT, Hruban RH. Solid-pseudopapillary tumors of the pancreas are genetically distinct from pancreatic ductal adenocarcinomas and almost always harbor beta-catenin mutations. Am. J. Pathol. 2002; 160:1361–1369.
90. Huang MY, Chen HC, Yang IP, Tsai HL, Wang TN, Juo SH, Wang JY. Tumorigenesis and tumor progression related gene expression profiles in colorectal cancer. Cancer Biomark. 2013; 13:269–279.
91. Kominami A, Fujino M, Murakami H, Ito M. beta-catenin mutation in ovarian solid pseudopapillary neoplasm. Pathol. Int. 2014; 64:460–464.
92. Thorne CA, Hanson AJ, Schneider J, Tahinci E, Orton D, Cselenyi CS, Jernigan KK, Meyers KC, Hang BI, Waterson AG, Kim K, Melancon B, Ghidu VP, et al. Small-molecule inhibition of Wnt signaling through activation of casein kinase 1alpha. Nat. Chem. Biol. 2010; 6:829–836.
93. Li B, Flaveny CA, Giambelli C, Fei DL, Han L, Hang BI, Bai F, Pei XH, Nose V, Burlingame O, Capobianco AJ, Orton D, Lee E, et al. Repurposing the FDA-approved pinworm drug pyrvinium as a novel chemotherapeutic agent for intestinal polyposis. PLoS ONE. 2014; 9:e101969.
94. Macartney KK, Baumgart DC, Carding SR, Brubaker JO, Offit PA. Primary murine small intestinal epithelial cells, maintained in long-term culture, are susceptible to rotavirus infection. J. Virol. 2000; 74:5597–5603.
95. Iwamoto T, Yamada K, Shimizu M, Totsuka M. Establishment of intestinal epithelial cell lines from adult mouse small and large intestinal crypts. Biosci Biotechnol Biochem. 2011; 75:925–929.