
INTRODUCTION
Senescence is a well-established barrier to tumorigenesis that prevents the proliferation of genomically damaged, but otherwise replication competent cells at risk of neoplastic transformation. Control of the G1 phase of the cell cycle is intrinsically linked with the maintenance of quiescent and senescent states in stem and somatic cells, respectively, and is primarily controlled by the INK4-CDK4/6-Rb pathway. The INK4 family of cell cycle inhibitors comprising of p16INK4A, p15INK4B, p18INK4C and p19INK4D (p16, p15, p18 and p19) inhibit CDK4 and CDK6, leading to the functional inactivation of RB [1]. Functional inactivation of this pathway is a common event in the development of most types of cancers [2], and also causes the loss of quiescent and senescent states of cells [3].
Unlike other INK4 family proteins, p16 is not expressed early, but is markedly increased with aging and senescence [4, 5]. Experiments in mice with a germline deletion of p16 reveal that p16 controls senescence in multiple organs during aging [6–9]. Importantly, p16 is deleted in ~50% of breast cancer cell lines, and p16 inactivation by DNA methylation occurs in ~30% of human breast cancers [2, 10, 11]. However, deleting p16 in mice or targeting DNA methylation within the murine p16 promoter does not result in mammary tumorigenesis but rather, these mice develop lymphomas and sarcomas with long latency [12–14]. How loss of p16 contributes to mammary tumorigenesis in vivo is not fully understood.
Breast cancer is heterogeneous with tumors that are both pathologically distinct and diverse in their responsiveness to treatment. Breast cancer is comprised of three main subtypes: HER2-positive, luminal, and basal-like cancers (BLBCs). Basal-like breast cancers (BLBCs) are ER-negative and more metastatic with few therapeutic options [15–18]. BRCA1 is a tumor suppressor, and its function has been linked with multiple pathways including DNA damage repair and oxidative stress regulation [19]. Functional loss of BRCA1 by germline or somatic mutation or by promoter methylation is associated with more than one third of basal-like breast cancers and cell lines [20–22]. Patients with a BRCA1 deficiency develop BLBCs that are enriched with tumor-initiating cells (TICs) and exhibit epithelial-mesenchymal transition (EMT) characteristics [15, 16]. EMT, a process in which epithelial cells lose many of their epithelial characteristics and acquire mesenchymal features, plays an important role in tumor heterogeneity and generation of TICs [23]. TICs are thought to drive clinical relapse and metastasis [17, 18].
BRCA1 deficiency in human and mouse mammary epithelial cells activates both the p16-RB and p53 pathways, inducing premature senescence [24–26]. Consistently, heterozygous germline deletion of Brca1 or specific deletion of Brca1 in mouse mammary epithelial cells rarely develop mammary tumors. About 10% of Brca1+/− or Brca1f/f;MMTV-cre mice develop mammary tumors by 18 months of age [24, 25, 27–29]. Loss of p53 or its downstream target, p21, in vivo as well as knockdown of p16 or its downstream target, Rb, in vitro partially rescues the premature senescence of Brca1-deficient cells [24–28, 30], suggesting that disruption of the p53 and p16-Rb pathway are required to overcome Brca1-deficiency induced senescence and induce breast cancers. Indeed, inactivation of the p53 pathway enhances mammary tumor incidence and shortens the time of tumor onset in Brca1-deficient mice [27, 28, 31, 32]. Notably, most human BLBCs with functional loss of BRCA1 have dysfunctional p16-RB and p53 pathways [33–36]. However, most genetic studies of Brca1 in mice co-mutate Brca1 with one of the genes in the p53 pathway [27, 28, 31, 32, 37]. It remains poorly understood whether p16 is involved in Brca1-deficient MEC senescence and tumorigenesis.
In this report, we generated p16 and Brca1 single and compound mutant mice to determine their function in controlling mammary epithelial cell (MEC) senescence and tumorigenesis.
RESULTS
p16 loss ameliorates Brca1 deficiency-induced senescence in MECs
We and others previously demonstrated that heterozygous germline mutation of Brca1 in human and mice leads to premature senescence in MECs [24, 26], though the molecular and cellular basis controlling Brca1-deficiency-induced senescence is not fully understood. To directly determine the role of Brca1 loss in mammary epithelial cell senescence and tumorigenesis, we used Brca1f/f;MMTV-Cre and Brca1f/−;MMTV-Cre mice (Brca1MGKO) mice as we previously described [25]. We compared Brca1MGKO mice to age-matched WT animals and found that mRNA levels of p16 were increased in Brca1MGKO mammaries relative to WT counterparts (Figure 1A). Taking into consideration the growth defects and increased SA-β-gal positivity in Brca1MGKO MECs relative to WT counterparts (see below), these data suggest that Brca1-deficiency induced senescence in the mammary epithelium correlates with increased expression of p16.
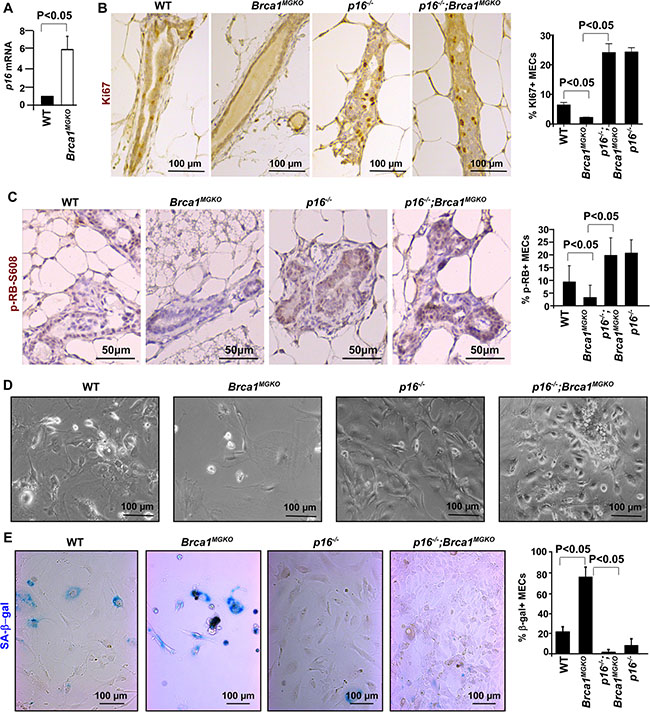
Figure 1: Loss of p16 rescues senescence caused by Brca1 deficiency. (A) RT-QPCR of mammary tissue from age-matched WT and Brca1MGKO mice. Data represent the mean ± SD from triplicates of three mice per genotype. (B, C) Immunohistochemical staining of Ki67 and pRB-s608 in mammary glands of the indicated genotypes. Results represent the mean ± SD of three animals and two animals per group respectively. (D) MECs of the indicated genotypes were isolated and cultured to analyze cell morphology. (E) SA-β-gal assay of MECs of the indicated genotypes. Results represent the mean ± SD of triplicates per genotype.
Considering that Brca1 loss induces premature senescence with an increase of p16 expression in MECs and that loss of p16 mitigates age-associated cellular senescence in compartments of the brain, pancreatic islets and blood [6–8], we were inspired to determine the role of p16 in controlling Brca1 deficiency-induced MEC senescence and tumorigenesis. To this end, we generated p16−/−;Brca1MGKO mice on a Balb/c-FVB-B6 mixed background. We found that the percentages of Ki67-positive MECs in p16−/− and p16−/−;Brca1MGKO mice (24.28 ± 2.1 and 24.06 ± 4.3) were significantly higher than those in WT mice (6.39 ± 1.3), which in turn were significantly higher than the percentage in Brca1MGKO (2.20 ± 0.2) mice (Figure 1B). We then directly examined Rb phosphorylation in MECs of different genotypes by using an antibody specifically recognizing Rb proteins phosphorylated at Ser608 by CDK4 and CDK6 [38, 39], the functional targets of p16. A consistent increase of pRb-Ser608 phosphorylation was detected in p16−/− mammary epithelia (9.86 ± 6.6% in WT to 21.64 ± 5.4% in p16−/−, Figure 1C), confirming the activation of CDK4 and/or CDK6. Consistent with our previous finding [24], 3.42 ± 5.0% pRb-Ser608 positive MECs were detected in Brca1MGKO females, which is significantly less than those in WT counterparts. However, p16−/−;Brca1MGKO mice had a significant higher percentage of positive MECs (20.69 ± 7.2%, Figure 1C) than Brca1MGKO mice. Together with the data derived from Ki67 staining, these results indicate that loss of p16 stimulates CDK4 and/or CDK6 activity toward the Rb protein in MECs, increasing their proliferation and rescuing the proliferative decline observed in Brca1-deficient MECs.
To further consolidate the role of loss of p16 and Brca1 in the regulation of MEC proliferation, we isolated and cultured primary MECs from virgin mice. We observed that Brca1MGKO MECs exhibited a large and flattened shape, a typical morphology of cellular senescence, while MECs from the other genotypes of mice were smaller and spindle-shaped (Figure 1D). Notably, most Brca1MGKO MECs exhibited strong, peri-nuclear staining of SA-β-gal whereas only a small population of WT MECs and very few p16−/− and p16−/−;Brca1MGKO MECs showed positive staining (Figure 1E). We then pulse-labeled primary MECs with bromodeoxyuridine (BrdU) for 15 hours and performed FACS analysis. Brca1MGKO MECs had increased G1 and decreased S phase cells relative to their WT counterparts (G1 phase cells, 29% vs 15%; S phase cells, 60% vs 68% Supplementary Figure S1). Importantly, MECs from p16−/− or p16−/−;Brca1MGKO mice displayed similar BrdU incorporation rates, 81% for p16−/− and 79% for p16−/;Brca1MGKO, which were significantly higher than their WT counterparts (Supplementary Figure S1). These data suggest that loss of Brca1 induces senescence in MECs, which is rescued by p16 loss.
Loss of p16 transforms Brca1-deficient MECs and induces mammary tumors
45% (n = 20) of p16−/− mice developed lymphoma and sarcoma in 24 months, which is consistent with previous reports [12, 13] (Table 1). Of the nine p16−/− tumors, two were detected in the mammary gland and were highly composed of lymphoma cells, as evidenced by their lymphocyte-like morphology, positivity for CD45 and CD31 by FACS analysis and negativity for Cdh1, an epithelial cell marker, by IHC (Supplementary Figure S2A, S2C and S2E). Interestingly, FACS analysis revealed that 1.7%–3.5% of the total tumor cell population was negative for CD45 and/or CD31 respectively (Supplementary Figure S2C), and IHC showed that less than 4% of sporadic tumor cells were epithelial-like and positively stained with Cdh1 (Supplementary Figure S2E), indicating that this tumor was comprised predominantly of lymphoma cells and that a small portion of cells originated from the mammary epithelium. These results confirm the predominant role of p16 in suppressing the development of lymphoma and sarcoma, and suggest that mammary tumorigenesis in p16 null mice may be masked by lymphomas and sarcomas.
Table 1: Spontaneous tumor development in WT and mutant female mice
Tumor |
Wt |
p16+/– |
p16–/– |
Brca1MGKO a |
p16+/–;Brca1MGKO |
p16–/–;Brca1MGKO |
|---|---|---|---|---|---|---|
11–24 m |
11–24 m |
11–24 m |
11–24 m |
11–24 m |
11–24 m |
|
Mammary Tumor |
0/9b |
0/6c |
0/20d |
0/8e |
4/9f (44%) |
5/8g (63%) |
Metastasis |
3/4h |
2/5i |
||||
Ck14 + tumor |
4/4 |
5/5 |
||||
EMT + tumor |
4/4 |
5/5 |
||||
Other tumors |
1/9j |
9/20k |
1/8l |
3/9m |
5/8n |
aBrca1MGKO, Brca1f/−; MMTV-Cre or Brca1f/f; MMTV-Cre mice.
bone mouse was 24 months of age, and the remaining mice were 11–20 months of age.
cone mouse was 24 months of age, and the remaining mice were 11–20 months of age.
dtwo mice developed tumors in mammary glands composed of lymphoma cells and 1%–4.0% epithelial-like cells respectively.
eone mouse was 23 months of age, and the remaining mice were 11–19 months of age.
ffour mice developed mammary tumors at 18, 18, 19 and 20 months of age, respectively. One mouse developed two different mammary tumors at two separate mammary glands. One mouse was 23 months of age, and the remaining mice were 14–20 months of age. One mouse was a breeder. Mammary tumor incidence, p16+/−;Brca1MGKO vs p16+/−, P = 0.103; p16+/−; Brca1MGKO vs Brca1MGKO, P = 0.082.
gfive mice developed mammary tumors at 11, 12, 16, 17 and 20 months of age, respectively. Two mice developed two different mammary tumors at two separate mammary glands. One mouse was 20 months of age, and the remaining mice were 11–17 months of age. One mouse was a breeder. Mammary tumor incidence, p16−/−; Brca1MGKO vs p16−/−, P = 0.0006; p16−/−;Brca1MGKO vs Brca1MGKO, P = 0.026; p16mt;Brca1MGKO (p16+/− Brca1MGKO and p16−/−;Brca1MGKO) vs Brca1MGKO, P = 0.022.
hlung metastasis from primary mammary tumors was detected in 3 mice, whose ages were 18, 18 and 20 months of age, respectively.
ilung metastasis from primary mammary tumors was detected in 2 mice, whose ages were 17 and 20 months of age, respectively.
jone mouse developed an ovarian tumor at 24 months of age.
knine mice developed sarcoma or lymphoma.
lone mouse developed lymphoma at 15 months of age.
mthree mice with mammary tumors also developed lymphomas.
none mouse developed pancreatic carcinoma at 16 months of age, and three mice with mammary tumors also developed lymphoma, sarcoma, lung adenoma, and hepatocellular carcinoma respectively.
We then followed the mammary tumor development in older mice and found that 63% of the p16−/−;Brca1MGKO mice (n = 8) and 44% of p16+/−;Brca1MGKO mice (n = 9) developed mammary tumors at 11–20 months and 16–23 months, respectively (Table 1, and Supplementary Table S1), whereas no p16−/−, p16+/− or Brca1MGKO mice did so at similar ages (Table 1). Median mammary tumor-free survival time in p16mt;Brca1MGKO mice (including p16−/−;Brca1MGKO and p16+/−;Brca1MGKO mice) was 18 months (Figure 2A). These results indicate that haploid or complete loss of p16 transforms Brca1-deficient mammary epithelial cells and induces mammary tumors.
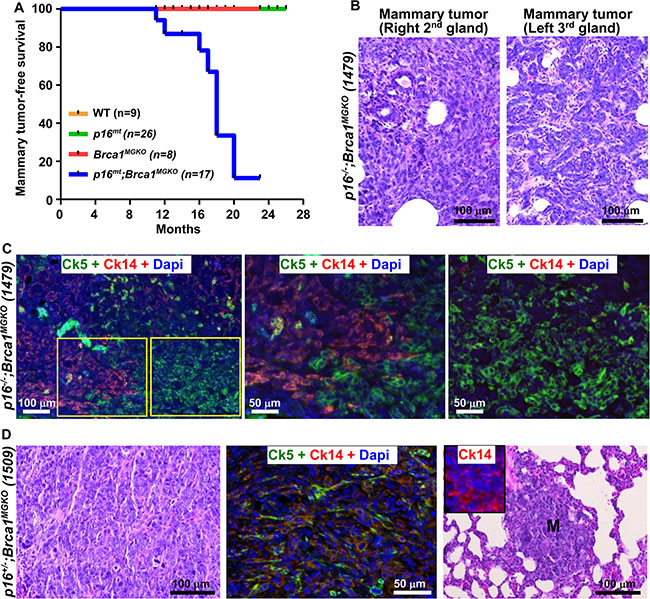
Figure 2: Characterization of primary tumors and distant metastases in mutant mice. (A) Mammary tumor-free survival curve. p16mt;Brca1MGKO includes: p16+/−;Brca1MGKO and p16−/−;Brca1MGKO mice. p16mt includes: p16+/− and p16−/− mice. Log-rank (Mantel-cox) test: p < 0.0001 (B) H.E. staining of tumors in a p16−/−;Brca1MGKO mouse (1479) at 11 months of age. Note this mouse developed two different mammary tumors. (C) Immunofluorescence staining of mammary tumors in mouse 1479 with Ck14 and Ck5. The boxed areas in the left panel are enlarged in the middle and right panels. Note the majority of tumor cells are positive for either Ck5 or Ck14. (D) Immunofluorescence and H.E. staining of mammary tumor in a p16+/−;Brca1MGKO mouse (1509) at 18 months of age. Note this tumor was comprised predominately of Ck5 and Ck14+ cells (middle panel) and developed a distant metastasis (M) to the lung (right panel), which also comprised of Ck14+ cells (inset in the right panel).
Depletion of both p16 and Brca1 leads to basal-like mammary tumors with EMT features
Mammary tumors developed in p16mt;Brca1MGKO mice were very aggressive and displayed typical morphological characteristics of highly malignant features (increased necrosis, spindle cells, nuclear-cytoplasm ratio, and mitotic indices) (Figure 2B, 2D, Supplementary Figure S2A, Supplementary Figure S3). 18% of the p16mt;Brca1MGKO mice (n = 17) developed two distinct mammary tumors in two separate mammary glands, demonstrating the ability of these mice to develop both intra- and inter- tumoral heterogeneity (Figure 2B, Supplementary Figure S3). Furthermore, 56% of the p16mt;Brca1MGKO mammary tumors (n = 9) metastasized to the lung in 17–20 months (Table 1, Figure 2D, Supplementary Table S1). All mammary tumors developed in p16mt;Brca1MGKO mice were positively stained with basal markers, Ck5 and Ck14 in 2–75% of total tumor cells (Figure 2C, 2D, Supplementary Figure S2B, Supplementary Figure S4, Supplementary Table S1). Further analysis revealed that most of these tumor cells expressed significantly reduced levels of Cdh1 protein when compared with levels in luminal cells of the mammary glands (Supplementary Figure S2E). Mammary tumors developed in p16mt;Brca1MGKO mice expressed 44% Gata3, and 54% Elf5, both of which are genes associated with luminal cell differentiation, relative to the tumor-free mammary tissues of the same mouse (Supplementary Figure S2D), confirming Brca1-deficiency impaired luminal differentiation during tumorigenesis, as we previously demonstrated [24, 25]. All mammary tumors derived from p16mt;Brca1MGKO mice were stained positively for vimentin (Vim), a mesenchymal marker, and Twist, an EMT transcription factor (Table 1, Supplementary Figure S2F). These data indicate that depletion of both p16 and Brca1 results in basal-like mammary tumors with activation of EMT, which is consistent with our previous finding that deletion of Brca1 activates EMT in mammary tumorigenesis [25].
We screened 43 human invasive breast cancers and selected 10 ER-negative samples with the lowest BRCA1 mRNA expression as previously described [25]. We compared tumor pathology and expression of CK5 and CK14 in these samples with mouse mammary tumors. We noticed that both the tumor cell morphology and expression pattern of CK5 and CK14 in p16;Brca1 double mutant mouse mammary tumors resembled human basal-like breast cancers that were ER-negative and expressed low BRCA1 (Supplementary Figure S4).
Given the function of Brca1 in DNA damage repair, we also evaluated the role of Brca1 loss in inducing DNA damage in tumor development. We determined the expression of γH2AX, a marker for DNA double-strand breaks, in spontaneous tumors from mutant mice. Since p16 single-mutant mice only developed lymphoma and sarcoma, we compared γH2AX expression in these tumors with mammary tumors. We found that the number of γH2AX-positive cells in p16mt;Brca1MGKO tumors was significantly greater than in p16 single-mutant tumors (4.5% ± 2.5% vs 0.84% ± 0.36%, p < 0.05, Supplementary Figure S2G, S2H), indicating a significant increase of cells with DNA damage in Brca1-deficient tumors.
Together, these results suggest that depletion of both p16 and Brca1 induces metastatic basal-like tumors that have an activated EMT program and enhanced DNA damage.
p16 and Brca1 double-mutant tumor cells are transplantable
A small portion of epithelial-like cells observed in two primary p16−/− lymphomas that developed in mammary glands were sporadic and cuboid, luminal-like cells that were negative for Ck14 and positive for Cdh1 (Supplementary Figure S2B and S2E). We transplanted 1 × 106 cells from one of, and 4 × 106 cells from the other one of the two p16−/− tumors that are mixed with lymphoma and epithelial-like cells into MFPs of NSG mice (three recipients per primary tumor). Tumors regenerated, like p16−/− primary tumors, were predominantly composed of lymphoma cells mixed with Cdh1-positive and Ck14-negative tumor cells (Figure 3, Supplementary Figure S2B, S2I, and data not shown). The Cdh1-positive cells in the regenerated tumors accounted for < 5% of total tumor cells, however, they aggregated together and formed a larger mass of epithelial-like tissue than in primary tumors (Supplementary Figure S2I and S2E). This result suggests that p16 loss stimulates luminal epithelial cell proliferation, possibly contributing to the development of premalignant lesions. These data are also consistent with our finding that loss of p18, another Ink4 family cell cycle inhibitor, promotes luminal epithelial cell proliferation and induces luminal tumorigenesis [40].
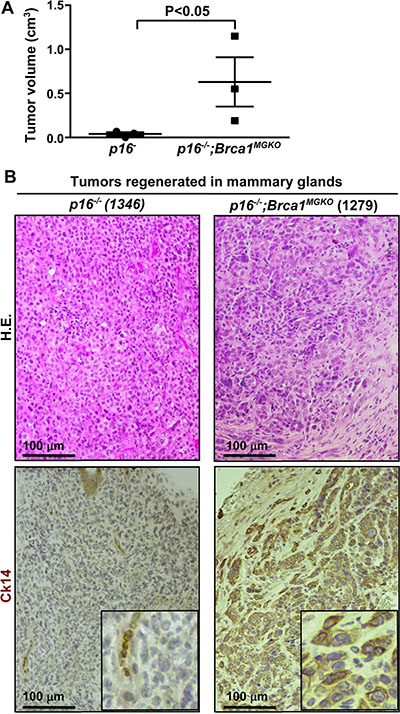
Figure 3: p16;Brca1 compound mutant tumor cells are enriched with TICs and capable of generating basal-like mammary tumors. (A) 1 × 106 cells from the tumors developed in the mammary glands of p16−/− and p16−/−;Brca1MGKO mice were transplanted into the left and right inguinal MFPs of female NSG mice, respectively. Generated tumor volume was measured 4 weeks after transplant. Results are representative of the mean ± SD of three tumors of each genotype. (B) Representative H.E. and immunostaining of Ck14 of regenerated mammary tumors.
We transplanted 1 × 106 cells from a p16−/−; Brca1MGKO mammary tumor, and 4 × 106 cells from a p16+/−; Brca1MGKO mammary tumor, respectively, into MFPs of NSG mice (three recipients per primary tumor). We found that p16mt;Brca1MGKO tumor cells regenerated significantly larger tumors than p16−/− cells with the same number of cells in the same time period (Figure 3A, and data not shown). Further analysis revealed that the generated p16mt;Brca1MGKO tumors resembled the pathology of the primary tumors and were positive for Ck14 recapitulating the phenotype observed in primary mammary tumors (Figure 3B). These data suggest that p16;Brca1 double-mutant mammary tumors are enriched with mammary tumor initiating cells.
Expression of p16 is lost in p16+/–;Brca1MGKO tumors due to promoter methylation
We determined the expression of p16 in p16+/−; Brca1MGKO mammary tumors. We found that p16 mRNA expression was retained in one tumor (number 1255), but clearly reduced in the other tumors when compared with tumor-free mammary tissues from the same mouse (Figure 4A). p16 mRNA levels in tumor 1255 were 8.5-fold more than tumor-free mammary tissues, which could be caused by a reduction in the function of the Rb or p53 pathways [41, 42]. We performed loss of heterozygosity (LOH) analysis for DNA extracted from p16+/−;Brca1MGKO mammary tumors and found that the remaining WT allele of p16 was retained in all four tumors (Figure 4B). These results suggest that epigenetic silencing of p16, likely by promoter methylation as observed in DMBA-induced p16+/− lymphomas and sarcomas [12], plays an important role in p16+/−;Brca1MGKO mammary tumorigenesis. To determine the methylation status of the p16+/−;Brca1MGKO mammary tumors, we performed methylation-specific PCR (MS-PCR) and detected methylation of the p16 promoter in three tumors with reduced expression of p16, but not in tumor 1255 in which p16 expression was not decreased (Figure 4C). Notably, we also detected p16 promoter methylation in a tumor-free, but premalignant lesion-containing mammary tissue in the mouse 1497 (Figure 4C), which explains why there was not a significant decrease in p16 expression between tumor vs tumor-free tissues in mouse 1497 (Figure 4A). We isolated and cultured primary cells from p16+/−;Brca1MGKO mammary tumors and found that p16 mRNA levels were significantly increased in the 1347 tumor cells treated with 5-aza-2′-deoxycytidine (DAC), a methylation inhibitor, but not in the 1255 tumor cells (Figure 4D), confirming the findings derived by MS-PCR in tumors. These results suggest that epigenetic silencing of p16 by promoter methylation plays an important role in the development of, at least, some of the p16+/−;Brca1MGKO mammary tumors.
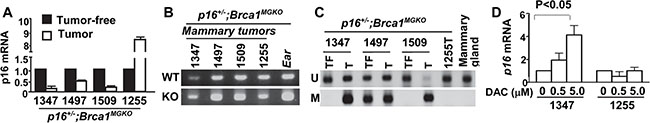
Figure 4: Promoter methylation silences p16 expression in p16+/–;Brca1MGKO tumors. (A) mRNA expression of p16 in p16+/−; Brca1MGKO mammary tumors was determined by q-RT-PCR. Corresponding tumor-free mammary tissues from the same mice were used as controls. Note the control mammary tissue for mouse 1497 tumor was from a premalignant lesion of the same mouse. Data are expressed as the mean of triplicate experiments. (B) LOH analysis of the p16 gene in p16+/−;Brca1MGKO mammary tumors. Ear DNA was used as a control. (C) Bisulphite-treated DNA from mammary tumors (T) or tumor-free mammary tissues (TF) from the same mice was analyzed for methylation of p16. U, unmethylated. M, methylated. A normal mammary gland from a WT mouse was used as a control. Note, no methylated p16 was detected in the mammary tumor developed in mouse 1255 in which the p16 mRNA level was not reduced relative to tumor-free mammary tissue of the same mouse. (D) p16 mRNA analysis in primary mammary tumor cells after treatment with DAC at 0, 0.5, or 5 μM for 72 hours.
DISCUSSION
In this article, we reported that disruption of Brca1 in the mammary epithelium results in premature senescence with an increase of p16 expression, which is rescued by p16 loss. We found that loss of p16 transforms Brca1-deficient mammary epithelial cells and induces mammary tumors, though p16 loss alone is not sufficient to induce spontaneous mammary tumorigenesis. We showed that mammary tumors deficient for both p16 and Brca1 are highly aggressive, metastatic, and enriched for TICs. We demonstrated that loss of p16 and Brca1 collaboratively induce basal-like mammary tumor development with the induction of EMT. To the best of our knowledge, this is the first genetic evidence directly showing that p16 which is frequently deleted and inactivated in human breast cancers, collaborates with Brca1 controlling mammary tumorigenesis.
The functions of BRCA1 have been linked with multiple pathways. BRCA1 deficiency causes chromosomal abnormalities, leading to the activation of DNA-damage checkpoint pathways and premature senescence [26–28, 30]. It has also been reported that BRCA1 deficiency in MECs impairs stability and activation of Nrf2, a key transcription factor in regulating antioxidant response, and leads to the accumulation of reactive oxygen species (ROS), along with the increase of p16 [43–45]. Interestingly, accumulation of ROS has been associated with cellular senescence [46], and Nrf2 activation restores ROS levels in Brca1-deficient MECs [43, 44]. These findings suggest that BRCA1 deficiency induced premature senescence in MECs, at least, partially resulted from accumulation of ROS. Our results that loss of p16 rescues the MEC senescence caused by Brca1 deficiency suggests that p16 loss may allow Nrf2 levels to accumulate in Brca1-deficient cells and suppress ROS, which remains to be determined.
The finding that loss of p16 rescues MEC senescence caused by Brca1 deficiency also suggests that p16 blocks these cells from entering an active cell cycle. These results indicate that, in addition to the p53-p21 pathway that is activated by BRCA1 loss [27, 28, 30], p16 is also a critical downstream target of BRCA1 in controlling mammary cell proliferation and senescence. In line with these data, it was recently reported in vitro that BRCA1 knockdown enhances the association of BRG1, a chromatin-remodeling factor that interacts with BRCA1, with the promoters of p16 and p21, leading to activation of their transcription and senescence [30]. More recently, it was found that human mammary epithelial cells from BRCA1-mutation carriers exhibit senescence, which is triggered by pRb pathway activation [26]. Since deregulation of p53 alone induces DNA damage, p16; Brca1 compound mutant mice and cells offer a unique opportunity to investigate the role of Brca1 in DNA damage repair under a genetically p53 intact background.
The function of p16 in breast cancer suppression has been extensively studied and confirmed in human breast cancer samples and cell lines [2, 47]. However, the role of p16 in suppressing mammary tumorigenesis in vivo is elusive. Previous findings [12–14] that mice lacking p16 or targeting DNA methylation within the p16 promoter rarely develop mammary tumors suggests that p16 loss alone is not sufficient for mammary tumorigenesis in vivo. Interestingly, loss of p16 increases MEC proliferation, rescues Brca1-deficiency induced MEC senescence, and induces mammary tumors in a Brca1-deficient background, suggesting that p16 collaborates with Brca1 to suppress mammary tumorigenesis. Though the remaining WT allele of p16 was retained in all p16+/−; Brca1MGKO mammary tumors, the p16 promoter was methylated and p16 mRNA was significantly reduced in most of these tumors, indicating that silencing of p16 by promoter methylation plays a role in the development of, at least, some of the p16+/−;Brca1MGKO mammary tumors. In line with these results are the clinical findings that methylation of BRCA1 and p16 is frequently detected in sporadic breast cancers and has a predictive value for tumor recurrence [48]. Together, our data not only support the function of p16 in suppression of Brca1-deficient mammary tumorigenesis, but also indicate that genetic mutation of p16 cooperates with epigenetic silencing of its transcription to promote tumorigenesis.
Of the four INK4 genes, p16 is frequently deleted and inactivated, and p18 expression is significantly downregulated in breast cancers ([1, 2, 10, 11, 34], and Pei XH, unpublished). The Rb family proteins consist of RB, p107, and p130, which are also frequently deleted and inactivated in breast cancers and are downstream targets of INK4 proteins [1, 2, 10, 11, 34]. RB is a major target for genomic disruption in BRCA1 mutant human breast cancers and loss of both RB and BRCA1 is a feature of basal-like breast cancers [34, 36, 49]. Deletion of both Rb and p107 in mouse epithelia results in mammary luminal tumor development [49]. We demonstrated that deletion of p18 in mice stimulates luminal progenitor cell proliferation, leading to mammary luminal tumorigenesis [40], and that depletion of Brca1 in p18 null mice converts luminal tumors into basal like tumors and activates EMT [24, 25]. We observed that depletion of p19 also stimulates mammary luminal cell proliferation [50]. In the present study, we report that loss of p16 increases MEC proliferation and induces mammary tumorigenesis in a Brca1- deficient background. More importantly, all p16;Brca1 compound mutant mammary tumors are poorly differentiated basal-like tumors with enriched TICs and activated EMT features. These findings suggest that the INK4-Rb pathway suppresses mammary luminal cell proliferation and tumorigenesis, and collaborates with Brca1 to control basal-like tumorigenesis and EMT.
MATERIALS AND METHODS
Mice
The generation of Brca1f/f;MMTV-Cre and Brca1f/–; MMTV-Cre mice in a Balb/c-B6 mixed background has been described previously [25]. Virgin Brca1f/f;MMTV-Cre and Brca1f/–;MMTV-Cre (Brca1MGKO) mammaries express < 20% of Brca1 protein and mRNA relative to the levels in Brca1f/+;MMTV-Cre, indicating depletion of Brca1 in the mammary epithelia [25]. p16 mutant mice in a FVB background were gifted by our collaborators, Dr. Norman Sharpless [12]. Virgin female mice were used in the study unless otherwise specified. All animal procedures were approved by the Institutional Animal Care and Use Committee at the University of North Carolina and University of Miami.
Histopathology, IHC, and qRT-PCR
Histopathology and IHC were performed as previously described [24, 25]. Primary antibodies used are as follows: Ki67 (Novocastra Laboratories, Newcastle upon Tyne, UK), gH2AX, phospho-RB (Cell Signaling), CK14 (Thermo Scientific), ERα, (Santa Cruz), Vim, Twist (Abcam), Cdh1 (BD Biosciences), CK5 (Covance prb-160p). Mammary tumors in which at least two EMT markers (decreased Cdh1, increased Vim or Twist) were detected in > 2% tumor cells are defined as EMT+ tumors, as we previously described [25]. The IACUC (Institutional Animal Care and Use Committee) at the University of Miami approved all procedures. QRT-PCR was carried out as previously described [24, 25].
Mammary cell preparation, cell cycle analysis, and mammary tumor cell transplantation
Mammary glands were dissected from virgin female mice at the indicated ages and genotypes. After mechanical dissociation, the tissue was processed as previously described [24, 25, 40]. MECs isolated from mice were cultured in in DMEM, with 10% FBS, 10 μg/ml insulin, 10 ng/ml EGF, 10 μg/ml Hydrocortisone. For cell cycle analysis, MECs were pulse-labeled with bromodeoxyuridine (BrdU) for 15 hours. Cells were then stained with propidium iodide (PI) and an antibody against BrdU and analyzed via flow cytometry as described [50]. For mammary tumor cell transplantation, 1 × 106 tumor cells were transplanted into MFPs of NSG mice as previously described [25]. Four weeks post-transplantation, newly generated tumors were dissected and analyzed.
LOH and methylation analysis
For LOH analysis, genomic DNA extracted from micro-dissected p16+/−;Brca1MGKO mammary tumor cells was analyzed with primers amplifying wild-type (WT) and knock-out (KO) allele of p16 as previously described [12, 24]. For methylation analysis, genomic DNA from p16+/−;Brca1MGKO mammary tumors and tumor-free mammary tissues of the same mice were treated with bisulfide and analyzed for p16 methylation with specific primers amplifying the unmethylated or methylated allele as described [12]. In addition, p16 mRNA levels in primary p16+/−;Brca1MGKO mammary tumor cells treated with DAC at the indicated concentrations for 72 hours were analyzed by q-RT-PCR.
Human breast cancer samples
Formalin fixed paraffin-embedded (FFPE) human breast cancer samples lacking patient-identifying information were obtained from the Tissue Bank Core Facility at the University of Miami. All samples obtained were non-treated invasive breast cancers with known ER status. The expression of BRCA1 in tumors was determined by microdissection-based RNA extraction and Q-RT-PCR as we previously described [25].
Statistical analysis
All data are presented as the mean ± SD for at least three repeated individual experiments for each group unless otherwise specified. Statistical analysis of mRNA expression, Ki67 positive cells, and tumor volume was performed using a two-tailed Student’s t-test. Statistical analysis of tumor incidence was performed using a two-tailed Fisher’s exact test. Statistical analysis of mammary tumor-free survival was performed using a Log-rank (Mantel-cox) test. P < 0.05 was considered statistically significant. Statistical analyses were conducted using Microsoft Excel and GraphPad Prism 5.
ACKNOWLEDGMENTS
We thank Drs. Norman Sharpless, Beverly Koller, Chuxia Deng, and Lothar Hennighausen for p16, Brca1 mutant and MMTV-cre mice, Ergonul A. Burcu for discussion on tumor pathology, the FACS core facility at University of Miami for cell sorting, the DVR core facility for animal husbandry.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
GRANT SUPPORT
This study was supported by DOD Idea Award (W81XWH-10-1-0302), DOD Idea Expansion Award (W81XWH-13-1-0282), Sylvester BFBCI Developmental Grant, IRG-98-277-13 from the American Cancer Society, and startup funds from the University of Miami Department of Surgery and Sylvester Cancer Center to Xin-Hai Pei.
REFERENCES
1. Pei XH, Xiong Y. Biochemical and cellular mechanisms of mammalian CDK inhibitors: a few unresolved issues. Oncogene. 2005; 24:2787–2795.
2. Sherr CJ, McCormick F. The RB and p53 pathways in cancer. Cancer Cell. 2002; 2:103–112.
3. Orford KW, Scadden DT. Deconstructing stem cell self-renewal: genetic insights into cell-cycle regulation. Nat Rev Genet. 2008; 9:115–128.
4. Zindy F, Quelle DE, Roussel MF, Sherr CJ. Expression of the p16INK4a tumor suppressor versus other INK4 family members during mouse development and aging. Oncogene. 1997; 15:203–211.
5. Krishnamurthy J, Torrice C, Ramsey MR, Kovalev GI, Al-Regaiey K, Su L, Sharpless NE. Ink4a/Arf expression is a biomarker of aging. J Clin Invest. 2004; 114:1299–1307.
6. Molofsky AV, Slutsky SG, Joseph NM, He S, Pardal R, Krishnamurthy J, Sharpless NE, Morrison SJ. Increasing p16INK4a expression decreases forebrain progenitors and neurogenesis during ageing. Nature. 2006; 443:448–452.
7. Janzen V, Forkert R, Fleming HE, Saito Y, Waring MT, Dombkowski DM, Cheng T, DePinho RA, Sharpless NE, Scadden DT. Stem-cell ageing modified by the cyclin-dependent kinase inhibitor p16INK4a. Nature. 2006; 443:421–426.
8. Krishnamurthy J, Ramsey MR, Ligon KL, Torrice C, Koh A, Bonner-Weir S, Sharpless NE. p16INK4a induces an age-dependent decline in islet regenerative potential. Nature. 2006; 443:453–457.
9. Sharpless NE, Sherr CJ. Forging a signature of in vivo senescence. Nat Rev Cancer. 2015; 15:397–408.
10. Kamb A, Gruis NA, Weaver-Feldhaus J, Liu Q, Harshman K, Tavtigian SV, Stockert E, Day RS, 3rd, Johnson BE, Skolnick MH. A cell cycle regulator potentially involved in genesis of many tumor types. Science. 1994; 264:436–440.
11. Herman JG, Merlo A, Mao L, Lapidus RG, Issa J PJ, Davidson NE, Sidransky D, Baylin SB. Inactivation of the CDKN2/p16/MTS1 gene is frequently associated with aberrant DNA methylation in all common human cancers. Cancer Res. 1995; 55:4525–4530.
12. Sharpless NE, Bardeesy N, Lee KH, Carrasco D, Castrillon DH, Aguirre AJ, Wu EA, Horner JW, DePinho RA. Loss of p16Ink4a with retention of p19Arf predisposes mice to tumorigenesis. Nature. 2001; 413:86–91.
13. Sharpless NE, Alson S, Chan S, Silver DP, Castrillon DH, DePinho RA. p16(INK4a) and p53 deficiency cooperate in tumorigenesis. Cancer Res. 2002; 62:2761–2765.
14. Yu DH, Waterland RA, Zhang P, Schady D, Chen MH, Guan Y, Gadkari M, Shen L. Targeted p16(Ink4a) epimutation causes tumorigenesis and reduces survival in mice. J Clin Invest. 2014; 124:3708–3712.
15. Foulkes WD. BRCA1 functions as a breast stem cell regulator. J Med Genet. 2004; 41:1–5.
16. Althuis MD, Fergenbaum JH, Garcia-Closas M, Brinton LA, Madigan MP, Sherman ME. Etiology of hormone receptor-defined breast cancer: a systematic review of the literature. Cancer Epidemiol Biomarkers Prev. 2004; 13:1558–1568.
17. Wright MH, Calcagno AM, Salcido CD, Carlson MD, Ambudkar SV, Varticovski L. Brca1 breast tumors contain distinct CD44+/CD24− and CD133+ cells with cancer stem cell characteristics. Breast Cancer Res. 2008; 10:R10.
18. Wicha MS. Cancer stem cell heterogeneity in hereditary breast cancer. Breast Cancer Res. 2008; 10:105.
19. Marks JR. Refining the role of BRCA1 in combating oxidative stress. Breast Cancer Res. 2013; 15:320.
20. Turner N, Tutt A, Ashworth A. Hallmarks of ‘BRCAness’ in sporadic cancers. Nat Rev Cancer. 2004; 4:814–819.
21. De Summa S, Pinto R, Sambiasi D, Petriella D, Paradiso V, Paradiso A, Tommasi S. BRCAness: a deeper insight into basal-like breast tumors. Ann Oncol. 2013; 8:viii13-viii21.
22. Elstrodt F, Hollestelle A, Nagel JH, Gorin M, Wasielewski M, van den Ouweland A, Merajver SD, Ethier SP, Schutte M. BRCA1 mutation analysis of 41 human breast cancer cell lines reveals three new deleterious mutants. Cancer Res. 2006; 66:41–45.
23. Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009; 119:1420–1428.
24. Bai F, Smith MD, Chan HL, Pei XH. Germline mutation of Brca1 alters the fate of mammary luminal cells and causes luminal-to-basal mammary tumor transformation. Oncogene. 2013; 32:2715–2725.
25. Bai F, Chan HL, Scott A, Smith MD, Fan C, Herschkowitz JI, Perou CM, Livingstone AS, Robbins DJ, Capobianco AJ, Pei XH. BRCA1 Suppresses Epithelial-to-Mesenchymal Transition and Stem Cell Dedifferentiation during Mammary and Tumor Development. Cancer Res. 2014; 74:6161–6172.
26. Sedic M, Skibinski A, Brown N, Gallardo M, Mulligan P, Martinez P, Keller PJ, Glover E, Richardson AL, Cowan J, Toland AE, Ravichandran K, Riethman H, et al. Haploinsufficiency for BRCA1 leads to cell-type-specific genomic instability and premature senescence. Nature communications. 2015; 6:7505.
27. Cao L, Li W, Kim S, Brodie SG, Deng CX. Senescence, aging, and malignant transformation mediated by p53 in mice lacking the Brca1 full-length isoform. Genes Dev. 2003; 17:201–213.
28. Cao L, Kim S, Xiao C, Wang RH, Coumoul X, Wang X, Li WM, Xu XL, De Soto JA, Takai H, Mai S, Elledge SJ, Motoyama N, et al ATM-Chk2-p53 activation prevents tumorigenesis at an expense of organ homeostasis upon Brca1 deficiency. EMBO J. 2006; 25:2167–2177.
29. Xu X, Wagner KU, Larson D, Weaver Z, Li C, Ried T, Hennighausen L, Wynshaw-Boris A, Deng CX. Conditional mutation of Brca1 in mammary epithelial cells results in blunted ductal morphogenesis and tumour formation. Nat Genet. 1999; 22:37–43.
30. Tu Z, Zhuang X, Yao YG, Zhang R. BRG1 is required for formation of senescence-associated heterochromatin foci induced by oncogenic RAS or BRCA1 loss. Mol Cell Biol. 2013; 33:1819–1829.
31. Deng CX, Scott F. Role of the tumor suppressor gene Brca1 in genetic stability and mammary gland tumor formation. Oncogene. 2000; 19:1059–1064.
32. Drost RM, Jonkers J. Preclinical mouse models for BRCA1-associated breast cancer. Br J Cancer. 2009; 101:1651–1657.
33. Jonsson G, Staaf J, Vallon-Christersson J, Ringner M, Gruvberger-Saal SK, Saal LH, Holm K, Hegardt C, Arason A, Fagerholm R, Persson C, Grabau D, Johnsson E, et al. The retinoblastoma gene undergoes rearrangements in BRCA1-deficient basal-like breast cancer. Cancer Res. 2012; 72:4028–4036.
34. Koboldt DC, Fulton RS, McLellan MD, Schmidt H, Kalicki-Veizer J, McMichael JF, Fulton LL, Dooling DJ, Ding L, Mardis ER, Wilson RK, Ally A, Balasundaram M, et al. Comprehensive molecular portraits of human breast tumours. Nature. 2012.
35. Stefansson OA, Jonasson JG, Olafsdottir K, Hilmarsdottir H, Olafsdottir G, Esteller M, Johannsson OT, Eyfjord JE. CpG island hypermethylation of BRCA1 and loss of pRb as co-occurring events in basal/triple-negative breast cancer. Epigenetics. 2011; 6:638–649.
36. Herschkowitz JI, He X, Fan C, Perou CM. The functional loss of the retinoblastoma tumour suppressor is a common event in basal-like and luminal B breast carcinomas. Breast Cancer Res. 2008; 10:R75.
37. Moynahan ME. The cancer connection: BRCA1 and BRCA2 tumor suppression in mice and humans. Oncogene. 2002; 21:8994–9007.
38. Kitagawa M, Higashi H, Jung HK, Suzuki-Takahashi I, Ikeda M, Tamai K, Kato J, Segawa K, Yoshida E, Nishimura S, Taya Y. The consensus motif for phosphorylation by cyclin D1-Cdk4 is different from that for phosphorylation by cyclin A/E-Cdk2. The EMBO journal. 1996; 15:7060–7069.
39. Zarkowska T, U S, Harlow E, Mittnacht S. Monoclonal antibodies specific for underphosphorylated retinoblastoma protein identify a cell cycle regulated phosphorylation site targeted by CDKs. Oncogene. 1997; 14:249–254.
40. Pei XH, Bai F, Smith MD, Usary J, Fan C, Pai SY, Ho IC, Perou CM, Xiong Y. CDK inhibitor p18(INK4c) is a downstream target of GATA3 and restrains mammary luminal progenitor cell proliferation and tumorigenesis. Cancer Cell. 2009; 15:389–401.
41. Zeng Y, Kotake Y, Pei XH, Smith MD, Xiong Y. p53 binds to and is required for the repression of Arf tumor suppressor by HDAC and polycomb. Cancer Res. 2011; 71:2781–2792.
42. Kotake Y, Cao R, Viatour P, Sage J, Zhang Y, Xiong Y. pRB family proteins are required for H3K27 trimethylation and Polycomb repression complexes binding to and silencing p16INK4alpha tumor suppressor gene. Genes Dev. 2007; 21:49–54.
43. Gorrini C, Baniasadi PS, Harris IS, Silvester J, Inoue S, Snow B, Joshi PA, Wakeham A, Molyneux SD, Martin B, Bouwman P, Cescon DW, Elia AJ, et al. BRCA1 interacts with Nrf2 to regulate antioxidant signaling and cell survival. J Exp Med. 2013; 210:1529–1544.
44. Gorrini C, Gang BP, Bassi C, Wakeham A, Baniasadi SP, Hao Z, Li WY, Cescon DW, Li YT, Molyneux S, Penrod N, Lupien M, Schmidt EE, et al. Estrogen controls the survival of BRCA1-deficient cells via a PI3K-NRF2-regulated pathway. Proc Natl Acad Sci USA. 2014; 111:4472–4477.
45. Cao L, Xu X, Cao LL, Wang RH, Coumoul X, Kim SS, Deng CX. Absence of full-length Brca1 sensitizes mice to oxidative stress and carcinogen-induced tumorigenesis in the esophagus and forestomach. Carcinogenesis. 2007; 28:1401–1407.
46. Fridman AL, Tainsky MA. Critical pathways in cellular senescence and immortalization revealed by gene expression profiling. Oncogene. 2008; 27:5975–5987.
47. Tlsty TD, Crawford YG, Holst CR, Fordyce CA, Zhang J, McDermott K, Kozakiewicz K, Gauthier ML. Genetic and epigenetic changes in mammary epithelial cells may mimic early events in carcinogenesis. J Mammary Gland Biol Neoplasia. 2004; 9:263–274.
48. Feng J, Hu LH, Lu J, Li YR, Xie F. [Diagnostic value of BRCA1 and p16 gene methylation in sporadic breast cancer]. Ai Zheng. 2009; 28:436–440.
49. Jiang Z, Deng T, Jones R, Li H, Herschkowitz JI, Liu JC, Weigman VJ, Tsao MS, Lane TF, Perou CM, Zacksenhaus E. Rb deletion in mouse mammary progenitors induces luminal-B or basal-like/EMT tumor subtypes depending on p53 status. J Clin Invest. 2010; 120:3296–3309.
50. Bai F, Chan HL, Smith MD, Kiyokawa H, Pei XH. p19Ink4d is a tumor suppressor and controls pituitary anterior lobe cell proliferation. Mol Cell Biol. 2014; 34:2121–2134.