
INTRODUCTION
Metastasis is the main cause of cancer-related deaths. Although some malignant tumors metastasize via the bloodstream most epithelial cancers, including breast tumors, first spread via lymphatic vessels to their regional lymph nodes and indeed the detection of tumor cells within the sentinel node has a main importance for patient prognosis [1, 2]. Expression of the lymphangiogenic growth factors by the tumor cells induces lymphangiogenesis, the growth and enlargement of lymphatic vessels, playing a crucial role in tumor dissemination [3–6]. Tumor lymphangiogenesis is mostly due to the proliferation and sprouting of pre-existing vessels, rather than to incorporation of circulating endothelial progenitor cells and is at least in part mediated by VEGF-C and to a lesser extent VEGF-D [3, 5, 7–11]. Lymph vessel density correlates with nodal status and is a prognostic factor in breast cancer [7, 12–15]. Tumor-associated macrophages can also produce lymphangiogenic factors contributing to vessels formation [16], thus showing the importance of the tumor microenvironment in this process. In addition, the lymphatic endothelial cells produce chemokines such as the stromal-derived factor 1 (or CXCL12), which bind CXCR4 receptors in the tumor cells [17, 18], facilitating their migration toward the lymphatic vessel [19].
NCoR (or Nuclear Corepressor-1) plays an important role in gene silencing. This corepressor associates with histone deacetylases (HDACs) and is recruited to target genes by interaction with nuclear receptors and other transcription factors, causing chromatin compaction and blocking transcription [20–23]. Through regulation of gene expression this corepressor could modulate cancer cell biology. Indeed, NCoR mutations have been found in breast tumors, and in these tumors frame-shift or nonsense inactivating mutations of the NCoR gene have been identified as driver mutations [24–26]. These observations support the findings that low NCoR expression is associated with invasive breast tumors [27, 28], a shorter relapse-free survival [29] and resistance to anti-estrogen treatment [30], suggesting the role of NCoR as a tumor suppressor. In agreement with this hypothesis, NCoR silences transcription of genes associated with metastatic growth and poor outcome in cancer patients, inhibiting tumor growth, invasion and metastatic potential in xenograft mouse models [31]. Furthermore, it has been demonstrated the existence of a positive auto-regulatory loop that maintains NCoR gene expression, suggesting that loss of NCoR expression can confer an advantage to the tumor cell, contributing to tumor progression even in the absence of NCoR gene mutations.
The actions of the thyroid hormones thyroxine (T4) and triiodothyronine (T3) are mediated by binding to the nuclear thyroid hormone receptors (TRs). Although the thyroid gland produces more T4, T3 is formed by deiodination of T4 in extrathyroidal tissues and is believed to be the active hormone since TRs show a higher binding affinity for T3 than for T4 [32]. TRs and particularly the TRβ isoform can have tumor suppressor actions. TRβ mutations, anomalous subcellular localization and biallelic inactivation of this gene by promoter methylation has been found in breast tumors [33–35]. Furthermore, expression of TRβ in breast cancer cells reduces tumor growth [36, 37]and has a strong inhibitory effect on invasion, extravasation, and metastasis formation in immunodeficient mice [38]. TRβ induces NCoR expression and this induction appears to be an essential mediator of the tumor suppressive and anti-metastatic actions of the receptor. Moreover, both NCoR and TRβ are downregulated in the more aggressive human estrogen receptor negative (ER−) breast tumors with respect to the ER+ tumors with a better prognosis, existing a positive correlation between transcript levels of the receptor and the corepressor [31].
In this work we tested the possibility that NCoR and TRβ could regulate the expression of VEGF genes and the growth of lymphatic vessels, thus regulating tumor invasion. We demonstrate that NCoR and TRβ repress transcription of the VEGF-C and VEGF-D genes in breast cancer cell lines and tumor xenografts. Furthermore, NCoR depletion increases lymph vessel density in the tumors and reverses the inhibitory effect of the receptor in lymphangiogenesis. The importance of our results is supported by the finding of a strong negative correlation between the mRNA levels of the lympangiogenic genes and either NCoR or TRβ in human breast tumors. This correlation is independent of the ER status, although lymphangiogenic genes are expressed at significantly higher levels in the ER- tumors. Since high VEGF-C and VEGF-D levels lead to a poor prognosis in breast cancer, our finding that NCoR and TRβ are potent inhibitors of these factors suggests that they may serve as novel therapeutic targets to inhibit lymphangiogenesis and breast tumor progression.
RESULTS
NCoR represses transcription of the VEGF-C and VEGF-D genes in breast cancer cell lines
To analyze a possible correlation between NCoR and VEGF-C and VEGF-D gene expression, we first measured their transcript levels in several human breast cancer cell lines, both ER+ and ER-. NCoR mRNA was significantly higher in the ER+ MCF-7 and ZR75–1 cells than in the very aggressive HCC-1954 cells and in the MDA-MB-231 cells, while VEGF-C and VEGF-D transcripts exhibited an opposite pattern, being lower in the ER+ positive cell lines (Figure 1A). Although other factors different from NCoR could be responsible for the negative association with lympangiogenic gene expression in these independently-derived cell lines, the inverse relationship observed suggested that NCoR could repress VEGF-C and VEGF-D gene transcription. Proximal promoter sequences appear to play an important role in the control of VEGF-C and VEGF-D transcription [39, 40]. To analyze if NCoR could bind to the regulatory region of these lymphangiogenic genes, we performed chromatin immunoprecipitation (ChIP) assays with an NCoR antibody and two different fragments of the 5´-flanking regions of these genes. Significant NCoR association to the -235/+13 region of the VEGF-C gene was observed in MCF-7 and ZR75-1 cells, while NCoR binding was much lower in MDA-MB-231 and HCC-1954 cells (Figure 1B). However, NCoR did not bind to the immediate upstream region of the VEGF-C promoter in any cell line. A very strong binding of the corepressor was found in the ER+ cells when the proximal -423/-119 region of the VEGF-D gene was analyzed and again NCoR binding to these sequences was very low in the ER- cells. However, NCoR was not recruited to the -608/-430 region of the VEGF-D gene, previously proposed to be necessary for VEFG-D transcription [39, 40] (Figure 1B).
To study the functionality of NCoR binding to the regulatory region of the lymphangiogenic genes, MDA-MB-231 and MCF-7 cells were transfected with a control siRNA or with an NCoR specific siRNA. Transfection of siNCoR very effectively reduced NCoR transcripts in the cells, and VEGF-C and VEGF-D gene expression was significantly increased upon NCoR depletion (Figure 1C). In accordance with the different levels of NCoR expression and promoter occupancy shown in panels A and B, this increase was stronger in MCF-7 cells and more moderate in MDA-MB-231 cells. These results show that the VEGF-C and VEGF-D genes are bona fide targets of NCoR in breast cancer cells.
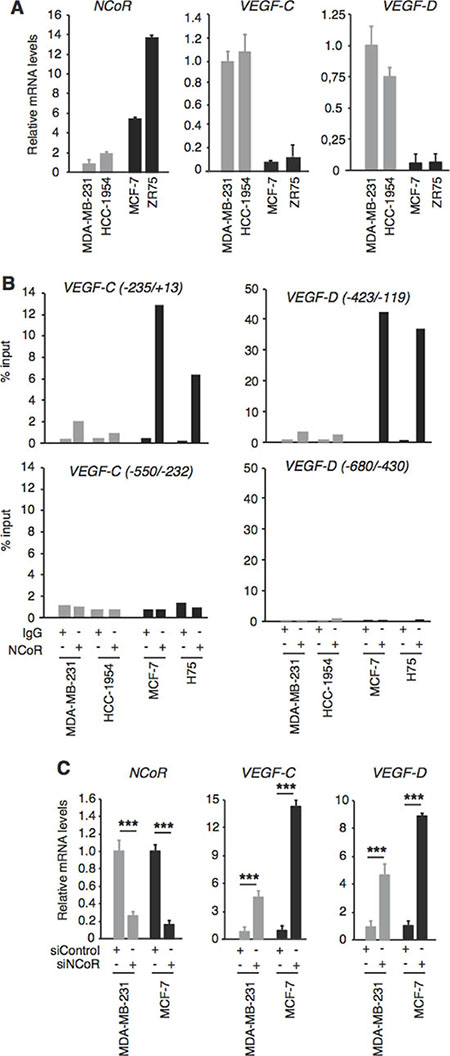
Figure 1: NCoR represses VEGF-C and VEGF-D gene transcription NCoR represses VEGF-C and VEGF-D gene transcription. (A) NCoR, VEGF-C and VEGF-D mRNA levels (means ± S.D) were measured by quantitative real-time PCR in the indicated human breast cancer cells lines and are expressed relative to the values obtained in MDA-MB-231 cells. (B) chromatin immunoprecipitation (ChIP) assays with NCoR antibody and control IgG were performed with the indicated fragments of the VEGF-C and VEGF-D gene promoters. Results obtained in two different experiments are shown. (C) levels of the indicated transcripts were determined in cells transfected with control or NCoR siRNAs 72 h before. Data (means ± SD) are expressed relative to the values obtained in cells transfected with siControl. Significance of t-test between cells transfected with siControl and siNCoR are indicated. ***P < 0.001.
TRβ silences VEGF-C and VEGF-D gene transcription
Since TRβ can increase NCoR mRNA and protein levels [31] and this corepressor silences VEGF-C and VEGF-D gene expression, we next examined the possibility that TRβ could reduce the expression of lymphangiogenic genes and the potential role of NCoR in this repression. With this purpose we first compared VEGF-C and VEGF-D transcripts in parental MDA-MB-231 cells and in cells expressing TRβ in a stable manner (from now on MDA and MDA-TRβ cells, respectively). As expected from the induction of NCoR expression by the receptor observed in several cell types [31], MDA-TRβ cells expressed higher NCoR protein and mRNA levels than the parental cells (Figure 2A and 2B), and also showed significantly lower levels of VEGF-C and VEGF-D transcripts. Silencing was observed in the absence of ligand, but incubation with T3 further reduced mRNA levels of lymphangiogenic genes (Figure 2B), showing the role of NCoR and TRβ as inhibitors of VEGFs gene expression in these cells. To analyze the role of NCoR in the repressive effect of TRβ, we next examined VEGF-C and VEGF-D mRNA levels in MDA and MDA-TRβ cells transfected with siControl or siNCoR (Figure 2C). NCoR depletion increased VEGF-C and VEGF-D transcripts both in parental and MDA-TRβ cells, strongly relieving the repressive effect of the unliganded TRβ and abolishing the inhibitory effect of T3 (Figure 2D). Therefore, NCoR appears to play a major role in lymphangiogenic gene silencing by TRβ. Since NCoR2 (or SMRT) could have redundant effects with NCoR in transcriptional repression, we conducted similar experiments in cells transfected with a specific SMRT siRNA. In contrast with NCoR, selective SMRT depletion (Figure 2E) did not increase VEGF-C and VEGF-D transcripts in MDA cells and was unable to relieve the inhibitory effects of TRβ in MDA-TRβ cells (Figure 2F), showing that SMRT does not participate in regulation of the lymphangiogenic genes in these cells. To further investigate the mechanism by which NCoR and TRβ regulate transcription of prometastatic genes, we conducted transient transfection studies with luciferase constructs containing the 5′-flanking region of the VEFG-C gene [39] in MDA and MDA-TRβ cells. As shown in Figure 3A, activity of the -1059/+206 promoter region was lower in the TRβ expressing cells and was further reduced in the presence of T3. Similar results were obtained with a shorter construct (−201/+206). However, no changes were observed when cells were transfected with the luciferase plasmid alone, indicating that the proximal promoter region that binds NCoR in ChIP assays also contains the response elements responsible for repression of VEGF-C gene transcription by TRβ. NCoR knock-down in cells transfected with the -201/+206 plasmid increased promoter activity in the parental cells, and almost totally reversed the inhibition by TRβ and T3 (Figure 3B), recapitulating the results obtained with the endogenous transcripts in Figure 2 and demonstrating again the important role of NCoR in VEGF-C gene silencing. In silico analysis of the proximal VEGF-C promoter sequences −231/+13 used in the ChIP assays revealed the existence of two putative hemisites that could bind the thyroid hormone receptor. ChIP assays with this region demonstrated that TRβ expression increased remarkably the association of NCoR with the VEGF-C gene and that this association was enhanced in T3-treated cells. TRβ also bound constitutively to the same region in MDA-TRβ cells and T3 further increased receptor recruitment to the promoter (Figure 3C). These results correlated with the lower transcriptional activity of the gene under these conditions. The more upstream –550/–232 sequences recruited neither NCoR nor TRβ, discarding their participation in repression by TRβ and confirming that the more proximal promoter sequences identified in the transient transfection assays appear to be sufficient to mediate gene repression by TRβ. Examination of he VEGF-D promoter region −423/−119, which binds NCoR, also predicted the existence of two hemisites that could act as binding motifs for TR (Figure 3D). Although NCoR bound strongly to this promoter fragment in MDA-TRβ cells and T3 increased this binding, the receptor was absent from this region. This result indicates that the hemisites do not act as TR binding sequences and that therefore NCoR is not recruited to this region by interaction with TRβ. In addition, the −608/−430 region of the VEGF-D gene has been described to contain and atypical hormone response element which could bind orphan nuclear receptors [40]. However, TRβ or NCoR association with this region was not observed in either MDA or MDA-TRβ cells, indicating that this region does not play a role in VEGF-D silencing by the corepressor.
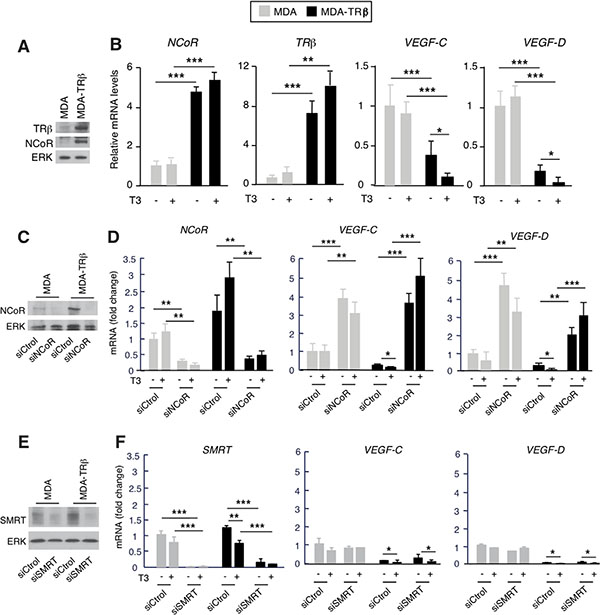
Figure 2: NCoR depletion increases VEGF-C and VEGF-D gene expression. (A) Western blot analysis of TRβ and NCoR in parental MDA-MB-231 cells and in cells stably expressing the receptor (MDA and MDA-TRβ, respectively). ERK was used as a loading control. (B) mRNA levels of the indicated genes were determined in cells treated in the presence and absence of 5 nM T3 for 36 h. (C) NCoR and ERK levels after 72 h of transfection with siControl or siNCoR. (D) Transcript levels of NCoR, VEGF-C and VEGF-D in cells transfected with siControl or siNCoR and treated with and without T3. (E) SMRT and ERK levels after 72 h of transfection with siControl or siSMRT. (F) Transcript levels of SMRT, VEGF-C and VEGF-D in cells transfected with siControl or siSMRT and treated with and without T3. All data are means ± S.D and are expressed relative to the values obtained in untreated parental cells transfected with the control siRNA. Significance of ANOVA post-test among the indicated groups is shown as * P < 0.05, **P < 0.01 and ***P < 0.001.
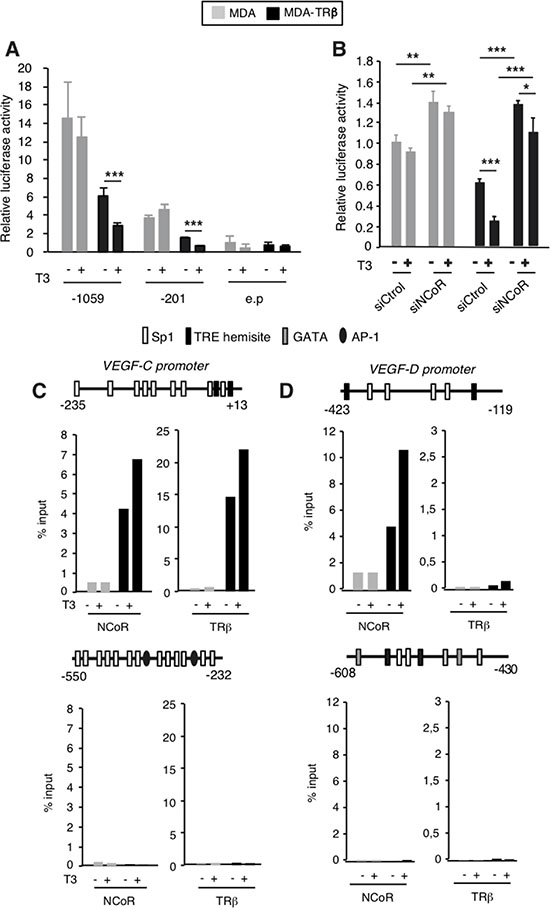
Figure 3: NCoR is involved in transcriptional inhibition of the VEGF-C and VEGF-D genes by TRβ. (A) Transient transfection assays in MDA and MDA-TRβ cells with reporter plasmids of the human VEGF-C promoter extending to nucleotides −1059 and −201 or the empty plasmid without promoter sequences (e.p). Luciferase activity (means ± S.D) was determined in cells treated for 36 h in the presence and absence of 5 nM T3 and is expressed relative to value obtained in the untreated cells transfected with the empty plasmid. Differences between untreated and T3-treated cells were calculated with the t-test and are indicated as ***P < 0.001. (B) similar experiments in cells cotransfected with the −201 plasmid and control or NCoR siRNAs. Luciferase activity (means ± S.D) was measured in cells treated with and without T3 and is expressed relative to that obtained in untreated MDA cells transfected with siControl. Statistically significant differences of the ANOVA post-test among groups of MDA and MDA-TRβ cells are indicated as *P < 0.05, **P < 0.01 and ***P < 0.001. (C) ChIP assays with the indicated fragments of the VEGF-C and VEGF-D promoters and NCoR and TRβ antibodies in cells treated in the presence and absence of T3 for 1 h. Schemes of the promoter fragments used showing the existence of putative binding sites for TR (TRE hemisites) and for other transcription factors are depicted. Results are expressed as % of the input after subtracting the values obtained after immunoprecipitation with control IgG that were always lower than 1% of the input. Data shown are the mean of two independent experiments.
NCoR and TRβ repress VEGF-C and VEGF-D expression in ER+ MCF-7 cells
To analyze if the silencing effect of TRβ in lymphangiogenic gene expression was restricted to the MDA cells or could be extended to other breast cancer cells, we next used MCF-7 cells stably expressing high levels of TRβ (MCF7-TRβ cells) [37] (Supplementary Figure S1A). As shown in Supplementary Figure S1B, TRβ expression also increased NCoR expression in MCF7 cells, while significantly reducing VEGF-C and VEGF-D mRNA levels. NCoR deletion also enhanced considerably VEGF-C and VEGF-D transcripts in MCF-7 cells and reversed the repressive effect of TRβ to a significant extent (Supplementary Figure S1C), while SMRT deletion did not affect expression of the lymphangiogenic genes (Supplementary Figure S1D). These results reproduce those obtained in MDA cells, indicating that NCoR, but not SMRT, silences VEGF-C and VEGF-D gene expression in breast cancer cells independently of the ER status, and that NCoR is an important element in TRβ-dependent repression of these genes.
NCoR and TRβ inhibit tumor lymphangiogenesis
To examine the effect of TRβ and NCoR in tumor lymphangiogenesis in vivo, we analyzed VEGF-C and VEGF-D gene expression as well as lymph vessel density in xenograft studies with MDA and MDA-TRβ cells transfected with siControl or siNCoR 72h before orthotopic inoculation into the fat mammary pad of nude mice. TRβ-expressing tumors were smaller and non invasive, but they became highly infiltrative in the absence of NCoR (Supplementary Figure S2). As expected from the existence of an autoregulatory loop that maintains NCoR gene expression [31], NCoR transcripts were still depleted in tumor xenografts formed by cells transfected with siNCoR more than one month before, while TRβ transcripts were not altered (Figure 4A). When VEGF-C and VEGF-D transcripts were measured, it was found that both genes were expressed al lower levels in tumors originated by MDA-TRβ cells than by the parental cells transfected with a control siRNA. However, NCoR depletion markedly enhanced lympangiogenic gene expression in both groups and under these conditions the repressive effect of TRβ on VEGF-C and VEGF-D expression was relieved, corroborating the results obtained in the cultured cells (Figure 4A). Transcript levels of mouse LYVE-1 (or lymphatic vessel endothelial hyaluronan receptor), a specific marker of lymphatic vessels [41], was also reduced in the tumors formed by MDA-TRβ cells with respect to the MDA cells and NCoR depletion increased LYVE-1 mRNA levels in parallel with the increased expression of the lymphangiogenic genes by the tumor cells (Figure 4B). Immunochemical detection of LYVE-1 showed that lymph vessels had a predominant peritumoral localization in the breast tumors formed by MDA cells and that they were very scarce in the tumors formed by the MDA-TRβ cells. However, vessel density increased significantly in NCoR-depleted tumors and the inhibitory effect of TRβ in tumor lymphangiogenesis was noticeably alleviated (Figure 4C).
Since not only the tumor cells but also the tumor microenvironment releases lymphangiogenic factors, we next examined mouse VEGF-C and VEGF-D expression in the xenografts (Figure 4D). Mouse NCoR levels were not altered by the human siRNA, showing the specificity of NCoR depletion exclusively in the tumor cells. However, host VEGF-C and VEGF-D expression was reduced in MDA-TRβ xenografts and increased when NCoR was depleted in the tumor cells. This indicates a reciprocal interplay between the tumor microenvironment and the tumor cells to collaboratively regulate tumor lymphangiogenesis.
Since TRβ expression reduces tumor size while tumors are bigger upon NCoR depletion (Supplementary Figure S2A), there was the possibility that the observed changes in lymphangiogenesis could be secondary to the different tumor size. Therefore, we next compared tumors formed by MDA and MDA-TRβ cells transfected with siControl or siNCoR when they reached the same volume (1 cm3). The results obtained confirmed again that NCoR depletion persisted for a very long time, since NCoR mRNA was significantly reduced both in MDA and MDA-TRβ cells originally transfected with siNCoR, whereas VEGF-C and VEGF-D transcripts were strongly induced, thus confirming the results obtained in the different size xenografts excised after one month (Supplementary Figure S3A). In addition, TRβ mRNA was not altered in the absence of NCoR, suggesting again that NCoR is the main mediator of the inhibitory effects of TRβ in lymphangiogenic gene repression. Moreover, LYVE-1 gene expression followed a similar pattern (Supplementary Figure S3B), and mouse VEGF-C and VEGF-D mRNAs were induced by NCoR depletion (Supplementary Figure S3C), suggesting again that the absence of this corepressor induces changes in the tumor cells that affect the tumor microenvironment.
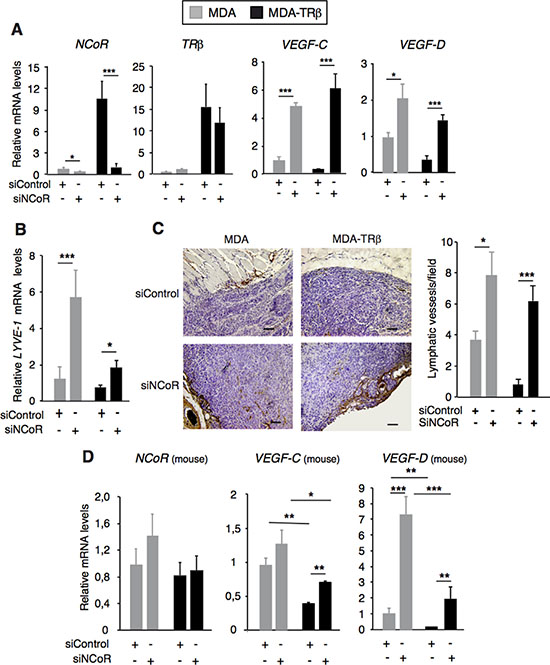
Figure 4: NCoR and TRβ inhibit tumor lymphangiogenesis. (A) NCoR, TRβ, VEGF-C and VEGF-D mRNA levels in xenografts of MDA and MDA-TRβ cells. Cells were transfected with siControl or siNCoR 72 h before orthotopical inoculation into nude mice and animals were sacrificed 4 weeks later. (B) relative mouse LYVE-1 mRNA levels in the different groups of tumors. (C) representative immunohistochemical staining of LYVE-1 in the tumors, showing the presence of peritumoral lymphatic vessels. Bars: 100 μM. The right panel shows the quantification of the number of lymphatic vessels/microscopic field (means ± S.E) in the tumors. (D) mouse NCoR, VEGF-C and VEGF-D transcripts (means ± S.E) in the xenografts. mRNA levels (means ± S.E) are expressed relative to the values obtained in MDA tumors transfected with siControl. Statistically significant differences between tumors generated from breast cancer cells transfected with siControl and siNCoR, analyzed by ANOVA and Bonferroni post-test, are indicated as * P < 0.05, **P < 0.01 and ***P < 0.001.
NCoR depletion increases the presence of tumor DNA in the sentinel node
As expression levels of the lymphangiogenic factors correlate with lymph node metastasis, the sentinel lymph nodes of the mice were dissected and the presence of breast tumor DNA was studied by means of quantification of human Alu sequences. As shown in Figure 5A, in parallel with the changes in VEGF-C and VEGF-D expression, the presence of tumor DNA in the sentinel node was decreased in animals inoculated with TR ®-expressing cells, while NCoR depletion resulted in a significant increase in the amount of tumor DNA reaching the node and in a partial reversion of the inhibitory effect of TR ®.
The lymphatic endothelial cells produce chemokines that can attract the tumor cells expressing their receptors, supporting their migration toward the lymphatic vessels. Interestingly, it has been previously shown that CXCR4 is an NCoR and TRβ target gene [31, 38]. Therefore, we next analyzed expression of this receptor and its ligand in the tumors. Confirming previous results, CXCR4 expression was reduced in the TRβ-expressing tumors and NCoR depletion resulted in a significant increase in its expression (Figure 5B). Remarkably, mouse CXCL12 gene expression followed a similar pattern (Figure 5B), suggesting again the importance of the cross-talk between the breast cancer cells and the cells in the microenvironment in the regulation of tumor spreading.
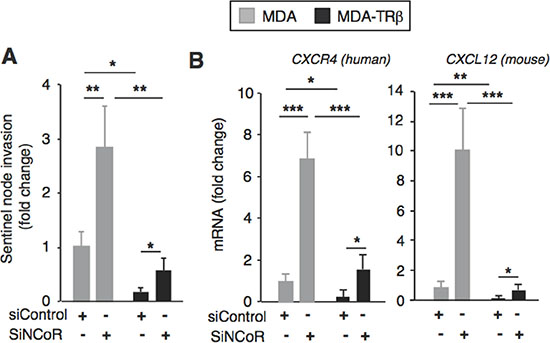
Figure 5: NCoR depletion increases chemokine expression and sentinel node invasion. (A) sentinel lymph nodes were excised from the mice shown in Figure 4 that were inoculated 4 weeks before with MDA and MDA-TRβ cells previously transfected with siControl or siNCoR. The presence of the breast cancer DNA in the mice lymph node was assessed by determination of human Alu sequences. (B) transcript levels of the human CXCR4 chemokine receptor and of the mouse CXCL12 chemokine, its ligand, in the tumors. Data (means ± S.E) are expressed relative to the values obtained in mice injected with the parental cells transfected with siControl. Statistically significant differences were analyzed by ANOVA followed by Bonferroni test and are shown as * P < 0.05, **P < 0.01 and ***P < 0.001.
Correlation of NCoR and TRβ with lymphangiogenic gene expression in human breast tumors
To examine whether or not the repressive role of NCoR in lymphangiogenesis could be also demonstrated in human tumors, we next performed immunohistochemical staining of NCoR and lymphatic vessels in samples from 6 ER+ and 5 ER- breast tumors. Figure 6A shows that NCoR staining was stronger in ER+ tumors than in ER- tumors in which the majority of the cells did not show nuclear staining with the NCoR antibody. These results confirm previous data with other tumor series in which transcript levels of NCoR were also reduced in ER- tumors [31]. In contrast, staining with the lymphatic vessel marker Podoplanin showed an opposite pattern with an increased number of peritumoral lymphatic vessels in the ER-tumors, suggesting that NCoR might also inhibit lymphangiogenesis in human breast tumors.
It has been shown that not only NCoR but also TRβ transcripts were markedly reduced in RNA samples from ER- tumors when compared with ER+ tumors [31]. To further explore the potential role of these molecules in tumor lymphangiogenesis, we next quantitated LYVE-1, VEGF-C and VEGF-D transcripts in the same tumor series finding that, as expected, expression of these genes was higher in the more aggressive ER- breast tumors (Figure 6B). To examine the possible existence of a negative correlation between NCoR or TRβ and lymphangiogenic gene expression, NCoR (Figure 6C) and TRβ (Figure 6D) mRNA levels were plotted against LYVE-1, VEGF-C and VEGF-D mRNAs. Statistical analysis showed that, indeed, there was a statistically significant negative correlation in all cases. The inverse relationship between transcript levels of NCoR and these genes was also found when ER+ and ER- tumors were considered separately (Supplementary Figure S4), and this also occurred when LYVE-1, VEGF-C and VEGF-D mRNAs were plotted against TRβ mRNA levels (Supplementary Figure S5). These results further indicate that NCoR is also a potent inhibitor of lympangiogenesis in human breast tumors and a downstream effector of TRβ in this process.
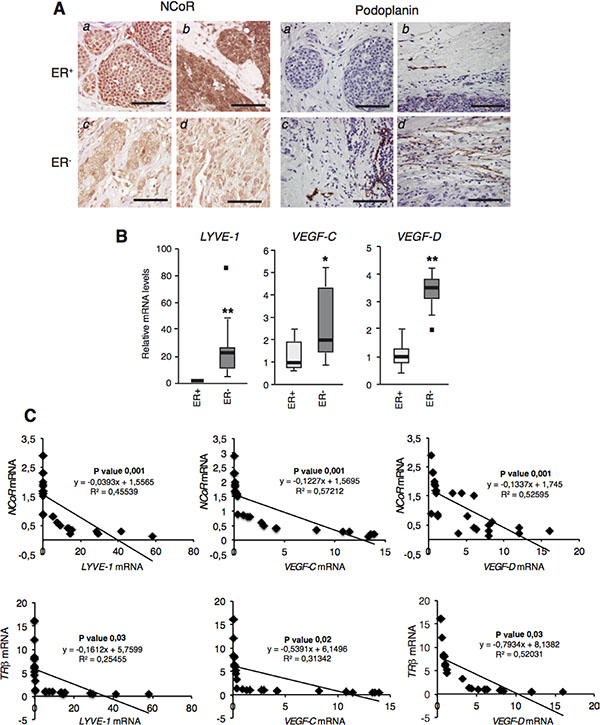
Figure 6: NCoR and TRβ levels correlate negatively with lymphangiogenic gene expresion in human breast tumors. (A) NCoR (left panels) and Podoplanin (right panels) immunohistochemistry of representative ER+ and ER- tumors. a: ER+ lobular tumor, b ER+ ductal tumor; c and d : ER- ductal tumors. Bars: 100 μM. (B) Whisker plot of LYVE-1, VEGF-C and VEGF-D mRNA levels in ER+ and ER- tumors. Data are mean ± S.E of 12 and 14 tumors, respectively. Outliers are shown by a black square. Significance of t-test between ER positive and negative tumors are indicated as * P < 0.05, **P < 0.01. (C) NCoR mRNA levels measured in Ref. were plotted against the corresponding LYVE-1, VEGF-C and VEGF-D mRNAs obtained in each sample. The p value and linear regression coefficient obtained are shown. (D) results with individual TRβ mRNAs obtained in Ref. 31, plotted against mRNAs of the lymphangiogenic genes.
DISCUSSION
Studies in lymphangiogenesis have shown the key role of two members of the VEGF family, VEGF-C and VEGF-D, which interact with the VEGFR-3 receptor not only in the development of the lymphatic system but also in promoting tumor lymphangiogenesis and lymphatic metastasis [2, 11]. Therefore, identification of the mechanisms that regulate expression of these genes may be important to understand the molecular basis of lymphatic vessel growth and for the potential development of novel therapeutic strategies for combating metastasis. Breast tumors are particularly interesting at this respect, since spreading through the lymphatic system is predominant in these tumors. In the present study, we show that the corepressor NCoR and the nuclear receptor TRβ can inhibit transcription of the VEGF-C and VEGF-D genes, acting as potent repressors of tumor lymphangiogenesis in breast cancer xenograft models and correlating negatively with the expression of lymphangiogenic genes in human breast tumors.
The following findings clearly show that VEGF-C and VEGF-D are bona fide target genes for NCoR repression: i, breast cancer cells expressing higher NCoR mRNA levels express lower levels of the lymphangiogenic genes; ii, NCoR associates with the regulatory region of these genes in ChIP assays and this association is stronger in cells presenting higher levels of the corepressor; iii, NCoR depletion with siRNA increases promoter activity of the VEGF-C gene in transient transfection studies and iv, transfection with NCoR siRNA increases endogenous transcript levels of VEGF-C and VEGF-D. In addition to NCoR, TRβ also represses expression of lymphangiogenic genes, as indicated by the finding that VEGF-C and VEGF-Dtranscripts are significantly lower in breast cancer cell lines stably expressing TRβ than in their corresponding parental cells that express very low receptor levels. The silencing effect of TRβ appears to be mediated, at least in part, by the elevated NCoR levels present in TRβ expressing cells. This is proved by the result that lymphangiogenic transcripts are significantly restored upon NCoR depletion. However, at least in the case of VEGF-C, TRβ is recruited to the same promoter region responsible for NCoR binding and could therefore directly down-regulate transcription. In the case of VEGF-D NCoR, but not TRβ, associates with proximal promoter sequences. Interestingly, in breast cancer cells neither TRβ nor NCoR bind to a region previously reported to mediate regulation by some orphan receptors and to be important for basal transcription [40].
The role of NCoR and TRβ as potent regulators of the VEGF-C and VEGF-D genes was confirmed in vivo using xenografts. Thus, TRβ expression resulted in reduced transcript levels of these genes in the tumors, while NCoR depletion had an opposite effect enhancing them significantly. Again, the suppressive effect of TRβ was significantly reversed in the absence of NCoR, reinforcing the idea that NCoR plays a critical role in lymphangiogenic gene silencing by the receptor. Moreover, although tumor size is an important parameter to predict lymph node involvement in breast cancer [42], this occurred independently of tumor size, showing that direct regulation of lymphangiogenic gene expression is not secondary to the differences in tumor growth caused by TRβ or NCoR. These results suggested to us that these molecules might also suppress tumor lymphangiogenesis. Accordingly, histologic analysis showed that NCoR depletion was sufficient for promoting tumor lymphangiogenesis, resulting in increased LYVE-1 gene expression and in increased density of LYVE-1–positive lymphatic vessels. Contrarily, TRβ-expressing tumors showed significantly reduced LYVE-1 gene expression and a very low number of lymphatic vessels, which reappeared when NCoR was knocked-down. These lymphatic vessels were mainly in the peritumoral area. While intratumoral vessels are normally considered as nonfunctional, the lymphatic vessels at the periphery of the tumor likely serve as conduits for the metastatic cells to reach the draining lymph nodes. This has led to the concept that a dense lymphatic vasculature in this area would increase the number of entry sites of the tumor cells to the vessels and consequently metastatic spreading [43]. Accordingly, we found an increased amount of tumor DNA reaching the sentinel node upon NCoR depletion in the tumors and a much lower amount, also partially reversed in the absence of the corepressor, when the tumor cells expressed TRβ. These changes are compatible with parallel alterations in the number of metastatic cells colonizing the draining nodes, but the possibility that metastatic lesions were still not present and that DNA travelled to the lymph nodes in exosomes or by other means cannot be dismissed. Once the metastatic cells reach the lymph node they may enter a latent stage or further disseminate to other lymph nodes, the blood vessels and more distant organs.
The tumor microenvironment, composed by endothelial cells, immune cells, fibroblasts, signaling molecules and the extracellular matrix provides signals to the tumor cells in the form of both cell-cell contacts and secreted factors. It is increasingly evident that crosstalk between cancer cells and cells of the neoplastic microenvironment is a crucial component of invasion and metastatic growth [44]. Particularly interesting are the interactions between lymphatic endothelial cells and tumor cells to promote cancer cells dissemination. Not only the tumor cells secrete VEGFs, but also the tumor environment and specifically tumor-associated macrophages can function as a second source of lymphangiogenic factors [45]. Of interest, we found that expression of mouse VEGF-C and VEGF-D transcripts is increased in the tumors generated by NCoR-deficient breast cancer cells. As mouse NCoR gene expression is not altered, this suggests that breast tumor cells with different NCoR levels send distinct, still unidentified, signals to the macrophages that are able to alter lymphangiogenic gene expression. On the other hand, tumor cells may activate lymphatic cells to secrete factors that help their transport into the lymphatic vessels. Among them, lymphatic endothelial cells secrete chemokines such as CXCL12 that can promote tumor cells expressing the cognate receptor CXCR4 to migrate toward the lymphatic vessels, promoting a lymphatic microenvironment that supports tumor growth [19]. We have confirmed that CXCR4, a marker and mediator of breast-cancer metastasis [18, 46, 47], is a target for repression by NCoR and TRβ [31, 38]. Importantly, our results indicate that NCoR and TRβ not only silence the expression of CXCR4 receptors in the breast tumor cells, but also reduce the production of its ligand, CXCL12, by the tumor environment. The CXCL12-CXCR4 chemokine pathway has been shown to enhance tumor lymphangiogenesis and to have additive effects with the VEGF-C pathway. Furthermore, targeting both the chemokine and VEGF-C results in a stronger inhibition of tumor lymphangiogenesis and lymph node metastasis in the breast cancer xenograft model used in our study [48]. Therefore, the increased expression of these genes in the absence of NCoR would create a highly favorable scenario for lymphatic dissemination of breast cancer cells.
Besides silencing of VEGFs and chemokine receptors, we cannot exclude the possibility that NCoR and the receptor might also have additional anti-lymphangiogenic effects via modulation of other genes. Thus, in addition to VEGFs, a growing number of additional factors including IGFs [49], HGF [50] and COX2 [51, 52] have been described to promote lymphangiogenesis. Interestingly, the HGF receptor c-Met or COX2 genes are TRβ and NCoR targets and the receptor induces the expression of IGF-BP3, an inhibitor of IGFs signaling [31, 38]. Therefore, it is likely that regulation of these factors might contribute to the anti-lymphangiogenic effects of TRβ and NCoR.
Lymphatic vessel density correlates with metastasis and poor outcome in most clinical studies in breast cancer and other tumors [3, 7, 8, 12, 14]. Accordingly, we found a higher number of lymphatic vessels in the more aggressive tumors. Furthermore, not only LYVE-1 mRNA but also VEGF-C and VEGF-D mRNAs were lower in ER+ tumors. It had been previously shown that NCoR gene expression was reduced in the ER- tumors [31] and, remarkably, there was a significant negative correlation, independent of the ER status, between NCoR and lymphangiogenic gene expression further validating that the corepressor is an important suppressor of lymphangiogenic genes transcription. A significant negative correlation between VEGF-C and VEGF-D and TRβ transcript levels was also found. This finding reinforces the hypothesis that TRβ is an upstream regulator of NCoR and suggests that the inhibitory effects of TRβ in human breast tumor lymphangiogenesis are most likely mediated by NCoR induction. These results suggest that NCoR and TRβ might be useful as novel biomarkers in breast cancer and as potential predictors of lymphatic dissemination. The development of therapeutic agents targeting lymphangiogenesis is being considered for the control of tumor growth and lymphatic metastasis. Thus, both the receptor and the corepressor might be novel therapeutic targets in both ER-positive and ER-negative tumors.
MATERIALS AND METHODS
Extended materials and methods are provided in Supplementary Materials and Methods. Animal and human studies were approved by the Ethics Committee of the Consejo Superior de Investigaciones Científicas. NCoR and SMRT were knocked-down in cells with specific siRNA SMART pools from Dharmacon. Experimental procedures for transfections, luciferase reporter assays, western blot, mRNA determination by real time PCR and chromatin immunoprecipitation assays have been published previously and are described together with the antibodies and primers used in Supplementary Materials and Methods. Tumor formation in nude mice was followed for 4 weeks after orthotopic Supplementary Materials and Methods inoculation into the mammary pad. Histology and immunohistochemistry was performed by standard procedures. Significance of ANOVA post-test or the Student t-test among the experimental groups indicated in the figures is shown as *P < 0.05, **P < 0.01 and ***P <0.001.
ACKNOWLEDGMENTS
We thank SY Cheng and H Peinado for cells, D Alitalo for plasmids, the IIB services for support and C Sanchez-Palomo for technical help. We want to particularly acknowledge the patients enrolled in this study for their participation and the Hospital Universitario Ramón y Cajal-IRYCIS Biobank integrated in the Spanish Hospital Platform Biobanks Network (RetBioH; www.redbiobancos.es) and HUVR-IBiS Biobank (Andalusian Public Health System Biobank and ISCIII-Red de Biobancos PT13/0010/0056) for their collaboration.
CONFLICTS OF INTEREST
The authors have declared that no conflicts of interest exists
FUNDING
This work was supported by Grants: BFU2014-53610-P and SAF2015-71878-REDT from Ministerio de Economía y Competitividad; S2011/BMD-2328, from the Comunidad de Madrid and RD12/0036/0030 from the Instituto de Salud Carlos III (to A.A), by a Grant from the European Thyroid Association (to O.M-I), by grants RD12 0036/0064 from Instituto de Salud Carlos III (to J.P) and SAF2014-56868-R (to M.S.S). M.S.S is also funded by the Asociación Española Contra el Cancer (AECC) and by the Melanoma Research Alliance (MRA). This work is partially supported by FEDER funds. O.M.I is supported by an AECC contract.
Authors’ contributions
O.MI, E.AM, AG, DO, EL and S.GR performed experiments and analyzed data. J.P provided samples and clinical data, JR performed and interpreted histology, MS.S analyzed data and AA conceived the study, supervised experiments and wrote the manuscript.
REFERENCES
1. Karaman S, Detmar M. Mechanisms of lymphatic metastasis. J Clin Invest. 2014; 124:922–928.
2. Stacker SA, Williams SP, Karnezis T, Shayan R, Fox SB, Achen MG. Lymphangiogenesis and lymphatic vessel remodelling in cancer. Nat Rev Cancer. 2014; 14:159–172.
3. He Y, Rajantie I, Pajusola K, Jeltsch M, Holopainen T, Yla-Herttuala S, Harding T, Jooss K, Takahashi T, Alitalo K. Vascular endothelial cell growth factor receptor 3-mediated activation of lymphatic endothelium is crucial for tumor cell entry and spread via lymphatic vessels. Cancer research. 2005; 65:4739–4746.
4. Mandriota SJ, Jussila L, Jeltsch M, Compagni A, Baetens D, Prevo R, Banerji S, Huarte J, Montesano R, Jackson DG, Orci L, Alitalo K, Christofori G, et al. Vascular endothelial growth factor-C-mediated lymphangiogenesis promotes tumour metastasis. The EMBO journal. 2001; 20:672–682.
5. Skobe M, Hawighorst T, Jackson DG, Prevo R, Janes L, Velasco P, Riccardi L, Alitalo K, Claffey K, Detmar M. Induction of tumor lymphangiogenesis by VEGF-C promotes breast cancer metastasis. Nature medicine. 2001; 7:192–198.
6. Stacker SA, Caesar C, Baldwin ME, Thornton GE, Williams RA, Prevo R, Jackson DG, Nishikawa S, Kubo H, Achen MG. VEGF-D promotes the metastatic spread of tumor cells via the lymphatics. Nature medicine. 2001; 7:186–191.
7. Gu Y, Qi X, Guo S. Lymphangiogenesis induced by VEGF-C and VEGF-D promotes metastasis and a poor outcome in breast carcinoma: a retrospective study of 61 cases. Clin Exp Metastasis. 2008; 25:717–725.
8. Hirakawa S, Brown LF, Kodama S, Paavonen K, Alitalo K, Detmar M. VEGF-C-induced lymphangiogenesis in sentinel lymph nodes promotes tumor metastasis to distant sites. Blood. 2007; 109:1010–1017.
9. Karpanen T, Egeblad M, Karkkainen MJ, Kubo H, Yla-Herttuala S, Jaattela M, Alitalo K. Vascular endothelial growth factor C promotes tumor lymphangiogenesis and intralymphatic tumor growth. Cancer research. 2001; 61:1786–1790.
10. Liersch R, Hirakawa S, Berdel WE, Mesters RM, Detmar M. Induced lymphatic sinus hyperplasia in sentinel lymph nodes by VEGF-C as the earliest premetastatic indicator. Int J Oncol. 2012; 41:2073–2078.
11. Zheng W, Aspelund A, Alitalo K. Lymphangiogenic factors, mechanisms, and applications. J Clin Invest. 2014; 124:878–887.
12. Bono P, Wasenius VM, Heikkila P, Lundin J, Jackson DG, Joensuu H. High LYVE-1-positive lymphatic vessel numbers are associated with poor outcome in breast cancer. Clin Cancer Res. 2004; 10:7144–7149.
13. El-Gohary YM, Metwally G, Saad RS, Robinson MJ, Mesko T, Poppiti RJ. Prognostic significance of intratumoral and peritumoral lymphatic density and blood vessel density in invasive breast carcinomas. Am J Clin Pathol. 2008; 129:578–586.
14. Nakamura Y, Yasuoka H, Tsujimoto M, Imabun S, Nakahara M, Nakao K, Nakamura M, Mori I, Kakudo K. Lymph vessel density correlates with nodal status, VEGF-C expression, and prognosis in breast cancer. Breast Cancer Res Treat. 2005; 91:125–132.
15. Van den Eynden GG, Van der Auwera I, Van Laere SJ, Trinh XB, Colpaert CG, van Dam P, Dirix LY, Vermeulen PB, Van Marck EA. Comparison of molecular determinants of angiogenesis and lymphangiogenesis in lymph node metastases and in primary tumours of patients with breast cancer. J Pathol. 2007; 213:56–64.
16. Schoppmann SF, Birner P, Stockl J, Kalt R, Ullrich R, Caucig C, Kriehuber E, Nagy K, Alitalo K, Kerjaschki D. Tumor-associated macrophages express lymphatic endothelial growth factors and are related to peritumoral lymphangiogenesis. Am J Pathol. 2002; 161:947–956.
17. Cabioglu N, Yazici MS, Arun B, Broglio KR, Hortobagyi GN, Price JE, Sahin A. CCR7 and CXCR4 as novel biomarkers predicting axillary lymph node metastasis in T1 breast cancer. Clin Cancer Res. 2005; 11:5686–5693.
18. Muller A, Homey B, Soto H, Ge N, Catron D, Buchanan ME, McClanahan T, Murphy E, Yuan W, Wagner SN, Barrera JL, Mohar A, Verastegui E, et al. Involvement of chemokine receptors in breast cancer metastasis. Nature. 2001; 410:50–56.
19. Kim M, Koh YJ, Kim KE, Koh BI, Nam DH, Alitalo K, Kim I, Koh GY. CXCR4 signaling regulates metastasis of chemoresistant melanoma cells by a lymphatic metastatic niche. Cancer research. 2010; 70:10411–10421.
20. Huang EY, Zhang J, Miska EA, Guenther MG, Kouzarides T, Lazar MA. Nuclear receptor corepressors partner with class II histone deacetylases in a Sin3-independent repression pathway. Genes & development. 2000; 14:45–54.
21. Perissi V, Jepsen K, Glass CK, Rosenfeld MG. Deconstructing repression: evolving models of co-repressor action. Nature reviews Genetics. 2010; 11:109–123.
22. Privalsky ML. The role of corepressors in transcriptional regulation by nuclear hormone receptors. Annual review of physiology. 2004; 66:315–360.
23. Yoon HG, Chan DW, Huang ZQ, Li J, Fondell JD, Qin J, Wong J. Purification and functional characterization of the human N-CoR complex: the roles of HDAC3, TBL1 and TBLR1. The EMBO journal. 2003; 22:1336–1346.
24. Banerji S, Cibulskis K, Rangel-Escareno C, Brown KK, Carter SL, Frederick AM, Lawrence MS, Sivachenko AY, Sougnez C, Zou L, Cortes ML, Fernandez-Lopez JC, Peng S, et al. Sequence analysis of mutations and translocations across breast cancer subtypes. Nature. 2012; 486:405–409.
25. CGAN. Comprehensive molecular portraits of human breast tumours. Nature. 2012; 490:61–70.
26. Stephens PJ, Tarpey PS, Davies H, Van Loo P, Greenman C, Wedge DC, Nik-Zainal S, Martin S, Varela I, Bignell GR, Yates LR, Papaemmanuil E, Beare D, et al. The landscape of cancer genes and mutational processes in breast cancer. Nature. 2012; 486:400–404.
27. Ciriello G, Sinha R, Hoadley KA, Jacobsen AS, Reva B, Perou CM, Sander C, Schultz N. The molecular diversity of Luminal A breast tumors. Breast Cancer Res Treat. 2013; 141:409–420.
28. Kurebayashi J, Otsuki T, Kunisue H, Tanaka K, Yamamoto S, Sonoo H. Expression levels of estrogen receptor-alpha, estrogen receptor-beta, coactivators, and corepressors in breast cancer. Clin Cancer Res. 2000; 6:512–518.
29. Green AR, Burney C, Granger CJ, Paish EC, El-Sheikh S, Rakha EA, Powe DG, Macmillan RD, Ellis IO, Stylianou E. The prognostic significance of steroid receptor co-regulators in breast cancer: co-repressor NCOR2/SMRT is an independent indicator of poor outcome. Breast Cancer Res Treat. 2008; 110:427–437.
30. Girault I, Lerebours F, Amarir S, Tozlu S, Tubiana-Hulin M, Lidereau R, Bieche I. Expression analysis of estrogen receptor alpha coregulators in breast carcinoma: evidence that NCOR1 expression is predictive of the response to tamoxifen. Clin Cancer Res. 2003; 9:1259–1266.
31. Martinez-Iglesias OA, Alonso-Merino E, Gomez-Rey S, Velasco-Martin JP, Martin Orozco R, Luengo E, Garcia Martin R, Ibanez de Caceres I, Fernandez AF, Fraga MF, Gonzalez-Peramato P, Varona C, Palacios J, et al. Autoregulatory loop of nuclear corepressor 1 expression controls invasion, tumor growth, and metastasis. Proc Natl Acad Sci USA. 2016; 113:E328–337.
32. Aranda A, Alonso-Merino E, Zambrano A. Receptors of thyroid hormones. Pediatric endocrinology reviews. 2013; 11:2–13.
33. Chandrasekar B, Mummidi S, Mahimainathan L, Patel DN, Bailey SR, Imam SZ, Greene WC, Valente AJ. Interleukin-18-induced human coronary artery smooth muscle cell migration is dependent on NF-kappaB- and AP-1-mediated matrix metalloproteinase-9 expression and is inhibited by atorvastatin. J Biol Chem. 2006; 281:15099–15109.
34. Conde I, Paniagua R, Zamora J, Blanquez MJ, Fraile B, Ruiz A, Arenas MI. Influence of thyroid hormone receptors on breast cancer cell proliferation. Ann Oncol. 2006; 17:60–64.
35. Silva JM, Dominguez G, Gonzalez-Sancho JM, Garcia JM, Silva J, Garcia-Andrade C, Navarro A, Munoz A, Bonilla F. Expression of thyroid hormone receptor/erbA genes is altered in human breast cancer. Oncogene. 2002; 21:4307–4316.
36. Aranda A, Martinez-Iglesias O, Ruiz-Llorente L, Garcia-Carpizo V, Zambrano A. Thyroid receptor: roles in cancer. Trends Endocrinol Metab. 2009; 20:318–324.
37. Park JW, Zhao L, Cheng SY. Inhibition of estrogen-dependent tumorigenesis by the thyroid hormone receptor beta in xenograft models. Am J Cancer Res. 2013; 3:302–311.
38. Martinez-Iglesias O, Garcia-Silva S, Tenbaum SP, Regadera J, Larcher F, Paramio JM, Vennstrom B, Aranda A. Thyroid hormone receptor beta1 acts as a potent suppressor of tumor invasiveness and metastasis. Cancer research. 2009; 69:501–509.
39. Chilov D, Kukk E, Taira S, Jeltsch M, Kaukonen J, Palotie A, Joukov V, Alitalo K. Genomic organization of human and mouse genes for vascular endothelial growth factor C. J Biol Chem. 1997; 272:25176–25183.
40. Schafer G, Wissmann C, Hertel J, Lunyak V, Hocker M. Regulation of vascular endothelial growth factor D by orphan receptors hepatocyte nuclear factor-4 alpha and chicken ovalbumin upstream promoter transcription factors 1 and 2. Cancer research. 2008; 68:457–466.
41. Banerji S, Ni J, Wang SX, Clasper S, Su J, Tammi R, Jones M, Jackson DG. LYVE-1, a new homologue of the CD44 glycoprotein, is a lymph-specific receptor for hyaluronan. J Cell Biol. 1999; 144:789–801.
42. Ravdin PM, De Laurentiis M, Vendely T, Clark GM. Prediction of axillary lymph node status in breast cancer patients by use of prognostic indicators. J Natl Cancer Inst. 1994; 86:1771–1775.
43. Tammela T, Alitalo K. Lymphangiogenesis: Molecular mechanisms and future promise. Cell. 2010; 140:460–476.
44. Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell. 2012; 21:309–322.
45. Moussai D, Mitsui H, Pettersen JS, Pierson KC, Shah KR, Suarez-Farinas M, Cardinale IR, Bluth MJ, Krueger JG, Carucci JA. The human cutaneous squamous cell carcinoma microenvironment is characterized by increased lymphatic density and enhanced expression of macrophage-derived VEGF-C. J Invest Dermatol. 2011; 131:229–236.
46. Minn AJ, Gupta GP, Siegel PM, Bos PD, Shu W, Giri DD, Viale A, Olshen AB, Gerald WL, Massague J. Genes that mediate breast cancer metastasis to lung. Nature. 2005; 436:518–524.
47. Zhang XH, Jin X, Malladi S, Zou Y, Wen YH, Brogi E, Smid M, Foekens JA, Massague J. Selection of bone metastasis seeds by mesenchymal signals in the primary tumor stroma. Cell. 2013; 154:1060–1073.
48. Zhuo W, Jia L, Song N, Lu XA, Ding Y, Wang X, Song X, Fu Y, Luo Y. The CXCL12-CXCR4 chemokine pathway: a novel axis regulates lymphangiogenesis. Clin Cancer Res. 2012; 18:5387–5398.
49. Bjorndahl M, Cao R, Nissen LJ, Clasper S, Johnson LA, Xue Y, Zhou Z, Jackson D, Hansen AJ, Cao Y. Insulin-like growth factors 1 and 2 induce lymphangiogenesis in vivo. Proc Natl Acad Sci USA. 2005; 102:15593–15598.
50. Kajiya K, Hirakawa S, Ma B, Drinnenberg I, Detmar M. Hepatocyte growth factor promotes lymphatic vessel formation and function. The EMBO journal. 2005; 24:2885–2895.
51. Lyons TR, Borges VF, Betts CB, Guo Q, Kapoor P, Martinson HA, Jindal S, Schedin P. Cyclooxygenase-2-dependent lymphangiogenesis promotes nodal metastasis of postpartum breast cancer. J Clin Invest. 2014; 124:3901–3912.
52. Timoshenko AV, Chakraborty C, Wagner GF, Lala PK. COX-2-mediated stimulation of the lymphangiogenic factor VEGF-C in human breast cancer. Br J Cancer. 2006; 94:1154–1163.