
INTRODUCTION
Ni is the most abundant element in the earth’s crust [1]. Due to its superior heat and electricity conductivity and high melting point [2], Ni and its alloys are widely used as catalysts and pigments across multiple global industries [3]. Widely used Ni salts exploited commercially include nickel chloride, sulphate, hydroxide, acetate, oxide and others.
Accelerated consumption of Ni-containing products induces discharge of Ni-pollutants into the environment. Excessive exposure to Ni may be harmful to human and animal health [4, 5]. Ni is one of the most commonly detected cutaneous allergens in children in the United States [6]. Epidemiological studies have associated Ni exposure with increased risk of nasal and lung cancer [2, 7], and the International Agency for Research on Cancer has classified Ni as an important human carcinogen [8]. Ni can accumulate in kidney, lung, bone, liver and heart, and exposure to Ni or Ni compounds can induce organ system-toxicity [9-12]. Gathwan, et al. [13] suggested that NiCl2 can induce hepatic DNA damage in mice. NiSO4 can induce apoptosis and oxidative stress in rat testes [14] and mouse liver [15]. The percentage of apoptotic cells is increased in porcine granulosa cells after exposure to 1,000 μmol/L NiCl2 [16]. Our previous findings also showed that dietary NiCl2 at 300+ mg/kg induces immunotoxicity, oxidative stress, apoptosis and cell cycle arrest in the kidney, spleen, small intestines, cecal tonsil and bursa of Fabricius of broiler chickens [17-25].
Apoptosis, or programmed cell death, plays a major role in homeostasis and development in multi-cellular organisms [26]. The caspase cascade promotes induction, transduction and amplification of intracellular apoptotic signaling [27, 28]. In the apoptotic pathway, caspases can be divided into initiators and executioners. The “initiator caspases” include caspase-2, 8, 9, 10 and 12, which are closely coupled to upstream pro-apoptotic signals. Initiator caspases cleave and activate “executioner” caspases, including caspase-3, 6 and 7, which ultimately induce apoptosis [29]. NiONPs increase caspase-3 activation and apoptosis rates in human bronchial epithelial cells [30]. Nickel acetate increases cyt-c and caspase-9, 3 and 6 protein levels in human proximal tubule cells [31]. Buschini, et al. [32] reported that exposure of p53-defective human leukemia cells (U937) to bis(S-citronellalthiosemicarbazonato)nickel(II) (Ni(tcitr)2) causes apoptosis via Bcl-2 downregulation, disruption of MMP and increased caspase-3 activity. Thus far, only Zhao, et al. [33] suggested that metallic nickel particles can induce Fas-mediated apoptosis in JB6 cells.
The precise mechanisms involved in Ni and Ni compound-induced apoptosis are presently still unclear. Mitochondria- and Fas-mediated caspase-dependent pathways are the main apoptosis regulatory mechanisms. Thus far, there have been no in vitro or in vivo systematic studies of mitochondria- and Fas-mediated caspase-dependent apoptosis induced by Ni and Ni compounds. The purpose of the present study was to investigate whether NiCl2 induced apoptosis in the broiler chicken kidney via caspase-dependent pathways. This study was designed to monitor apoptosis in the kidney and to elucidate possible NiCl2-induced apoptosis mechanisms, including mitochondria- and Fas-mediated caspase-dependent pathways.
RESULTS
Pathological changes in animals
Clinical observations were performed as previously described [34]. From 14 to 42 d, broiler feed intake in the three NiCl2-treated groups, except the 300 mg/kg group at 14 d, began to decline compared to controls, From 21 to 42 d, broilers in the three NiCl2-treated groups exhibited signs of depression and showed reduced appetites and growth. A few broilers showed polypnea. No unexpected deaths occurred during the experiment.
There were no macroscopic changes in the three NiCl2-treated groups during the experiment compared to controls. However, relative kidney weights were lower (P<0.05 or P<0.01) in the three NiCl2-treated groups than in the control group at 28 and 42 d (Figure 1A).
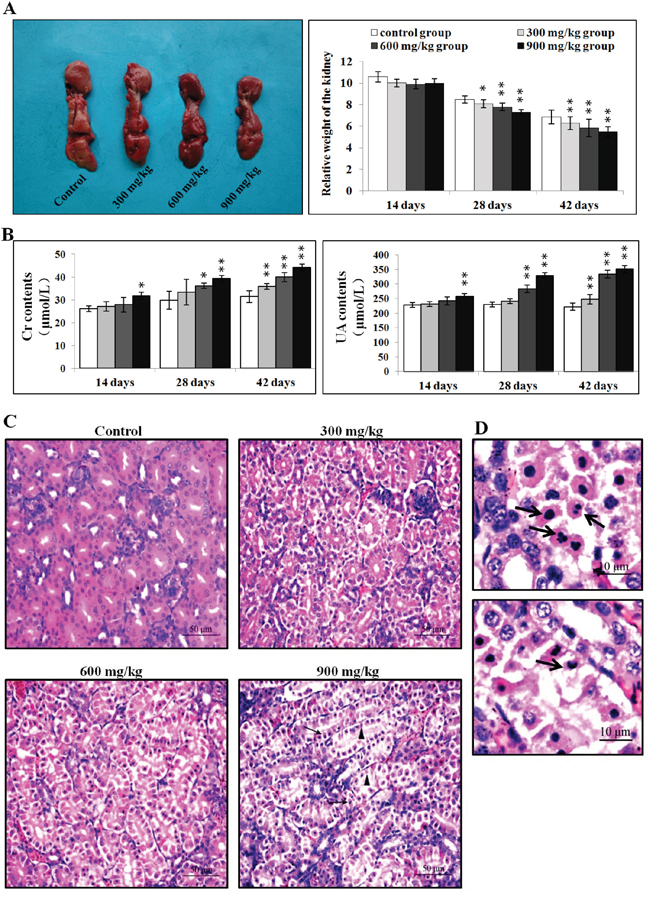
Figure 1: Pathological changes in the kidney. A. Changes in kidney size and relative weight at 42 d. B. Changes in serum creatinine and uric acid levels. C. Histopathological changes in the kidney at 42 d. Control group: no changes observed; 300 mg/kg group: tubular cells show granular and vacuolar degeneration, few necrotic tubular cells and apoptotic tubular cells are observed; 600 mg/kg group: tubular cells show marked granular and vacuolar degeneration, some necrotic tubular cells and apoptotic tubular cells are observed; 900 mg/kg group: Many necrotic tubular cells (▲) and apoptotic tubular cells (↑) are observed. (H·E ×400). D. Morphological changes in apoptotic cells. Apoptotic cell cytoplasm was intensely eosinophilic, and nuclei were shrunken, dense, ring-shaped or crescentic. Some apoptotic cell nuclei were cracked into two or multiple apoptotic bodies (↑). (H·E ×1000). Data are presented as means ± standard deviation (n=5). *P<0.05, **P<0.01 compared with the control group.
Creatinine and uric acid levels
Serum creatinine and uric acid levels were higher (P<0.05 or P<0.01) in the 900 mg/kg group at 14 d, in the 600 and 900 mg/kg groups from 28 to 42 d, and in the 300 mg/kg group at 42 d as compared to the control group (Figure 1B).
Histopathological changes
NiCl2 induced dose- and time-dependent histopathological changes in the kidney, including tubular granular degeneration, vacuolar degeneration, necrosis and apoptosis. Small particles and variably-sized vacuoles appeared in the cytoplasm of degenerated cells (Figure 1C) [20]. Necrotic cells exhibited karyorrhexis, karyolysis and hypochromatosis. The cytoplasm of apoptotic cells was eosinophilic (Figure 1D) [20]. Nuclei were shrunken, dense, ring-shaped and crescentic. Apoptotic bodies were observed.
NiCl2 increased apoptosis in the kidney
After NiCl2 treatment, renal cells were labeled using Annexin V-FITC and PI to discriminate live (Annexin V-FITC- and PI-), early apoptotic (Annexin V-FITC+ and PI-), late apoptotic (Annexin V-FITC+ and PI+) and primary/secondary necrotic cells (Annexin V-FITC− and PI+). Apoptotic cells (early apoptotic + late apoptotic cells) were more prevalent in the 600 mg/kg and 900 mg/kg groups at 14 d compared to the control group (P<0.05 or P<0.01) (Figure 2), and increased in the three NiCl2-treated groups from 28 to 42 d (P<0.05 or P<0.01).
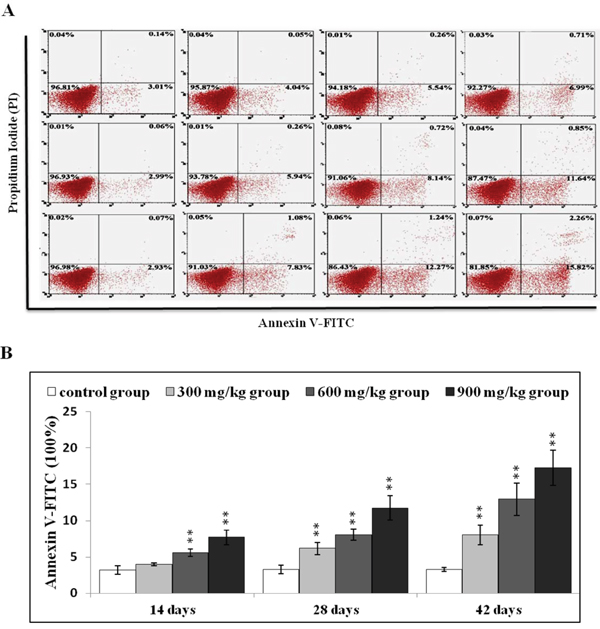
Figure 2: NiCl2 induces apoptosis in the kidney. A. Representative flow cytometry apoptosis analysis diagram. B. Changes in numbers of annexin V positive cells in the kidney Data are presented with the mean ± standard deviation (n=5). *P<0.05, **P<0.01 compared with the control group.
Mitochondria-mediated caspase-dependent apoptosis
In our previous study, NiCl2 disrupted MMP and increased AIF and Endo G release from the mitochondria to the cytosol [35]. In the present study, we assessed expression changes in cyt-c, Smac/Diablo (referred to hereafter as Smac), HtrA2/Omi (referred to hereafter as HtrA2) and their downstream proteins, including XIAP, apaf-1, PARP and caspase-3, 6, 7 and 9 via qRT-PCR analysis.
Cyt-c expression increased (P<0.05 or P<0.01) in the three NiCl2-treated groups from 14 to 42 d compared to controls (Figure 3). Smac and HtrA2 levels were higher (P<0.05 or P<0.01) in the 600 mg/kg and 900 mg/kg groups from 14 to 28 d and in the three NiCl2-treated groups at 42 d. HtrA2 expression increased (P<0.05) in the 300 mg/kg groups at 28 d. XIAP expression decreased (P<0.05) in the 600 mg/kg and 900 mg/kg groups at 14 d and in the three NiCl2-treated groups from 28 to 42 d. Apaf-1 and PARP levels were higher (P<0.05 or P<0.01) in the three NiCl2-treated groups from 14 to 42 d.
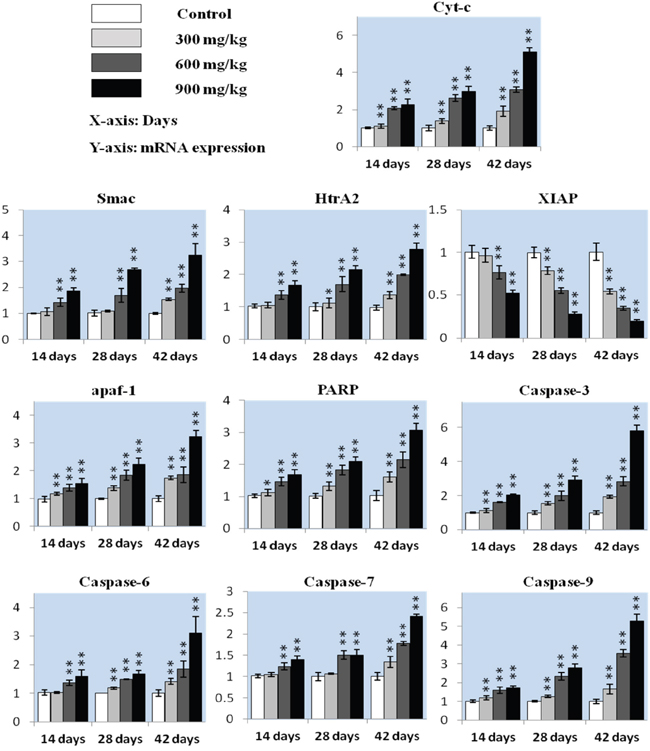
Figure 3: Cyt-c, Smac, HtrA2, XIAP, apaf-1, PARP caspase-3, caspase-6, caspase-7 and caspase-9 mRNA in the kidney. Data are presented as means ± standard deviation (n=5). *P<0.05, **P<0.01 compared with the control group.
Caspase-3 expression increased (P<0.05 or P<0.01) in the three NiCl2-treated groups from 14 to 42 d. Caspase-6 and caspase-7 levels were higher (P<0.05 or P<0.01) in the 600 mg/kg and 900 mg/kg groups from 14 to 28 d and in the three NiCl2-treated groups at 42 d compared to controls. Caspase-6 expression increased (P<0.05) in the 300 mg/kg groups at 28 d, and caspase-9 expression was higher (P<0.05 or P<0.01) in the three NiCl2-treated groups from 14 to 42 d.
Cyt-c, caspase-9, caspase-3 and PARP protein levels were assessed via immunohistochemical staining. Cyt-c protein levels increased (P<0.05 or P<0.01) in the three NiCl2-treated groups from 14 to 42 d (Figure 4). Caspase-9 protein was higher (P<0.01) in the 900 mg/kg group at 14 d, in the 600 and 900 mg/kg groups at 28 d, and in the three NiCl2-treated groups at 42 d compared to controls. Caspase-3 and PARP protein levels increased (P<0.05 or P<0.01) in the 600 and 900 mg/kg groups at 14 d and in the three NiCl2-treated groups from 28 to 42 d.
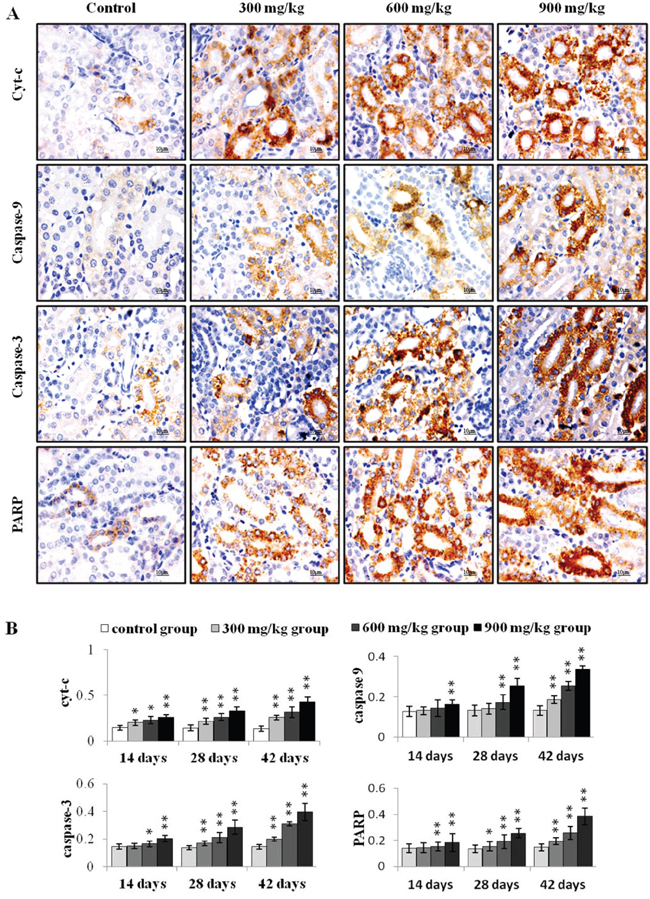
Figure 4: Immunohistochemical analysis of cyt-c, caspase-9, caspase-3 and PARP in the kidney. A. Representative immunohistochemical staining results for cyt-c, caspase-9, caspase-3 and PARP in the kidney at 42 d. B. Quantification of cyt-c, caspase-9, caspase-3 and PARP protein levels in the kidney. Data are presented as means ± standard deviation (n=5×5). *P<0.05, **P<0.01 compared with the control group.
Fas-mediated caspase-dependent apoptosis
We measured whether the Fas-mediated caspase-dependent apoptotic pathway played a role in NiCl2-induced apoptosis. Fas and FasL mRNA levels were increased (P<0.05 or P<0.01) in the 600 mg/kg and 900 mg/kg groups from 14 to 28 d and in the three NiCl2-treated groups from 14 to 42 d compared with controls (Figure 5). Caspase-8 expression was higher (P<0.05 or P<0.01) in the 900 mg/kg groups at 14 d and in the three NiCl2-treated groups from 28 to 42 d. Caspase-10 expression increased (P<0.05 or P<0.01) in the 600 and 900 mg/kg groups at 42 d. Bid expression was higher (P<0.05 or P<0.01) in the 600 mg/kg and 900 mg/kg groups from 14 to 42 d and in the 300 mg/kg groups at 42 d.
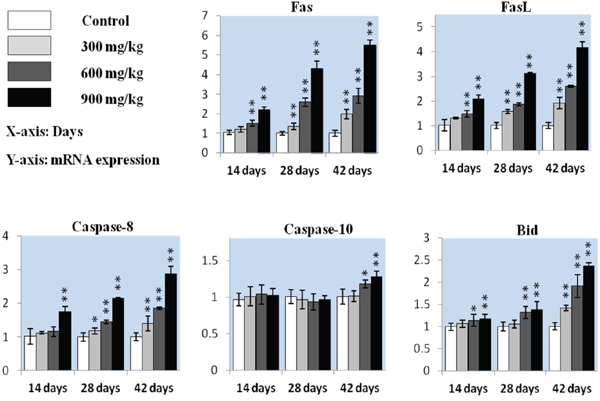
Figure 5: Fas, FasL, caspase-8, caspase-10 and Bid mRNA in the kidney. Data are presented as means ± standard deviation (n=5). *P<0.05, **P<0.01 compared with the control group.
Caspase 8 protein levels were higher (P<0.05 or P<0.01) in the 900 mg/kg group at 14 d, in the 600 and 900 mg/kg groups at 28 d, and in the three NiCl2-treated groups at 42 d compared to controls (Figure 6). Caspase 10 protein increased (P<0.05 or P<0.01) in the 600 and 900 mg/kg groups at 42 d.
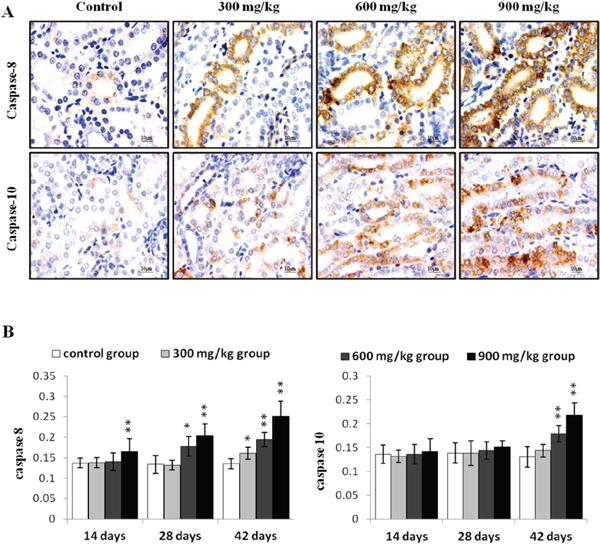
Figure 6: Immunohistochemical analysis of caspase-8 and caspase-10 in the kidney. A. Representative immunohistochemical staining results for caspase-8 and caspase-10 in the kidney at 42 d. B. Quantification of caspase-8 and caspase-10 protein levels in the kidney. Data are presented as means ± standard deviation (n=5×5). *P<0.05, **P<0.01 compared with the control group.
Ni residue in the kidney
Renal Ni accumulation was greater (P<0.05 or P<0.01) in the three NiCl2-treated groups at 42 d compared to controls [20].
DISCUSSION
In this study, we found that NiCl2 induced time- and dose-dependent apoptosis and functional injury in the kidney. Histopathological lesions, functional damage and apoptosis are consistent with Ni accumulation, indicating that Ni accumulation is a direct cause of renal injury. In our previous studies, TUNEL and histopathological results showed that NiCl2 induces apoptosis in the kidney [20, 21], thymus, spleen and cecal tonsil [17, 25, 36]. Our results are consist with those of Zheng, et al. [37] who showed that NiSO4 induces JNK-mediated oxidative stress and apoptosis in Carassius auratus liver. Ni compounds can increase apoptosis in HepG2 cells [38], normal rat kidney cells [39], and human neutrophils and lymphocytes [40, 41]. Based on these and other findings, a number of groups have focused on the potential intrinsic and extrinsic apoptotic signaling pathways induced by Ni and Ni compounds.
In the intrinsic apoptosis pathway, e.g., the mitochondria-mediated caspase-dependent apoptotic pathway, the mitochondria plays a key role in apoptosis [42]. The death signal disrupts the mitochondrial membrane and pro-apoptotic proteins, including cyt-c, Smac, HtrA2, AIF and Endo G, are released from the mitochondria into the cytosol [43-47]. We previously found that NiCl2 induces mitochondrial-mediated caspase-independent apoptosis via MMP damage and increased AIF and EndoG expression [35]. Our present study showed that cyt-c, Smac and HtrA2 levels were also increased after NiCl2-induced MMP disruption [21]. Zhao, et al. [33] reported that cyt-c and AIF release from the mitochondria into the cytoplasm increased after exposure to NiNPs and Ni fine particles. Cyt-c can activate the caspase-dependent apoptosis pathway [44].
The results of this study showed that NiCl2 increased apaf-1, caspase-9, caspase-3, caspase-6, caspase-7 and PARP expression and decreased XIAP levels. Increased transcription of these genes is the most probable explanation for activation of mitochondria-mediated caspase-dependent apoptosis. NiCl2 also increased cyt-c, caspase-9, caspase-3 and PARP protein levels. NiNPs and Ni fine particles reportedly increase caspase-3, 6 and 9 protein levels and activation in JB8 and A431 cells [33, 48]. Nickel ferrite nanoparticles increase caspase-3 and caspase-9 activation and expression in HepG2 and MFC-7 cells [49], and NiSO4 increases hepatic caspase-3 activity in Carassius auratus [37]. Patel, et al. [50] suggested that NiCl2 increases caspae-3 and caspase-7 protein levels in human lung epithelial cells. Caspase-3 activity also increases in NiNP-treated cells in a dose- and time-dependent fashion [51]. NiONPs increase numbers of Annexin V positive cells and caspase-3 activation [30]. NiCl2 treatment promotes MMP disruption and cyt-c release into the cytosol, which in turn cleaves and activates caspase-9 [44]. Activated caspase-9 can cleave and activate caspase-3, 6 and 7, inducing PARP cleavage and apoptosis [52, 53]. XIAP suppresses apoptosis by directly inhibiting caspase-3 and -9 [54, 55]. Smac and HtrA2 reportedly promote apoptosis by inhibiting IAP activity [56]. Increased Smac and HtrA2 and decreased XIAP expression also contribute to apoptosis.
In this study, increased Fas, FasL, caspase-8, caspase-10 and Bid transcription was the most probable explanation for the activation of Fas-mediated (extrinsic) caspase-dependent apoptosis. Our results showed that NiCl2 increased caspase-8 and caspase-10 protein levels, two important components of the Fas-mediated caspase-dependent apoptosis pathway. In this pathway, FasL combined with the Fas receptor to activate caspase-8 and 10 [57]. Activated caspase-8 and 10 could directly cleave and activate caspase-3, 6 and 7, leading to apoptosis [58]. Our results are consistent with those of Zhao, et al. [33] who found that metallic Ni particles increased Fas, FADD and caspase-8 expression in JB6 cells. Moreover, caspase-8 and caspase-10 can also cleave the Bcl-2 family member Bid to tBid. tBid can bind to Bax, inducing MMP disruption and cyt-c release [59]. Therefore, NiCl2-medicated Bid cleavage may create a crucial connection between intrinsic and extrinsic apoptosis. In Figure 7, we summarize the possible mechanism of NiCl2-induced tubular apoptosis via mitochondria- and Fas-mediated caspase-dependent apoptotic pathways.
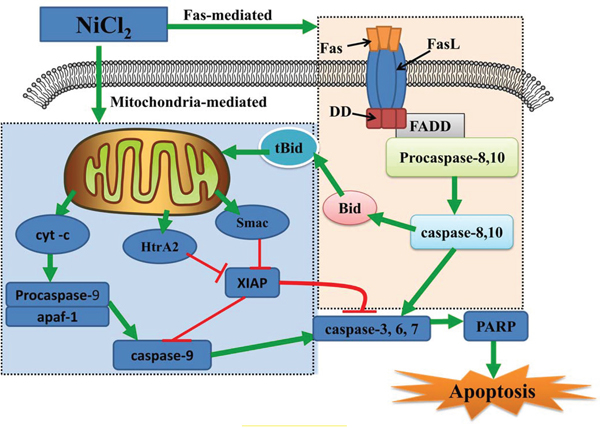
Figure 7: Schematic diagram of NiCl2-induced mitochondria- and Fas-mediated caspase-dependent apoptosis. NiCl2 increases the cyt-c, Smac and HtrA2 release from the mitochondria into the cytosol. Cyt-c cleaves and activates caspase-9, which in turn cleaves and activates downstream caspases, such as caspase-3, 6 and 7. Caspase-3, 6 and 7 cleave PARP, which then induces apoptosis. Concurrently, Smac and HtrA2 inhibit XIAP expression, also contributing to apoptosis. NiCl2 also promotes Fas and Fas ligand interactions, leading to activation of caspase-8 and casapase-10. Activated caspase-8 and 10 can directly cleave and activate downstream effector proteases, such as caspase-3, 6 and 7, leading to apoptosis.
In conclusion, the present study showed that dietary NiCl2 at 300+ mg/kg induces tubular apoptosis in broiler chickens, involving both mitochondria- and Fas-mediated caspase-dependent apoptotic pathways. Our results provide novel insights into Ni and Ni compound toxicology in vitro and in the broiler chicken kidney in vivo.
MATERIALS AND METHODS
Experimental design
Two hundred and eighty one-day-old healthy broiler chickens (Chia Tai Group, Wenjiang, Sichuan, China) were divided into four groups (N=70). All experimental procedures involving broiler chickens were approved by Animal Care and Use Committee, Sichuan Agricultural University (Approval No: 2012-024). Chickens were housed in cages with electrical heaters and provided with feed and water, as well as the experimental diets, ad libitum for 42 d. A corn-soybean meal formulated by the National Research Council [60] was the control diet, and NiCl2 (NiCl2·6H2O, Cheng Du Kelong Chemical Co., Ltd., Chengdu, China) was mixed into this basal diet to produce experimental diets containing 300, 600 or 900 mg/kg NiCl2. These three doses were chosen based on the results of previous studies. Ling and Leach reported that dietary NiCl2 concentrations of 300 mg/kg or more resulted in reduced growth rates. Mortality and anemia were observed in chicks receiving 1100 mg/kg Ni [61]. Weber and Reid observed a growth reduction at 700 mg/kg or more NiSO4 or nickel acetate [62]. Chicks fed more than 250–300 mg/kg Ni exhibited depressed growth and reduced feed intake [63]. Bersenyi, et al. [64] reported that supplementation with 500 mg/kg NiCl2 reduced weight gain by 10% and feed intake by 4%, and reduced feed conversion efficiencies by 5% in growing broiler cockerels.
Macroscopic kidney examination
At 14, 28 and 42 d, five chickens in each group were euthanized and necropsied. Kidneys were observed and weighed after dissecting connective tissue around the organ. Relative kidney weight was calculated using the following formula: Relative weight = organ weight (g)/body weight (kg).
Clinical pathological kidney examination
At 14, 28 and 42 d, five broiler chickens in each group were phlebotomized from the jugular vein to collect serum. Non-anticoagulative blood samples were clotted for 15 min at room temperature and then centrifuged at 3000 rpm for 15 min. Serum creatinine and uric acid were detected by biochemical methods following the manufacturer’s instructions (creatinine, C011-1; uric acid, C012-1; Nanjing Jiancheng Bioengineering Institute of China, Nanjing, China).
Histopathological kidney examination
Histopathological examination of the kidney was performed as previously described [20].
Apoptosis analysis by flow cytometry
At 14, 28 and 42 d, five broilers in each group were used to assess apoptosis in the kidney by flow cytometry as described by Tang, et al. [17]. Briefly, broilers in each subsample were humanely killed, and kidneys were immediately ground to form a cell suspension, which was filtered through a 300-mesh nylon screen. Cells were washed twice with ice-cold PBS (pH 7.2–7.4), and then suspended in PBS at 1×106 cells/mL. A total of 100 μL of the cell suspension was transferred to a 5-mL culture tube. Cells were stained with 5 μL Annexin V-FITC (Cat: 51-65874X, BD, USA) and 5 μL of PI (Cat: 51-66211E, BD, USA) at 25ºC for 15 min in the dark. Finally, 400 μL of 1× binding buffer was added to each tube and cells were analyzed by flow cytometry (BD FACSCalibur) within 1 h of preparation. Results were analyzed using the Mod Fit LT for Mac V3.0 program.
Quantitative real-time PCR
Kidneys were taken at 14, 28 and 42 d from five broilers in each group and stored in liquid nitrogen. They were then homogenized in liquid nitrogen using a mortar and pestle. Total RNA was isolated using RNAiso Plus (9108/9109, Takara, Japan). RNA was reverse transcribed to cDNA using the Prim-Script™ RT reagent Kit (RR047A, Takara, Japan) according to the manufacturer’s protocol. cDNA was used as a template for qRT-PCR analysis. Sequences for target apoptosis-related genes were obtained from the NCBI database. Oligonucleotide primers were designed using Primer 5 software and synthesized at Takara (Dalian, China; Table 1).
Table 1: qRT-PCR primers
Gene symbol |
Accession number |
Primer |
Primer sequence (5’-3’) |
Product size |
Tm (°C) |
|---|---|---|---|---|---|
cyt-c |
NM001079478 |
Forward |
TGTCCAGAAATGTTCCCAGTGC |
138bp |
60 |
XIAP |
NM204588 |
Forward |
CTAAACAACGAACAGCATCCAAGG |
146bp |
58 |
HtrA2 |
XM423666 |
Forward |
CATCCAGACAGACGCCGCTAT |
145bp |
62 |
Smac |
XM415152 |
Forward |
TCCCAGAAGGCAGAGACCAAG |
118bp |
62 |
apaf1 |
XM416167 |
Forward |
AAGGGCATAAGGAAGCAATCAA |
156bp |
61 |
caspase-9 |
AY057940 |
Forward |
CGAAGGAGCAAGCACGACAG |
130bp |
61 |
caspase-3 |
NM204725 |
Forward |
TGGCCCTCTTGAACTGAAAG |
139bp |
62 |
caspase-6 |
AF469049 |
Forward |
TCAGAGGAGACAAGTGCCAGAGT |
107bp |
59 |
caspase-7 |
XM421764 |
Forward |
CCGAAGTCCTCACTCAGTAACCA |
137bp |
58 |
Fas |
NM001199487 |
Forward |
TGTTCGTCATCACCGTCTATCG |
133bp |
60 |
FasL |
AJ890143 |
Forward |
AGATCGCATCCCTCCAGCTC |
135bp |
59 |
caspase-8 |
NM204592 |
Forward |
TGGGAAAGTGGACAAGAGCCT |
146bp |
59 |
caspase-10 |
XM421936 |
Forward |
GCAGCGTTCAGAAGACCACAA |
141bp |
61 |
Bid |
NM204522 |
Forward |
AGTGGAAGGACTTGCCAGAGC |
162bp |
60 |
PARP |
NM205263 |
Forward |
AAGCTCCGAACTGATATTAAGGTGG |
172bp |
56 |
β-actin |
L08165 |
Forward |
TGCTGTGTTCCCATCTATCG |
178bp |
62 |
qRT-PCR reactions (25 ul each) included 12.5 ul SYBR® Premix Ex Taq™II (DRR820A, Takara, Japan), 1 ul forward and 1 ul reverse primer, 8.5 ul of RNAase-free water (RT12102, Tiangen, China) and 1 ul of cDNA. A Bio Rad C1000 Thermal Cycler (Bio Rad, USA) was used to perform qRT-PCR reactions. The PCR procedure consisted of 95ºC for 3 min followed by 44 cycles of 95ºC for 10 s, Tm of a specific primer pair for 30 s, and then 95ºC for 10 s, 72ºC for 10 s. Melting curve analysis showed only one peak for each PCR product.
Chicken β-actin was used as an internal reference housekeeping gene. Gene expression values from control group subsamples at 14, 28 and 42 d were used to calibrate gene expression in experimental subsamples. Expression fold changes were calculated using the 2-ΔΔCT method [65].
Immunohistochemistry
Five broilers in each group were humanely sacrificed for gross examination at 14, 28 and 42 d. Kidneys were collected and fixed in 4% paraformaldehyde, dehydrated in ethanol and embedded in paraffin. As described by Wu, et al. [25], renal slices were dewaxed in xylene, rehydrated through a graded series of ethanol solutions and washed in distilled water and PBS. Endogenous peroxidase activity was blocked by incubation with 3% H2O2 in methanol for 15 min. Slices were subjected to antigen retrieval by microwaving in 0.01 M sodium citrate buffer pH 6.0. Additional washing in PBS was performed before 30 min of incubation at 37°C in 10% normal goat serum (Boster, Wuhang, China). Slices were incubated overnight at 4°C with caspase-9 (1:100) antibody, which could recognize both pro- and cleaved-caspase-9 (ab32539, Abcam, USA), cleaved-caspase-3 (1:2000) antibody (9664, CST, USA), cleaved-caspase-8 (1:100) antibody (9496, CST, USA), caspase-10 (1:100) antibody, which could recognize both the pro- and cleaved-caspase-10 (sc-6184, Santa Cruz, USA), cyt-c (1:100) antibody (sc-13560, Santa Cruz, USA) or cleaved-PARP (1:50) antibody (5625, CST, USA). After washing in PBS, slices were exposed to 1% biotinylated goat anti-mouse IgG secondary antibody (Boster, Wuhang, China) for 1 h at 37ºC, and then incubated with streptavidin-biotin complex (SABC; Boster, Wuhang, China) for 30 min at 37°C. To visualize the immunoreaction, slices were immersed in diaminobenzidine hydrochloride (DAB; Boster, Wuhang, China). Slices were monitored microscopically and stopped by immersion in distilled water as soon as brown staining was visible. Slices were lightly counterstained with hematoxylin, dehydrated in ethanol, cleared in xylene and mounted.
Protein expression was quantitatively assessed using a computer-supported imaging system connected to a light microscope (Olympus, Shimadzu, Japan) with an objective magnification of ×1000. Staining intensity for each protein was quantified using Image-pro Plus 5.1 (Madia Cybernetics, MD, USA). Five sections were measured from each group and five fields from each section were measured and averaged [25].
Renal Ni quantification by GFAAS
Five broilers in each group were humanely killed at 42 d, and kidneys were immediately removed, weighed, dried and collected for determination of Ni. Ni concentrations in the kidney were measured by GFAAS as previously described [20].
Statistical analysis
All treatment groups were compared to their respective controls. Significant differences among the three treatment groups and the control group were analyzed by one-way ANOVA. Results are presented as means ± standard deviation (M ± SD). All tests were performed using SPSS 16.0 for Windows.
Abbreviations
Abbreviations appeared in the text are listed in the Table 2.
Table 2: Abbreviations appeared in the text
Abbreviation |
Name |
Abbreviation |
Name |
|---|---|---|---|
Ni |
Nickel |
MMP |
mitochondrial membrane potential |
NiCl2 |
Nickel Chloride |
Cr |
creatinine |
qRT-PCR |
Quantitative Real Time-polymerase Chain Reaction |
UA |
uric acid |
cytochrome c |
cyt-c |
PS |
phosphatidylserine |
HtrA2/Omi |
High-temperature-requirement protein A2 |
AIF |
apoptosis inducing factor |
Smac/Diablo |
second mitochondria-derived activator of caspase |
Endo G |
endonuclease G |
apaf-1 |
apoptotic peptidase activating factor 1 |
TUNEL |
terminal deoxynucleotidyl transferase 2'-deoxyuridine 5'-triphosphate dUTP nick end-labeling |
PARP |
poly (ADP-ribose) polymerase |
JNK |
c-Jun N-terminal kinase |
XIAP |
X-linked inhibitor of apoptosis protein |
HepG2 |
human hepatocellular carcinoma |
FasL |
Fas ligand |
PI3K |
phosphoinositide-3-kinase |
Bid |
BH3 interacting domain death agonist |
Akt |
serine-threonine kinase |
NiSO4 |
Nickel Sulfate |
NiNPs |
nickel nanoparticles |
IARC |
International Agency for Research on Cancer |
FCE |
feed conversion efficiency |
NiONPs |
NiO nanoparticles |
PI |
propidium iodide |
Ni(tcitr)2 |
bis (S-citronellalthiosemicarbazonato) Ni (II) |
GFAAS |
Graphite Furnace Atomic Absorption Spectrometry |
ACKNOWLEDGMENTS
The study was supported by the program for Changjiang scholars and innovative research team in university (IRT 0848) and the Shuangzhi project of Sichuan Agricultural University (03570327; 03571198).
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
Author contributions
Hongrui Guo and Hengmin Cui designed the experiments. Hongrui Guo, Bangyuan Wu, Kejie Chen and Jie Deng carried out the experiments. Hongrui Guo, Hengmin Cui, Jing Fang, Zhicai Zuo, Junliang Deng, Xun Wang, Ling Zhao, Bangyuan Wu, Kejie Chen and Jie Deng analyzed and interpreted data. Hongrui Guo and Hengmin Cui concluded the scientific findings and wrote and revised the manuscript.
REFERENCES
1. Poonkothai M VBS. Nickel as an essential element and a toxicant. Inter J Environ Sci. 2012; 1:285-288.
2. Zhao J, Shi X, Castranova V, Ding M. Occupational toxicology of nickel and nickel compounds. J Environ Pathol Toxicol Oncol. 2009; 28:177-208.
3. Cempel M, Nikel G. Nickel: A Review of Its Sources and Environmental Toxicology. Pol J Environ Stud. 2006; 15:375-382.
4. Kienle C, Kohler HR, Gerhardt A. Behavioural and developmental toxicity of chlorpyrifos and nickel chloride to zebrafish (Danio rerio) embryos and larvae. Ecotoxicol Environ Saf. 2009; 72:1740-1747.
5. Attig H, Dagnino A, Negri A, Jebali J, Boussetta H, Viarengo A, Dondero F, Banni M. Uptake and biochemical responses of mussels Mytilus galloprovincialis exposed to sublethal nickel concentrations. Ecotoxicol Environ Saf. 2010; 73:1712-1719.
6. Tuchman M, Silverberg JI, Jacob SE, Silverberg N. Nickel contact dermatitis in children. Clin Dermatol. 2015; 33:320-326.
7. Forti E, Salovaara S, Cetin Y, Bulgheroni A, Tessadri R, Jennings P, Pfaller W, Prieto P. In vitro evaluation of the toxicity induced by nickel soluble and particulate forms in human airway epithelial cells. Toxicol In Vitro. 2011; 25:454-461.
8. Kasprzak K. Nickel carcinogenesis. Mutat Res-Fund Mol M. 2003; 533:67-97.
9. Das KK, Das SN, Dhundasi SA. Nickel, its adverse health effects & oxidative stress. Indian J Med Res. 2008; 128:412-425.
10. Spears JW, Harvey RW, Samsell LJ. Effects of dietary nickel and protein on growth, nitrogen metabolism and tissue concentrations of nickel, iron, zinc, manganese and copper in calves. J Nutr. 1986; 116:1873-1882.
11. Das KK, Dasgupta S. Effect of nickel on testicular nucleic acid concentrations of rats on protein restriction. Biol Trace Elem Res. 2000; 73:175-180.
12. Ragsdale SW. Nickel biochemistry. Curr Opin Chem Biol. 1998; 2:208-215.
13. Gathwan KH, Al-Karkhi IHT, Jaffar Al-Mulla EA. Hepatic toxicity of nickel chloride in mice. Res Chem Intermed. 2012; 39:2537-2542.
14. Su L, Deng Y, Zhang Y, Li C, Zhang R, Sun Y, Zhang K, Li J, Yao S. Protective effects of grape seed procyanidin extract against nickel sulfate-induced apoptosis and oxidative stress in rat testes. Toxicol Mech Methods. 2011; 21:487-494.
15. Liu CM, Zheng GH, Ming QL, Chao C, Sun JM. Sesamin protects mouse liver against nickel-induced oxidative DNA damage and apoptosis by the PI3K-Akt pathway. J Agr Food Chem. 2013; 61:1146-1154.
16. Krockova J, Massanyi P, Sirotkin AV, Lukac N, Kovacik A. Nickel-induced structural and functional alterations in porcine granulosa cells in vitro. Biol Trace Elem Res. 2013; 154:190-195.
17. Tang K, Guo H, Deng J, Cui H, Peng X, Fang J, Zuo Z, Wang X, Wu B, Li J, Yin S. Inhibitive Effects of Nickel Chloride (NiCl2) on Thymocytes. Biol Trace Elem Res. 2015; 164:242-252.
18. Wu B, Cui H, Peng X, Fang J, Zuo Z, Deng J, Wang X, Huang J. Toxicological effects of nickel chloride on the cytokine mRNA expression and protein levels in intestinal mucosal immunity of broilers. Environ Toxicol. 2015; 30:1309-1321.
19. Guo H, Wu B, Cui H, Peng X, Fang J, Zuo Z, Deng J, Wang X, Deng J, Yin S, Li J, Tang K. NiCl2-down-regulated antioxidant enzyme mRNA expression causes oxidative damage in the broiler(')s kidney. Biol Trace Elem Res. 2014; 162:288-295.
20. Guo H, Deng H, Cui H, Peng X, Fang J, Zuo Z, Deng J, Wang X, Wu B, Chen K. Nickel chloride (NiCl2)-caused inflammatory responses via activation of NF-kappaB pathway and reduction of anti-inflammatory mediator expression in the kidney. Oncotarget. 2015; 6:28607-28620. doi: 10.18632/oncotarget.5759.
21. Guo H, Cui H, Peng X, Fang J, Zuo Z, Deng J, Wang X, Wu B, Chen K, Deng J. Modulation of the PI3K/Akt Pathway and Bcl-2 Family Proteins Involved in Chicken's Tubular Apoptosis Induced by Nickel Chloride (NiCl(2)). Int J Mol Sci. 2015; 16:22989-23011.
22. Guo H, Cui H, Peng X, Fang J, Zuo Z, Deng J, Wang X, Wu B, Chen K, Deng J. Dietary NiCl2 causes G2/M cell cycle arrest in the broiler's kidney. Oncotarget. 2015; 6:35964-35977. doi: 10.18632/oncotarget.5934.
23. Yin S, Cui H, Peng X, Fang J, Zuo Z, Deng J, Wang X, Wu B, Guo H. Toxic effect of NiCl2 on development of the bursa of Fabricius in broiler chickens. Oncotarget. 2016; 7:125-139. doi: 10.18632/oncotarget.6591.
24. Wu B, Guo H, Cui H, Peng X, Fang J, Zuo Z, Deng J, Wang X, Huang J. Pathway underlying small intestine apoptosis by dietary nickel chloride in broiler chickens. Chemico-Biological Interactions. 2016; 243:91-106.
25. Wu B, Cui H, Peng X, Fang J, Zuo Z, Deng J, Huang J. Dietary nickel chloride induces oxidative stress, apoptosis and alters Bax/Bcl-2 and caspase-3 mRNA expression in the cecal tonsil of broilers. Food Chem Toxicol. 2014; 63:18-29.
26. Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007; 35:495-516.
27. Fan T-J, Han L-H, Cong R-S, Liang J. Caspase Family Proteases and Apoptosis. Acta Bioch Bioph Sin. 2005; 37:719-727.
28. Lam M, Bhat MB, Nunez G, Ma J, Distelhorst CW. Regulation of Bcl-xl channel activity by calcium. Journal of Biological Chemistry. 1998; 273:17307-17310.
29. Tait SWG, Green DR. Mitochondria and cell death: outer membrane permeabilization and beyond. Nature Reviews Molecular Cell Biology. 2010; 11:621-632.
30. Duan WX, He MD, Mao L, Qian FH, Li YM, Pi HF, Liu C, Chen CH, Lu YH, Cao ZW, Zhang L, Yu ZP, Zhou Z. NiO nanoparticles induce apoptosis through repressing SIRT1 in human bronchial epithelial cells. Toxicol Appl Pharmacol. 2015; 286:80-91.
31. Wang YF, Shyu HW, Chang YC, Tseng WC, Huang YL, Lin KH, Chou MC, Liu HL, Chen CY. Nickel (II)-induced cytotoxicity and apoptosis in human proximal tubule cells through a ROS- and mitochondria-mediated pathway. Toxicol Appl Pharmacol. 2012; 259:177-186.
32. Buschini A, Pinelli S, Pellacani C, Giordani F, Ferrari MB, Bisceglie F, Giannetto M, Pelosi G, Tarasconi P. Synthesis, characterization and deepening in the comprehension of the biological action mechanisms of a new nickel complex with antiproliferative activity. J Inorg Biochem. 2009; 103:666-677.
33. Zhao J, Bowman L, Zhang X, Shi X, Jiang B, Castranova V, Ding M. Metallic nickel nano- and fine particles induce JB6 cell apoptosis through a caspase-8/AIF mediated cytochrome c-independent pathway. Journal of nanobiotechnology. 2009; 7:2.
34. Yin S, Guo H, Cui H, Peng X, Fang J, Zuo Z, Deng J, Wang X, Tang K, Li J. Nickel Chloride (NiCl2) Induces Histopathological Lesions via Oxidative Damage in the Broiler's Bursa of Fabricius. Biol Trace Elem Res. 2016; 171:214-223.
35. Guo H, Cui H, Peng X, Fang J, Zuo Z, Deng J, Wang X, Wu B, Chen K, Deng J. Modulation of the PI3K/Akt Pathway and Bcl-2 Family Proteins Involved in Chicken’s Tubular Apoptosis Induced by Nickel Chloride (NiCl2). Int J Mol Sci. 2015; 9:22989-23011.
36. Huang J, Cui H, Peng X, Fang J, Zuo Z, Deng J, Wu B. The Association between Splenocyte Apoptosis and Alterations of Bax, Bcl-2 and Caspase-3 mRNA Expression, and Oxidative Stress Induced by Dietary Nickel Chloride in Broilers. Int J Environ Res Public Health. 2013; 10:7310-7326.
37. Zheng GH, Liu CM, Sun JM, Feng ZJ, Cheng C. Nickel-induced oxidative stress and apoptosis in Carassius auratus liver by JNK pathway. Aquat Toxicol. 2014; 147:105-111.
38. Ahamed M, Ali D, Alhadlaq HA, Akhtar MJ. Nickel oxide nanoparticles exert cytotoxicity via oxidative stress and induce apoptotic response in human liver cells (HepG2). Chemosphere. 2013; 93:2514-2522.
39. Chen C-Y, Lin T-K, Chang Y-C, Wang Y-F, Shyu H-W, Lin K-H, Chou M-C. Nickel (II)-induced oxidative stress, apoptosis, G2/M arrest, and genotoxicity in normal rat kidney cells. J Toxicol Environ Health A. 2010; 73:529-539.
40. Freitas M, Barcellos-de-Souza P, Barja-Fidalgo C, Fernandes E. Nickel induces apoptosis in human neutrophils. Biometals. 2013; 26:13-21.
41. Chen CY, Wang YF, Huang WR, Huang YT. Nickel induces oxidative stress and genotoxicity in human lymphocytes. Toxicol Appl Pharmacol. 2003; 189:153-159.
42. Martinou JC, Youle RJ. Mitochondria in apoptosis: Bcl-2 family members and mitochondrial dynamics. Dev Cell. 2011; 21:92-101.
43. Du C, Fang M, Li Y, Li L, Wang X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell. 2000; 102:33-42.
44. Jiang X, Wang X. Cytochrome C-mediated apoptosis. Annu Rev Biochem. 2004; 73:87-106.
45. Duckett CS. IAP proteins: sticking it to Smac. Biochem J. 2005; 385:e1-2.
46. Garrido C, Galluzzi L, Brunet M, Puig PE, Didelot C, Kroemer G. Mechanisms of cytochrome c release from mitochondria. Cell Death Differ. 2006; 13:1423-1433.
47. Vande Walle L, Lamkanfi M, Vandenabeele P. The mitochondrial serine protease HtrA2/Omi: an overview. Cell Death Differ. 2008; 15:453-460.
48. Alarifi S, Ali D, Alakhtani S, Al Suhaibani ES, Al-Qahtani AA. Reactive oxygen species-mediated DNA damage and apoptosis in human skin epidermal cells after exposure to nickel nanoparticles. Biol Trace Elem Res. 2014; 157:84-93.
49. Ahamed M, Akhtar MJ, Alhadlaq HA, Khan MA, Alrokayan SA. Comparative cytotoxic response of nickel ferrite nanoparticles in human liver HepG2 and breast MFC-7 cancer cells. Chemosphere. 2015; 135:278-288.
50. Patel E, Lynch C, Ruff V, Reynolds M. Co-exposure to nickel and cobalt chloride enhances cytotoxicity and oxidative stress in human lung epithelial cells. Toxicol Appl Pharmacol. 2012; 258:367-375.
51. Ahamed M. Toxic response of nickel nanoparticles in human lung epithelial A549 cells. Toxicol In Vitro. 2011; 25:930-936.
52. Zhang N, Chen Y, Jiang R, Li E, Chen X, Xi Z, Guo Y, Liu X, Zhou Y, Che Y, Jiang X. PARP and RIP 1 are required for autophagy induced by 11'-deoxyverticillin A, which precedes caspase-dependent apoptosis. Autophagy. 2011; 7:598-612.
53. Boulares AH, Yakovlev AG, Ivanova V, Stoica BA, Wang G, Iyer S, Smulson M. Role of poly(ADP-ribose) polymerase (PARP) cleavage in apoptosis. Caspase 3-resistant PARP mutant increases rates of apoptosis in transfected cells. J Biol Chem. 1999; 274:22932-22940.
54. Deveraux QL, Reed JC. IAP family proteins—suppressors of apoptosis. Genes Dev. 1999; 13:239-252.
55. Hasegawa T, Suzuki K, Sakamoto C, Ohta K, Nishiki S, Hino M, Tatsumi N, Kitagawa S. Expression of the inhibitor of apoptosis (IAP) family members in human neutrophils: up-regulation of cIAP2 by granulocyte colony-stimulating factor and overexpression of cIAP2 in chronic neutrophilic leukemia. Blood. 2003; 101:1164-1171.
56. Wu G, Chai J, Suber TL, Wu JW, Du C, Wang X, Shi Y. Structural basis of IAP recognition by Smac/DIABLO. Nature. 2000; 408:1008-1012.
57. Scott FL, Stec B, Pop C, Dobaczewska MK, Lee JJ, Monosov E, Robinson H, Salvesen GS, Schwarzenbacher R, Riedl SJ. The Fas-FADD death domain complex structure unravels signalling by receptor clustering. Nature. 2009; 457:1019-1022.
58. Lavrik IN, Krammer PH. Regulation of CD95/Fas signaling at the DISC. Cell Death Differ. 2012; 19:36-41.
59. Esposti MD. The roles of Bid. Apoptosis. 2002; 7:433-440.
60. NRC, 1994. Nutrient Requirements of Poultry. 9th Revised Ed. National Academy Press Washington, DC.
61. Ling J, Leach R. Studies on nickel metabolism: interaction with other mineral elements. Poultry Sci. 1979; 58:591-596.
62. Weber CW, Reid BL. Nickel toxicity in growing chicks. J Nutr. 1968; 95:612-616.
63. Szilagyi M, Szentmihalyi S, Anke M. Changes in some of the biochemical parameters in Ni and Mo deficient animals [goat, sheep, pig, chicken, rat]. Proceeding (Hungary). 1981; 1:257-260.
64. Bersényi A, Fekete SG, Szilágyi M, Berta E, Zöldág L, Glávits R. Effects of nickel supply on the fattening performance and several biochemical parameters of broiler chickens and rabbits. Acta Vet Hungarica. 2004; 52:185-197.
65. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001; 25:402-408.