
INTRODUCTION
Colorectal cancer (CRC) is the third most common cancer with nearly 1.4 million new cases in 2012. Based on the lesions within colon or rectum, CRC varies in terms of biological characteristics and carcinogenic mechanisms [1]. Several intracellular signaling pathways such as Ras/Raf/MEK/ERK, PI3k/Akt, Wnt/β-catenin and p53 are frequently dysregulated in CRC [2–4]. Notably, p53 missense or nonsense mutations constantly occur in approximately 40-50% of sporadic CRC, causing therapeutic resistance and poor prognosis [5].
p53 is a key tumor suppressor regarded as the “guardian of the genome”. Once activated in response to a variety of stress, p53 elicits potent inhibition of tumorigenesis, leading to cell cycle arrest, apoptosis, senescence and autophagy [6–8]. However, mutant p53 not only abrogates tumor suppressor functions of wild-type p53 but also acquires an oncogenic “gain-of-function” defined as the ability for malignant proliferation, invasion, metastasis and anti-apoptotic effect [9]. Therefore, a promising strategy to treat cancer with mutant p53 via restoring wild-type p53 function has been pursued. Several small molecules such as PRIMA-1, APR-246 (PRIMA-1Met), CP-31398, MIRA have been shown to restore wild-type activity to mutant p53 and induce massive p53-dependent apoptosis [10–13].
PRIMA-1 is one of chemical compounds originally identified by screening a library of low-molecular-weight compounds [10]. PRIMA-1Met, as methylated analog of PRIMA-1, could enhance killing efficacy. Biochemical analysis demonstrated that either PRIMA-1 or its methylated version is converted into active product named methylene quinuclidinone (MQ), which in turn covalently binds to several cysteine (Cys) residues in the DNA-binding domains of p53 and restores functional conformation of wild-type p53 [14]. Recent reports revealed other targets of PRIMA-1Met such as oxidosqualene cyclase and selenoprotein thioredoxin reductase 1 (TrxR1) which are sufficient to suppress cancer cell growth irrespective of p53 mutation status [15, 16]. Therefore, identifying potential targets of PRIMA-1Met is important to the development of new cancer therapy approaches.
The mitogen-activated protein kinase (MAPK) pathway Ras/Raf/MEK/ERK is often aberrantly activated in CRC and plays a critical role in multiple cellular processes including proliferation, transformation, apoptosis and senescence [17]. Generally, constitutive activation of MAPK cascades is stimulated by mutant Ras because activating K-Ras mutations occur in approximately 37-45% CRC [18]. So far, no K-Ras mutant inhibitors are available for clinical practice [19, 20]. MEK is a core downstream effector of Ras/Raf kinases and appears as a promising alternative for therapeutic strategy targeting Ras-activated MAPK pathway [21].
In the present study, we reported that PRIMA-1Met attenuated colorectal cancer cell growth irrespective of mutant p53 status. In p53 null or p53 wild-type HCT116 colorectal cancer cells, we demonstrated that PRIMA-1Met effectively inhibited cell proliferation and anchorage independent growth. Furthermore, we showed that PRIMA-1Met directly bound MEK kinase and suppressed its activity both in vitro and in cells. Moreover, in vivo animal experiments confirmed that PRIMA-1Met inhibited MEK activity to suppress the growth of colorectal cancer xenografts.
RESULTS
PRIMA-1Met inhibits the proliferation and growth of CRC cells independent of p53 status
The chemical structure of PRIMA-1Met was shown in Figure 1A. To evaluate p53-independent efficacy of PRIMA-1Met, we selected a series of CRC cell lines representative of different TP53 heterogeneity, including TP53wt (HCT116wt and LOVO), TP53mut (SW480, DLD-1 and HT29) and TP53neg (HCT116neg). p53 protein expression level and genotypes of these cell lines were presented (Supplementary Figure S1 and Supplementary Table S1). MTS assay showed that the treatment of different cells with 25 uM PRIMA-1Met for 12 h or 24 h led to similar suppression of cell growth, indicating that CRC cells with different TP53 status were generally sensitive to PRIMA-1Met (Figure 1B). Meanwhile, we treated the cells with different concentrations of PRIMA-1Met for 24 h. MTS assay showed that PRIMA-1Met reduced the viability of all six CRC cell lines in a dose-dependent manner (Figure 1C). Notably, the inhibitory effect of PRIMA-1Met at concentrations less than 50 umol/L showed no significant difference among the cells (Figure 1C). However, high dosage of PRIMA-1Met (75 uM) triggered apoptosis, especially in cells harboring mutant TP53 (Supplementary Figure S2A-S2D).
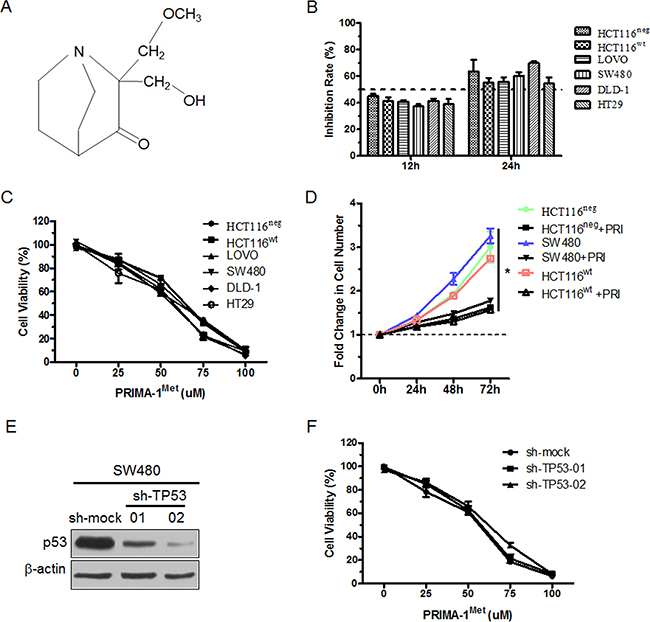
Figure 1: PRIMA-1Met inhibited the proliferation of CRC cells with different p53 status. A. Chemical structure of PRIMA-1Met. B. CRC cells with different p53 status were treated with 25 uM PRIMA-1Met for 12 or 24 h. The cell viability was measured by MTS assay. C. Six colorectal cancer cells were exposed to PRIMA-1Met with various concentrations for 24 h and cell viability was measured by MTS assay. D. 1×106 HCT116wt, HCT116neg or SW480 cells were treated with 25 uM PRIMA-1Met or control. Growth curve was measured by counting cell number at indicated time point. *P<0.05. PRI: PRIMA-1Met. E. SW480 cells were transiently transfected with sh-TP53 or sh-mock and p53 expression was analyzed by Western blot analysis. F. SW480 cells transfected with sh-mock or sh-TP53 were treated with various concentrations of PRIMA-1Met for 24 h and cell viability was measured by MTS assay. All data were shown as mean ± SEM from triplicate experiments.
Next we examined the effect of PRIMA-1Met on CRC cell growth, and the results showed that PRIMA-1Met inhibited the growth of three cell lines: TP53wt HCT116wt, TP53mut SW480, and TP53neg HCT116neg (Figure 1D), confirming that the inhibitory effect of PRIMA-1Met on CRC cell growth is independent of TP53 status.
To further investigate whether the effect of PRIMA-1Met on cell proliferation is dependent on TP53, we compared the effects of PRIMA-1Met in SW480 or DLD-1 cells transfected with sh-mock or sh-TP53 plasmid (Figure 1E and Supplementary Figure S3A). The viability of sh-mock or sh-TP53 cells exhibited no significant difference (Figure 1F and Supplementary Figure S3B). Taken together, these results suggest that the inhibitory effects of PRIMA-1Met on CRC cell proliferation and growth are independent of p53 status.
PRIMA-1Met inhibits CRC cell colony formation and EGF-induced cell transformation independent of p53 status
Next, we determined the effect of PRIMA-1Met on anchorage-independent growth of cancer cells. Colony formation assay showed that PRIMA-1Met markedly inhibited anchorage-independent growth of HCT116wt, SW480 and HCT116neg cells in a concentration dependent manner (Figure 2A–2C). In particular, PRIMA-1Met at 50 uM significantly decreased more than 70% of colony formation in all 3 cell lines compared to control (P<0.05). Even at the lower concentration of 25 uM, PRIMA-1Met caused approximate 30% decrease of colony formation in soft agar. Our data showed no significant differences in dose-dependent inhibition by PRIMA-1Met among TP53wt, TP53mut and TP53neg cells.
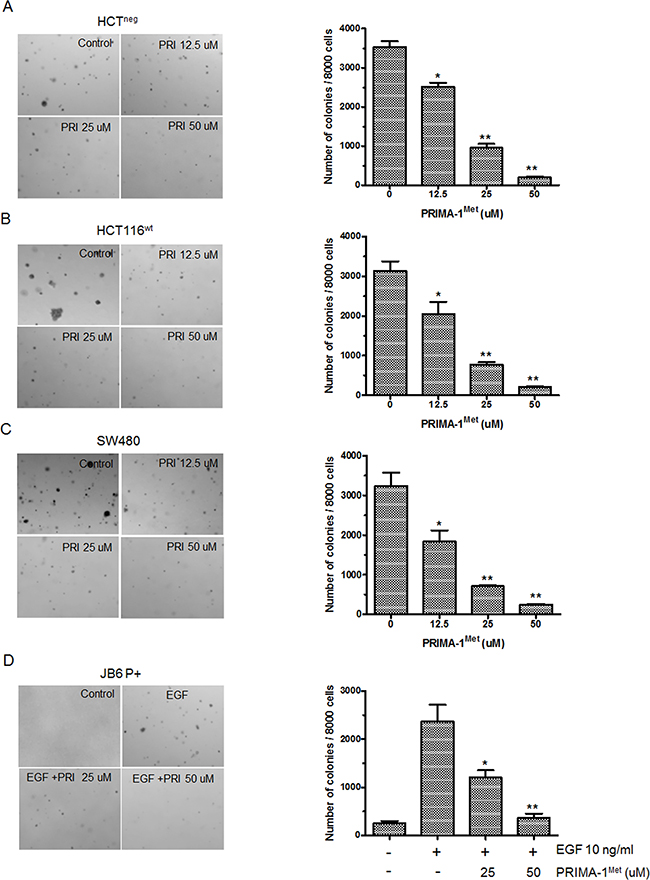
Figure 2: PRIMA-1Met inhibited anchorage-independent growth of colorectal cancer cells and EGF-induced neoplastic transformation of JB6 P+ cells. A-C., Representative images of colony formation. Cells were seeded in soft agar with the addition of different concentration of PRIMA-1Met (0, 12.5, 25, 50 uM) and cultured for 7 days (SW480 and HCT116neg) or 10 days (HCT116wt), then colonies were counted. Data were shown as mean ± SEM from triplicate experiments. **P<0.01 vs. vehicle group. PRI: PRIMA-1Met. D. JB6 P+ cells were cultured with or without EGF (10 ng/ml) and different concentrations of PRIMA-1Met (0, 25, 50 uM) for 7 days, then colonies were counted. Data were shown as mean ± SEM from triplicate experiments. * P<0.05; **, P<0.01 vs. EGF single group. PRI: PRIMA-1Met.
We further accessed whether PRIMA-1Met could inhibit EGF-promoted neoplastic cell transformation in immortalized normal cells. We found that PRIMA-1Met significantly inhibited EGF induced anchorage-independent growth of JB6 P+, an immortalized normal skin cell line with impaired p53 function (Figure 2D). These data strongly suggest that p53 status is not indispensable for PRIMA-1Met mediated inhibition of anchorage-independent growth and EGF-induced cell transformation.
Kinase profiling identifies PRIMA-1Met as a potential MEK inhibitor
To understand the mode of action of anti-tumor activity of PRIMA-1Met, we performed in vitro kinase profiling assay using Millipore’s Kinase Profiler (Table 1). The results showed that MEK1 or MEK2 kinase activity was significantly suppressed by PRIMA-1Met at the concentration of 25 uM. At the same concentration, PRIMA-1Met had little effect on EGFR, MAPK1/2 and RSK1/2, which were key kinases in Ras/Raf/MEK/ERK pathway. These data suggest that MEK is a strong candidate target of PRIMA-1Met compared to other screened kinases.
Table 1: Kinase profiling results of PRIMA-1Met
Kinase |
Activity |
Kinase |
Activity |
|---|---|---|---|
ALK |
90 |
JNK3(h) |
96 |
AMPKα1(h) |
94 |
KDR(h) |
93 |
Aurora-A(h) |
83 |
MAPK1(h) |
75 |
Aurora-B(h) |
91 |
MAPK2(h) |
80 |
B-Raf (h) |
80 |
MEK1(h) |
35 |
CDK1/cyclinB(h) |
94 |
MEK2(h) |
48 |
CDK2/cyclinA(h) |
102 |
mTOR(h) |
86 |
CDK3/cyclinE(h) |
73 |
p70S6K(h) |
90 |
C-MET(h) |
60 |
PDK1(h) |
76 |
C-RAF(h) |
92 |
PKBα(h) |
110 |
cSRC(h) |
67 |
PKBβ(h) |
100 |
EGFR(h) |
95 |
PKCα(h) |
88 |
FGFR1(h) |
83 |
PKCδ(h) |
92 |
GSK3α(h) |
105 |
Rsk1(h) |
88 |
GSK3β(h) |
110 |
Rsk2(h) |
79 |
IGF-1R(h) |
92 |
Src(T341M)(h) |
110 |
IKKα(h) |
94 |
TGFBR1 (h) |
93 |
IKKβ(h) |
90 |
PI3K (p110α/p85α) (h) |
97 |
JNK1α1(h) |
70 |
PI3K (p110 β/p85α) (h) |
90 |
JNK2α2(h) |
84 |
PI3K (p110δ/p85α) (h) |
94 |
Kinase screening assay was performed according to Millipore’s Kinase Profiler protocols. Briefly, the reaction concentration of PRIMA-1Met was set at 100 μM and the concentration of ATP at 10 μM. Scores for each kinase were represented as the percent (%) of kinase activity after PRIMA-1Met treatment.
PRIMA-1Met inhibits MEK kinase activity in vitro and in cells
To confirm that PRIMA-1Met could inhibit MEK activity, we conducted in vitro kinase assay by using inactive ERK1 as a substrate of MEK and MEK inhibitor AZD6244 as a positive control. We found that the phosphorylation of ERK was potently decreased by PRIMA-1Met in a dose dependent manner (Figure 3A).
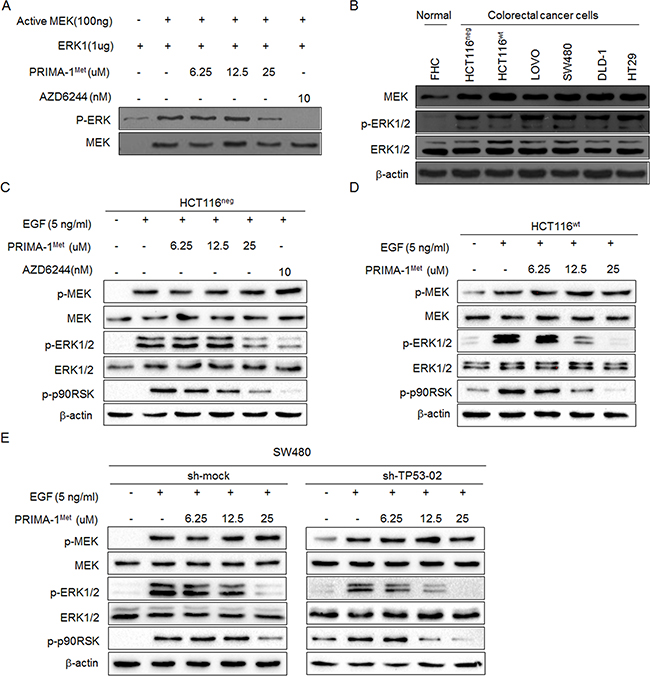
Figure 3: PRIMA-1Met inhibited kinase activity of MEK in vitro and in cells. A. Representative results of in vitro kinase assay. Inactive ERK1 protein was used as a substrate and mixed with active MEK kinase and different doses of PRIMA-1Met. The phosphorylation level of ERK1 (Thr202/Tyr204) was detected by Western blot analysis. Total MEK was used as loading control and MEK inhibitor AZD6244 was used as a positive control. B. Western blot analysis of MEK protein expression level in six colorectal cancer cell lines and normal colon epithelial cell line FHC. The activation of MEK signaling was detected in HCT116neg C., HCT116wt D. and SW480 E. cells. After starvation in serum-free medium for 24 h, cells were treated with different doses of PRIMA-1Met for 12 h and then treated with 5 ng/ml EGF for 30 min. AZD6244 was used as a positive control. Data were representative results from three independent experiments.
Next, we detected MEK kinase activity in colorectal cancer cells and found that its activity was significantly elevated in six colorectal cancer cells compared with normal colon cells (Figure 3B). When colon cancer cell lines HCT116wt and HCT116neg were treated with PRIMA-1Met, The phosphorylation of downstream molecules of MEK such as ERK1/2 and RSK2 was dramatically attenuated with PRIMA-1Met concentration more than 25 uM (Figure 3C, 3D). However, there were no significant differences in MEK inhibition between HCT116wt and HCT116neg cells, indicating that p53 status does not interfere with PRIMA-1Met induced MEK inactivity.
To further exclude p53-dependent regulatory mechanisms, we depleted the expression of mutant p53 in SW480 cells by shRNA and found that depletion of p53 showed minimal effect on the attenuation of MEK activity after PRIMA-1Met treatment (Figure 3E). Collectively, these data confirm that PRIMA-1Met inhibits MEK activity independent of p53 status.
PRIMA-1Met binds MEK directly in vitro and ex vivo
To better understand PRIMA-1Met-induced inhibition of MEK activity, we wondered whether PRIMA-1Met could interact with MEK. We docked PRIMA-1Met to MEK monomer using automated docking tool AutoDock 4 [22]. The results showed that PRIMA-1Met bound to the ATP-binding pocket of MEK1 monomer (Figure 4A). The oxygen atom of PRIMA-1Met could form hydrogen bond with the sulfur group of Cys207 and Met143, and the carboxylate group of Asp208 (Figure 4B).
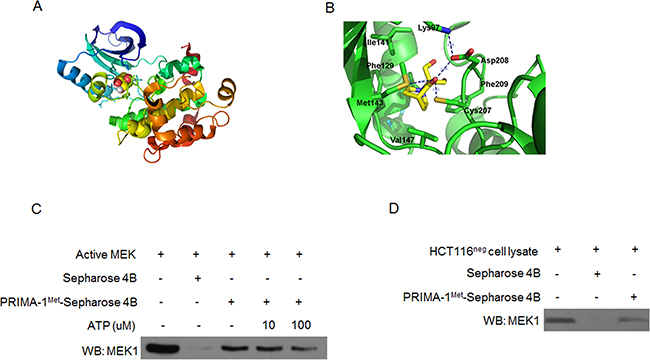
Figure 4: PRIMA-1Met specifically binds MEK in vitro and in vivo. A. Computational binding model of PRIMA-1Met in proposed ATP-binding pocket of MEK1 monomer. PRIMA-1Met was shown as colored sphere and MEK1 monomer as cartoon. B. The proposed binding-position of hydrogen bonds between PRIMA-1Met and MEK1 was indicated. C. Pull-down assay was conducted in vitro by incubating 600 ug HCT116neg cell lysates with PRIMA-1Met-Sepharose 4B beads followed by Western blot analysis. D. In vivo ATP competitive binding assay was conducted by incubating 300 ng active MEK1 protein with 100 ul PRIMA-1Met-Sepharose 4B beads and various concentrations of ATP (0, 10, 100 uM) followed by Western blot analysis. Sepharose 4B only beads were used as negative control. Data shown were representative of three independent experiments.
Next we performed ATP competitive binding assay by using PRIMA-1Met-conjugated Sepharose 4B beads in vitro. Compared with Sepharose 4B beads alone, PRIMA-1Met-conjugated Sepharose 4B beads could pull-down MEK and their affinity was impaired by the presence of ATP (Figure 4C). Furthermore, immunoprecipitation assay in HCT116neg cells confirmed the binding of PRIMA-1Met to endogenous MEK (Figure 4D). Collectively, these data provide strong evidence that PRIMA-1Met could directly bind MEK in ATP-binding pocket in vitro and in cells.
MEK is indispensable for the anti-tumor activity of PRIMA-1Met in vitro
To confirm that PRIMA-1Met attenuated cancer cell proliferation and anchorage-independent growth via the inhibition of MEK kinase activity, we used shRNA to knockdown MEK1 expression in HCT116neg or HCT116wt cells (Figure 5A). The depletion of MEK abrogated PRIMA-1Met-induced suppression of anchorage-independent growth of HCT116neg and HCT116wt cells, and the magnitude of growth reduction was correlated with the efficiency of MEK1 knockdown (Figure 5B).
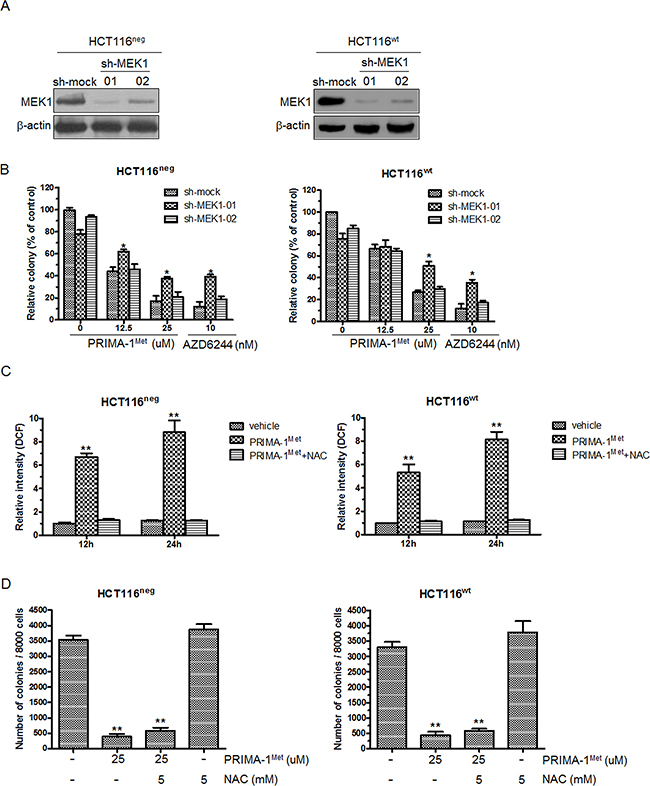
Figure 5: Knockdown of MEK1 decreases the sensitivity of colorectal cancer cells to PRIMA-1Met. A. HCT116neg or HCT116wt cells were transfected with sh-mock or sh-MEK1. Knockdown efficiency was evaluated by Western blot analysis. B. HCT116neg or HCT116wt cells transfected with sh-mock or sh-MEK1 were treated with 12.5 and 25 uM PRIMA-1Met for 7 days and colony formation in soft agar was assessed. Data were shown as mean ± SEM. AZD6244 was used as a positive control. *P<0.05. C. HCT116neg cells were treated with 25 uM PRIMA-1Met in the presence or absence of NAC (5 mM) for 12 or 24 h and stained with 20 uM DCFHDA for 30 min. The mean fluorescent intensity was measured by flow cytometer and normalized to vehicle group. Data were shown as mean ± SEM from triplicate experiments. **P<0.01 vs. vehicle group. D. Cells were seeded in soft agar with the addition of 25 uM PRIMA-1Met alone or in combination of 5mM NAC and cultured for 7 days. Then colony formation was counted. Data represented mean ± SEM. **P<0.01 vs. control.
PRIMA-1Met could induce reactive oxygen species (ROS) to mediate p53-independent cell apoptosis [16]. Thus it is possible that PRIMA-1Met inhibits CRC cell growth by promoting ROS-induced cell apoptosis. To test this possibility, we measured endogenous ROS in HCT116neg and HCT116wt cells treated with 25 uM PRIMA-1Met. PRIMA-1Met induced massive production of ROS and this was significantly inhibited by ROS scavenger NAC (Figure 5C). However, NAC had little effect on PRIMA-1Met induced attenuation of anchorage-independent growth of HCT116neg and HCT116wt cells (Figure 5D). These results indicate that MEK but not ROS predominantly contributes to p53-independent anti-tumor activity of PRIMA-1Met.
PRIMA-1Met suppresses CRC growth in xenograft mouse model by inhibiting MEK activity
To confirm that the anti-tumor activity of PRIMA-1Met is mediated by MEK in vivo, we established subcutaneous xenograft nude mice model implanted with HCT116neg or HCT116wt cells. Once the tumor xenografts grew to approximately 50 mm3, the mice were randomly assigned to three groups: vehicle (treated with PBS); lower dose of PRIMA-1Met (20 mg/kg); and higher dose of PRIMA-1Met (100 mg/kg). PRIMA-1Met markedly reduced tumor size in both HCT116neg and HCT116wt derived xenografts in a dose dependent manner (Figure 6A). Compared to vehicle group, PRIMA-1Met (20 mg/kg or 100 mg/kg) significantly inhibited average tumor volume (P < 0.05). In addition, body weight was not significantly affected by PRIMA-1Met (Figure 6B), indicating that treatment dose of PRIMA-1Met was well tolerated by the mice.
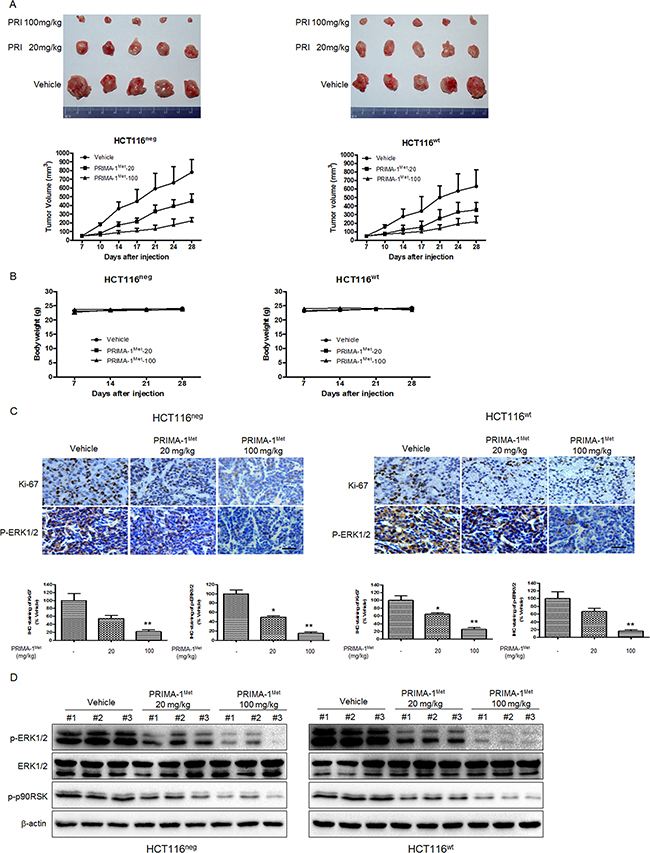
Figure 6: PRIMA-1Met suppresses tumor growth in vivo by inhibiting MEK activity. A. Mice bearing HCT116neg or HCT116wt tumors were treated with PBS, PRIMA-1Met (20 mg/kg or 100 mg/kg). Tumor volume was measured twice a week and data were represented as mean ± SEM. *P<0.05; **P<0.01 vs. vehicle group. B. The body weight of mice in each group was measured and recorded once a week. Data were shown as mean ± SEM. C. Immunohistochemical staining of Ki-67 and phosphorylated ERK1/2 in paraffin embedded sections of tumor xenografts. Scale bar: 50 um. *P<0.05; **P<0.01. D. The levels ofp-ERK1/2, T-ERK1/2 and p-p90RSK in the dissected xenografts were determined by Western blot analysis.
Immunohistochemical analysis of tumor samples showed that the phosphorylation of ERK1/2 was substantially reduced in treatment groups compared to vehicle group. Additionally, PRIMA-1Met inhibited HCT116neg and HCT116wt cell proliferation because fewer cells were positively stained with proliferation marker Ki-67 compared to vehicle group (Figure 6C). Western blot analysis of tumor samples showed that the phosphorylation of ERK1/2 was substantially suppressed in treatment groups compared to vehicle group in a dose dependent manner (Figure 6D). Taken together, our data indicate that PRIMA-1Met inhibits CRC growth in vivo independent of p53 status by suppressing MEK activity.
DISCUSSION
Tumor suppressor p53 is encoded by TP53 gene which is frequently mutated in half of all human cancers, including breast cancer, multiple myeloma, lung cancer and colorectal cancer [23]. The most mutations in TP53 such as Y220C or R175H are clustered within the DNA-binding domain and thus impair transcription activity of p53, resulting in the inactivation of p53 targets such as p21, Bax, PUMA and Noxa [24]. As a consequence, loss of p53 tumor suppressor activity prevents cancer cells from p53 mediated cell senescence, cell cycle arrest and apoptosis. Moreover, mutant p53 acquires gain-of-function to promote malignant phenotypes of cancer [25]. Thus it is urgent to develop novel anti-cancer strategies targeting mutant p53.
PRIMA-1 and its methylated form PRIMA-1Met were originally isolated from the low-molecular-weight compounds library for the ability to restore sequence-specific DNA binding and transcription-dependent apoptotic function to mutant p53 in vitro and in vivo [10]. Although PRIMA-1Met has demonstrated preferential inhibitory effect on cancer cells harboring mutant p53, recent studies suggest p53-independent activity of PRIMA-1Met. Saha et al. found that PRIMA-1Met induced apoptosis via p53-independent Noxa induction [26]. Grinter et al. found that PRIMA-1Met exerted anti-cancer effect by the inhibition of oxidosqualene cyclase (OSC), a key enzyme involved in cholesterol synthetic pathway [15]. Because p53 family members p63 and p73 have similar sequence homology to p53 in DNA binding domains, PRIMA-1Met was found to restore mutant p63 and p73 with apoptotic function irrespective of p53 status [27]. Meanwhile, PRIMA-1Met could induce ROS generation and cause p53-independent massive cell death by converting TrxR1 to NADPH oxidase or impairing GSH metabolism [16, 28]. These observations prompted us to evaluate p53 independent ant-tumor activities of PRIMA-1Met.
In the present study, we first used PRIMA-1Met to treat six colorectal cancer cell lines with different p53 status. We found that PRIMA-1Met inhibited the proliferation and growthof all cell lines in a dose and time dependent manner, and high dose of PRIMA-1Met triggered cell apoptosis regardless of p53 status. In addition, we found that treatment with 25 or 50 uM PRIMA-1Met caused G2/M arrest in HCT116neg cells (Supplementary Figure S4), consistent with previous study [29]. Moreover, SW480 and DLD-1 cell lines with depleted mutant p53 were as sensitive as their parental cells to PRIMA-1Met treatment, demonstrating that PRIMA-1Met could suppress CRC cell growth in a p53-independent manner.
Kinase signaling pathways, such as Ras/Raf/MEK and PI3-K/Akt/mTOR cascades, are generally hyperactivated and contribute to cancer cell proliferation, migration, invasion and chemoresistance [30]. Therefore, we performed kinase profiling assay by screening total 40 kinases and identified MEK kinase as a potential target that mediates p53-independent anti-tumor activity of PRIMA-1Met. MEK kinase and its downstream effectors are consistently activated owing to aberrant mutant activation of Ras and B-Raf in colorectal cancer [31]. Once activated, MEK1 and MEK2 form heterodimer kinase and consecutively mediate downstream ERK1/ERK2 phosphorylation. Several selective and allosteric MEK inhibitors that bind non-ATP competitive site, such as Trametinib, Cobimetinib, MEK162, AZD6244 and PD-0325901, have shown promise in preclinical and clinical studies [32–34]. In contrast, we speculated that PRIMA-1Met directly binds MEK in the ATP-binding pocket based on the results of automated docking analysis, pull-down binding and ATP competitive assay. Thus the mode of action of PRIMA-1Met in inhibiting MEK activity seems to be different from that of MEK inhibitors binding non-ATP competitive site. Further studies are needed to compare the efficacy of PRIMA-1Met and other MEK inhibitors.
By detecting the phosphorylation of MEK substrate ERK, we showed that PRIMA-1Met potently inhibited MEK kinase activity in CRC cells in a dose dependent manner. More importantly, knockdown of mutant p53 in SW480 did not affect the inhibition of MEK activity by PRIMA-1Met, confirming that PRIMA-1Met could inhibit MEK kinase activity in cells with different p53 status. Moreover, in vivo study showed that PRIMA-1Met inhibited MEK activity, leading to significant tumor regression in HCT116neg and HCT116wt xenograft model compared with vehicle group. These in vitro and in vivo data indicate that PRIMA-1Met is a novel MEK1 inhibitor.
In this study we depleted endogenous MEK1 in CRC cells to investigate whether MEK1 is indispensable for anti-tumor activity of PRIMA-1Met since many small molecules that targeting the main target have off targets [35, 36]. As expected, the depletion of MEK1 abrogated PRIMA-1Met induced suppression of CRC cell growth. To avoid the confounding potential of ROS, which could be induced by PRIMA-1Met and mediate p53-independent cancer cell apoptosis [16], we further assessed ROS level and found that ROS production was induced after PRIMA-1Met treatment. However, ROS scavenger NAC only slightly compensated for PRIMA-1Met induced CRC cell growth suppression. Collectively, these results demonstrate that direct binding to MEK and inhibiting its activity is the main, if not exclusive, mechanism by which PRIMA-1Met suppresses CRC independent of p53 status.
In conclusion, we provide the first lines of evidence that PRIMA-1Met inhibits colorectal cancer cell proliferation and tumor growth predominately by directly targeting MEK in p53 independent manner. Consequently, targeting both MEK and mutant p53 by PRIMA-1Met may allow us to initiate new strategy for the treatment of malignant colorectal cancer that harbors both hyper-activated Ras/Raf/MEK signaling and p53 mutation.
MATERIALS AND METHODS
Cell culture and transfection
HCT116neg, HCT116wt, LOVO, DLD-1, SW480, HT-29 and JB6 P+ mouse skin epidermal cells were obtained from American Type Culture Collection (ATCC). HCT116neg, HCT116wt and HT-29 cells were cultured in McCoy’s 5a and other colorectal cancer cells were cultured in RPMI-1640 supplemented with 10% fetal bovine serum (FBS) and 1% antibiotic. JB6+t mouse skin epidermal cells were cultured in Minimum Essential Medium (MEM) supplemented with 5% FBS and 1% antibiotic. Cells were cultured at 37°C in a 5% CO2 incubator. Lentivirus shRNAs pLKO.1-sh-mock, pLKO.1-sh-p53 (#1:TRCN0000003756, #2:TRCN0000003758) and pLKO.1-sh-MEK (#1:TRCN0000002314, #2:TRCN0000002315) were ordered from Thermo Scientific (Huntsville, AL, USA), and co-transfected with packaging vectors (pMD2.0G and psPAX from Addgene) into HEK-293T package cells by using Lipofectamine 2000 (Thermo Scientific, MA, USA). 36 hours after transfection, cell supernatants were harvested, filtered and used to infect target cells. 24 hours after infection, cells were supplemented with 8 ug/ml puromycin to select stable cell lines and RNAi efficiency was confirmed by immunoblotting. PRIMA-1Met was purchased from Tocris Bioscience (R&D Systems, MN, USA). AZD6244 was purchased from Array BioPharma (Boulder, CO, USA).
MTS assay
The cells were seeded (1×103/well) in 96-well plates and cultured overnight. Cells were treated with different concentrations of PRIMA-1Met and cell proliferation inhibition was measured by MTS assay (Promega, WI, USA) according to the manufacturer’s instructions.
Anchorage-independent cell growth assay
A total of 8,000 cells were suspended in 1 ml solidified Basal Medium Eagle containing 10% FBS and added to 0.33% top agarose with various concentrations of PRIMA-1Met. A base layer of 0.6% agarose was placed at the bottom of top agar. Three weeks after seeding, colonies were counted in low-power field of light microscope using the Image-Pro Plus Software (vs.4) program (Media Cybernetics).
Kinase profiling
Kinase profile assay was performed according to Millipore’s Kinase Profiler protocols. The concentration of PRIMA-1Met and ATP for each kinase analysis was set at 100 uM and 10 uM, respectively.
Molecular modeling
Briefly, the PDB files of compound PRIMA-1Met was generated from the PRODRG server [37]. The X-ray crystallographic structures of MEK-1/2 (PDB code: 1S9I [38]) were downloaded from Protein Data Bank and prepared by using the Protein Preparation Wizard in Maestro v9.2. Hydrogen atoms were added to the complexes consistently with a pH of 7 and water molecules were deleted. Ligand docking the compound to MEK-1/2 monomer was accomplished using the automated docking tool AutoDock 4. Ten independent runs were carried out to yield ten AutoDock docking solutions, which were ranked by the calculated energy favorable scores. Top hits based on hydrogen-bond interactions were considered as top-ranked conformations of docked complex.
In vitro kinase assay
Active MEK1 (100 ng) and its inactive substrate ERK1 (1 ug) were purchased from Millipore (Temecula, CA, USA), mixed in the presence of various doses of PRIMA-1Met, and incubated in 100 uM ATP and ×1 kinase buffer (10 mM MgCl2, 1 mM EDTA, 50 mM Tris-HCl pH 7.5, 0.01% Brij35, 1 mM DTT; Cell Signaling Technology) at 30°C for 30 min. Then the samples were resolved in ×6 SDS loading buffer. Phosphorylated ERK1 and total MEK protein level were analyzed by Western blot analysis.
In vitro ATP competitive assay and pull-down binding assay
PRIMA-1Met-conjugated Sepharose 4B beads (GE Healthcare, Pittsburgh, PA, USA) were prepared according to the manufacturer’s instruction. The ATP competition assay was conducted by incubating 300 ng active MEK1 protein with various concentrations of ATP (0, 10, 100 uM) at 4°C for 2 h, followed by incubation with 100 ul PRIMA-1Met-conjugated Sepharose 4B beads or Sepharose 4B beads only for another 2 h in ×1 reaction buffer (150 mM NaCl, 50 mM Tris-HCl pH 7.5, 5 mM EDTA, 0.01% NP40, 1 mM DTT, 0.02 mM phenylmethysulfonyl fluoride, 2 ug/ml bovine serum albumin and ×1 protease inhibitor mixture). For pull-down binding assay, HCT116neg cell lysates (600 ug) were incubated with 100 ul PRIMA-1Met-conjugated Sepharose 4B beads or Sepharose 4B beads only overnight at 4°C, then the beads were washed with ×1 wash buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 1 mM DTT, 0.02 mM phenylmethylsulfonyl fluoride and 0.01% NP40). The proteins that bound to PRIMA-1Met-conjugated Sepharose 4B beads were resolved by 10% SDS-PAGE and detected by Western blot analysis.
Measurement of ROS
HCT116neg and HCT116wt cells were cultured in six-well plate and grown to subconfluency. Cells were treated with PRIMA-1Met in the presence or absence of NAC for indicated time. Then cells were stained with 20 uM DCFHDA for 30 min and fluorescence was analyzed using flow cytometer in the FITC channel. The fluorescence intensity of 15,000 cells was measured by WinMDI 2.8 software and normalized to vehicle group.
Western blot analysis
Cells were lysed with RIPA buffer (150 mM NaCl, 50 mM Tris-HCl pH 8.0, 0.25% SOD, 0.1% sodium dodecyl sulfate, 1 mM EDTA, 1% NP40 and ×1 protease/phosphatase inhibitor cocktail). Lysates from mouse xenograft were prepared by homogenizing tumor mass in RIPA buffer. Proteins were separated by SDS-PAGE and transferred to PVDF membranes (Amersham Pharmacia Biotech). Membranes were blocked with 5% non-fat milk and then probed with appropriate primary antibody overnight at 4ºC. Then membranes were washed in PBS three times and incubated with horseradish peroxidase-conjugated secondary antibody. Protein bands were visualized using an enhanced chemiluminescence reagent kit (Amersham). Antibodies against cleaved caspase 3, total MEK1/2, phosphorylated MEK1/2 (Ser217/221), phosphorylated ERK1/2 (Thr202/Tyr204), phosphorylated p90RSK (Thr359/Ser363) and b-actin were purchased from Cell Signaling (Beverly, MA, USA). Antibodies against total ERK1/2, total MEK1/2, TP53, p21 and Ki-67 were from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Xenograft mouse model
Athymic nude mice (6-9 weeks old) were provided by SLAC Laboratory Animal Co. Ltd. (Shanghai, China). Briefly, cells in logarithmic phase were harvested and suspended in cold PBS at 5×106/mL. 1×106 cells (in 200 μl of PBS) were injected subcutaneously into right flank of each mouse. Once the tumor volume reached around 50 mm3, mice were randomly divided into three groups (n=5 each): (i) vehicle group; (ii) 20 mg/kg PRIMA-1Met-treated group; (iii) 100 mg/kg PRIMA-1Met-treated group. PRIMA-1Met or vehicle (PBS) was intraperitoneally injected daily for 5 days, stopped for 2 days and reinjected for additional 5 days. Tumor volume was measured twice a week based on the following formula: volume = (Length×Width2)/2. Mice body weight was measured and recorded once a week. Mice were euthanized on day 28 and tumor tissues were dissected for further analysis.
Immunohistochemistry analysis
The tumor samples were fixed in 10% neutral formalin solution and embedded in paraffin. Then sections were cut into fragments of 6 um, deparaffinized and hydrated. Tissues on slides were permeabilized by 0.5% Triton X-100 and incubated overnight at 4ºC with primary antibodies against Ki67 and phosphorylated ERK1/2. The slides were washed by PBS for 5 times and incubated with 1:200 dilution of secondary biotinylated antibody for 2 h at room temperature. All slides were developed using horseradish peroxidase-streptavidin (Santa Cruz Biotechnology, CA, USA). Images were taken under microscope and signal densities were analyzed using the Image-Pro Plus Software (Media Cybernetics).
Statistical analysis
All data were presented as mean values ± SD or SE as indicated. The Student’s t test (two groups only) or a one-way ANOVA (more than two groups) was used for the comparison. p < 0.05 was accepted as statistical significance.
ACKNOWLEDGMENTS
This work was supported by Grant No. 81201779 (Hua Xiong) from the National Natural Science Youth Foundation; Grant No. 81502118 (Yanmei Zou) from the National Natural Science Youth Foundation; Grant No. 2014CFB250 (Yanmei Zou) from the Natural Science Foundation of Hubei Province; Grant No. 81372434 (Huihua Xiong) from the National Natural Science Foundation. The funding source has no role in the writing, submission and publication of this manuscript.
CONFLICTS OF INTEREST
No potential conflicts of interest were disclosed.
REFERENCES
1. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Jr., Kinzler KW. Cancer genome landscapes. Science. 2013; 339:1546-1558.
2. Meador CB, Pao W. Old Habits Die Hard: Addiction of BRAF-Mutant Cancer Cells to MAP Kinase Signaling. Cancer discovery. 2015; 5:348-350.
3. Medico E, Russo M, Picco G, Cancelliere C, Valtorta E, Corti G, Buscarino M, Isella C, Lamba S, Martinoglio B, Veronese S, Siena S, Sartore-Bianchi A, et al. The molecular landscape of colorectal cancer cell lines unveils clinically actionable kinase targets. Nature communications. 2015; 6:7002.
4. Ebi H, Corcoran RB, Singh A, Chen Z, Song Y, Lifshits E, Ryan DP, Meyerhardt JA, Benes C, Settleman J, Wong KK, Cantley LC, Engelman JA. Receptor tyrosine kinases exert dominant control over PI3K signaling in human KRAS mutant colorectal cancers. The Journal of clinical investigation. 2011; 121:4311-4321.
5. Li XL, Zhou J, Chen ZR, Chng WJ. P53 mutations in colorectal cancer - molecular pathogenesis and pharmacological reactivation. World J Gastroenterol. 2015; 21:84-93.
6. Hainaut P, Wiman KG. 30 years and a long way into p53 research. Lancet Oncol. 2009; 10:913-919.
7. Duffy MJ, Synnott NC, McGowan PM, Crown J, O'Connor D, Gallagher WM. p53 as a target for the treatment of cancer. Cancer treatment reviews. 2014; 40:1153-1160.
8. Zhang C, Lin M, Wu R, Wang X, Yang B, Levine AJ, Hu W, Feng Z. Parkin, a p53 target gene, mediates the role of p53 in glucose metabolism and the Warburg effect. Proc Natl Acad Sci U S A. 2011; 108:16259-16264.
9. Freed-Pastor WA, Prives C. Mutant p53: one name, many proteins. Genes Dev. 2012; 26:1268-1286.
10. Bykov VJ, Issaeva N, Shilov A, Hultcrantz M, Pugacheva E, Chumakov P, Bergman J, Wiman KG, Selivanova G. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat Med. 2002; 8:282-288.
11. Bykov VJ, Issaeva N, Zache N, Shilov A, Hultcrantz M, Bergman J, Selivanova G, Wiman KG. Reactivation of mutant p53 and induction of apoptosis in human tumor cells by maleimide analogs. J Biol Chem. 2005; 280:30384-30391.
12. Zache N, Lambert JM, Wiman KG, Bykov VJ. PRIMA-1MET inhibits growth of mouse tumors carrying mutant p53. Cell Oncol. 2008; 30:411-418.
13. Foster BA, Coffey HA, Morin MJ, Rastinejad F. Pharmacological rescue of mutant p53 conformation and function. Science. 1999; 286:2507-2510.
14. Lambert JM, Gorzov P, Veprintsev DB, Soderqvist M, Segerback D, Bergman J, Fersht AR, Hainaut P, Wiman KG, Bykov VJ. PRIMA-1 reactivates mutant p53 by covalent binding to the core domain. Cancer Cell. 2009; 15:376-388.
15. Grinter SZ, Liang Y, Huang SY, Hyder SM, Zou X. An inverse docking approach for identifying new potential anti-cancer targets. J Mol Graph Model. 2011; 29:795-799.
16. Peng X, Zhang MQ, Conserva F, Hosny G, Selivanova G, Bykov VJ, Arner ES, Wiman KG. APR-246/PRIMA-1MET inhibits thioredoxin reductase 1 and converts the enzyme to a dedicated NADPH oxidase. Cell Death Dis. 2013; 4:e881.
17. Sebolt-Leopold JS, Herrera R. Targeting the mitogen-activated protein kinase cascade to treat cancer. Nature reviews Cancer. 2004; 4:937-947.
18. Saif MW, Shah M. K-ras mutations in colorectal cancer: a practice changing discovery. Clin Adv Hematol Oncol. 2009; 7:45-53, 64.
19. Surade S, Blundell TL. Structural biology and drug discovery of difficult targets: the limits of ligandability. Chem Biol. 2012; 19:42-50.
20. Chan DA, Giaccia AJ. Harnessing synthetic lethal interactions in anticancer drug discovery. Nat Rev Drug Discov. 2011; 10:351-364.
21. Engelman JA, Chen L, Tan X, Crosby K, Guimaraes AR, Upadhyay R, Maira M, McNamara K, Perera SA, Song Y, Chirieac LR, Kaur R, Lightbown A, et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med. 2008; 14:1351-1356.
22. Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J Comput Chem. 2009; 30:2785-2791.
23. Petitjean A, Achatz MI, Borresen-Dale AL, Hainaut P, Olivier M. TP53 mutations in human cancers: functional selection and impact on cancer prognosis and outcomes. Oncogene. 2007; 26:2157-2165.
24. Vousden KH, Prives C. Blinded by the Light: The Growing Complexity of p53. Cell. 2009; 137:413-431.
25. Muller PA, Vousden KH. Mutant p53 in cancer: new functions and therapeutic opportunities. Cancer Cell. 2014; 25:304-317.
26. Saha MN, Jiang H, Yang Y, Reece D, Chang H. PRIMA-1Met/APR-246 displays high antitumor activity in multiple myeloma by induction of p73 and Noxa. Mol Cancer Ther. 2013; 12:2331-2341.
27. Rokaeus N, Shen J, Eckhardt I, Bykov VJ, Wiman KG, Wilhelm MT. PRIMA-1(MET)/APR-246 targets mutant forms of p53 family members p63 and p73. Oncogene. 2010; 29:6442-6451.
28. Tessoulin B, Descamps G, Moreau P, Maiga S, Lode L, Godon C, Marionneau-Lambot S, Oullier T, Le Gouill S, Amiot M, Pellat-Deceunynck C. PRIMA-1Met induces myeloma cell death independent of p53 by impairing the GSH/ROS balance. Blood. 2014; 124:1626-1636.
29. Russo D, Ottaggio L, Foggetti G, Masini M, Masiello P, Fronza G, Menichini P. PRIMA-1 induces autophagy in cancer cells carrying mutant or wild type p53. Biochim Biophys Acta. 2013; 1833:1904-1913.
30. Samatar AA, Poulikakos PI. Targeting RAS-ERK signalling in cancer: promises and challenges. Nat Rev Drug Discov. 2014; 13:928-942.
31. Zhu F, Zykova TA, Kang BS, Wang Z, Ebeling MC, Abe Y, Ma WY, Bode AM, Dong Z. Bidirectional signals transduced by TOPK-ERK interaction increase tumorigenesis of HCT116 colorectal cancer cells. Gastroenterology. 2007; 133:219-231.
32. Carlino MS, Todd JR, Gowrishankar K, Mijatov B, Pupo GM, Fung C, Snoyman S, Hersey P, Long GV, Kefford RF, Rizos H. Differential activity of MEK and ERK inhibitors in BRAF inhibitor resistant melanoma. Mol Oncol. 2014; 8:544-554.
33. Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, Sosman J, Hamid O, Schuchter L, Cebon J, Ibrahim N, Kudchadkar R, Burris HA, 3rd, Falchook G, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med. 2012; 367:1694-1703.
34. Hatzivassiliou G, Haling JR, Chen H, Song K, Price S, Heald R, Hewitt JF, Zak M, Peck A, Orr C, Merchant M, Hoeflich KP, Chan J, et al. Mechanism of MEK inhibition determines efficacy in mutant KRAS- versus BRAF-driven cancers. Nature. 2013; 501:232-236.
35. O'Connor KA, Roth BL. Finding new tricks for old drugs: an efficient route for public-sector drug discovery. Nat Rev Drug Discov. 2005; 4:1005-1014.
36. Wauson EM, Guerra ML, Barylko B, Albanesi JP, Cobb MH. Off-target effects of MEK inhibitors. Biochemistry. 2013; 52:5164-5166.
37. Schuttelkopf AW, van Aalten DM. PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr D Biol Crystallogr. 2004; 60:1355-1363.
38. Ohren JF, Chen H, Pavlovsky A, Whitehead C, Zhang E, Kuffa P, Yan C, McConnell P, Spessard C, Banotai C, Mueller WT, Delaney A, Omer C, et al. Structures of human MAP kinase kinase 1 (MEK1) and MEK2 describe novel noncompetitive kinase inhibition. Nat Struct Mol Biol. 2004; 11:1192-1197.