
Introduction
ATM is a serine/threonine protein kinase that is rapidly activated by several stressors including nuclear replication stress [1], oxidative stress [2] [3] and mitochondrial dysfunction [4] [5]. Activation of ATM by DNA damage triggers cell cycle checkpoints, including G1/S and G2/M [6] [7], and DNA repair or apoptosis, preventing the transmission of mutations to daughter cells. The absence of ATM in humans and mice increases the incidence of tumors of predominantly lymphoid origin, with most tumors appearing in Atm-/- mice being thymic lymphomas [8] [9].
MAPK7 (also named ERK5) is a member of the mammalian mitogen-activated protein kinase family with a kinase domain homologous to that of MAPK3 (ERK1) and MAPK1 (ERK2), and a unique C-terminal domain that determines its cellular location and activity [10]. Deletion of Mapk7 in mice leads to embryonic lethality due to defective vascular and cardiac development [11-13]. MAPK7 controls the expression of genes that regulate angiogenesis and cardiovascular development [14], is necessary for endothelial homeostasis [15] [16] and contributes to B-cell survival [17] and erythropoiesis in mice [18].
MAPK7 is deregulated in numerous types of cancer [19], including lymphoma [20]. MAPK7 may also mediate chronic inflammation-dependent tumorigenesis in the skin [21] [22]. The tumorigenicity of MAPK7 has been related to its capacity to interact with promyelocytic leukemia protein and inhibit its tumor suppressor activity [23]. By blocking the interaction of promyelocytic leukemia protein with MDM2, active MAPK7 might facilitate the inhibition of tumor suppressor p53 by MDM2 [24]. Based on the use of the MAPK7 inhibitor XMD8-92, it has been shown that MAPK7-dependent inhibition of p53 contributes to tumor growth in a xenograft model [23] [24], though XMD8-92 could have another cellular target besides MAPK7 [25].
In contrast to the role of ATM in triggering cell cycle checkpoints upon DNA damage, MAPK7 activity peaks at G2/M and is required for progression through this cell cycle phase [26]. The data describing opposing roles of ATM and MAPK7 in cell cycle regulation and tumor growth suggest that these could be functionally linked. To test this hypothesis, and as Atm-/- mice show a strong predisposition to thymic lymphomas, we eliminated MAPK7 in the hematopoietic system of Atm-/- mice and assessed the time of tumor appearance. This approach can yield information on the role played by MAPK7 during the spontaneous progression of a cell toward malignancy. We show that loss of Mapk7 in the hematopoietic system of Atm-/- mice increases the DNA damage response in thymocytes and delays death by spontaneous tumor development in Atm-/- mice.
Results
Absence of MAPK7 in the hematopoietic system delays death by spontaneous tumor development in Atm-/- mice
To determine whether Mapk7 and Atm are functionally linked in tumorigenesis, and given that Atm-/- mice spontaneously develop thymic lymphomas, we removed Mapk7 in the hematopoietic system of Atm-/- mice, and assessed the spontaneous rate of tumor development in Atm-/- and in Atm/Mapk7-double null mice. To this end, Atm+/- mice were crossed to Mapk7loxP/loxPvav-cre mice (hereafter referred to as Mapk7hemat-/- mice; Supplementary Figure 1), which lack Mapk7 in hematopoietic cells [27] [28] or to control Mapk7loxP/loxP mice (referred to as wild-type mice), and double heterozygous F1 progeny were crossed to obtain F2 Mapk7hemat-/-Atm-/- mice and Mapk7loxP/loxPAtm-/- mice (referred to as Atm-/- mice).
Most Mapk7hemat-/-Atm-/- mice grew to adulthood and showed the low body weight characteristic of Atm-/- mice (data not shown). Genotyping of young offspring from 11 (Mapk7hemat-/-Atm+/-) x (Mapk7loxP/loxPAtm+/-) crosses and 9 Mapk7hemat-/-Atm+/- mice intercrosses revealed no statistical differences in the frequency of mice homozygous for Atm (p = 0.35 by chi-squared analysis) as compared with the expected Mendelian frequency (Supplementary Figure 2). However, mice with null homozygosis for both Atm and Mapk7 were born at a lower frequency than expected (p = 0.024), suggesting that the absence of both genes in the hematopoietic system produces a developmental drawback. However, the possibility that it is the presence of Cre recombinase, rather than the absence of MAPK7 what affects the birth rate of Atm-/- mice cannot be excluded.
We then assessed the survival of these adult mice. In agreement with previous studies, we did not observe spontaneous tumor formation in Mapk7hemat-/- mice [17] [18] [27] [29] and no mice died during the first year of life (Figure 1A). In contrast, the median survival time of Atm-/- mice was 153 days of age (Figure 1A) with ~80% of these mice having died at 300 days of age. Interestingly, the median survival time of Mapk7hemat-/-Atm-/- mice was 281 days of age (Figure 1A). Thus, Mapk7hemat-/-Atm-/- mice showed a significant increase in survival compared to Atm-/- mice (p = 0.02 at day 320, log-rank test), indicating that MAPK7 deficiency in the hematopoietic system delays death by spontaneous tumor development in Atm-/- mice and suggesting an antagonistic interaction between the MAPK7- and ATM-signaling pathways affecting thymic lymphoma development.
Most tumors appearing in Atm-/- and Mapk7hemat-/-Atm-/- mice were thymic lymphomas. However, older mice (~1 year or more) presented tumors of different types or died of unknown causes associated with a wasting syndrome (Supplementary Table I). The longer survival of Mapk7hemat-/-Atm-/- mice might facilitate the emergence of other disorders linked to the absence of ATM [30]. The thymic tumor cells were clonal, showed either an immature phenotype with a CD4+CD8+ signature or were single-positive (SP) thymocytes (Supplementary Figure 3).
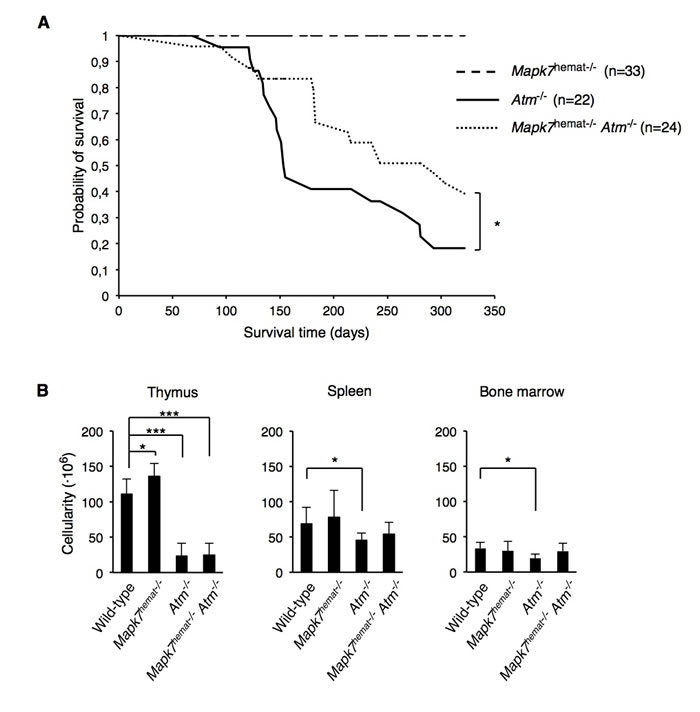
Figure 1: Absence of MAPK7 delays tumorigenesis in Atm-/- mice. A. Kaplan-Meier survival curves of Mapk7hemat-/- (n = 33), Atm-/- (n = 22) and Mapk7hemat-/-Atm-/- (n = 24) mice over 320 days. P values were obtained by the log-rank test. B. Total cellularity of wild-type (n ≥ 5), Mapk7hemat-/- (n = 7), Atm-/- (n = 7) and Mapk7hemat-/-Atm-/- (n ≥ 8), in the indicated organs.
Normal erythropoiesis in Mapk7hemat-/-Atm-/- mice and impaired B cell development in Atm-/- and Mapk7hemat-/-Atm-/-mice
Some hematopoietic-specific traits of Atm-/- mice, such as reduced thymocyte cellularity, were also evident in Mapk7hemat-/-Atm-/- mice, whereas Mapk7hemat-/- mice presented a small increase (Figure 1B). Interestingly, Atm-/- mice, but not Mapk7hemat-/-Atm-/- mice, showed also reduced cellularity in the bone marrow and spleen. Absence of Atm or Mapk7 leads to hematopoietic impairment [17] [18] [30], we therefore analyzed in more detail the erythroid and B cell development in the bone marrow in 4- to 8-week-old mice.
The generation of erythropoietic precursors (cells CD71+ter119+) diminished in the bone marrow of Mapk7hemat-/- mice but not in Atm-/- mice (Figure 2A) [18]. Interestingly, the erythropoiesis impairment observed in Mapk7hemat-/- mice disappeared in Mapk7hemat-/-Atm-/- mice, indicating that the absence of Atm compensates for the absence of Mapk7 during erythropoiesis in bone marrow of young mice (Figure 2A, bottom).
The analysis of B cell progenitors in the bone marrow showed that generation of the B cell precursor B220lowIgM+ was impaired in the three genotypes: Mapk7hemat-/-, Atm-/- and Mapk7hemat-/-Atm-/- mice (Figure 2B) [17] [18]. Moreover, the generation of the B cell precursor B220highIgM+ was also impaired in Mapk7hemat-/- and Mapk7hemat-/-Atm-/- mice. The combined absence of Mapk7 and Atm did not have an additive effect on decreasing further the bone marrow B220lowIgM+ or B220highIgM+ pools.
In the lymph nodes and spleen, the percentage of mature B cells was not affected by the absence of Atm and/or Mapk7. Regarding T cells, while CD8+ T cells were not altered, a decrease in the percentage of CD4+ T cells was observed in the lymph nodes of Atm-/- mice and in the spleen of mice lacking both Mapk7 and Atm (Supplementary Figure 4).
These results indicate that these two kinases play a different role in erythroid and B cell development, with both kinases contributing to B cell development, but only Mapk7 being necessary for normal erythroid development in young mice.
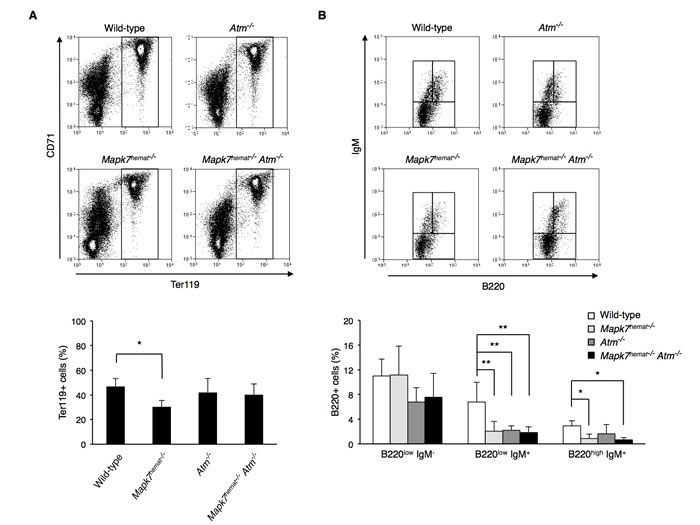
Figure 2: Normal erythropoiesis in Mapk7hemat-/-Atm-/-mice and impaired B cell development in Atm-/- and Mapk7hemat-/-Atm-/- mice. A. Representative flow cytometry profiles representing the erythroid population in the bone marrow. Dot plots representing erythroid precursor distribution based on ter119 and CD71 expression (top). Percentage of ter119+ population in wild-type (n = 3), Mapk7hemat-/- (n = 7), Atm-/- (n = 3) and Mapk7hemat-/-Atm-/- (n = 6) bone marrow (bottom). B. Representative flow cytometry profiles representing B cell precursor distribution (B220lowIgM-, B220lowIgM+, B220highIgM+) in the bone marrow population (top). Percentage of bone marrow B cell precursors in wild-type (n = 3), Mapk7hemat-/- (n = 7), Atm-/- (n = 3) and Mapk7hemat-/-Atm-/- (n = 6) mice.
Equivalent thymic cellular distribution in Atm-/- and Mapk7hemat-/-Atm-/- mice
We then assessed whether Mapk7 and Atm are functionally linked during T cell development, by performing a flow cytometry analysis on thymocytes isolated from 4- or 5-week-old mice. The percentage of SP CD4+ thymocytes decreased in mice strains lacking Atm, whereas the percentage of CD4-CD8- double-negative (DN), CD4+CD8+ double-positive (DP) and SP CD8+ thymocytes did not change (Figure 3A) [9] [31]. When the absolute cell numbers were considered, the DP and SP CD4+ thymocytes decreased in mice strains lacking Atm (Figure 3B) [31]. Atm-/- mice and Mapk7hemat-/-Atm-/- mice presented a similar number of DN, DP and SP thymocytes (Figure 3B).
Thymocytes at DN stage start the recombination of V(D)J gene segments, a process that ends at the DP stage and requires the activity of ATM for proper repair of the DNA double-strand breaks (DSB) produced during DNA recombination [31] [32]. The lower number of DP and mature SP CD4+ thymocytes in mice strains lacking Atm suggests that a partial arrest of thymocyte maturation during the DN stage is taking place in these mice, probably as a result of the incapacity to resolve the DSB generated during DNA recombination [33].
As the main proliferative population, the DN thymocytes were analyzed in more detail. The DN pool can be fractionated in progressively maturing DN1 to DN4 subsets according to the expression of the IL-2 receptor alpha chain (CD25) and CD44 (Figure 3C, 3D). Thymocyte precursors initiate the recombination of tcrg, tcrd and tcrb mainly at the CD4−CD8−CD44−CD25+ (DN3) stage, and completion of this process and expression of pre-TCR leads to differentiation into CD4−CD8−CD44−CD25− (DN4) cells. Neither the percentage of DN populations nor the number of cells in each DN pool did show differences between wild-type and the other genotypes, or between Atm-/- mice and Mapk7hemat-/-Atm-/- mice (Figure 3C, 3D). Only Mapk7hemat-/- mice and Mapk7hemat-/-Atm-/- mice did show a different number of CD4−CD8−CD44+CD25- (DN1) and DN4 cells (Figure 3D).
Taken together, these results indicate that deletion of Mapk7 in Atm-/- mice did not change the distribution of major thymic populations.
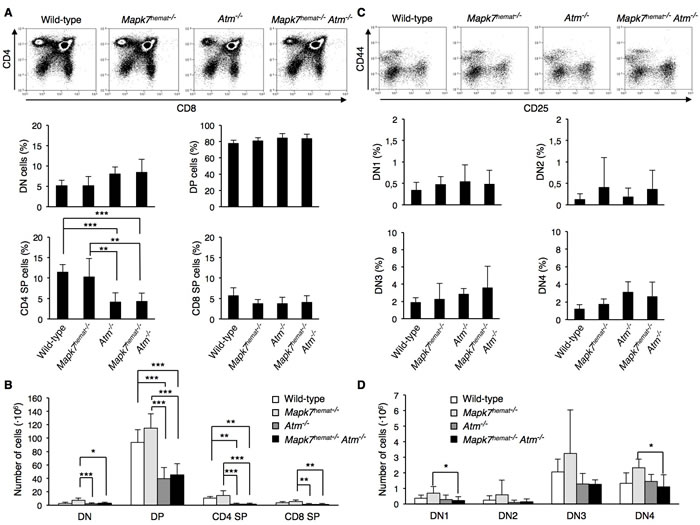
Figure 3: Equivalent thymic cellular distribution in Atm-/- and Mapk7hemat-/-Atm-/- mice. A. Representative flow cytometry profiles representing thymic cell distribution based on CD4 and CD8 expression (top). Percentage of thymic cell subsets (bottom). Thymocytes were isolated from 4- or 5-week-old wild-type (n = 6), Mapk7hemat-/- (n = 7), Atm-/- (n = 7) and Mapk7hemat-/-Atm-/- (n = 8) mice, stained with anti-CD4-APC and anti-CD8-FITC, and then analyzed by flow cytometry. Data represent mean +/- SD of CD4-CD8- DN, CD4+CD8+ DP and SP CD4+ and CD8+ cell subsets. B. Number of thymocytes in each developmental stage, for the same mice analyzed in A.. C. Representative flow cytometry profiles of thymic DN population based on CD44 and CD25 expression (top). Percentage of DN thymocyte cell subsets (bottom). Thymocytes were isolated from 4- and 5-week-old wild-type (n = 6), Mapk7hemat-/- (n = 7), Atm-/- (n = 7) and Mapk7hemat-/-Atm-/- (n = 8) mice, stained with anti-CD4-APC, anti-CD8-alexa 488, anti-CD44-PerCP and anti-CD25-PE, and then analyzed by flow cytometry. Data represent mean +/- SD of CD44+CD25- (DN1), CD44+CD25+ (DN2), CD44-CD25+ (DN3) and CD44-CD25- (DN4) subsets. D. Number of thymocytes in each DN cell subset for the same mice analyzed in C..
Increased G2/M cell cycle phase accumulation in DN thymocytes of Mapk7hemat-/-Atm-/- mice
Although no major changes between the thymocyte distribution of 4- to 5-week old Atm-/- mice and Mapk7hemat-/-Atm-/- mice could be found, the delay in thymic lymphoma development in the latter suggest that the proliferative capacity of thymocytes or their apoptotic response to DNA damage has changed. Cell cycle distribution was assessed in total thymocytes, and a significant increase in G2/M fraction in Mapk7hemat-/-Atm-/- mice was observed in comparison to wild-type mice (Supplementary Figure 5A). As Atm-/- thymocytes fail to progress through the DN3 stage [33] and DN thymocytes are the thymic population with proliferative activity, we analyzed the cell cycle distribution of DN thymocytes. Interestingly, DN thymocytes from Mapk7hemat-/-Atm-/- mice presented a higher percentage of cells in the G2/M-phase of the cell cycle (Figure 4A). Besides DN thymocytes, no differences in cell cycle distribution were detected among the different genotypes in DP, CD4+ SP and CD8+ SP thymocyte subsets (Suplementary Figure 5B), indicating a specific effect of the combined deficiency of Mapk7 and Atm in DN thymocyte proliferation. The increase in the G2/M fraction of DN thymocytes in Mapk7hemat-/-Atm-/- mice did not increase the number of DN cells (Figure 3), suggesting that the lack of Mapk7 in Atm-/- DN thymocytes could be inducing a partial G2/M arrest or an increase in thymocyte apoptosis in these mice. We were unable to detect changes in apoptosis, as analyzed by annexin V staining, which was similar in the three genotypes investigated (Figure 4B).
In addition to a deficit in the DNA damage response, the absence of Atm leads to mitochondrial dysfunction and oxidative stress [34] [35]. In order to ascertain whether the delay in thymic lymphoma growth observed in Mapk7hemat-/-Atm-/- mice was also related to changes in mitochondrial function, we analyzed the mitochondrial content and membrane potential in thymocytes. No major differences were observed between Atm-/- and Mapk7hemat-/-Atm-/- mice in mitochondrial content or membrane potential (Figure 4C). Furthermore and related to the alterations in mitochondria, Atm-/- mice of approximately 6 months of age have been described to exhibit an increase in reactive oxygen species (ROS) [30]. Quantitation of ROS in thymocytes of 4- to 5-week old mice by staining with dihydrorhodamine-123 did not show major differences between control Mapk7hemat-/-, Atm-/- and Mapk7hemat-/-Atm-/- mice (Figure 4C). These results suggest that neither differences in mitochondrial activity nor ROS content were responsible for the delay in thymic lymphoma observed in Mapk7hemat-/-Atm-/- mice.
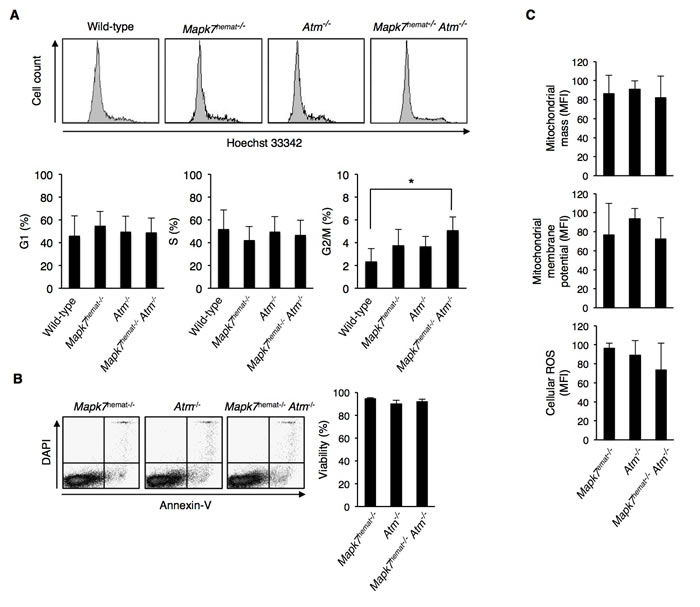
Figure 4: Increased G2/M cell cycle phase accumulation in DN thymocytes of Mapk7hemat-/-Atm-/- mice. A. Representative flow cytometry profiles of DNA content in DN thymocytes based on Hoechst staining (top). Percentage of DN thymocytes in each cell cycle phase (bottom). Thymocytes were fixed in 1% paraformaldehyde, labeled with anti-CD4 and anti-CD8 antibodies as indicated in Figure 3 and stained with Hoechst 33342. B. Percentage of live thymocytes measured immediately after thymic extraction. Representative dot plots (left); quantification of FACS staining (right, n ≥ 3 for each genotype). Apoptosis was measured by staining with annexin V and DAPI followed by flow cytometry analysis. C. The mitochondrial mass, membrane potential and cellular ROS in thymocytes were determined by staining with mitoTracker-green, mitoTracker-red and dihydrorhodamine-123, respectively, and analyzed by flow cytometry (n ≥ 3 for each genotype). The MFI in arbitrary units is shown.
Absence of Mapk7hemat-/-Atm-/- mice restores the DNA damage response to ionizing radiation
Tumor susceptibility in Atm-/- mice is caused by deficiencies in the DNA damage response; therefore, we investigated the effect of Mapk7 loss on DNA damage signaling. The DSB caused by ionizing radiation activate ATM, which phosphorylates several key participants in the DNA damage response, including histone H2AX, which enables recruitment of DNA repair complexes [36] [37]. Exposure of control Mapk7hemat-/- mice to 6-Gy ionizing radiation led to phosphorylation of H2AX, indicating the presence of DNA damage in thymocytes harvested 2 hours after the mice were exposed to radiation (Figure 5A). However, thymocytes isolated from irradiated Atm-/- mice showed a deficit in this signaling response (Figure 5A), consistent with published data [38]. Conversely, loss of Mapk7 in Atm-/- mice restored the capacity of thymocytes to respond to DNA damage caused by ionizing radiation, via phosphorylation of H2AX (Figure 5A). In untreated mice, phosphorylation of H2AX in thymocytes presented no differences between the different genotypes. These results indicate that the deficient DNA damage response in Atm-/- mice can be restored by loss of Mapk7.
Phosphorylation of H2AX promotes the recruitment of checkpoint proteins and initiation of the G2/M checkpoint [39]. We therefore determined the cell cycle phases in thymocytes from irradiated mice. Ionizing radiation did not induce a change in the cell cycle profile of Atm-/- thymocytes, indicating radioresistant DNA synthesis (Figure 5B) [40] [41]. Interestingly, thymocytes from irradiated Mapk7hemat-/-Atm-/- mice showed a partial G2/M arrest (Figure 5B). This indicates that in Atm-/- thymocytes, the presence of MAPK7 inhibited the G2/M checkpoint triggered by radiation-dependent DNA damage, and this G2/M checkpoint was partially restored in Mapk7hemat-/-Atm-/- thymocytes.
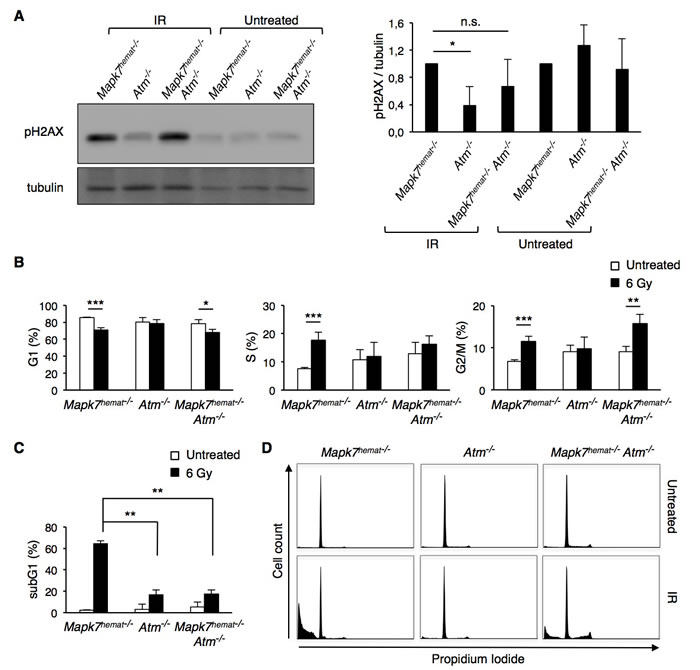
Figure 5: Absence of Mapk7 in Atm-/- mice restores the DNA damage response to ionizing irradiation. Mice were treated with 6 Gy and two hours later, thymocytes were isolated and lysed in SDS-buffer or fixed for cell cycle analysis. A. Phosphorylation of H2AX (pH2AX) in thymocytes isolated from irradiated (IR) mice. Representative Western blot with pH2AX and tubulin as loading control (left); pH2AX:tubulin ratio normalized to control Mapk7hemat-/- (right; n ≥ 3 for each genotype). n.s., not significant. B. Cell cycle distribution of live thymocytes isolated from the same mice evaluated in A., as analyzed by PI staining. C. Cell death of the same thymocyte samples evaluated in B. (sub-G1 fraction). D. Representative flow cytometry profiles of DNA content in thymocytes based on PI staining. The quantitation of these results is represented in B. for live thymocytes and in C. for dead cells.
As anticipated, irradiation of control Mapk7hemat-/- mice provoked an early massive apoptosis of thymocytes (Figure 5C). Although the early apoptotic response of thymocytes from irradiated Atm-/- mice and Mapk7hemat-/-Atm-/-mice was similar (Figure 5C, 5D), we further examined the apoptosis of these cells after culturing them at 37ºC for 20h and staining them with annexin V and DAPI. Whereas the survival of irradiated control Mapk7hemat-/- thymocytes was markedly low, a substantial percentage of thymocytes from Atm-/- mice survived after mice irradiation and cell culture (Figure 6). Importantly, the loss of Mapk7 in Atm-/- mice partially restored the apoptotic response of thymocytes to DNA damage (Figure 6). Taken together, these results indicate that loss of Mapk7 in Atm-/- mice restores the DNA damage-signaling pathway in thymocytes, leading to G2/M arrest and apoptosis when mice are subjected to irradiation.
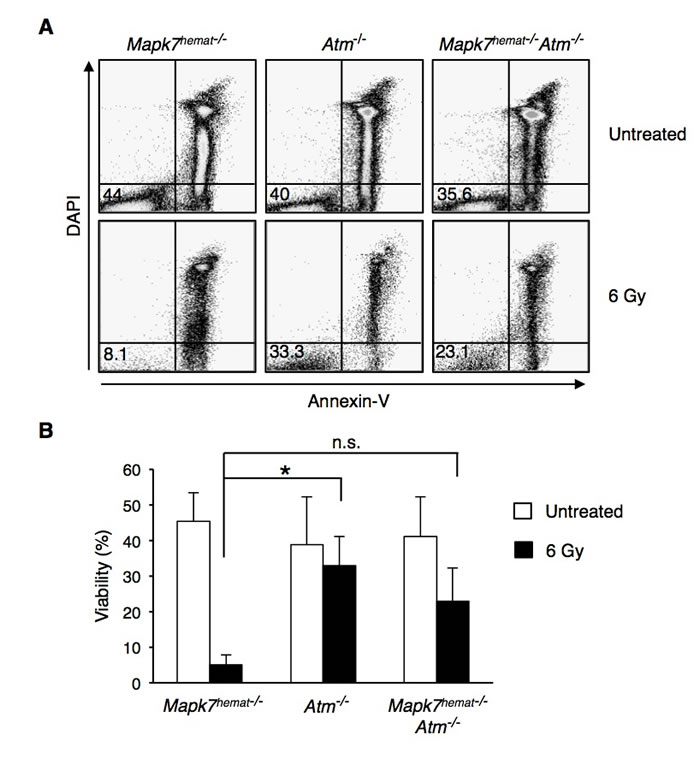
Figure 6: Absence of Mapk7 in Atm-/- mice restores the apoptotic response to ionizing irradiation. Percentage of live thymocytes after isolation from irradiated mice (6 Gy) and culturing for 20 h. Representative flow cytometry profiles A. and quantification of FACS staining (percentage of live cells) B.. Cell death was measured by staining with annexin V and DAPI followed by flow cytometry (n ≥ 3 for each genotype). n.s., not significant.
Human cancers with ATM mutations do not show increased Mapk7 mRNA content
The data presented above suggest that in absence of Atm, an increase in the expression or function of MAPK7 could be advantageous for oncogenic transformation. To test this hypothesis, we queried The Cancer Genome Atlas (TCGA) clinical database for the expression of MAPK7 mRNA in human cancers presenting mutated ATM. The results indicate that there is not a statistically significant relationship between the presence of ATM mutations and MAPK7 mRNA content in human cancer (Supplementary Figure 6).
Discussion
ATM is activated by DNA damage and is necessary during T cell development to repair DSB generated during Tcr recombination [32]. Developing T cells in Atm-/- mice present unprotected DNA ends that recombine abnormally [42], leading to immunodeficiency and genetic instability, and ultimately this can provoke T cell lymphoma. However, Atm-/- mice lacking recombinase activating genes are free of tumors [43].
In this study, we have shown that the loss of Mapk7 in Atm-/- mice delays tumorigenesis, indicating a functional interaction between Mapk7 and Atm. Upon DNA damage, ATM phosphorylates several key participants in the DNA damage response, including H2AX, which enables recruitment of DNA repair complexes [44] and the arrest of DNA replication. In contrast, Atm-/- cells show impaired H2AX phosphorylation and radioresistant DNA replication (the present study) [40] [41]. The results from irradiated mice reported here indicate that when Mapk7 is eliminated in Atm-/- mice, H2AX phosphorylation is restored, together with partial G2/M arrest and increased apoptosis of thymocytes. The phosphorylation of H2AX in Mapk7hemat-/-Atm-/- thymocytes is probably responsible for restitution of the G2/M checkpoint [39]. The reported activities of MAPK7 related to cell cycle control include its increased activity at G2/M, facilitating this cell cycle transition [26]. Loss of Mapk7 might result in stronger activation of the G2/M checkpoint upon DNA damage, as observed in the present study. Restoration of the DNA damage response and the G2/M checkpoint in Mapk7hemat-/-Atm-/- thymocytes reduces the generation of DNA instability and probably contributes to the delay of thymic lymphoma in Mapk7hemat-/-Atm-/- mice.
During the V(D)J recombination process, ATM, DNA-PKc and H2AX function in a common DNA repair pathway [45]. ATM regulates hairpin-sealed DNA end-processing by phosphorylation of DNA-PKc [46], whereas H2AX has a specific role in preservation of the structural integrity of broken DNA ends during V(D)J recombination in murine lymphocytes [47]. The phosphorylation of H2AX in cells under ionizing radiation is mediated by both ATM and DNA-PKc [48]. Therefore, the phosphorylation of H2AX in thymocytes from irradiated Mapk7hemat-/-Atm-/- mice, but not from irradiated Atm-/- mice, suggests that in absence of ATM, MAPK7 inhibits DNA-PKc. These data suggest that a normal function of MAPK7, regulated by ATM, may be to inhibit DNA-PKc kinase activity in DN thymocytes, reducing the DNA damage response and apoptosis during tcr recombination. If unrestrained by ATM, this MAPK7 activity in developing T cells could therefore lead to genetic instability.
Due to damaged DNA, Atm-/- thymocytes cannot mature efficiently and their number diminish after the DN stage, showing a shortage of DP and maturing SP CD4+ T thymocytes (the present study and [9]). Restitution of the DNA damage response in Mapk7hemat-/-Atm-/- thymocytes did not alleviate arrest at the DN stage, suggesting that although undetected, increased apoptosis of potentially oncogenic DN thymocytes in Mapk7hemat-/-Atm-/- mice contributes to delaying thymic lymphoma development.
We have recently reported that MAPK7 is necessary for normal erythropoiesis in mice, contributing to proper dNTP metabolism in highly proliferative erythroid progenitors [18]. Loss of Mapk7 in the hematopoietic compartment gives rise to extramedullary erythropoiesis, splenomegaly and an increased mutation rate in erythrocyte precursors, probably due to an imbalance of dNTPs [18]. Contrary to Mapk7hemat-/- mice, erythropoiesis was restored in the bone marrow of Mapk7hemat-/-Atm-/- mice (the present study), indicating that the absence of Atm compensates for the absence of Mapk7 during erythropoiesis in young mice, and suggesting that the erythroid dNTP pool is normalized in Mapk7hemat-/-Atm-/- mice. This is consistent with increased cellular dNTP levels during replication stress in Atm-/- mice [49].
B cell development in the bone marrow was impaired in the three genotypes studied here, though the spleen and lymph nodes contained a normal number of mature B cells. MAPK7 is necessary in B cells to signal BAFF-surviving stimuli [17], whereas in Atm-/- mice the impairment in B cell development is related to a deficient V(D)J recombination [50]. Curiously, Mapk7hemat-/-Atm-/- mice did not show a stronger alteration of B cell precursors, suggesting that both genes are acting through the same pathway.
In addition to its role in the DDR response, ATM is activated by oxidative stress and is needed to maintain cellular homeostasis in front of oxidative and mitochondrial stressors [2-5] [30]. In the present study, we analyzed whether Atm-/- thymocytes presented evidences of oxidative and mitochondrial stress, but no alterations were observed. We hypothesize that a reason why no dysfunctions were detected is that our study on oxidative and mitochondrial activity was carried out with thymocytes from 4- to 5-week-old mice, which were younger than the ones used in other studies [5]. In addition, the genetic background of mice used in the present study could lie behind the apparent mitochondrial normality of Atm-/- thymocytes [51].
In an attempt to confirm that the development of cancer in absence of Atm is favoured by MAPK7 function, we examined the MAPK7 mRNA content in human cancers presenting ATM mutations. However, no statistically significant relationship was found between the amount of MAPK7 mRNA and the presence of mutated ATM. These data do not confirm that MAPK7 provides an advantage to ATM-dependent tumorigenesis, though they do not formally exclude this possibility. MAPK7 function could be beneficial to the oncogenic process when this is initiated by a particular type of ATM mutation. In addition, although we used mRNA data, the mRNA content and the activity of the expressed MAPK7 are distantly related. Therefore, the prospect of using functional data (i.e. MAPK7 activity) will provide a closer view to the MAPK7 - ATM relationship in human cancer.
Taken together, these data establish a functional relationship between Atm and Mapk7 during bone marrow erythropoiesis and suggest a non-redundant role of MAPK7 in thymic precursors during repair of tcr recombination-dependent DSB, facilitating mitigation of an excessive DNA damage response and apoptosis. In absence of MAPK7, thymocytes from irradiated Atm-/- mice partially recover the G2/M checkpoint, progress with increased difficulty through cell cycle and ultimately show increased apoptosis. These results suggest that MAPK7 inhibitors could be of therapeutic value in lymphoma caused by absence of ATM by re-establishing cell cycle checkpoints in cancer cells.
Materials and Methods
Ethics statement
Investigation has been conducted in accordance with the ethical standards and according to national and international guidelines and has been approved by the authors’ institutional review board (the Departament d’Agricultura, Ramaderia, Pesca, Alimentació i Medi Natural of Generalitat de Catalunya).
Mice
Atm+l- mice were kindly provided by Óscar Fernández-Capetillo (National Cancer Research Centre, Madrid, Spain) [52]. Mapk7loxP/loxP mice were kindly provided by Cathy Tournier (University of Manchester, Manchester, U.K.) [29]. Mapk7loxP/loxPvav-cre mice which lack Mapk7 in hematopoietic cells are described elsewhere [27] [28]. Mapk7loxP/loxPvav-cre mice were on C57BL/6 background whereas Atm+/- mice had a mixed background (129S6, CD-1, C57BL/6). Atm+/- mice were crossed to Mapk7loxP/loxPvav-cre mice (referred to as Mapk7hemat-/-mice) or to control Mapk7loxP/loxP mice (referred to as wild-type mice), and double heterozygous F1 progeny were crossed to obtain F2 Mapk7hemat-/-Atm-/- mice and Mapk7loxP/loxPAtm-/- mice (referred to as Atm-/- mice). The Mapk7hemat-/- mice and wild-type mice, used in experiments were F1 crossings, in order to keep the same genetic background for all mice. Experiments were conducted on 4- to 8-week-old mice. To genotype mice, the following oligonucleotides were used: to detect floxed Mapk7, forward 5’-tccatgctgttagtcctttgg-3’ and reverse 5’-agcggctgtgaagagtgaat-3’ (floxed amplicon = 260bp, wild-type amplicon = 200bp). Presence of Vav-Cre was analyzed with the oligonucleotides: forward 5’-cgagtgatgaggttcgcaag-3’ and reverse 5’-atcttcaggttctgcgggaa-3’. Wild-type Atm was detected with oligonucleotides: forward 5’-gctgccatacttgatccatg-3’ and reverse 5’- tccgaatttgcaggagttg-3’ (wild-type amplicon = 147bp). To detect neomycin gene insertion in Atm the following oligonucleotides were used: forward 5’-cttgggtggagaggctattc-3’ and reverse 5’-aggtgagatgacaggagatc-3’ (neomycin amplicon = 280bp).
Cell culture and reagents
For cell preparations, single cell suspensions were isolated from thymus, spleen and bone marrow of mice. The suspension was counted to determine the organ cellularity, and then mature red blood cells of spleen and bone marrow suspensions were discarded from the suspension by overlaying cells on a solution of 16% iodixanol, 0.63% NaCl and 10 mM Hepes and centrifuged at 900x g for 30 minutes. The mononuclear fraction of the density gradient was collected, washed with PBS, counted and stained or fixed. Cell preparations were cultured in RPMI 1640 medium (ref. BE12-702F, Lonza) supplemented with 9% FBS, penicillin/streptomycin, 2 mM L-glutamine. Mitotracker Green or Red (Life Technologies) was used at a concentration of 50nM for 15 to 30 minutes at 37ºC.
Western blot
Cells were lysed in Laemmli buffer and loaded on 10% or 15% acrylamide gels. Transfer to nitrocellulose membranes and immunoblotting was performed as previously described [18]. Incubation with primary antibody (mouse anti-H2AX phosphorylated S139, Biolegend #613401; rabbit anti-ERK5, Cell Signaling #3372; mouse anti-beta actin, Sigma-Aldrich #A2228; mouse anti-tubulin, Sigma-Aldrich #T9026) was performed overnight at 4ºC under agitation. The following day, after washing, the membrane was incubated with the secondary antibody-HRP conjugated (rabbit anti-mouse, Sigma-Aldrich #A9044; goat anti-rabbit, Sigma-Aldrich #A0545) for 1 hour under agitation at room temperature. Western blots were quantified on a Luminescent Image Analyzer LAS-3000 (Fujifilm). Densitometry analysis was performed using ImageJ software.
Immunostaining and flow cytometry analysis
For extracellular labeling, thymocytes, splenocytes, lymph node cells and bone marrow cells were stained with saturating amounts of primary antibodies conjugated to fluorochromes (anti-CD4 APC, Biolegend #100516; anti-CD8 AlexaFluor 488, Biolegend #100706; anti-CD44 PerCP, Biolegend #103032; anti-CD25 PE, Immunotools #22150254; anti CD71 PE, BD #553267; anti-Ter119 FITC, Biolegend #116205; anti-B220 FITC, Immunotools #22159453; anti-IgM PerCP, Santa Cruz #sc-45105) in PBS + 2% FBS for 25 minutes at 4ºC. Cells were then washed with PBS and analyzed using a Gallios flow cytometer (Beckman Coulter, Inc., Fullerton, CA). 4’,6-diamidino-2-phenylindole (DAPI) was added to the samples in order to discard the dead cells. Changes in the mitochondrial mass, membrane potential and cellular ROS were determined by incubating the cells for 30 min at 37ºC with mitoTracker-green, mitoTracker-red or dihydrorhodamine-123, respectively, and analyzed by flow cytometry. The mean fluorescence intensity (MFI) was represented. Fluorescence was collected on the logarithmic scale. The cell population was selected by gating in a forward scatter vs. side scatter dot plot, excluding aggregates and cell debris, and by excluding the DAPI-positive population.
Cell cycle analysis
In order to determine the cell cycle of irradiated thymocytes, 1*106 cells were fixed in 70% ethanol and stored at -20ºC. Samples were washed once with PBS and incubated in 0.1% triton X100, 0.02% RNAse A and 0.0005% propidium iodide (PI) in PBS for 30 minutes at room temperature in the dark. Samples were analyzed in a Coulter XL cytometer and a subsequent cell cycle profile analysis was performed using cytomation summit software.
For cell cycle analysis of thymocyte subsets, 1*106 cells were fixed in 1% paraformaldehyde, and stained with anti-CD4 and anti-CD8 antibodies for 30 minutes at 4ºC. Cells were then washed with PBS and incubated for 30 minutes at room temperature with Hoescht 33342 in the dark. Samples were analyzed in a Gallios flow cytometer and cell cycle profile was analyzed with the Watson Pragmatic model.
Apoptosis assay
Apoptosis was assessed in thymocytes by incubation with annexin V FITC (diluted 1:20, Immunotools) for 15 minutes. Then, PI was added and cells were analyzed by flow cytometry. Samples were analyzed using a Gallios flow cytometer.
Statistics
Data are represented as the mean and standard deviation (S.D.) of biological replicates. Statistical analysis was performed in GraphPad Prism (La Jolla, CA, USA). One-way Analysis of variance with Bonferroni post-test was used for multiple comparisons. Differences were considered significant when *p < 0.05, **p < 0.01, ***p < 0.001; n.s. not significant.
Acknowledgments
We wish to thank the technical staff at the FACS Facility and the staff at the Animal Facility in the Faculty of Psychology for their support. We wish to thank to Cristina Gutierrez and Ignasi Modolell from the Institut Català d’Oncologia for their assistance with X-ray irradiation of mice. The results shown in supplementary figure 6 are based upon data generated by the TCGA Research Network: http://cancergenome.nih.gov/.
Conflicts of Interest
No potential conflicts of interest were disclosed.
Grant Support
This study was supported by grant FBG300412 from the Bosch Gimpera Foundation, University of Barcelona (to M.R.). X.R-C. and M.A-I. were supported by a predoctoral fellowship from the Bosch Gimpera Foundation (FBG300412). FX.S. is funded by the Spanish Ministerio de Economía y Competitividad (SAF2014-59872-P).
Author contributions
MAI, AGJ and EE conceived and designed most experiments.
AGJ and MAI performed most experiments.
MAI, AGJ, XRC and EE analyzed and interpreted the data.
XRC conducted the bioinformatics analysis.
FXS designed the experiments to study mitochondrial and oxidative activities.
CPVG did the histological analysis of mice.
MAI, XRC and EE wrote the manuscript and MR revised the manuscript.
References
1. Aird KM, Worth AJ, Snyder NW, Lee JV, Sivanand S, Liu Q, Blair IA, Wellen KE, Zhang R. ATM couples replication stress and metabolic reprogramming during cellular senescence. Cell Rep. 2015, 11:893-901.
2. Alexander A, Cai S-L, Kim J, Nanez A, Sahin M, MacLean KH, Inoki K, Guan K-L, Shen J, Person MD, Kusewitt D, Mills GB, Kastan MB, Walker CL. ATM signals to TSC2 in the cytoplasm to regulate mTORC1 in response to ROS. Proc Natl Acad Sci USA. 2010, 107:4153-4158.
3. Guo Z, Kozlov S, Lavin MF, Person MD, Paull TT. ATM activation by oxidative stress. Science. 2010, 330:517-521.
4. Valentin-Vega YA, MacLean KH, Tait-Mulder J, Milasta S, Steeves M, Dorsey FC, Cleveland JL, Green DR, Kastan MB. Mitochondrial dysfunction in ataxia-telangiectasia. Blood. 2012, 119:1490-1500.
5. D’Souza AD, Parish IA, Krause DS, Kaech SM, Shadel GS. Reducing Mitochondrial ROS Improves Disease-related Pathology in a Mouse Model of Ataxia-telangiectasia. Mol Ther. 2012, 21:42-48.
6. Shackelford RE, Innes CL, Sieber SO, Heinloth AN, Leadon SA, Paules RS. The Ataxia telangiectasia gene product is required for oxidative stress-induced G1 and G2 checkpoint function in human fibroblasts. J Biol Chem. 2001, 276:21951-21959.
7. Carson CT, Schwartz RA, Stracker TH, Lilley CE, Lee DV, Weitzman MD. The Mre11 complex is required for ATM activation and the G2/M checkpoint. EMBO J. 2003, 22:6610-6620.
8. Boder E. Ataxia-telangiectasia: some historic, clinical and pathologic observations. Birth Defects Orig Artic Ser. 1975, 11:255-270.
9. Barlow C, Hirotsune S, Paylor R, Liyanage M, Eckhaus M, Collins F, Shiloh Y, Crawley JN, Ried T, Tagle D, Wynshaw-Boris A: Atm-deficient mice: a paradigm of ataxia telangiectasia. Cell 1996, 86:159-171.
10. Kasler HG, Victoria J, Duramad O, Winoto A. ERK5 is a novel type of mitogen-activated protein kinase containing a transcriptional activation domain. Mol Cell Biol. 2000, 20:8382-8389.
11. Regan CP, Li W, Boucher DM, Spatz S, Su MS, Kuida K. Erk5 null mice display multiple extraembryonic vascular and embryonic cardiovascular defects. Proc Natl Acad Sci USA. 2002, 99:9248-9253.
12. Sohn SJ, Sarvis BK, Cado D, Winoto A. ERK5 MAPK regulates embryonic angiogenesis and acts as a hypoxia-sensitive repressor of vascular endothelial growth factor expression. J Biol Chem. 2002, 277:43344-43351.
13. Yan L, Carr J, Ashby PR, Murry-Tait V, Thompson C, Arthur JSC. Knockout of ERK5 causes multiple defects in placental and embryonic development. BMC Dev Biol. 2003, 3:11.
14. Sohn SJ, Li D, Lee LK, Winoto A. Transcriptional regulation of tissue-specific genes by the ERK5 mitogen-activated protein kinase. Mol Cell Biol. 2005, 25:8553-8566.
15. Le N-T, Takei Y, Izawa-Ishizawa Y, Heo K-S, Lee H, Smrcka AV, Miller BL, Ko KA, Ture S, Morrell C, Fujiwara K, Akaike M, Abe J-I. Identification of activators of ERK5 transcriptional activity by high-throughput screening and the role of endothelial ERK5 in vasoprotective effects induced by statins and antimalarial agents. J Immunol. 2014, 193:3803-3815.
16. Komaravolu RK, Adam C, Moonen J-RAJ, Harmsen MC, Goebeler M, Schmidt M. Erk5 inhibits endothelial migration via KLF2-dependent down-regulation of PAK1. Cardiovasc Res. 2015, 105:86-95.
17. Jacque E, Schweighoffer E, Tybulewicz VLJ, Ley SC. BAFF activation of the ERK5 MAP kinase pathway regulates B cell survival. J Exp Med. 2015, 212:883-892.
18. Angulo-Ibáñez M, Rovira-Clavé X, Granados-Jaén A, Downs B, Kim YC, Wang SM, Reina M, Espel E. Erk5 contributes to maintaining the balance of cellular nucleotide levels and erythropoiesis. Cell Cycle. 2015, 14:3864-3876.
19. Lochhead PA, Gilley R, Cook SJ. ERK5 and its role in tumour development. Biochem Soc Trans. 2012, 40:251-256.
20. Nagel S, Burek C, Venturini L, Scherr M, Quentmeier H, Meyer C, Rosenwald A, Drexler HG, MacLeod RAF. Comprehensive analysis of homeobox genes in Hodgkin lymphoma cell lines identifies dysregulated expression of HOXB9 mediated via ERK5 signaling and BMI1. Blood 2007, 109:3015-3023.
21. Wu L, Chen X, Zhao J, Martin B, Zepp JA, Ko JS, Gu C, Cai G, Ouyang W, Sen G, Stark GR, Su B, Vines CM, Tournier C, Hamilton TA, Vidimos A, Gastman B, Liu C, Li X. A novel IL-17 signaling pathway controlling keratinocyte proliferation and tumorigenesis via the TRAF4-ERK5 axis. J Exp Med. 2015, 212:1571-1587.
22. Finegan KG, Perez-Madrigal D, Hitchin JR, Davies CC, Jordan AM, Tournier C. ERK5 is a critical mediator of inflammation-driven cancer. Cancer Res. 2015, 75:742-753.
23. Yang Q, Deng X, Lu B, Cameron M, Fearns C, Patricelli MP, Yates JR, Gray NS, Lee J-D. Pharmacological inhibition of BMK1 suppresses tumor growth through promyelocytic leukemia protein. Cancer Cell. 2010, 18:258-267.
24. Yang Q, Liao L, Deng X, Chen R, Gray NS, Yates JR, Lee JD. BMK1 is involved in the regulation of p53 through disrupting the PML-MDM2 interaction. Oncogene. 2013, 32:3156-3164.
25. Lin ECK, Amantea CM, Nomanbhoy TK, Weissig H, Ishiyama J, Hu Y, Sidique S, Li B, Kozarich JW, Rosenblum JS. ERK5 kinase activity is dispensable for cellular immune response and proliferation. Proc Natl Acad Sci USA. 2016, doi/10.1073/pnas.1609019113.
26. Cude K, Wang Y, Choi H-J, Hsuan S-L, Zhang H, Wang C-Y, Xia Z. Regulation of the G2-M cell cycle progression by the ERK5-NFkappaB signaling pathway. J Cell Biol. 2007, 177:253-264.
27. Rovira-Clavé X, Angulo-Ibáñez M, Tournier C, Reina M, Espel E. Dual role of ERK5 in the regulation of T cell receptor expression at the T cell surface. J Leukoc Biol. 2016, 99:143-152.
28. Stadtfeld M, Graf T. Assessing the role of hematopoietic plasticity for endothelial and hepatocyte development by non-invasive lineage tracing. Development. 2005, 132:203-213.
29. Ananieva O, Macdonald A, Wang X, Mccoy CE, Mcilrath J, Tournier C, Arthur JSC. ERK5 regulation in naïve T-cell activation and survival. Eur J Immunol. 2008, 38:2534-2547.
30. Ito K, Hirao A, Arai F, Matsuoka S, Takubo K, Hamaguchi I, Nomiyama K, Hosokawa K, Sakurada K, Nakagata N, Ikeda Y, Mak TW, Suda T. Regulation of oxidative stress by ATM is required for self-renewal of haematopoietic stem cells. Nature. 2004, 431:997-1002.
31. Vacchio MS, Olaru A, Livak F, Hodes RJ. ATM deficiency impairs thymocyte maturation because of defective resolution of T cell receptor alpha locus coding end breaks. Proc Natl Acad Sci USA. 2007, 104:6323-6328.
32. Bredemeyer AL, Sharma GG, Huang C-Y, Helmink BA, Walker LM, Khor KC, Nuskey B, Sullivan KE, Pandita TK, Bassing CH, Sleckman BP. ATM stabilizes DNA double-strand-break complexes during V(D)J recombination. Nature. 2006, 442:466-470.
33. Isoda T, Takagi M, Piao J, Nakagama S, Sato M, Masuda K, Ikawa T, Azuma M, Morio T, Kawamoto H, Mizutani S. Process for immune defect and chromosomal translocation during early thymocyte development lacking ATM. Blood. 2012, 120:789-799.
34. Ambrose M, Goldstine JV, Gatti RA. Intrinsic mitochondrial dysfunction in ATM-deficient lymphoblastoid cells. Hum Mol Genet. 2007, 16:2154-2164.
35. Eaton JS, Lin ZP, Sartorelli AC, Bonawitz ND, Shadel GS. Ataxia-telangiectasia mutated kinase regulates ribonucleotide reductase and mitochondrial homeostasis. J Clin Invest. 2007, 117:2723-2734.
36. Paull TT, Rogakou EP, Yamazaki V, Kirchgessner CU, Gellert M, Bonner WM. A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr Biol. 2000, 10:886-895.
37. Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998, 273:5858-5868.
38. Burma S, Chen BP, Murphy M, Kurimasa A, Chen DJ. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J Biol Chem. 2001, 276:42462-42467.
39. Fernandez-Capetillo O, Chen HT, Celeste A, Ward I, Romanienko PJ, Morales JC, Naka K, Xia Z, Camerini-Otero RD, Motoyama N, Carpenter PB, Bonner WM, Chen J, Nussenzweig A. DNA damage-induced G2-M checkpoint activation by histone H2AX and 53BP1. Nat Cell Biol. 2002, 4:993-997.
40. Houldsworth J, Lavin MF. Effect of ionizing radiation on DNA synthesis in ataxia telangiectasia cells. Nucleic Acids Res. 1980, 8:3709-3720.
41. Falck J, Mailand N, Syljuåsen RG, Bartek J, Lukas J. The ATM-Chk2-Cdc25A checkpoint pathway guards against radioresistant DNA synthesis. Nature. 2001, 410:842-847.
42. Bowen S, Wangsa D, Ried T, Livak F, Hodes RJ. Concurrent V(D)J recombination and DNA end instability increase interchromosomal trans-rearrangements in ATM-deficient thymocytes. Nucleic Acids Res. 2013, 41:4535-4548.
43. Callén E, Jankovic M, Difilippantonio S, Daniel JA, Chen HT, Celeste A, Pellegrini M, McBride K, Wangsa D, Bredemeyer AL, Sleckman BP, Ried T, Nussenzweig M, Nussenzweig A. ATM prevents the persistence and propagation of chromosome breaks in lymphocytes. Cell. 2007, 130:63-75.
44. Fernandez-Capetillo O, Lee A, Nussenzweig M, Nussenzweig A. H2AX: the histone guardian of the genome. DNA Repair. 2004, 3:959-967.
45. Riballo E, Kühne M, Rief N, Doherty A, Smith GCM, Recio M-J, Reis C, Dahm K, Fricke A, Krempler A, Parker A, Jackson SP, Gennery A, Jeggo PA, Löbrich M. A Pathway of Double-Strand Break Rejoining Dependent upon ATM, Artemis, and Proteins Locating to -H2AX Foci. Mol Cell. 2004, 16:715-724.
46. Jiang W, Crowe JL, Liu X, Nakajima S, Wang Y, Li C, Lee BJ, Dubois RL, Liu C, Yu X, Lan L, Zha S. Differential Phosphorylation of DNA-PKcs Regulates the Interplay between End-Processing and End- Ligation during Nonhomologous End-Joining. Mol Cell. 2015, 58:172-185.
47. Helmink BA, Tubbs AT, Dorsett Y, Bednarski JJ, Walker LM, Feng Z, Sharma GG, McKinnon PJ, Zhang J, Bassing CH, Sleckman BP. H2AX prevents CtIP-mediated DNA end resection and aberrant repair in G1-phase lymphocytes. Nature. 2010, 469:245-249.
48. Stiff T, O’Driscoll M, Rief N, Iwabuchi K, Löbrich M, Jeggo PA. ATM and DNA-PK function redundantly to phosphorylate H2AX after exposure to ionizing radiation. Cancer Res. 2004, 64:2390-2396.
49. Aird KM, Zhang R. ATM in senescence. Oncotarget. 2015, 6:14729-14730. doi: 10.18632/oncotarget.4411.
50. Huang C-Y, Sharma GG, Walker LM, Bassing CH, Pandita TK, Sleckman BP. Defects in coding joint formation in vivo in developing ATM-deficient B and T lymphocytes. J Exp Med. 2007, 204:1371-1381.
51. Puccini J, Dorstyn L, Kumar S. Genetic background and tumour susceptibility in mouse models. Cell Death Differ. 2013, 20:964-4.
52. Cuadrado M, Martinez-Pastor B, Murga M, Toledo LI, Gutierrez-Martinez P, Lopez E, Fernandez-Capetillo O. ATM regulates ATR chromatin loading in response to DNA double-strand breaks. J Exp Med. 2006, 203:297-303.