
Introduction
Skeletal muscle is the most abundant tissue in mammal’s body, constituting approximately 40-50% of the body mass. In addition to its primary role in posture and movement, skeletal muscle performs a number of critical functions, such as the regulation of energy and glucose metabolisms [1, 2]. Notably, the differentiation and fiber type composition of skeletal muscle are critical for these functions. During skeletal muscle development, myoblasts derived from the differentiation of myotome progress along the myogenic pathway through cellular proliferation, terminal differentiation and fusion into multinucleated myofibers [3]. The muscle specific transcription factors, MyoD and the myocyte enhancer factor 2 (MEF2) families, which proteins result in reprogramming expression of genes involved in muscle differentiation, govern the skeletal myogenesis [4, 5]. In addition, skeletal muscle is a highly contractile and dynamic tissue with striking plasticity. The composition of diverse muscle fiber subtypes defined by metabolic activity and specific myosin heavy chain (MyHC) isoforms is closely relevant with muscle plasticity and function. Although the fiber type specification varies between different species, on the basis of specific MyHC isoform expression, in mammal adult skeletal muscle fibers are prevalently classified as type I (slow oxidative muscle fibers), type IIa (fast oxidative muscle fibers), type IIx/d (intermediate muscle fibers), and type IIb (fast glycolytic muscle fibers) [6]. Both the skeletal muscle differentiation and fiber type composition are regulated in response to changes in environment, physical activity or pathological conditions [7]. Multiple transcription factors are involved in these two programs by effecting a change in expression of certain specific genes.
In mammals, transcription factors of the forkhead box protein O (FoxO) family consists of four members: FoxO1 (FKHR), FoxO3 (FKHRL1), FoxO4 (AFX), and FoxO6, of which proteins play considerable roles in a diverse sets of cellular physiological functions. As transcription factors, they can perform their functions through binding to downstream gene promoter or tethering to the target site by protein-protein interaction with other transcription factors [8]. FoxO family members have been shown to regulate various cellular functions, including proliferation, survival, cell cycle, metabolism, and muscle atrophy [9-12]. In addition, FoxO proteins have also been implicated in myoblast, preadipocyte, and endothelial cell differentiation [13]. FoxO1-null mice died during embryonic development, while FoxO3- or FoxO4-null mice could survive [14-17], indicating that FoxO1 may be the most important factor in mammal’s life among this family. The ability of FoxO is largely dependent on posttranscriptional modifications [18], such as phosphorylation [19]. Phosphorylated FoxO1 would be excluded from nucleus and thus lost its capacity of binding to target regulatory elements [20]. Previous studies have revealed that FoxO1 provokes myotube fusion of primary mouse myoblasts [21, 22]. But more recent researches support FoxO1 as an inhibitor of muscle differentiation [9, 13, 19]. Moreover, FoxO1 is believed to play a role in skeletal muscle fiber type specification [23-26]. FoxO1 transgenic mice showed a significant decrease in skeletal muscle mass and impaired skeletal muscle function, accompanied with the reduced expression of slow fiber genes [23, 24]. Reciprocally, FoxO1 conditional deletion in the soleus muscle resulted in a reduction of slow fiber and an increase of fast fiber formation [25]. These findings present controversial roles of FoxO1 in muscle fiber type composition.
Here, we summarize the most recent advances on the roles of FoxO1 in the regulation of skeletal muscle differentiation and fiber type specification. The molecular mechanism responsible for FoxO1 regulation in these two processes will be detailed in this review.
Upstream regulation of FoxO1 in skeletal muscle differentiation
There are plenty of researches showed that FoxO1 is widely expressed in various tissues, such as liver, fat, and skeletal muscle [27, 28], especially richly expressed in stem cells and adult skeletal muscle [27, 29]. Its function on muscle differentiation has been reported in considerable studies (Table 1). Several signals, including phosphatidylinositol 3-kinase (PI3K)/Akt pathway, insulin, insulin receptor substrate-1 (IRS-1) and Rho/ROCK signaling, have been demonstrated to mediate FoxO1 transcriptional activity through a phosphorylation-mediated nuclear exclusion event during myogenic differentiation (Figure 1).
Table 1: FoxO1 functions on different myoblast differentiation stage
Myoblasts mold |
Functions on myoblast differentiation |
Conclusion |
Year |
Reference |
|
Inhibits myoblast early differentiation |
Required for myoblast fusion |
||||
Mouse primary myoblasts |
Yes |
FoxO1 is required for mouse primary myoblast fusion |
2003 |
[21] |
|
C2C12 myoblasts |
Yes |
An active form of FoxO1 mutant inhibits C2C12 cell differentiation whereas an inactive mutant FoxO1 can partially restore inhibition of C2C12 cell differentiation regulated by wortmannin |
2003 |
[33] |
|
C2C12 myoblasts |
Yes |
Inactivation of Rho/ROCK signaling is crucial for myoblast fusion and nuclear translocation of FoxO1 |
2004 |
[49] |
|
C2C12 myoblasts |
Yes |
Negative-feedback loop between FoxO1a and cGKI fine-tunes the progress of muscle cell fusion process |
2005 |
[76] |
|
C2C12 myoblasts |
Yes |
Interaction between FoxO1 and Notch1 inhibits myoblast differentiation through promoting corepressor clearance and recruiting the coactivator of Csl, leading activation of Hes family, which is considered to be a myoblast differentiation repressor |
2007 |
[25] |
|
C2C12-RasV12/C2C12-RasV12C40 myoblasts |
Yes |
Nuclear exclusion of FoxO1 is required for C2C12-RasV12C40 myoblast differentiation |
2008 |
[43] |
|
C2C12 myoblasts |
Yes |
FoxO1 negatively regulates myoblast differentiation through degradation of mTOR pathway components |
2008 |
[52] |
|
L6 myoblasts |
Yes |
Inhibition of FoxO1 transcriptional activity or nuclear exclusion of FoxO1 suppresses L6-mIRS1 cell differentiation |
2011 |
[22] |
|
C2C12 myoblasts |
Yes |
PAX3/FOXO1A and PAX7/FOXO1A suppress myogenesis through inhibiting transcriptional activity of MyoD-target genes |
2013 |
[70] |
|
Rhabdomyosarco-ma cells |
Yes |
PAX3-FOXO1 fusion protein inhibits rhabdomyosarcoma cell differentiation through upregulating JARID2 |
2014 |
[74] |
|
C2C12 myoblasts |
Yes |
Insulin triggers FoxO1 nuclear exclusion and protein degradation to reverse inhibited myogenesis by FoxO1 |
2014 |
[35] |
|
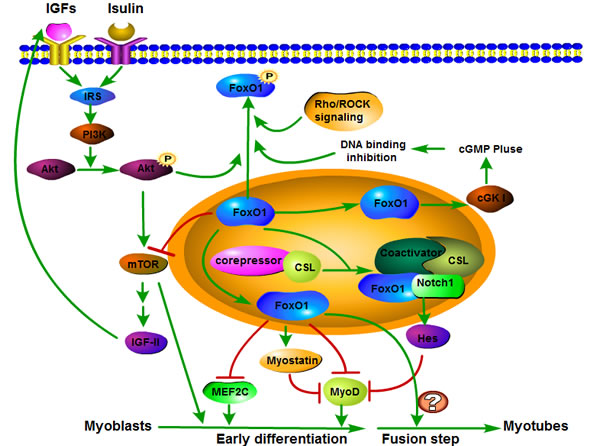
Figure 1: FoxO1 signaling pathway involved in skeletal muscle differentiation. The FoxO1 upstream signals including IGFs, insulin and IRS regulate FoxO1 transcriptional activity through phosphorylating FoxO1 in a PI3K-Akt dependent manner. Phosphorylated FoxO1 will be excluded from nucleus and thus loses its capacity of binding to target regulatory elements. In addition, other signals, such as cGKI and Rho/ROCK signaling, directly mediate FoxO1 transcriptional activity by phosphorylation. Myostatin, MEF2C, MyoD and mTOR are downstream factors of FoxO1. FoxO1 negatively regulates myoblast early differentiation through promoting myostatin and inhibiting MEF2C, MyoD and mTOR. Then the decrease of MEF2C, MyoD and mTOR delays myoblast early differentiation. In addition, the relationship among FoxO1, mTOR, IGF-II and PI3K/Akt pathway presents a feedback loop that can preferably fine-tune the regulation of muscle differentiation. Moreover, FoxO1 can inhibit early step of myoblast differentiation through interacting with Notch signaling and promoting corepressor clearance and recruiting the coactivator of Csl, leading activation of Hes family, which is considered to be a myoblast differentiation repressor. Notably, although FoxO1 suppresses the early muscle differentiation process, FoxO1 is required for myoblast terminal differentiation fusion into myotubes. However, the molecular mechanism in which FoxO1 is required for myotube fusion has remained poorly understood.
Akt pathway
In established cell lines, activation of FoxO1 induces cell cycle arrest followed by apoptosis. However, activation of PI3K/Akt pathway removes the effect through phosphorylating FoxO1 [30-32]. Subsequently, phosphorylated FoxO1 was shuttled out of nucleus to the cytoplasm and lost the regulatory function. FoxO1 gain-of-function mutant inhibited myoblast differentiation in C2C12 cells and blocked myotube fusion that was induced by constitutively active Akt, whereas the dominant-negative FoxO1 led to a slight but striking increase in the expression of differentiation markers, myogenin and MyHC [33]. Moreover, the inactive form of FoxO1 can be able to partly but not completely rescue the repression of differentiation mediated by wortmannin, a specific inhibitor of PI3K/Akt pathway, to block the PI3K/Akt ability to phosphorylate FoxO1 [33, 34]. These data revealed FoxO1 as a key effector of the PI3K/Akt pathway in skeletal muscle differentiation. In addition, inactive FoxO1 cannot fully reverse the suppression of differentiation caused by wortmannin which may account for the multiple downstream effectors of PI3K/Akt pathway in differentiation process [33].
Insulin
A study conducted by Wu et al. [35] revealed that overexpression of FoxO1 dramatically inhibited C2C12 myoblasts differentiation. Previous studies demonstrated insulin and IGF-I as crucial regulators of FoxO1 subcellular localization and intense potentiators of myogenic differentiation through a PI3K-Akt dependent way [36]. FoxO1 inhibited the generation of insulin-positive cells [37] and insulin treatment significantly restored the inhibiting myogenic differentiation caused by FoxO1 [35]. Moreover, treatment with LiCl, a strong stimulator of myogenesis and an activator of Wnt signaling that cooperates with insulin to promote myogenesis [38, 39], parallelly significantly restored FoxO1 inhibiting function on myogenic differentiation [40]. Simultaneous application of insulin and LiCl can distinctly overcome the inhibiting effect caused by active FoxO1 and promote myoblast differentiation synergistically [35, 40].
In addition, nuclear exclusion of FoxO1 blocks the transcriptional regulatory roles on its target genes [41]. FoxO1 is located in both cytoplasm and nucleus in proliferating myoblasts, while it is exported to the nucleus and no more transported into nucleus when myoblasts fused to form multinucleated myotubes. Series of studies revealed that nuclear exclusion of FoxO1 is a crucial step for early myogenic differentiation [42, 43]. Insulin dramatically reduced the FoxO1 nuclear level and the stability of FoxO1 protein [44, 45], suggesting that insulin triggered FoxO1 nuclear exclusion and protein degradation to reverse inhibited myogenesis by FoxO1. Taken together, FoxO1 suppressed muscle differentiation via directly reducing the expression of differentiation markers and repressing the promoter activity of myogenic genes, including MyoD and MEF2C, whereas this repression could be fully removed by LiCl and insulin [35, 46]. These data fully indicated that FoxO1 transcriptional activity is suppressed by insulin and IGF signaling.
IRS-1
On the other hand, compared to the proposition that FoxO1 negatively regulated muscle cell differentiation, conflicting observations held that FoxO1 nuclear accumulation was required for muscle cell fusion. A study conducted by Hakuno et al. [22] reported that expression of a FoxO1 dominant-negative mutant, lacking 256 N-terminus residues including Akt phosphorylation sites and transcriptional activation domain, resulted in significantly decreased myogenic marker expression including myogenin and MyHC in L6 cells, indicating that the myogenic differentiation was suppressed by this FoxO1 dominant-negative mutant form [22]. In addition, the constitutive expression of IRS-1 could inhibit myoblast fusion, accompanied with excluding FoxO1 from the cells nuclei to cytosol. This cytosolic localization was correlated with FoxO1 phosphorylation in a PI3k-Akt-dependent manner. It is noteworthy that FoxO1 is localized in the nucleus during myoblast differentiation, where it exists in active form [22]. These results suggest that inhibition of FoxO1 transcriptional activity or excluding FoxO1 from the cells nuclei is at least one of the reasons why L6-mIRS1 cell differentiation is suppressed, indicating that FoxO1 transcriptional activity is required for L6 cell fusion. Thus, a pattern in which the inhibition of myogenic differentiation is at least partially caused by FoxO1 exclusion from the nuclei by IRS-1 overexpression is speculated.
Rho/ROCK signaling
Rho GTPases are molecular switches that modulate a variety of cytoskeleton-dependent cell functions [47]. Rho and its effector, the Rho-associated kinase ROCK, also play important roles in skeletal muscle differentiation. Both Rho and ROCK were high in proliferating myoblasts but decreased during differentiation. Several reports have showed Rho to be a negative regulator of muscle differentiation. For example, no multinucleated myotubes were observed in rat L6 myoblasts transfected with an active Rho mutant even under differentiation conditions [48]. Moreover, constitutive activation of Rho or ROCK resulted in a defect in myoblast fusion but did not abrogate the expression of early differentiation markers, MyoD and myogenin [49], in association with FoxO1 cytoplasmic retention. In addition, inactivation of ROCK was required for the nuclear accumulation of FoxO1 before the onset of myoblast terminal differentiation and then highly promoted myoblast fusion. This result is further supported by observations that FoxO1 is localized in the nucleus during myoblast terminal differentiation [21, 22]. Thus, these observations are abundantly revealed that Rho and ROCK may negatively regulate myoblast fusion but not the earlier steps of differentiation and the nuclear accumulation of FoxO1 is required for myoblast fusion. Notably, FoxO1 is a direct substrate of ROCK and ROCK directly phosphorylates FoxO1 in C2C12 cells, leading the FoxO1 shuttled out of nucleus. Thus, it appears to be that down-regulation of Rho/ROCK signaling is essential for FoxO1 nuclear translocation and myoblast fusion in vitro, providing a novel regulatory role of Rho/ROCK signaling regulating FoxO1 localization in myogenic differentiation.
Mechanisms of FoxO1 in the regulation of skeletal muscle differentiation
Although above researches believe FoxO1 is crucial for terminal myogenic differentiation, more studies mentioned below support FoxO1 as a negative regulator of muscle cell differentiation at early stage (Table 1). For example, Hribal et al. [33] showed that C2C12 myoblast differentiation was closely related to the increased FoxO1 phosphorylation and that differentiation appeared to require FoxO1 inhibition, similar to what has been revealed in thymocytes [50] and adipocytes [51]. These conflicting findings enough suggest that FoxO1 have dual roles in muscle differentiation according to the step of differentiation program (Figure 1). Here, we will declare the possible molecular pathway for FoxO1 mediating skeletal muscle differentiation.
mTOR pathway
The result from Wu et al. [52] confirmed a FoxO1 active mutant blocked C2C12 myoblasts differentiation at an early myogenesis stage and demonstrated that a regulatory loop between FoxO1 and the mammalian target of rapamycin (mTOR) pathway in myogenic differentiation course. mTOR, a most important regulator of cellular processes including growth, survival, proliferation and differentiation, has been found to be essential for the differentiation of C2C12 myoblasts by regulating the expression of IGF-II [53-56]. The autocrine production of IGF-II, which is critically participated in skeletal muscle differentiation as well as adult muscle regeneration, is upregulated by mTOR pathway at the transcriptional level. Moreover, as a major mediator of myogenic signaling, the PI3K/AkT pathway is a downstream of IGF-II signal [53]. Since FoxO1 is a downstream target of PI3K/AkT signaling [30], the IGF-II-PI3K/Akt-FoxO1-mTOR regulatory loop appears to be a major mediator of skeletal muscle differentiation.
FoxO1 can reduce the nutrient-dependent production of IGF-II and consequently breaks the process of myogenesis that relies on the IGF-II autocrine actions through decreasing the protein levels of mTOR. Activation of an inducible mutant of FoxO1 induces proteasome-dependent degradation of mTOR pathway components that are required for differentiation, including mTOR, raptor, S6 proteinkinase 1 and tuberous sclerosis complex 2, and then attenuates IGF-II expression at the transcriptional activation level [53]. In addition, when the active FoxO1 inhibits the myocyte differentiation, treatment of exogenous IGF-II could completely rescue this inhibition of myogenesis process from FoxO1 [53]. Thus, FoxO1 appears to exert the inhibitory function on muscle differentiation by reducing the IGF-II expression through degradation of mTOR which has been identified as a critical regulator of IGF-II transcription. Therefore, degradation of mTOR pathway components by FoxO1 provides a regulatory mechanism specific to the differentiation process. Overall, the relationship among FoxO1, mTOR, IGF-II and PI3K/Akt pathway presents a feedback loop that can preferably fine-tune the regulation of muscle differentiation.
Notch pathway
The study by Kitamura and coworkers has reported that FoxO1 is required for the inhibitory effect of Notch on myoblast differentiation and the ability to control myogenesis of FoxO1 is mediated through interaction with Notch [25]. Several reports have proved that active Notch signaling inhibits C2C12 and 10T/2 myoblasts differentiation, similar to the effect of active FoxO1 on cellular differentiation, via suppressing MyoD transcription [25, 57-59]. In addition, FoxO1 ablation simulates Notch1 ablation in mice [15, 60]. These data suggest that FoxO1 and Notch1 not only have a certain similarity function but also may have a further corelation on muscle differentiation. Csl is a DNA-binding protein and an identified Notch downstream effector [61]. Moreover, Hes1, another prototypical effector of Notch and also a Csl downstream target gene [62], has been considered to be a myoblast differentiation repressor by suppressing MyoD transcriptional level [53]. Kitamura et al. [25] demonstrated that through promoting corepressor clearance and recruiting the coactivator of Csl, FoxO1 physically and functionally interacted with Notch, resulting in activation of Hes1, thus inhibiting the myogenic progress. In this study, the Notch1 decoy partly rescued FoxO1 inhibition of myoblast differentiation. Likewise, FoxO1 small interfering RNA (siRNA) also rescued the inhibitory effect of Notch1 on myoblast differentiation and myosin expression. In addition, the authors also found that the DNA-binding protein Csl binds to FoxO1 through FoxO1 N terminal domain interacts with Csl N terminal and then binds to a consensus sequence in the Hes1 promoter [25, 61]. Since the Hes1 promoter contains no forkhead binding sites, FoxO1 cannot bind to it directly but through binding Csl element of Hes1 in differentiating C2C12 cells. Compendiously, FoxO1 functions to repress muscle differentiation via accompanying with constitutive binding to the Csl-binding site in the Hes1 promoter. Active FoxO1 and Notch1 increased the promoter activity and expression of Hes1, respectively, while FoxO1 siRNA inhibited this increase. These dada suggest that FoxO1 is required for Csl/Notch interaction. Meanwhile, in this study, the authors also showed that both FoxO1 and Notch1 binding to Hes1 promoter are dependent on Csl. Notch1 binding to Hes1 promoter is dependent on FoxO1 and the myoblast differentiation inhibited by Notch1 overexpression is rescued by inhibiting FoxO1. In addition, expression analyses found that overexpression of Notch1 or FoxO1 decreased MyoD expression, while Notch1 decoy or FoxO1 siRNA partially rescued the decrease of MyoD expression [25]. Thus, the findings present a mechanism by which FoxO1-Notch-Csl converge in a synergistic manner to suppress myoblast differentiation process in vivo.
Myostatin
Myostatin, a secreted factor that belongs to the transforming growth factor (TGF)-β superfamily, plays an important role in modulating skeletal muscle type formation [63], cell growth and differentiation [64]. Myostatin is preferentially expressed in skeletal muscle [63]. It has been reported to be a potent negative regulator of myoblast proliferation and differentiation [64, 65]. Myostatin lost function leads to heavy muscle growth due to hyperplasia [66]. In contrast, increased expression of myostatin results in cachectic muscle wasting [67]. A previous study revealed that myostatin suppressed myoblast differentiation through down-regulating expression and activity of MyoD [64], whereas MyoD expression were increased in myostatin-/- mice muscle. Allen et al. [68] demonstrated that myostatin is an additional target of FoxO1, since FoxO1 can directly bind and increase the activity of myostatin promoter to upregulate myostatin mRNA expression. Treatment of TGF-β greatly potentiated FoxO1-mediated inhibition of myoblast differentiation [68]. Together, these results suggest that FoxO1 could repress myogenic differentiation through stimulating the expression of myostatin.
MyoD
Liu et al. [69] revealed that blocking FoxO1 expression through RNA oligonucleotide increased the myogenic factor MyoD level and skeletal muscle mass of the mice while decreased expression of the muscle negative regulator myostatin both in C2C12 cell line and in vivo models. The chimeric transcription factors PAX3/FOXO1 and PAX7/FOXO1 suppress myogenic differentiation, similar to the effect of dominant-negative versions of Pax3 or Pax7 constructs on terminal differentiation of satellite cells, through inhibiting transcriptional activation of MyoD target genes, including myogenin, muscle creatine kinase (MCK) and p21 while the transcriptional activity of MyoD is not perturbed [70]. Moreover, silencing the PAX3-FOXO1 fusion gene promotes myogenic differentiation in Rhabdomyosarcomas (RMS) cell lines [71]. Furthermore, transgenic PAX3-FOXO1 in mice disrupts normal myogenesis in the developing somites [72]. In addition, JARID2 is required for cardiac myocytes and RMS cell lines differentiation [73, 74]. Knockdown JARID2 results in decrease of cell proliferation and facilitates myogenic differentiation [74]. Walters et al. [74] reported that JARID2 is a direct transcriptional target of the PAX3-FOXO1 fusion protein. Constitutive overexpression of PAX3-FOXO1 leads to an increase of both the RNA and protein levels of JARID2.
cGKI
The cyclic GMP-dependent protein kinase I (cGKI), which regulates cytoskeleton remodeling by phosphorylating the vasodilator-stimulated phosphoprotein and receding its activity [75], is a demonstrated direct transcriptional target of FoxO1a [76]. Moreover, excessive cell fusion was observed in cGKI-/- primary myoblasts [76]. In addition, cGKI is an identified regulator of FoxO1a activity and also promotes re-localization of FoxO1a out from the nucleus during muscle cell fusion. In muscle cell differentiation progress, FoxO1a directly activates transcription of cGKI. In turn, cGKI reduces the FoxO1a function in directing muscle cell fusion by phosphorylating FoxO1a, thus abolishes the binding ability of FoxO1a to its response elements [76]. This result suggests the negative-feedback loop between FoxO1a and cGKI fine-tunes the progress of muscle cell fusion process.
On the other hand, Bois and Grosveld [21] found that FoxO1 was mainly distributed in the cytoplasm in proliferating mouse primary myoblasts, while located in the nucleus by nuclear translocation from cytoplasm to nucleus in differentiated myoblasts through a non-Akt-dependent but phosphorylation-mediated nuclear exclusion event, suggesting that other kinases steer FoxO1a transcriptional activity during myogenic differentiation. Indeed, the above mentioned Rho-associated kinase ROCK and cGKI have been demonstrated to be mediators for FoxO1 transcriptional activity. Moreover, this result is consistent with the discovery that FoxO1 is localized in the nucleus during myoblast differentiation [22]. FoxO1 phosphorylation seems to decrease the primary myoblast terminal differentiation ability and a dominant-active dephosphorylated FoxO1 significantly upregulates the rate and extent of myotube formation [21, 22]. But expression of a dominant-negative FoxO1 mutant restricts myotube fusion [21]. Furthermore, inhibiting FoxO1 transcriptional activity suppressed myoblast fusion without affecting cell survival, whereas FoxO1 activation had no effect on apoptosis index of primary myoblast [21]. It is worth noting that these indispensable roles of FoxO1 on myotube formation is after the initiation of differentiation.
So, a question emerged in front of us. What precise functions do FoxO1 play in muscle differentiation? In the myoblast differentiation process, MyoD as the earliest marker of differentiation, activated the expression of myogenic specific genes to initiate the myogenic program. After a later stage of myogenic differentiation, myoblast fusion is started to form multinucleated myotubes and this is a critical step triggering terminal differentiation in a series of these events. Taken together with the before mentioned observations, FoxO1 is tightly and selectively associated with the myoblast differentiation, according to the step of differentiation program [21]. Also of note, FoxO1 is required for myoblast terminal differentiation fusion into myotubes, but at early or middle stage, FoxO1 inhibits differentiation process (Figure 1). Although numerous studies have elaborated the dual roles of FoxO1 in different stages of myoblast differentiation, the molecular mechanism of FoxO1 for myotube fusion remains poorly understood.
Role of FoxO1 in skeletal muscle fiber type specification
Skeletal muscle consists of heterogeneous specialized muscle fibers that differ in their biochemical and metabolic properties. It is this diversity of myofibers that enables different type of muscles to accomplish a variety of functions. The total number of muscle fibers is invariable prior to hatching or birth of animals, whereas the formation of the muscle fiber types could be regulated during muscle development and the muscle fiber type composition could fortunately be altered along the animal’s life [77]. A striking characteristic of myofiber is the ability to remodel and transform from another type in response to environmental demands. The fiber-type switch occurs in a sequential reversible way: I ↔ IIa ↔ IIx/d ↔ IIb [78]. Many genes and signaling pathways have been reported to participate in the muscle fiber type specification and transition in developing embryos or adult muscles.
Most recent studies reported that FoxO1 plays a critical role in skeletal muscle type specification. The study of Kamei et al. [23] showed that transgenic mice specifically overexpressing FoxO1 in skeletal muscle using the skeletal muscle α-actin promoter weighed less than the wildtype control mice and significantly reduced the muscle mass and the size of both type I and type II fibers. This data suggests that FoxO1 may be implicated in the breakdown of muscle fibers. Meanwhile, it has been reported that FoxO1 transgenic overexpression in mice led to the decrease of type I fiber-related gene expression and presented a marked decrease in the number of type I fibers whereas the type II fiber isoform genes did not alter, accompanied with downregulation of muscle anti-fatigue ability [23]. It was also reported that in controlled conditions, FoxO1 was closely related to muscle fiber type distribution and expressed much higher in fast twitch fiber enriched muscles than in slow muscles [79-81]. In addition, a study conducted by Yuan et al. [79] demonstrated that endurance swimming exercise program induced a fast-to-slow fiber type transition, accompanied with a decrease of FoxO1 expression in both fast and slow muscles, indicating that this conversion may result from suppression of FoxO1 expression. Accordingly, this study revealed a constitutively active form of FoxO1 changed the muscle fiber type composition, accompanied by a slow- to fast-twitch fiber transition in C2C12 myoblast. These findings appear to show that FoxO1 may negatively regulate type I fiber formation but positively regulate type II. However, a contrasting finding conducted by Kitamura et al. [25] supported that FoxO1 conditional deletion in skeletal muscle using chimeric myogenin-FoxO1 transgenics decreased slow-twitch fibers and reduced expression of type I fiber genes while type II fiber genes increased. These data suggest that conditional ablation of FoxO1 decreased formation of myogenin-containing muscle fibers and changed fiber type distribution increasing MyoD-containing fibers, because myogenin is the predominant myogenic factor in slow fibers while MyoD in fast fibers [82]. This fiber-type switch in myogenin-FoxO1 mice could be accounted for the inhibitory effect of FoxO1 on MyoD expression [25]. When the inhibitory effect was removed, the formation of fast fibers was increased, potentially at the expense of slow fibers.
Mechanisms of FoxO1 in the regulation of skeletal muscle fiber type specification
FoxO1 regulates skeletal muscle fiber type specification via regulating the formation of slow and fast fibers through or in association with several factors and signaling pathways, such as PGC1α, MEF2C, CaMK and calcineurin pathway (Figure 2).
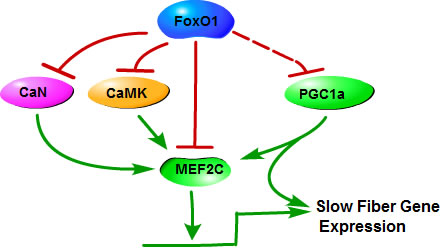
Figure 2: Mechanisms of FoxO1 in the regulation of slow skeletal muscle fiber gene expression. FoxO1 downregulates calcineurin (CaN), CaMK and MEF2C expression, leading to a decrease of MEF2C that can increase the transcriptional activation of slow fiber genes, to inhibit slow fiber genes expression. In addition, PGC1α induces fiber-type switching from glycolytic toward oxidative fibers. FoxO1 may interact with PGC1α to inhibit certain functions of PGC1α, inhibiting expression of slow fiber gene.
PGC1α
Peroxisome proliferator-activated receptor-γ coactivator-1 (PGC1α), which co-activates PPARγ in muscle [1, 83] and is a required metabolic transcriptional coactivator for oxidative metabolism, mitochondrial biogenesis, and slow-twitch fiber formation, is a direct co-activator of FoxO1 [1, 84-86]. Several reports showed that insulin decreases the basal PGC1α promoter activity and Akt overexpression similar to the effect of insulin [33]. FoxO1 could directly bind to the three insulin response sequences (IRSs) addressed in the PGC1α promoter to upregulate the promoter activity of PGC1α in liver HepG2 cells [84]. However, insulin and Akt inactivate FoxO1 and inhibit the FoxO1-increased PGC1α promoter activity directly through phosphorylation of putative Akt sites in FoxO1, further suppressing the combining capacity of FoxO1 to PGC1α promoter in liver.
Previous study showed that PGC1α is abundantly expressed in skeletal muscle [87] and is remarkably induced by endurance exercise [88]. Notably, a study conducted by Lin et al. [87] showed that PGC1α is richly expressed in type I fibers and that drives the formation of slow-twitch muscle fibers. In PGC1α transgenic mice, type-II-rich muscles showed redder and better antifatigue ability than control mice and genes related mitochondrial oxidative metabolism were widely activated. Moreover, a fiber-type switching from glycolytic toward oxidative fibers was observed in transgenic PGC1α mice [87, 88]. It is widely known that type I fibers are more dependent on oxidative metabolism than type II, and this scenario may be due to the particular myofibrillar proteins and mitochondrial content much higher in type I fibers than in type II [89].
Given that PGC1α has the potential for differentiation of type I fibers and control the glycolytic to oxidative fiber-type switching, FoxO1 might be implied in these progresses. FoxO1 and PGC1α present diverse actions in different fiber type muscle. FoxO1 mRNA level was increased in both the soleus and plantaris muscle, whereas PGC1α mRNA level was decreased in the soleus but not in the plantaris muscle under the condition of hindlimb unloading [90]. Furthermore, the oxidative enzyme activity and the percentage of type I fibers were reduced and the percentages of glycolytic fibers were increased in the soleus muscle, but not in the plantaris muscle during hindlimb unloading [90]. These data sufficiently suggested that both FoxO1 and PGC1α affected slow fibers more efficient than fast fibers. Regrettably, to date, we have not yet found studies verifying that FoxO1 directly physically interacts with PGC1α in skeletal muscle. But, Kamei et al. [23] showed that FoxO1 had a discrepant function with PGC1α on skeletal muscle type fiber gene: the expression of type I fiber genes, including troponin I (slow) and myoglobin, were decreased in FoxO1 transgenic mice, whereas increased in PGC1α transgenic mice. These data suggest that, although FoxO1 may facilitate PGC1α expression in muscle, decrease of type I fiber genes mediated by FoxO1 may be not directly linked to PGC1α gene expression, but through that FoxO1 protein interacts with PGC1α protein to inhibit certain functions of PGC1α [86]. After all, FoxO1 itself is a transcription factor.
MEF2C and CaMK
The key regulator of muscle development, MEF2C transcription factor, is preferentially expressed in slow, oxidative myofibers and selectively active in slow oxidative fibers [91, 92]. Several reports suggest that overexpression of active form MEF2C in transgenic mice promotes slow-fiber formation and enhances running endurance through responding to calcium-dependent signaling pathway that stimulates the transformation of fast, glycolytic fibers into slow, oxidative fibers [93], whereas inactivation of MEF2C results in a severe decrease of type I fibers and losses of fiber transformation [5, 94]. Moreover, it has been conclusively suggested that MEF2C is a necessary upstream transcriptional activator of myofiber identity and troponins in skeletal muscle [95]. In addition to MEF2C, signals generated by CaMK (calmodulin-dependent kinase), downstream molecule of calcium signaling, also facilitate type I fiber gene expression. CaMK increases the transactivating function of MEF2C [92]. Although to our knowledge, FoxO1 has not been shown to be directly involved in the decision of fiber-type composition via MEF2C and CaMK, coincidentally enough, expression levels of both MEF2C and CaMK are significantly reduced in skeletal muscle of FoxO1 transgenic mice, indicating that the downregulation of type I fiber genes suppressed by FoxO1 may, in part, contribute to FoxO1 inhibiting the expression of MEF2C and CaMK.
Calcineurin pathway
A study conducted Yuan et al. [79] found that overexpression of FoxO1 induced the formation of fast-twitch fibers and altered the proportion of muscle fiber type composition, along with a decrease of muscle oxidative capacity and a slow-oxidative to fast-glycolytic fiber type transformation in C2C12 myotubes. Treatment with resveratrol, which inhibited the endogenous FoxO1 activity, led to an increase of type I fiber related gene troponin (slow) and myoglobin [79] and prevented TNF-α-induced muscle atrophy [96], whereas constitutively active FoxO1 mutant significantly blocked the resveratrol-induced increased expression of these two genes. Most interestingly, addition of resveratrol could also block the FoxO1-induced slow to fast-twitch fiber transition.
It is generally known that the calcineurin pathway is a master chief regulatory pathway stimulating slow fiber-selective gene expression and slow-twitch fiber formation [97]. Several reports suggest that FoxO1 protein decreased calcineurin phosphatase activity in the cardiomyocytes [16, 98]. In turn, overexpression of calcineurin inhibited FoxO factors protein levels and precluded myotube atrophy [99]. Moreover, slow-twitch oxidative fibers present greater resistance than fast-twitch glycolytic fibers. Given the function of calcineurin on slow-twitch oxidative fiber formation and the interrelation between FoxO1 and calcineurin mentioned before, FoxO1 might be implied in calcineurin pathway controlling muscle fiber conversion. Coincidentally enough, Yuan et al. [79] revealed that a constitutively active FoxO1 mutant caused a significant decrease of endogenous calcineurin phosphatase activity and even significantly decreased the mRNA level of MCIP1.4 (modulatory calcineurin interacting protein, exon 4 isoform), a target of the calcineurin pathway. However, the mRNA level of MCIP1.4 in FoxO1-infected myoblasts was strikingly increased by resveratrol addition, suggesting that FoxO1 inhibition could reverse the negative effect on the target of calcineurin pathway expression. In summary, these results suggest that FoxO1 promotes slow-to-fast fiber-type switch and decreases muscle oxidative capacity at least, in part, through suppressing calcineurin pathway.
Conclusions
FoxO1 is a critical transcription factor that plays an important role in skeletal muscle differentiation and fiber type specification. Although the inhibitory effect of FoxO1 on early stage of skeletal muscle differentiation by affecting several signaling including mTOR pathway, Notch pathway, myostatin, MyoD and cGKI has been known, the precise mechanisms on how FoxO1 is critical for myoblast fusion into myotube remain largely unknown. Moreover, FoxO1 is closely related to muscle fiber type specification. FoxO1 may negatively regulate type I fiber formation through inhibiting expression of MEF2C, CaMK and calcineurin or through suppressing certain functions of PGC1α, thus controlling the specification of muscle fiber type. This review has highlighted molecular mechanisms of FoxO1 in the regulation of skeletal muscle differentiation and fiber type specification. An understanding in molecular mechanisms of FoxO1 in muscle may develop new therapeutic approaches that can be used to prevent myopathies, such as muscle atrophy, in spite of great challenges that remain to be conquered.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No. 31472110).
Conflicts of Interest
None declared.
References
1. P. de Lange, M. Moreno, E. Silvestri, A. Lombardi, F. Goglia, A. Lanni. Fuel economy in food-deprived skeletal muscle: signaling pathways and regulatory mechanisms. FASEB J. 2007; 21: 3431-3441.
2. A. Eijkelenboom, B.M. Burgering. FOXOs: signalling integrators for homeostasis maintenance. Nature reviews. Nat Rev Mol Cell Biol. 2013; 14: 83-97.
3. M. Buckingham. Skeletal muscle formation in vertebrates. Curr Opin Genet Dev. 2001; 11: 440-448.
4. G.T. Pipes, E.E. Creemers, E.N. Olson. The myocardin family of transcriptional coactivators: versatile regulators of cell growth, migration, and myogenesis. Genes Dev. 2006; 20: 1545-1556.
5. M.J. Potthoff, E.N. Olson. MEF2: a central regulator of diverse developmental programs. Development. 2007; 134: 4131-4140.
6. D. Pette, R.S. Staron. Myosin isoforms, muscle fiber types, and transitions. Microsc Res Tech. 2000; 50: 500-509.
7. K. Lee, E. Ochi, H. Song, K. Nakazato. Activation of AMP-activated protein kinase induce expression of FoxO1, FoxO3a, and myostatin after exercise-induced muscle damage. Biochem Biophys Res Commun. 2015; 466: 289-294.
8. H.H. Zhao, R.E. Herrera, E. Coronado-Heinsohn, M.C. Yang, J.H. Ludes-Meyers, K.J. Seybold-Tilson, Z. Nawaz, D. Yee, F.G. Barr, S.G. Diab. Forkhead homologue in rhabdomyosarcoma functions as a bifunctional nuclear receptor-interacting protein with both coactivator and corepressor functions. J Biol Chem. 2001; 276: 27907-27912.
9. D. Gross, A. van den Heuvel, M. Birnbaum. The role of FoxO in the regulation of metabolism. Oncogene. 2008; 27: 2320-2336.
10. G. Milan, V. Romanello, F. Pescatore, A. Armani, J.H. Paik, L. Frasson, A. Seydel, J. Zhao, R. Abraham, A.L. Goldberg, B. Blaauw, R.A. DePinho, M. Sandri. Regulation of autophagy and the ubiquitin-proteasome system by the FoxO transcriptional network during muscle atrophy. Nat Commun. 2015; 6: 6670.
11. Z.Q. Zou, J. Xu, L. Li, Y.S. Han. Down-regulation of SENCR promotes smooth muscle cells proliferation and migration in db/db mice through up-regulation of FoxO1 and TRPC6. Biomed Pharmacother. 2015; 74: 35-41.
12. D. Lee, A.L. Goldberg. SIRT1 protein, by blocking the activities of transcription factors FoxO1 and FoxO3, inhibits muscle atrophy and promotes muscle growth. J Biol Chem. 2013; 288: 30515-30526.
13. D.N. Gross, M. Wan, M.J. Birnbaum. The role of FOXO in the regulation of metabolism. Curr Diab Rep. 2009; 9: 208-214.
14. D.H. Castrillon, L. Miao, R. Kollipara, J.W. Horner, R.A. DePinho. Suppression of ovarian follicle activation in mice by the transcription factor Foxo3a. Science. 2003; 301: 215-218.
15. T. Hosaka, W.H. Biggs, D. Tieu, A.D. Boyer, N.M. Varki, W.K. Cavenee, K.C. Arden. Disruption of forkhead transcription factor (FOXO) family members in mice reveals their functional diversification. Proc Natl Acad Sci U S A. 2004; 101: 2975-2980.
16. Y.G. Ni, K. Berenji, N. Wang, M. Oh, N. Sachan, A. Dey, J. Cheng, G. Lu, D.J. Morris, D.H. Foxo transcription factors blunt cardiac hypertrophy by inhibiting calcineurin signaling. Circulation. 2006; 114: 1159-1168.
17. K. Maiese. FoxO proteins in the nervous system. Anal Cell Pathol. 2015; 569392.
18. P. Urbanek, L.O. Klotz. Posttranscriptional regulation of FOXO expression: microRNAs and beyond. Br J Pharmacol. 2016; doi: 10.1111/bph.13471.
19. D. Accili, K.C. Arden. FoxOs at the crossroads of cellular metabolism, differentiation, and transformation. Cell. 2004; 117: 421-426.
20. M. Kortylewski, F. Feld, K.D. Krüger, G. Bahrenberg, R.A. Roth, H.G. Joost, P.C. Heinrich, I. Behrmann, A. Barthel. Akt modulates STAT3-mediated gene expression through a FKHR (FOXO1a)-dependent mechanism. J Biol Chem. 2003;278: 5242-5249.
21. P.R. Bois, G.C. Grosveld. FKHR (FOXO1a) is required for myotube fusion of primary mouse myoblasts. EMBO J. 2003; 22: 1147-1157.
22. F. Hakuno, Y. Yamauchi, G. Kaneko, Y. Yoneyama, J. Nakae, K. Chida, T. Kadowaki, K. Yamanouchi, M. Nishihara, S. Takahashi. Constitutive expression of insulin receptor substrate (IRS)-1 inhibits myogenic differentiation through nuclear exclusion of Foxo1 in L6 myoblasts. PLoS One. 2011; 6: e25655.
23. Y. Kamei, S. Miura, M. Suzuki, Y. Kai, J. Mizukami, T. Taniguchi, K. Mochida, T. Hata, J. Matsuda, H. Aburatani. Skeletal muscle FOXO1 (FKHR) transgenic mice have less skeletal muscle mass, down-regulated Type I (slow twitch/red muscle) fiber genes, and impaired glycemic control. J Biol Chem. 2004; 279: 41114-41123.
24. L. Lin, K. Chen, W. Abdel Khalek, J.L. Ward, 3rd, H. Yang, B. Chabi, C. Wrutniak-Cabello, Q. Tong. Regulation of skeletal muscle oxidative capacity and muscle mass by SIRT3. PLoS One. 2014; 9: e85636.
25. T. Kitamura, Y.I. Kitamura, Y. Funahashi, C.J. Shawber, D.H. Castrillon, R. Kollipara, R.A. DePinho, J. Kitajewski, D. Accili. A Foxo/Notch pathway controls myogenic differentiation and fiber type specification. J Clin Invest. 2007; 117: 2477-2485.
26. A.M. Sanchez, R.B. Candau, H. Bernardi. FoxO transcription factors: their roles in the maintenance of skeletal muscle homeostasis. Cell Mol Life Sci. 2014; 71:1657-1671.
27. W.J. Pang, T.Y. Yu, L. Bai, Y.J. Yang, G.S. Yang. Tissue expression of porcine FoxO1 and its negative regulation during primary preadipocyte differentiation. Mol Biol Rep. 2009; 36: 165-176.
28. A. Yamashita, Y. Hatazawa, Y. Hirose, Y. Ono, Y. Kamei. FOXO1 delays skeletal muscle regeneration and suppresses myoblast proliferation. Biosci Biotechnol Biochem. 2016; 80: 1531-1535.
29. P. Levinger, M.K. Caldow, J.R. Bartlett, J.M. Peake, C. Smith, D. Cameron-Smith, I. Levinger. The level of FoxO1 and IL-15 in skeletal muscle, serum and synovial fluid in people with knee osteoarthritis: a case control study. Osteoporos Int. 2016; 27: 2137-2143.
30. T.N. Stitt, D. Drujan, B.A. Clarke, F. Panaro, Y. Timofeyva, W.O. Kline, M. Gonzalez, G.D. Yancopoulos, D.J. Glass. The IGF-1/PI3K/Akt pathway prevents expression of muscle atrophy-induced ubiquitin ligases by inhibiting FOXO transcription factors. Mol Cell. 2004; 14: 395-403.
31. A. Wagatsuma, M. Shiozuka, Y. Takayama, T. Hoshino, K. Mabuchi, R. Matsuda. Effects of ageing on expression of the muscle-specific E3 ubiquitin ligases and Akt-dependent regulation of Foxo transcription factors in skeletal muscle. Mol Cell Biochem. 2016; 412: 59-72.
32. X. Li, H. Liu, H. Wang, L. Sun, F. Ding, W. Sun, C. Han, J. Wang. Follistatin could promote the proliferation of duck primary myoblasts by activating PI3K/Akt/mTOR signalling. Biosci Rep. 2014; 34: 609-620.
33. M.L. Hribal, J. Nakae, T. Kitamura, J.R. Shutter, D. Accili. Regulation of insulin-like growth factor-dependent myoblast differentiation by Foxo forkhead transcription factors. J Cell Biol. 2003; 162: 535-541.
34. S. Wilton, I. Skerjanc. Factors in serum regulate muscle development in P19 cells. In Vitro Cell Dev Biol Anim. 1999; 35: 175-177.
35. Y.J. Wu, Y.H. Fang, H.C. Chi, L.C. Chang, S.Y. Chung, W.C. Huang, X.W. Wang, K.W. Lee, S.L. Chen. Insulin and LiCl synergistically rescue myogenic differentiation of FoxO1 over-expressed myoblasts. PLoS One. 2014; 9: e88450.
36. A. Pirskanen, J.C. Kiefer, S.D. Hauschka. IGFs, insulin, Shh, bFGF, and TGF-1 interact synergistically to promote somite myogenesis in vitro. Dev Biol. 2000; 224: 189-203.
37. R. Bouchi, K.S. Foo, H. Hua, K. Tsuchiya, Y. Ohmura, P.R. Sandoval, L.E. Ratner, D. Egli, R.L. Leibel, D. Accili. FOXO1 inhibition yields functional insulin-producing cells in human gut organoid cultures. Nat Commun. 2014; 5: 4242.
38. A. Rochat, A. Fernandez, M. Vandromme, J.P. Moles, T. Bouschet, G. Carnac, N.J. Lamb. Insulin and wnt1 pathways cooperate to induce reserve cell activation in differentiation and myotube hypertrophy. Mol Biol Cell. 2004; 15: 4544-4555.
39. K. Okada, A.T. Naito, T. Higo, A. Nakagawa, M. Shibamoto, T. Sakai, A. Hashimoto, Y. Kuramoto, T. Sumida, S. Nomura, M. Ito, T. Yamaguchi, T. Oka et al. Wnt/beta-Catenin Signaling Contributes to Skeletal Myopathy in Heart Failure via Direct Interaction With Forkhead Box O. Circ Heart Fail. 2015; 8: 799-808.
40. A. Litwiniuk, B. Pijet, M. Pijet-Kucicka, M. Gajewska, B. Pajk, A. Orzechowski. FOXO1 and GSK-3 Are Main Targets of Insulin-Mediated Myogenesis in C2C12 Muscle Cells, PLoS One. 2016; 11: e0146726.
41. J. Nakae, V. Barr, D. Accili. Differential regulation of gene expression by insulin and IGF-1 receptors correlates with phosphorylation of a single amino acid residue in the forkhead transcription factor FKHR. EMBO J. 2000; 19: 989-996.
42. C. De Alvaro, I. Nieto-Vazquez, J.M. Rojas, M. Lorenzo. Nuclear exclusion of Forkhead Box O and Elk1 and activation of nuclear factor-B are required for C2C12-RasV12C40 myoblast differentiation. Endocrinology. 2008; 149: 793-801.
43. S.D Gopinath, A,E Webb, A. Brunet, T.A Rando. FOXO3 promotes quiescence in adult muscle stem cells during the process of self-renewal. Stem cell reports. 2014; 2: 414-426.
44. D.R. Calnan, A. Brunet. The FoxO code. Oncogene. 2008; 27: 2276-2288.
45. H. Matsuzaki, H. Daitoku, M. Hatta, K. Tanaka, A. Fukamizu. Insulin-induced phosphorylation of FKHR (Foxo1) targets to proteasomal degradation. Proc Natl Acad Sci U S A. 2003; 100: 11285-11290.
46. W.H. Biggs, J. Meisenhelder, T. Hunter, W.K. Cavenee, K.C. Arden. Protein kinase B/Akt-mediated phosphorylation promotes nuclear exclusion of the winged helix transcription factor FKHR1. Proc Natl Acad Sci U S A. 1999; 96: 7421-7426.
47. S. Etienne-Manneville, A. Hall. Rho GTPases in cell biology. Nature. 2002; 420: 629-635.
48. M. Meriane, P. Roux, M. Primig, P. Fort, C. Gauthier-Rouviere. Critical activities of Rac1 and Cdc42Hs in skeletal myogenesis: antagonistic effects of JNK and p38 pathways. Mol Biol Cell. 2000; 11: 2513-2528.
49. T. Nishiyama, I. Kii, A. Kudo. Inactivation of Rho/ROCK signaling is crucial for the nuclear accumulation of FKHR and myoblast fusion. J Biol Chem. 2004; 279: 47311-47319.
50. H. Leenders, S. Whitfield, C. Benoist, D. Mathis. Role of the forkhead transcription family member, FKHR, in thymocyte differentiation. Eur J Immunol. 2000; 30: 2980-2990.
51. J. Nakae, T. Kitamura, Y. Kitamura, W.H. Biggs, K.C. Arden, D. Accili. The forkhead transcription factor Foxo1 regulates adipocyte differentiation. Dev Cell. 2003; 4: 119-129.
52. A.L. Wu, J.H. Kim, C. Zhang, T.G. Unterman, J. Chen. Forkhead box protein O1 negatively regulates skeletal myocyte differentiation through degradation of mammalian target of rapamycin pathway components. Endocrinology. 2008; 149: 1407-1414.
53. E. Erbay, I.-H. Park, P.D. Nuzzi, C.J. Schoenherr, J. Chen. IGF-II transcription in skeletal myogenesis is controlled by mTOR and nutrients. J Cell Biol. 2003; 163: 931-936.
54. E. Erbay, J. Chen. The mammalian target of rapamycin regulates C2C12 myogenesis via a kinase-independent mechanism. J Biol Chem. 2001; 276: 36079-36082.
55. L. Shu, X. Zhang, P.J. Houghton. Myogenic differentiation is dependent on both the kinase function and the N-terminal sequence of mammalian target of rapamycin. J Biol Chem. 2002; 277: 16726-16732.
56. A.E. Webb, A. Brunet. FOXO transcription factors: key regulators of cellular quality control. Trends Biochem Sci. 2014; 39: 159-169.
57. E. Hirsinger, P. Malapert, J. Dubrulle, M.C. Delfini, D. Duprez, D. Henrique, D. Ish-Horowicz, O. Pourquié. Notch signalling acts in postmitotic avian myogenic cells to control MyoD activation. Development. 2001; 128: 107-116.
58. I.M. Conboy, T.A. Rando. The regulation of Notch signaling controls satellite cell activation and cell fate determination in postnatal myogenesis. Dev Cell. 2002; 3: 397-409.
59. K. Kuroda, S. Tani, K. Tamura, S. Minoguchi, H. Kurooka, T. Honjo. Delta-induced Notch signaling mediated by RBP-J inhibits MyoD expression and myogenesis. J Biol Chem. 1999; 274: 7238-7244.
60. L.T. Krebs, Y. Xue, C.R. Norton, J.R. Shutter, M. Maguire, J.P. Sundberg, D. Gallahan, V. Closson, J. Kitajewski, R. Callahan. Notch signaling is essential for vascular morphogenesis in mice. Genes Dev. 2000; 14: 1343-1352.
61. E.C. Lai. Keeping a good pathway down: transcriptional repression of Notch pathway target genes by CSL proteins. EMBO Rep. 2002; 3: 840-845.
62. D.A. Ross, P.K. Rao, T. Kadesch. Dual roles for the Notch target gene Hes-1 in the differentiation of 3T3-L1 preadipocytes. Mol Cell Biol. 2004; 24: 3505-3513.
63. A. Hennebry, C. Berry, V. Siriett, P. O’Callaghan, L. Chau, T. Watson, M. Sharma, R. Kambadur. Myostatin regulates fiber-type composition of skeletal muscle by regulating MEF2 and MyoD gene expression. Am J Physiol Cell Physiol. 2009; 296: C525-C534.
64. B. Langley, M. Thomas, A. Bishop, M. Sharma, S. Gilmour, R. Kambadur. Myostatin inhibits myoblast differentiation by down-regulating MyoD expression. J Biol Chem. 2002; 277: 49831-49840.
65. R. Vivar, C. Humeres, C. Munoz, P. Boza, S. Bolivar, F. Tapia, S. Lavandero, M. Chiong, G. Diaz-Araya. FoxO1 mediates TGF-beta1-dependent cardiac myofibroblast differentiation. Biochim Biophys Acta. 2016; 1863: 128-138.
66. A.C. McPherron, A.M. Lawler, S.J. Lee. Regulation of skeletal muscle mass in mice by a new TGF-beta superfamily member. Nature. 1997; 387: 83-90.
67. T.A. Zimmers, M.V. Davies, L.G. Koniaris, P. Haynes, A.F. Esquela, K.N. Tomkinson, A.C. McPherron, N.M. Wolfman, S.J. Lee. Induction of cachexia in mice by systemically administered myostatin. Science. 2002; 296: 1486-1488.
68. D.L. Allen, T.G. Unterman. Regulation of myostatin expression and myoblast differentiation by FoxO and SMAD transcription factors. Am J Physiol Cell Physiol. 2007; 292: C188-C199.
69. C. Liu, Z. Yang, C. Liu, R. Wang, P. Tien, R. Dale, L. Sun. Effect of RNA oligonucleotide targeting Foxo-1 on muscle growth in normal and cancer cachexia mice. Cancer Gene Ther. 2007; 14: 945-952.
70. F. Calhabeu, S. Hayashi, J. Morgan, F. Relaix, P. Zammit. Alveolar rhabdomyosarcoma-associated proteins PAX3/FOXO1A and PAX7/FOXO1A suppress the transcriptional activity of MyoD-target genes in muscle stem cells. Oncogene. 2013; 32: 651-662.
71. K. Kikuchi, K. Tsuchiya, O. Otabe, T. Gotoh, S. Tamura, Y. Katsumi, S. Yagyu, S. Tsubai-Shimizu, M. Miyachi, T. Iehara. Effects of PAX3-FKHR on malignant phenotypes in alveolar rhabdomyosarcoma. Biochem Biophys Res Commun. 2008; 365: 568-574.
72. F.G. Finckenstein, E. Davicioni, K.G. Osborn, W.K. Cavenee, K.C. Arden, M.J. Anderson. Transgenic mice expressing PAX3-FKHR have multiple defects in muscle development, including ectopic skeletal myogenesis in the developing neural tube. Transgenic Res. 2006; 15: 595-614.
73. J. Jung, T.G. Kim, G.E. Lyons, H.R.C. Kim, Y. Lee. Jumonji regulates cardiomyocyte proliferation via interaction with retinoblastoma protein. J Biol Chem. 2005; 280: 30916-30923.
74. Z.S. Walters, B. Villarejo-Balcells, D. Olmos, T.W. Buist, E. Missiaglia, R. Allen, B. Al-Lazikani, M.D. Garrett, J. Blagg, J. Shipley. JARID2 is a direct target of the PAX3-FOXO1 fusion protein and inhibits myogenic differentiation of rhabdomyosarcoma cells. Oncogene. 2014; 33: 1148-1157.
75. T. Gudi, J.C. Chen, D.E. Casteel, T.M. Seasholtz, G.R. Boss, R.B. Pilz. cGMP-dependent protein kinase inhibits serum-response element-dependent transcription by inhibiting rho activation and functions. J Biol Chem. 2002; 277: 37382-37393.
76. P.R. Bois, V.F. Brochard, A.V. Salin-Cantegrel, J.L. Cleveland, G.C. Grosveld. FoxO1a-cyclic GMP-dependent kinase I interactions orchestrate myoblast fusion. Mol Cell Biol. 2005; 25: 7645-7656.
77. B. Picard, L. Lefaucheur, C. Berri, M.J. Duclos. Muscle fibre ontogenesis in farm animal species. Reprod Nutr Dev. 2002; 42: 415-431.
78. D. Pette, R.S. Staron. Myosin isoforms, muscle fiber types, and transitions. Microsc Res Tech. 2000; 50: 500-509.
79. Y. Yuan, X.E. Shi, Y.G. Liu, G.S. Yang. FoxO1 regulates muscle fiber-type specification and inhibits calcineurin signaling during C2C12 myoblast differentiation. Mol Cell Biochem. 2011; 348: 77-87.
80. M. Azad, N. Khaledi, M. Hedayati. Effect of acute and chronic eccentric exercise on FOXO1 mRNA expression as fiber type transition factor in rat skeletal muscles. Gene. 2016; 584: 180-184.
81. X.E. Shi, Z.Y. Song, Q.M. Yang, Y.G. Liu, G.S. Yang. Correlation of forkhead box transcription factor O1 and myosin heavy chain isoforms in porcine skeletal muscle. Genet Mol Res. 2014; 13: 10231-10240.
82. S.M. Hughes, J.M. Taylor, S.J. Tapscott, C.M. Gurley, W.J. Carter, C.A. Peterson. Selective accumulation of MyoD and myogenin mRNAs in fast and slow adult skeletal muscle is controlled by innervation and hormones. Development. 1993; 118: 1137-1147.
83. E. Castillero, N. Alamdari, Z. Aversa, A. Gurav, P.O. Hasselgren. PPARbeta/delta regulates glucocorticoid- and sepsis-induced FOXO1 activation and muscle wasting. PLoS One. 2013; 8: e59726.
84. H. Daitoku, K. Yamagata, H. Matsuzaki, M. Hatta, A. Fukamizu. Regulation of PGC-1 promoter activity by protein kinase B and the forkhead transcription factor FKHR. Diabetes. 2003; 52: 642-649.
85. C. Kang, L. Li Ji. Role of PGC-1 signaling in skeletal muscle health and disease. Ann N Y Acad Sci. 2012; 127: 110-117.
86. P. Puigserver, J. Rhee, J. Donovan, C.J. Walkey, J.C. Yoon, F. Oriente, Y. Kitamura, J. Altomonte, H. Dong, D. Accili. Insulin-regulated hepatic gluconeogenesis through FOXO1-PGC1 interaction. Nature. 2003; 423: 550-555.
87. J. Lin, H. Wu, P.T. Tarr, C.Y. Zhang, Z. Wu, O. Boss, L.F. Michael, P. Puigserver, E. Isotani, E.N. Olson. Transcriptional co-activator PGC1 drives the formation of slow-twitch muscle fibres. Nature. 2002; 418: 797-801.
88. A.P. Russell, J. Feilchenfeldt, S. Schreiber, M. Praz, A. Crettenand, C. Gobelet, C.A. Meier, D.R. Bell, A. Kralli, J.-P. Giacobino. Endurance training in humans leads to fiber type-specific increases in levels of peroxisome proliferator-activated receptor- coactivator-1 and peroxisome proliferator-activated receptor- in skeletal muscle. Diabetes. 2003; 52: 2874-2881.
89. M.W. Berchtold, H. Brinkmeier, M. Müntener. Calcium ion in skeletal muscle: its crucial role for muscle function, plasticity, and disease. Physiol Rev. 2000; 80: 1215-1265.
90. F. Nagatomo, H. Fujino, H. Kondo, H. Kondo, H. Suzuki, M. Kouzaki, I. Takeda, A. Ishihara. PGC1 and FOXO1 mRNA levels and fiber characteristics of the soleus and plantaris muscles in rats after hindlimb unloading. Histol Histopathol. 2011; 26: 1545-1553.
91. H. Wu, E.N. Olson. Activation of the MEF2 transcription factor in skeletal muscles from myotonic mice. J Clin Invest. 2002; 109: 1327-1333.
92. H. Wu, F.J. Naya, T.A. McKinsey, B. Mercer, J.M. Shelton, E.R. Chin, A.R. Simard, R.N. Michel, R. Bassel-Duby, E.N. Olson. MEF2 responds to multiple calcium-regulated signals in the control of skeletal muscle fiber type. EMBO J. 2000; 19: 1963-1973.
93. H. Wu, B. Rothermel, S. Kanatous, P. Rosenberg, F.J. Naya, J.M. Shelton, K.A. Hutcheson, J.M. DiMaio, E.N. Olson, R. Bassel-Duby. Activation of MEF2 by muscle activity is mediated through a calcineurin-dependent pathway. EMBO J. 2001; 20: 6414-6423.
94. M.J. Potthoff, H. Wu, M.A. Arnold, J.M. Shelton, J. Backs, J. McAnally, J.A. Richardson, R. Bassel-Duby, E.N. Olson. Histone deacetylase degradation andMEF2 activation promote the formation of slow-twitch myofibers. J Clin Invest. 2007; 117: 2459-2467.
95. R. Bassel-Duby, E.N. Olson. Signaling pathways in skeletal muscle remodeling. Annu Rev Biochem. 2006; 75: 19-37.
96. D.T. Wang, Y. Yin, Y.J. Yang, P.J. Lv, Y. Shi, L. Lu, L.B. Wei. Resveratrol prevents TNF-alpha-induced muscle atrophy via regulation of Akt/mTOR/FoxO1 signaling in C2C12 myotubes. Int Immunopharmacol. 2014; 19: 206-213.
97. F.J. Naya, B. Mercer, J. Shelton, J.A. Richardson, R.S. Williams, E.N. Olson. Stimulation of slow skeletal muscle fiber gene expression by calcineurin in vivo. J Biol Chem. 2000; 275: 4545-4548.
98. Y.G. Ni, N. Wang, D.J. Cao, N. Sachan, D.J. Morris, R.D. Gerard, M. Kuro-o, B.A. Rothermel, J.A. Hill. FoxO transcription factors activate Akt and attenuate insulin signaling in heart by inhibiting protein phosphatases. Proc Natl Acad Sci U S A. 2007; 104: 20517-20522.
99. E. Lara-Pezzi, N. Winn, A. Paul, K. McCullagh, E. Slominsky, M.P. Santini, F. Mourkioti, P. Sarathchandra, S. Fukushima, K. Suzuki. A naturally occurring calcineurin variant inhibits FoxO activity and enhances skeletal muscle regeneration. J Cell Biol. 2007; 179: 1205-1218.