
INTRODUCTION
Human mast cells (MCs) are non-circulating, tissue-resident cells derived from CD34+ hematopoietic stem cells in the bone marrow (BM) [1]. Mature MCs are found in most vascularized organs, surrounding blood vessels and nerves [2]. MC and their progenitors express the receptor for stem cell factor (SCF), KIT (CD117), a transmembrane type III tyrosine kinase receptor (RTK) [3]. Binding of SCF to KIT governs most if not all the aspects of human MC biology, from proliferation to differentiation, migration, activation and survival [4, 5].
Mastocytosis are a heterogeneous group of diseases characterized by accumulation of abnormal (neoplastic) MCs in one or several organs, affecting both children and adults [6]. In adults, most patients present with a systemic involvement (systemic mastocytosis; SM). According to the World Health Organization (WHO), SM is classified into four major categories. Indolent SM (ISM) has a good prognosis, usually requires only symptomatic therapies, and ISM patients have a nearly normal life expectancy [6, 7]. The three other categories, collectively termed advanced SM, share a poor prognostic. Advanced SM categories include SM with an associated hematologic neoplasm (SM-AHN), aggressive SM (ASM), and MC leukemia (MCL) [8]. All categories of SM are characterized by an accumulation of abnormal MCs in BM and in other extra-cutaneous organs [9, 10].
WHO criteria for the diagnostic of SM include one major criterion and four minor criteria. The diagnosis of SM is established if at least the major and one minor criterion or at least three minor criteria are fulfilled [8, 11]. The major diagnostic criterion is defined by the presence of aggregates of at least 15 MCs identified by tryptase staining in BM and/or other extra-cutaneous organ biopsies. Minor criterions are: i) presence of 25% MCs with atypical morphology in BM smears [12], ii) aberrant immunophenotype of MCs (expression of CD2 and/or CD25) [13], iii) presence of an activating point mutation in codon 816 of the KIT gene in BM, peripheral blood (PB) or other extracutaneous organs [14, 15], and iv) increased level of serum tryptase (> 20 ng/mL) [16]. If SM is diagnosed, the next step is to evaluate its aggressiveness according to the presence of B-findings (borderline benign) and C-finding (consider cytoreduction). B- and C-findings correspond respectively to high MC burden and to organ dysfunction leading to the use of cytoreductive therapy [7]. The absence of B- and C-findings reflects an ISM [6, 7]. By contrast, the presence of at least one C-findings directs the diagnosis towards an advanced form of SM (ASM, SM-AHN or MCL). ASM is diagnosed when MCs in BM smears represent less than 20% of total nucleated cells [8, 17]. MCL is diagnosed when MCs in BM smears represent > 20% of total nucleated cells (with or without circulating neoplastic MCs in the bloodstream) [8, 17].
KIT is crucially involved in the pathophysiology of SM as the majority of patients carry KIT mutations, notably KIT D816V [18]. This mutation, found in > 85% of all patients with SM, activates constitutively KIT [15]. Activated KIT induces sustained proliferative and anti-apoptotic signaling in neoplastic MCs [18]. This KIT mutant receptor is resistant to most type I tyrosine kinase inhibitors (TKIs) targeting the wild-type receptor (KIT WT), such as imatinib [19]. By contrast, the mutant is sensitive to multikinase inhibitors such as midostaurin (PKC412) [20]. Thus, midostaurin is currently under clinical trials to treat Advanced SM [21]. However, this drug seems not sufficient to induce long-lasting complete responses in ASM and MCL [21]. There is thus a need to identify novel therapies for these diseases. To this purpose, relevant preclinical in vivo models of SM may be very useful. Unfortunately, the present lack of in vivo models mimicking at the best human KIT D816V+ advanced SM hampers pathophysiological studies and the development of new therapeutics. Only a few mouse models have been previously developed. In 2005, Zappulla et al. described a transgenic murine model using the primate chymase promoter as a driver of human KIT D816V mutation [22]. The authors reported the development of a SM-like disease within 12 to 24 months in only 30% of transgenic mice [22]. A few years after, Gerbaulet et al. described another transgenic mouse model expressing kit D814V mutation (murine homolog to human KIT D816V) [23]. However, this model has a limited utility because of the variety of diseases observed, ranging from perinatal lethality to pure MC hyperplasia, and of a very long period of latency (52 weeks) [23]. Besides, two xenograft mouse models have been described: one consisted in the injection of the leukemic HMC-1.2 cell line in SCID mice, giving rise to solid tumors [24], while the second consisted in injecting P815 mastocytoma cells in DBA-2 mice [25]. In this latter model, a severe mortality was observed within 9 days in injected animals [25]. These models are not useful for preclinical studies because of drawbacks such as the low incidence of disease and/or the kinetic of disease appearance and progression, which is either very short or too long. Thus, to date, no relevant in vivo models of KIT D816V+ advanced SM are available. In addition, none of the existing models allows monitoring of disease progression in real time.
We have recently reported on a new tumorigenic KIT D816V+ human MC line, termed ROSAKIT D816V, which presents most of the characteristics of neoplastic MCs found in ASM/MCL [26]. ROSAKIT D816V cell line was obtained by stable lentiviral transfection of a construct encoding for the KIT D816V mutant gene in the parental, non tumorigenic human mast cell line, ROSA KIT wild-type (ROSAKIT WT) [26]. In the present study, we have stably transduced ROSAKIT D816V cells with a construct encoding the naturally secreted Gaussia princeps luciferase (Gluc). This allowed us to obtain a new human neoplastic MC line, termed ROSAKIT D816V-Gluc. Gluc reporter is a highly sensitive luciferase allowing simultaneously the quantification of cells engraftment by measuring its activity in peripheral blood (PB) and in vivo imaging system (IVIS) [27]. ROSAKIT D816V-Gluc cells were then injected in NOD-SCID IL-2R γ-/- (NSG) mice, giving rise to a unique humanized in vivo model of KIT D816V+ advanced SM. In this model, disease progression and localization of neoplastic MCs were monitored in vivo and ex vivo using Gluc as a reporter [27].
RESULTS
Generation and characterization of ROSAKIT D816V-Gluc cells secreting luciferase
We transduced the human ROSAKIT D816V MC line stably expressing the KIT D816V mutant and GFP [26] with a lentiviral vector expressing Gluc and CFP, LV-Gluc-CFP. Cells expressing Gluc (ROSAKIT D816V-Gluc cells) were sorted by selection of GFP+/CFP+ cells (Figure 1A) and cultured in the same medium than their parental cell line. As shown in Figure 1B, the introduction of the lentiviral vector expressing Gluc and CFP into ROSAKIT D816V cells did not induce a significant change in their proliferation rate. In addition, the transduction did not change the phenotypic characteristics of the cells, as evidenced by the similar morphology of ROSAKIT D816V and ROSAKIT D816V-Gluc cells when observed on cytospin preparation after May Grünwald Giemsa (MGG) staining (Figure 1C). In addition, both cell lines expressed equally KIT (CD117) (Figure 1D). Besides, allele specific RT-PCR experiments confirmed that the two MC lines expressed the KIT D816V mutant at the same level (Figure 2A). Of note, as already described for ROSAKIT D816V cells [26], the KIT receptor was constitutively phosphorylated in ROSAKIT D816V-Gluc cells in the absence of SCF (Figure 2B). This level of phosphorylation was comparable in the two KIT D816V+ MC lines, and slightly higher in ROSAKIT WT cells stimulated by SCF (Figure 2B). We then determined the level of Gluc activity in the cell-free supernatants of both KIT D816V+ cell lines cultured at various concentrations, using the Gluc substrate coelenterazine (CTZ). Gluc acitivity was detected at a significant level in ROSAKIT D816V-Gluc cells supernatants after 24h of incubation, even at low cell concentrations (102 cells per mL). As expected, there was no Gluc activity detectable in ROSAKIT D816V cell supernatants, at any cell concentration (Figure 2C). Moreover, we observed that increasing the number of ROSAKIT D816V-Gluc cells increased Gluc activity in a virtually linear manner (Figure 2C). Finally, in order to demonstrate that the transduction of by LV-Gluc-CFP did not change the sensitivity of the cells to TKIs, we assessed the effects of Imatinib or of Midostaurin on the proliferation of both KIT D816V+ cell lines. As expected, both cell lines were equally resistant to Imatinib (Figure 2D) and similarly sensitive to the antiproliferative effects of Midostaurin. The IC50 for Midostaurin was found at 70 nM for ROSAKIT D816V cells and at 94 nM for ROSAKIT D816V-Gluc cells (Figure 2E).
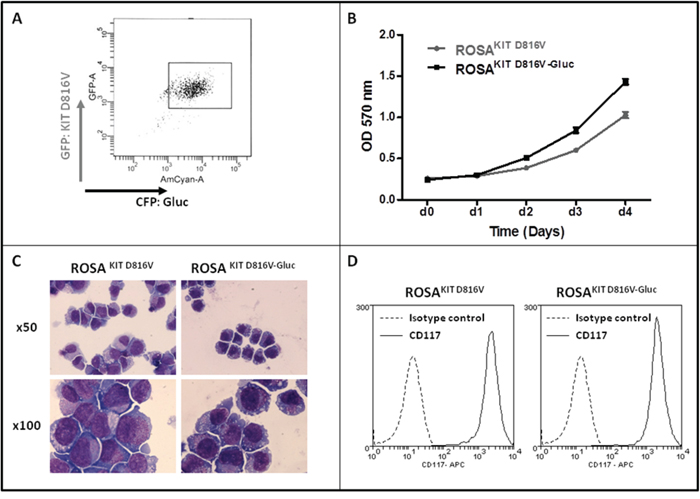
Figure 1: Generation of ROSAKIT D816V-Gluc cell line and comparison with the parental ROSAKIT D816V cell line. A. ROSAKIT D816V-Gluc GFP+/CFP+ sorted cells using flow cytometry. B. Comparison of ROSAKIT D816V and ROSAKIT D816V-Gluc cell proliferation by the use of the MTT method. Cells were seeded in 96-well plate for 5 days (1 plate/day). At each day (d0 - d4), 20 μL of MTT were added in each well and the cells were incubated for 3 additional hours at 37°C. After adding 100 μL of acidified isopropanol, the number of living cells was measured for each condition by reading the absorbance at 570 nm. Data are presented as the mean ± SD (n = 3). C. Phenotypic comparison of the cytological aspect of ROSAKIT D816V and ROSAKIT D816V-Gluc. MGG-stained cytospin preparations of ROSAKIT D816V (left panel) and ROSAKIT D816V-Gluc (right panel). Magnification is x50 (top), x100 (bottom). D. Comparative expression of CD117 (KIT) by in ROSAKIT D816V (left) and ROSAKIT D816V-Gluc (right) analyzed by flow cytometry.
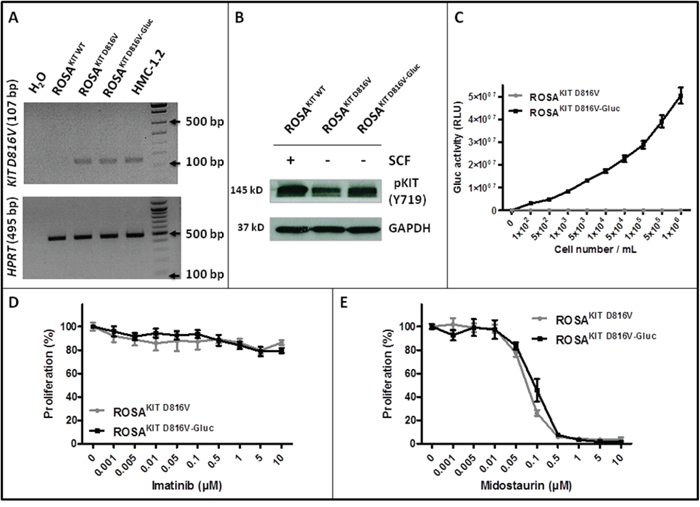
Figure 2: Effects of transduction of ROSAKIT D816V cells with LV-Gluc-CFP on KIT activation and response to TKIs. A. Analysis of KIT D816V gene expression in ROSAKIT D816V-Gluc cell line using agarose gel electrophoresis of conventional RT-PCR products. Reactions were compared to the levels of expression of mRNA for HPRT. The image is an inverted form of the original picture. B. Detection of spontaneous phosphorylation of KIT receptor by western blotting in ROSAKIT D816V-Gluc. Cell lysates from ROSAKIT WT cells stimulated with SCF, or from unstimulated ROSAKIT D816V and ROSAKIT D816V-Gluc cells, were subjected to electrophoresis and stained with antibody against p-KIT Y719. An anti-human glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as a loading control. The lanes were run on the same gel. C. Gluc activity in cell culture supernatants of ROSAKIT D816V (grey line) and ROSAKIT D816V-Gluc (black line). Signals were calculated as RLU; relative luminescent units. Data present the mean ± SD (n = 3). D, E. Effect of Imatinib or Midostaurin on the proliferation of ROSAKIT D816V (grey line) and ROSAKIT D816V-Gluc cells (black line). Each cell lines were seeded for 72h in 96-well plate in the presence of various concentrations (0.001 - 10 μM) of Imatinib (D) or Midostaurin (E). Data are presented as the mean ± SD (n = 3) and are expressed as percent of proliferation in each condition relative to the control (untreated cells) considered as 100% proliferation.
Analysis of the level of engraftment of ROSAKIT D816V-Gluc cells in vivo in NSG mice
Various amounts of ROSAKIT D816V-Gluc cells were injected intravenously in irradiated NSG mice. We then measured Gluc activity in the plasma of grafted mouse four, eight and ten weeks after cell injection. Interestingly, at 4 weeks of engraftment, the Gluc activity in plasma was found linearly increased in relationship with increasing numbers of injected cells (R2 = 0.97) (Figure 3A). However, although Gluc activity increased in a time-dependent manner, there was no apparent difference between groups injected with 5x106 or 10x106 cells at 8 and 10 weeks (Figure 3A). Compared with the level of Gluc activity determined in plasma, Gluc intensity measured after 10 weeks of engraftment by IVIS was found heterogeneous among groups, particularly in the groups injected with 5x106 or 10x106 cells at 10 weeks (Figure 3B and 3C).
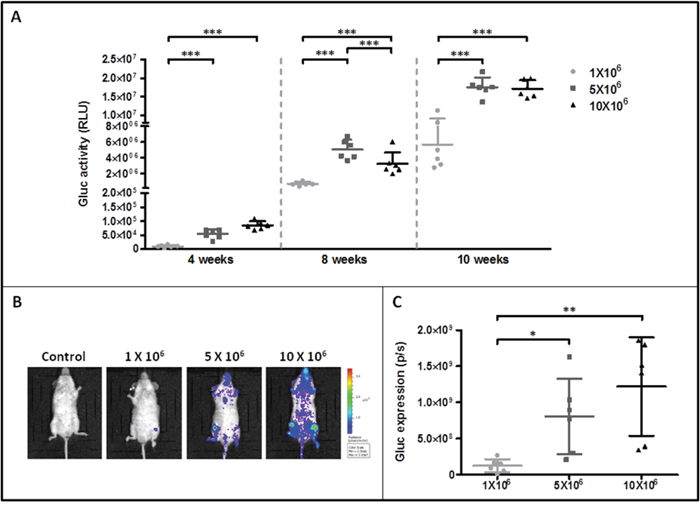
Figure 3: Monitoring of ROSAKIT D816V-Gluc cells engraftment in injected mice by measurement of Gluc activity in blood and by IVIS. A. Gluc activity in plasma of mice after various times of engraftment. Different numbers of ROSAKIT D816V-Gluc cells [1x106 (●), 5x106 (■) and 10x106 (▲)] were injected intravenously in mice and engraftment was monitored over time (4, 8 and 10 weeks). Each point represents an individual mouse. B. IVIS showing localization of Gluc in injected mice after 10 weeks. Indicated numbers of ROSAKIT D816V-Gluc cells were IV injected in mice (n = 6). Units in rainbow color scales are photons per second per cm2 per steradian (p/sec/cm2/sr). Results shown are from one representative mouse for each group. C. Total relative units (RLU) per second were calculated for Gluc intensity shown in (B) by ROI analysis after 10 weeks of engraftment in the three groups of mice. Each point represents an individual mouse.
To further investigate the disease progression in the three groups of xenografted mice, PB samples were analyzed for the presence of ROSAKIT D816V-Gluc cells by quantifying the percentage of hCD45+/hCD117+ cells using flow cytometry. The percentage of ROSAKIT D816V-Gluc cells in PB was found very low at 4 weeks (around 0.007 % in the 1x106 cells group, 0.12 % in the 5x106 cells group and 0.17% in the 10x106 cells group). This percentage increased at 10 weeks in a dose (slightly > 0.45% in the 1x106 cells group, > 1 % in the 5x106 cells group and 1.5 % in the 10x106 cells group at 10 weeks) and time-dependent manner (Figure 4A). The same phenomenon was observed when calculating the absolute numbers of ROSAKIT D816V-Gluc cells in PB at different time periods and after injection of different numbers of cells (Table 1).
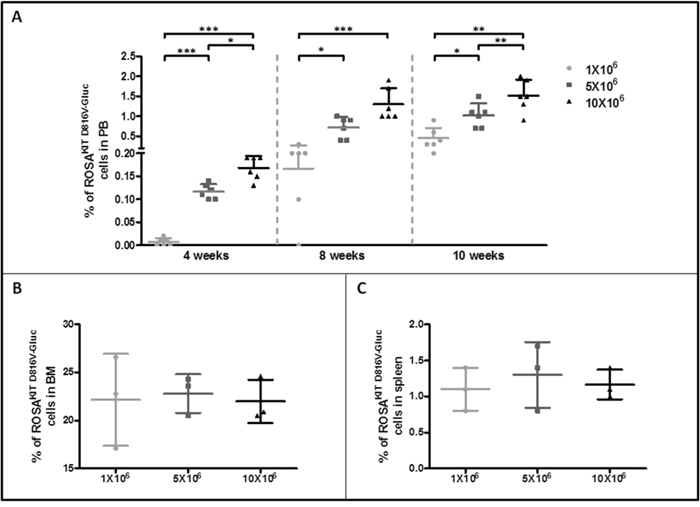
Figure 4: Evaluation of ROSAKIT D816V-Gluc cells numbers by flow cytometry in peripheral blood, bone marrow and spleen. A. Percentage of ROSAKIT D816V-Gluc cells in PB after increasing times of engraftment (4, 8 and 10 weeks). The percentage of ROSAKIT D816V-Gluc cells was determined in the three groups of mice: 1x106 (●), 5x106 (■) and 10x106 (▲) by flow cytometry using monoclonal antibodies against human CD45 and human CD117. B, C. Percentages of ROSAKIT D816V-Gluc cells after 10 weeks of engraftment in BM and spleen respectively in the three groups of mice: 1x106 (●), 5x106 (■) and 10x106(▲). Each point represents an individual mouse.
Table 1: Absolute numbers of ROSAKIT D816V-Gluc cells in PB of NSG mice injected with different amounts of cells
ROSAKIT D816V-Gluc cell number in PB (x106/mL) |
|||
Groups |
4 weeks |
8 weeks |
10 weeks |
1x106 (n=6) |
0 |
0.006 ± 0.004 |
0.012 ± 0.004 |
5x106 (n=6) |
0.004 ± 0.001*** |
0.021 ± 0.007* |
0.025 ± 0.009* |
10x106 (n=6) |
0.003 ± 0.001*** |
0.042 ± 0.013*** |
0.039 ± 0.017* |
Data are presented as mean ± SD and paired t-test was performed in comparison to the group receiving 1x106 cells.
Engraftment of ROSAKIT D816V-Gluc cells in vivo in NSG mice leads to an advanced SM phenotype
To evaluate the level of ROSAKIT D816V-Gluc cells engraftment in BM and spleen, thus the severity of the disease, mice (n=3 per group) were euthanized in each group 10 weeks after engraftment. BM cells were counted and the percentage of hCD45+/CD117+ cells was determined in each group. As expected, and in line with the human situation for ASM/MCL, the percentage of ROSAKIT D816V-Gluc cells in BM was found much higher (around 22 %) than in spleen (1.1 %) and in PB (1.0 %). There was no difference in cell percentages between the three groups of mice in BM as well as in spleen (Figure 4B and 4C, respectively) and this was confirmed by calculation of the absolute number of ROSAKIT D816V-Gluc cells in affected tissues (Table 2). Interestingly, the percentage of ROSAKIT D816V-Gluc cells in BM reached a plateau at 10 weeks regardless the number of ROSAKIT D816V-Gluc cells injected (Figure 4B and Table 2). All in all, the high percentages of neoplastic MCs found in BM, together with a moderate infiltration of the spleen and of the PB, might indicate that diseased animals suffer of a pathology similar to an aleukemic MCL, as observed in some human patients [28].
Table 2: Absolute numbers of ROSAKIT D816V-Gluc cells in affected tissues of NSG mice injected with different amounts of cells after 10 weeks of engraftment
ROSAKIT D816V-Gluc cell number (x106/mL) |
|||
Groups |
PB |
Spleen |
BM |
1x106 (n=3) |
0.012 ± 0.004 |
0.856 ± 0.13 |
4.509 ± 0.690 |
5x106 (n=3) |
0.025 ± 0.009* |
1.091 ± 0.294 |
3.618 ± 0.669 |
10x106 (n=3) |
0.032 ± 0.007* |
1.134 ± 0 |
4.323 ± 0.802 |
Data are presented as mean ± SD and paired t-test was performed in comparison to the group receiving 1x106 cells.
We then studied in detail the organs engrafted by ROSAKIT D816V-Gluc cells, using immunohistochemical (IHC) staining with antibodies directed against human tryptase and human CD45. Analysis of BM sections of mice euthanized at 10 weeks revealed the presence of fibrosis, especially in the group injected with 10x106 cells, as well as the presence of large clusters of tryptase and hCD45 positive cells (Figure 5), consistent with the pathological appearance of BM biopsies in human patients with advanced SM [29]. Whatsoever, tryptase+ and hCD45+ cells were detected at a high level in BM sections but in a lower extent in spleen (Figure 6A) and liver sections (Figure 6B). In the last two organs, a few clusters of MCs and isolated MCs were observed in all the organ sections (Figure 6A and 6B).
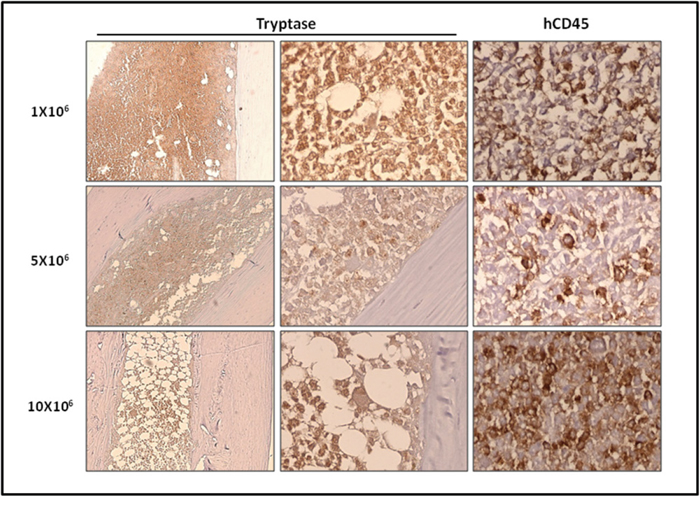
Figure 5: Localization of primary engrafted ROSAKIT D816V-Gluc cells in mice BM by IHC detection of tryptase and hCD45. Ten weeks after engraftment, BM sections from the three groups of mice were stained by IHC with an anti-human tryptase antibody (left and middle panels) antibody and an anti-human CD45 antibody (right panel). Staining was visualized by Histomouse Kit, showing human MCs in brownish staining. Magnification is x10 (left panel) and x50 (middle and right panels). Results are from one representative mouse from each group.
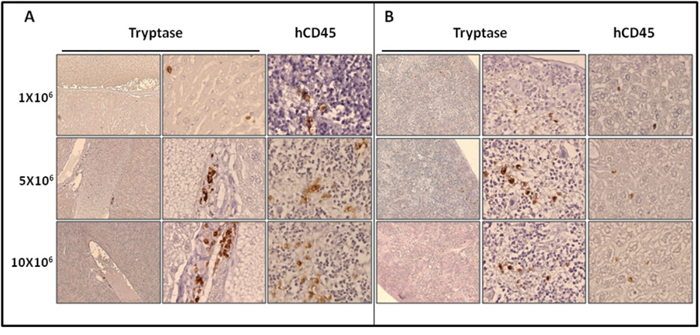
Figure 6: Localization of primary engrafted ROSAKIT D816V-Gluc cells in mice spleen and liver by IHC detection of tryptase and hCD45. Ten weeks after engraftment, spleen A. and liver B. sections from the three groups of mice were stained by IHC with an anti-human tryptase (left and middle panels) antibody and an anti-human CD45 (right panel) showing human MCs in brownish staining. Magnification is x10 (left panel) and x50 (middle and right panels). Results are from one representative mouse from each group.
To further confirm the relevance of our in vivo model of SM towards the human pathology, we measured serum human tryptase levels in PB of xenotransplanted animals at 10 weeks. Consistent with the observation that tryptase serum levels are elevated in SM patients [30], significant levels of human tryptase (ranging from > 7.0 to > 11.0 μg/L) were measured in all xenotransplanted mice compared to the negative control, where the level of tryptase was found undetectable (< 1 μg/L) (Table 3). Interestingly, increased levels of serum tryptase paralleled increased numbers of cells injected at the beginning of the experiments (Table 3).
Table 3: Serum tryptase levels in NSG mice injected with different numbers of ROSAKIT D816V-Gluc cells
Groups |
Tryptase level (μg/L) |
Negative control |
< 1.00 |
1x106 (n=3) |
7.29 ± 1.00 |
5x106 (n=3) |
9.17 ± 0.31* |
10x106 (n=3) |
11.65 ± 2.56 |
Data are presented as mean ± SD and paired t-test was performed in comparison to the group receiving 1x106 cells.
Finally, after 12 weeks of transplantation, signs of health deterioration appeared in mice injected respectively with 5x106 or 10x106 ROSAKIT D816V-Gluc cells, such as a decrease in water and food consumption accompanied with bristly hair, prostration and paralysis. This clinical phenotype further confirmed that the mice suffered from an aggressive form of the disease.
Xeno-transplanted ROSAKIT D816V-Gluc cells have enhanced grafting capabilities
In order to appreciate if ROSAKIT D816V-Gluc cells were able to retain their grafting capabilities in a secondary transplant model, we grafted naive irradiated animals with a low number of human cells purified from the BM of previously xenotransplanted mice. Five weeks after the secondary transplantation, the efficacy of engraftment was assessed by measurement of Gluc activity in plasma, assessment of whole body by IVIS, and by measurement of the percentage of hCD45+/hCD117+ cells in PB. As previously seen in the experiments of first transplantation, we observed that Gluc activity increased in a time-dependent manner in the plasma (Figure 7A). In addition, as compared to non-injected control mouse, Gluc was detected by IVIS as soon as 5 weeks following the xenotransplantation in the majority of the mice injected with previously engrafted ROSAKIT D816V-Gluc cells. The bioluminescence was found localized mainly in femur and feet (Figure 7B). Of note, after 9 weeks of secondary transplantation, Gluc expression was considerably increased when measured by IVIS (Figure 7B). Interestingly, in mice xenografted with pre-transplanted ROSAKIT D816V-Gluc cells, the percentage of hCD45+/hCD117+ cells in PB, as well as the level of Gluc activity, were found greater after the secondary transplantation of 5x105 cells than after the primary transplantation of 10x106 cells, suggesting that the first transplantation selected more “mice adapted” neoplastic MCs.
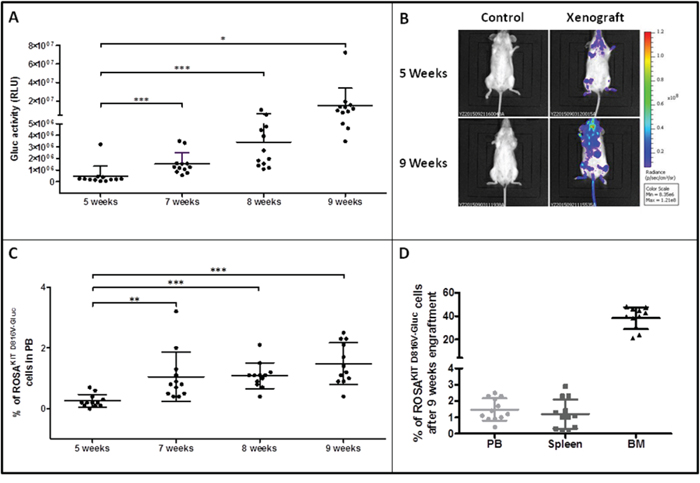
Figure 7: Reconstitution capacity of the human ROSAKIT D816V-Gluc cells in NSG mice after secondary transplantation. Ten weeks after engraftment in primary recipient mice, human cells were isolated from BM samples and injected into secondary recipient mice (n = 12). Engraftment was assessed 5, 7, 8 and 9 weeks after injection. A. Time-dependent increase of Gluc activity in plasma after secondary transplantation with ROSAKIT D816V-Gluc cells. Each point represents an individual mouse. B. In vivo imaging of Gluc activity in secondary injected mice (right lane) after 5 weeks (top lane) and 9 weeks (bottom lane) versus negative control (left lane). Results shown are from one representative mouse. C. Time-dependent increase in the percentages of ROSAKIT D816V-Gluc cells in PB after secondary transplantation. D. Percentage of ROSAKIT D816V-Gluc cells after secondary transplantation in PB (●), spleen (■) and BM (▲). The percentage of hCD45+/hCD117+ was measured using flow cytometry in C and D.
To evaluate more precisely the progression of the disease in the mice injected with previously transplanted cells, we measured the percentage of ROSAKIT D816V-Gluc cells in PB, BM and spleen after 5, 7, 8 and 9 weeks of secondary transplantation through the quantification of hCD45+/hCD117+ cells by flow cytometry. We observed that the percentage of ROSAKIT D816V-Gluc cells in PB increased significantly in a time-dependent manner (Figure 7C). Besides, at 9-weeks period of engraftment, the percentage of ROSAKIT D816V-Gluc cells was found much higher in BM (around 35 %) than in spleen (around 1.5 %) or in PB (1.5 %) (Figure 7D). This observation was confirmed after IHC staining of human CD45 in BM, spleen, and liver (Figure 8). Finally, to confirm that secondary transplantation of the human KIT D816V+ MC line did not alter the expression of the mutant KIT gene, we explored the expression of KIT D816V mutant on BM cells of secondary transplanted mice using allele-specific RT-PCR. As shown on Figure 9A, BM cells of the two injected mice expressed the KIT D816V mutant, exactly as did the ROSAKIT D816V cell line. As expected, no expression of the KIT mutant was detected in control, KIT mutant-negative ROSAKIT WT cells (Figure 9A).
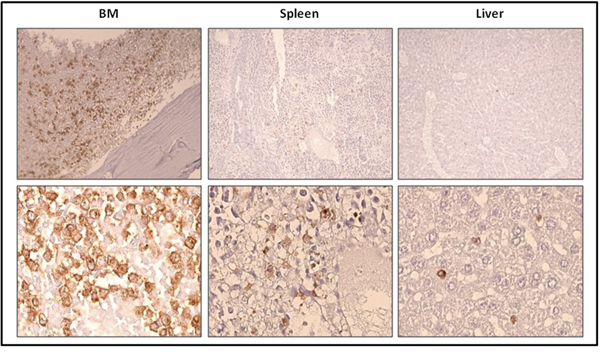
Figure 8: Localization of previously xenotransplanted ROSAKIT D816V-Gluc cells in mice BM using IHC detection of hCD45 after secondary transplantation. BM sections (left panel), spleen sections (middle panel), and liver sections (right panel) stained by IHC with anti-human CD45 antibody showing human cells in brownish staining. Magnification is x10 (top lane) and x50 (bottom lane). Each result shown is from one representative mouse having received previously xenotransplanted ROSAKIT D816V-Gluc cells.
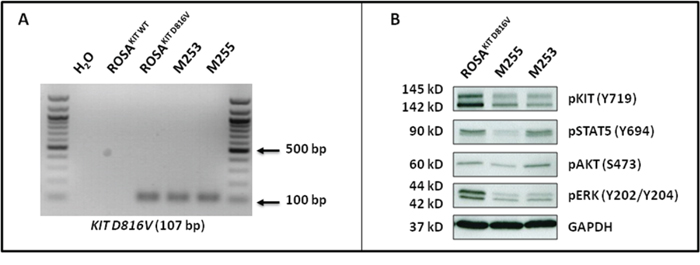
Figure 9: ROSAKIT D816V-Gluc cells express in vivo KIT D816V mutant leading to constitutive phosphorylation of KIT and of downstream signaling pathways after secondary transplantation. A. Expression of the KIT D816V gene in BM samples of injected mice using allele-specific RT-PCR. KIT D816V allele-specific PCR was performed using ROSAKIT D816V cells as a positive control. The image is an inverted form of the original picture. B. Spontaneous phosphorylation of the KIT receptor and of its downstream signaling pathways (STAT5, AKT and ERK) revealed by western blotting in ROSAKIT D816V cells and BM cells of secondary transplanted mice. An anti-human glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as a loading control. The lanes were run on the same gel.
Xenotransplanted ROSAKIT D816V-Gluc cells have advanced SM-like alterations in signaling pathways
In KIT D816V+ neoplastic MCs found in the BM of SM patients, and particularly those with advanced SM, KIT is found spontaneously activated, but also other intracellular substrates are aberrantly activated (phosphorylated), such as signal transducer and activator of transcription 5 (STAT5), AKT and extracellular signal-regulated kinases 1/2 (ERK1/2) [31, 32]. Those signaling molecules have been found increasingly phosphorylated in parallel with the aggressiveness of the disease and resistance of the neoplastic MCs to TKIs [31, 32]. To further confirm the similarity of our in vivo model of KIT D816V+ advanced SM with the human situation, we analyzed the spontaneous phosphorylation state of KIT, STAT5, AKT and ERK in BM cells isolated from secondary injected mice, as compared to the ROSAKIT D816V parental cell line. As shown in Figure 9B, the constitutive activation of the KIT receptor was accompanied by aberrant phosphorylation of KIT-downstream signaling pathways such as AKT, STAT5 and ERK. These findings are consistent with those reported for neoplastic MCs in advanced SM patients [31].
DISCUSSION
SM are rare neoplasms characterized by a specific, recurrent and almost always acquired molecular defect inducing constitutive activation of the KIT receptor [33]. The most frequent defect found in all categories of SM is the KIT D816V point mutation [18]. KIT D816V mutant is found in > 90% of all ISM cases and in up to 80% of advanced SM patients [34]. Recently, we have established a unique human MC line, termed ROSAKIT D816V, which is considered as the first relevant in vitro model of human neoplastic MCs bearing only the KIT D816V mutant [26]. We reported that this KIT D816V+ MC line is highly tumorigenic in immunocompromised mice, by contrast to its parental cell line presenting with a wild type KIT (ROSAKIT WT) [26]. However, to the best of our knowledge, there is no in vivo model of engraftment of human KIT D816V+ MCs where of the disease progression can be monitored repeatedly by a simple, non-stressing technique. Thus, we took advantage of the availability of our ROSAKIT D816V cell line to generate a new in vivo model of engraftment of human KIT D816V+ neoplastic MCs in NSG mice, using the Gluc reporter system, that allows to monitor the disease progression by a simple measurement in blood samples, in real time, without stressing the animals [27, 35].
In the first step of our work, we generated ROSAKIT D816V cells expressing the secreted Gaussia princeps luciferase [27, 35], termed ROSAKIT D816V-Gluc cells. We then confirmed by the measurement of Gluc activity in the cell supernatants that ROSAKIT D816V-Gluc cells effectively express Gluc, as previously reported for other cell models [27]. In addition, we demonstrated that lentiviral infection of ROSAKIT D816V cells with the Gluc-containing construct did not alter their morphology, the KIT D816V expression, the constitutive phosphorylation of the KIT receptor, and their SCF-independence. In addition, we demonstrated that the ROSAKIT D816V-Gluc cells retained the same sensitivity (or resistance) to different TKIs than the parental ROSAKIT D816V cell line, making it a very attractive tool to screen for new targeted drugs in vitro and in vivo. Particularly, as previously described in other neoplastic MC models [20], we confirmed the resistance of our KIT D816V+ MC model towards Imatinib, while the cells remained sensitive to Midostaurin.
With the aim to establish a new in vivo model of KIT D816V+ SM in which the progression of the disease could be easy to follow accurately, we injected various numbers of ROSAKIT D816V-Gluc cells in previously irradiated NSG mice. Expression of Gluc by injected cells allowed us to check and monitor accurately not only the cell engraftment but also the progression of SM in NSG mice. Indeed, we observed that, after 4 weeks following the cell transplantation, Gluc activity in the PB of xenografted animals displayed a linear relationship with the number of cells injected, as previously described for other in vivo models [27]. However, in our study, there was no apparent difference between the 5x106 and the 10x106 groups at 8 and 10 weeks, suggesting that over a given number of cells injected, saturation is reached (Figure 3A). Since Gluc blood assay complements IVIS, which has the ability to localize the signal in organs [36], we analyzed Gluc expression in the whole body of the injected mice. Quantification through ROI analysis revealed that Gluc intensity was heterogeneous among groups. Comparing the two methods, measurement of Gluc activity in plasma seemed thus more reliable than measurement of Gluc intensity by IVIS to reflect the total MC burden in the animals. In conclusion, Gluc assay in PB provides both a sensitive and quantitative assessment of engraftment, complementing IVIS, which has the ability to localize the signal (thus the grafted cells) in different organs of the animals.
Furthermore, we showed that ROSAKIT D816V-Gluc cells can engraft all injected NSG mice, not only at the BM level but also in spleen, liver and PB, giving rise to a SM-like disease nearing the one found in humans, according to the WHO criteria for the diagnosis of SM [37]. The percentage of ROSAKIT D816V-Gluc cells increased in a time- and number of cells injected-dependent manner, suggesting that the disease progression can be accelerated or slowed by varying the number of cells injected. As expected, the percentage of ROSAKIT D816V-Gluc cells in BM was found much higher than in spleen and PB, suggesting that ROSAKIT D816V-Gluc mainly accumulated in BM, as evidenced on BM sections stained for hMC tryptase and hCD45. However, the percentage of ROSAKIT D816V-Gluc cells in BM was not strictly parallel to the increase of total ROSAKIT D816V-Gluc cell number in mice, meaning that the cell number reached a plateau in the BM, while the cells continued to proliferate in other tissues. Whatsoever, the percentage of neoplastic human MCs reached 20% or above in the BM of injected animals. This criterion is diagnostic for MCL [8]. In addition, all injected mice presented a high level of serum tryptase (ranging from > 7.0 ng/mL for mice injected with 1x106 cells to > 11.0 ng/mL for mice injected with 10x106 cells), although not exceeding the threshold of 20 ng/mL, a value established for SM diagnosis in humans [8]. This may be explained by the relativity low content of tryptase of the ROSAKIT D816V-Gluc cells as compared to neoplastic MCs found in the BM of SM patients (data not shown). In addition, one has to keep in mind that, contrasting to the human situation where tryptase is already detectable at several ng/mL in the bloodstream of healthy individuals, the level of human tryptase in healthy mice is equal to zero. Thus, any increase in human tryptase level in PB of our mice reflects MCs engraftment. Interestingly, the level of tryptase in plasma was significantly correlated with the level of Gluc in plasma at 10 weeks (p value = 0.007 - R square = 0.67), indicating that measuring Gluc activity in plasma is a reliable method to monitor disease progression in our in vivo model. Of note, while quantification of serum tryptase level required more than 50 μL of plasma, only 5 μL of blood or plasma samples are needed to measure Gluc activity.
All in all, our in vivo model of primary xenotransplantation fits well with the one of an advanced (ASM/MCL-like) SM disease, a fact further confirmed by the rapid deterioration of the health status of the mice after 12 weeks of engraftment. In line with this observation, it has to be underlined that the mean overall survival of MCL patients is usually very short in the absence of treatment, being of 6 months or less [38].
In addition, we have demonstrated that the transplantation of ROSAKIT D816V-Gluc cells from primary engrafted NSG mice into secondary recipient increased their capacity of engraftment. This indicates that ROSAKIT D816V-Gluc cells were adapted to the mouse microenvironment upon the first transplantation, explaining thus the acceleration of secondary engraftment. Such phenomenon has been previously shown in serial transplantations of other types of leukemic cells isolated from patients [39]. Interestingly, this enhanced engraftment capability of previously xenotransplanted ROSAKIT D816V-Gluc cells might enable us to use these cells to achieve larger lots of mice for preclinical studies.
Of note, the in vivo model presented here is a unique model paralleling human KIT D816V+ advanced SM disease, with many advantages over previously published in vivo models which did not really reproduce the clinical and biological characteristics of advanced SM. In fact, injection of the human leukemic MC line HMC-1.2 exhibiting two KIT mutations, KIT V560G and KIT D816V - which is unusual in SM patients - in SCID mice, gave rise to solid tumors in 6 weeks to 5 months [24]. More recently, we have described a model of mice transgenic for KIT D816V, which expressed the human KIT D816V mutant specifically in MCs under the control of baboon chymase promoter [22]. However, only one third of the transgenic mice developed an ISM-like disease, with a moderate MC hyperplasia, after 12 to 24 months of latency [22]. In this model, the low disease penetrance and the delay of disease appearance made drug studies problematic, in addition to the fact that the KIT D816V mutant was expressed in murine MCs. In 2006, Demehri et al. have developed a murine model based on IV injection of P815 cells, a mouse mastocytoma cell line harboring kit D814Y mutant (analogue to KIT D816Y mutant in human) in DBA/2 mice [25]. This model gave rise to an ASM/MCL-like disease after 6-9 days, with a severe mortality at 9 days, making in vivo drug studies impossible to perform in a so short time. More recently, another transgenic mice model have been developed by Gerbaulet et al. using the Cre/loxP recombination system allowing conditional expression of the Kit D814V mutant (murine homolog of human KIT D816V mutation) under the control of the Kit promoter. In this model, the development of a SM-like disease followed a slow kinetics (around 52 weeks), and mice developed colitis associated with mucosal MC accumulation [23]. Thus, this model is far from nearing human KIT D816V+ advanced SM as it deals with a Kit D814V mutant expressed in murine MCs [23].
Furthermore, compared to previous models of SM, our in vivo model nears at the best the biological characteristics of neoplastic MCs found in vivo in advanced SM patients. Particularly, the presence of KIT D816V mutant, together with the constitutive activation of KIT, but also of STAT5, AKT and ERK in these cells mimic the situation observed in such patients where STAT5, AKT and ERK have been found increasingly phosphorylated in parallel with the aggressiveness of the disease and its resistance to TKIs [31, 32]. Our model will thus help to test targeted therapies, alone or in combination, directed not only against the KIT D816V mutant but also against other oncogenic signaling involved in the progression of the disease and in the resistance to TKIs in vivo.
In conclusion, the secretion of luciferase (Gluc) by our newly generated human KIT D816V+ Gluc+ neoplastic MC line greatly facilitates the assessment of engraftment in mice and allows the accurate monitoring of disease progression. Moreover, although MC engraftment is observed within 4 weeks in 100% of injected mice, health deterioration is observed after 12 weeks or even later, depending on the number of cells injected, allowing thus i) to increase or decrease disease aggressiveness by increasing or decreasing the number of injected cells and ii) sufficient time between the development of the disease and its fatal outcome, which is a critical requisite for easy preclinical drug studies. Such additional studies are ongoing in our laboratory on this model.
MATERIALS AND METHODS
Cell culture
The human parental, SCF-dependent non-tumorigenic MC line, ROSAKIT WT was cultured exactly as previously described, in the presence of 10% of supernatant of Chinese hamster ovary (CHO) cells transfected with the murine SCF gene, used as a source of SCF [26]. The human neoplastic, SCF-independent tumorigenic MC line ROSAKIT D816V was obtained by stable lentiviral transduction of a construct encoding for the human KIT D816V gene and GFP in the parental ROSAKIT WT cell line, as already described [26] and was cultured in the absence of SCF, exactly as previously described [26]. The human MCL cell line HMC-1.2 harboring both KIT V560G and KIT D816V mutations (kindly provided by Dr. J. H. Butterfield; Mayo Clinic, Rochester, MN) was cultured exactly as already described [40].
Transduction of ROSAKIT D816V cells with LV-Gluc-CFP construct
Lentivirus vector construction and production of LV-Gluc-CFP were performed as previously described [27, 35]. Briefly, the Gaussia luciferase cDNA was cloned into lentivirus vector under the control of the strong constitutive cytomegalovirus (CMV) promoter to produce LV-Gluc. In another vector, cDNA for Gluc and the optimized cyan fluorescent protein (CFP) separated by IRES (LV-Gluc-CFP) were cloned under the CMV promoter. Lentivirus particles were produced by transfection of 293T cells as previously described [27]. ROSAKIT D816V cells were then transduced overnight with lentiviral particles of LV-Gluc-CFP in order to produce ROSAKIT D816V-Gluc CFP+ cells. The efficacy of transduction was measured after 7 days by flow cytometry. ROSAKIT D816V-Gluc cells expressing both GFP and CFP were then sorted using a BD FACSAria (Beckton Dickinson Biosciences).
Morphological and phenotypical analysis of the ROSAKIT D816V-Gluc cell line
In order to ensure that lentiviral transduction of ROSAKIT D816V cells with LV-Gluc-CFP construct have not affected the morphology of the cells, May-Grünwald Giemsa (MGG)-stained cytospin preparations of ROSAKIT D816V and ROSAKIT D816V-Gluc cells were examined using a Zeiss Axiophot microscope (Carl Zeiss). In addition, the expression of KIT (CD117) was analyzed on both cell lines cells by direct immunofluorescence using a FACSCalibur (BD Biosciences). Briefly, 2x105 untreated cells were incubated with APC (allophycocyanin)-labeled monoclonal antibody directed against human CD117 (BioLegend). Isotype control was used for each experiment, and at least 10,000 events were recorded on a BD FACSCalibur.
Allele specific RT-PCR for the detection of the KIT D816V mutant
Total RNA from 5x106 ROSAKIT WT cells, ROSAKIT D816V cells, ROSAKIT D816V-Gluc cells, HMC1.2 cells, or from 5x105 human cells purified from BM of mice previously xenografted with ROSAKIT D816V-Gluc cells was isolated using TRIzol (Life Technologies) or using Nucleospin RNA (Machery Nagel) according to the manufacturer’s protocol. RNA concentration and purity was determined using a Nanovue plus spectrophotometer (GE Healthcare). Starting from these RNA samples, first strand cDNA synthesis was conducted using the iScript Select cDNA synthesis kit (Biorad) or using Superscript II reverse transcriptase (Invitrogen) according to the manufacturer’s instructions. PCR was performed using primer sequences designed as described by Kristensen et al. [41]: a forward primer 5’-AGAGACTTGFCAGCCAGAAAA-3’, and a reverse primer 5’-TTAACCACATAATTAGAATCATTCTTGATCA-3’ for KIT D816V. Reactions were compared to levels of the hypoxanthine phospho-ribo-transferase (HPRT) amplified using the following primers: HPRT F; 5’-ATGGACAGGACTGAACGTCTTGC-3’; and HPRT R; 5’-GACACAAACATGATTCAAATCCCTGA-3’. Complementary DNA were amplified using the following cycle conditions: 95°C for 10 min, followed by 40 cycles of 95°C for 15 s, 60°C for 1 min and 72°C for 1 min. All PCR experiments included a no template control (water). The amplified PCR products were visualized by staining with ethidium bromide (Electran) after 1.5 % agarose gel electrophoresis in Tris–acetate–EDTA buffer (Euromedex).
Western blot analysis for KIT signaling
Western blots were performed on cell lysates from ROSAKIT WT cells stimulated with SCF, or from unstimulated ROSAKIT D816V and ROSAKIT D816V-Gluc cells, using an antibody against phosphorylated KIT Y719 (Cell Signaling Technology), in order to visualize the phosphorylation state of KIT. Western blots were also performed on lysates of mouse BM cells using antibodies against phosphorylated KIT Y719, phosphorylated STAT5 Y694, phosphorylated AKT S473 and phosphorylated ERK Y202/204 (all from Cell Signaling Technology). Proteins were visualized with horseradish peroxidase-conjugated secondary antibodes and chemoluminescent substrate (Promega). A mouse monoclonal IgG1 anti-human glyceraldehyde-3-phosphate dehydrogenase (GAPDH) Ab (Santa Cruz Biotechnology) was used as a loading control.
In vitro gluc activity assay
To measure Gluc activity in cell supernatant, ROSAKIT D816V cells expressing or not Gluc were seeded in 24-well plates at various concentrations (from 100 to 1x106 cells/mL) and incubated for 24 hours at 37°C and 5 % CO2. Twenty microliters of conditioned medium were assayed using 100 μL (5 μg/mL) water-soluble coelenterazine (CTZ) (Nanolight Technologies) in full white plates. Photon counts were acquired immediately using a luminometer (PerkinElmer - EnSpire®).
Analysis of the effects of TKIs on cell proliferation
To ensure that lentiviral transduction of ROSAKIT D816V cells with LV-Gluc-CFP construct has not influenced the sensitivity of the cells towards different TKIs, the effect of Imatinib (Sequoia Research) and of Midostaurin (Novartis Pharmaceutical Corporation) on cell proliferation was measured using the 3-(4, 5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) test. ROSAKIT D816V and ROSAKIT D816V–Gluc cells were incubated with Imatinib or Midostaurin (1 nM to 10 μM) in 96 well plates (Falcon) at 37°C for 72 hours. After incubation, 20 μl of MTT (5 mg/mL) (Life technologies) were added in each well and the plates were incubated for 3 additional hours at 37°C. Then, 100 μl of acidified isopropanol were added to each well to dissolve formazan crystals. The number of living cells was then measured in each condition by reading the absorbance at 570 nm using a plate reader (Thermo-Labsystems). Results were expressed in % of living cells as compared to the control (untreated cells).
Xenogeneic transplantation of ROSAKIT D816V-Gluc cells in NOD-SCID IL-2R γ-/- (NSG) mice
NSG mice were bred and maintained under specific pathogen-free conditions at the animal facility of Gustave Roussy Institute. Animal experiments were performed in accordance with guidelines established by the Institutional Animal Committee.
Increasing amounts of ROSAKIT D816V-Gluc cells (1x106, 5x106 or 10x106) were injected to mice (3 groups; 6 mice per group) 24 h after irradiation at 2.5 Gy from a cesium-137 source. Engraftment was assessed after 4, 8 and 10 weeks using quantitative measurement of Gluc activity in plasma, and quantitative measurement of hCD45+/hCD117+ cells in PB using flow cytometry. After 10 weeks, in vivo bioluminescence imaging was performed on engrafted mice. KIT wild type (ROSAKIT WT control cells) were not included in this study as a control because they hardly - if not at all - graft in mice, as described elsewhere [26].
Secondary transplantations
Secondary transplantations were performed using human cells isolated from the BM of mice previously injected with ROSAKIT D816V-Gluc cells. Briefly, after 10 weeks of engraftment, one femur and two tibias were recovered from each animal and flushed in 1 mL of PBS. BM cells were then purified using EasySep™ Mouse/Human Chimera Isolation Kit (Stem Cell) according to the manufacters’ instructions. Purified BM cells were then pooled, and 5x105 cells were IV injected to each NSG mouse (n = 12) 24 h after their irradiation at 2.5 Gy from a cesium-137 source.
Flow cytometry analysis for the detection of xenografted cells
ROSAKIT D816V-Gluc cells were quantified in mouse PB, BM and spleen samples by flow cytometry using PE-labeled monoclonal antibodies (mAb) directed against human CD117 (Beckman Coulter), PE-Cy7- labeled hCD45 and APC-labeled mCD45 (BD Biosciences). Engrafted ROSAKIT D816V-Gluc cells were defined as hCD45+/CD117+ cells.
Measurement of gluc activity ex vivo in peripheral blood
Blood samples were collected in EDTA pre-coated vials (KABE Labortecknik). Gluc activity was determined on 5 μl of plasma by adding 100 μl of water soluble CTZ (50 μg/mL) followed by immediate acquisition of photon counts using a luminometer (PerkinElmer - EnSpire) as previously described by Morse and Tannous [42].
In vivo bioluminescence imaging
Mice were anesthetized using isoflurane and Gluc imaging was performed immediately after IV injection of 100 μl water soluble CTZ (4 mg/kg body weight) using a CCD camera (IVIS 50) and analyzed with Living Image® Software as previously described [42]. Quantitative analysis of Gluc intensity was performed by measuring luminescence signal intensity using region of interest (ROI) setting of the Living Image® software. ROIs were placed around the total area of body mice. ROI measurements were expressed as total flux (p/s).
Histopathology and immunohistochemistry (IHC) analysis
Deparaffinized sections of BM, spleen and liver processed for heat-induced antigen retrieval were incubated with a mouse anti-human CD45 mAb (Dako) or with a mouse anti-human MC tryptase (Dako). Staining was visualized by Histomouse Kit (Zymed). The sections were then counterstained with hematoxylin and examined using a Zeiss Axiophot microscope (Carl Zeiss).
Serum tryptase measurement
Total serum tryptase was measured using fluorescent enzyme-linked immunoassay that detects specifically human tryptase, following the manufacturer’s recommendations (Unicap Pharmacia) [43]. The detection limit of this assay is 1 ng/mL, and in human healthy controls, serum tryptase levels range between less than 1 and 15 ng/mL, with a median of approximately 5 ng/mL [44].
Statistical analysis
Statistical analysis was performed using GraphPad Prism 5.03 software. Data were analyzed using a two tailed Student’s t test. The levels of significance were as follow: *, p<0.05; **, p<0.01; ***, p<0.001.
ACKNOWLEDGMENTS
We thank Dr. Patrice Dubreuil for providing us with the KIT D816V-GFP plasmid construct and the murine SCF-producing CHO-KL cell line. We would like to express our appreciation to all the people belonging to the platform of animal studies at Gustave Roussy Institute for excellent animal colony care. We also thank the imaging and cytometry platform (PFIC) and pathology service at Gustave Roussy Institute.
CONFLICTS OF INTEREST
The authors declare no competing financial interest.
GRANT SUPPORT
This study was supported in part by Deciphera Pharmaceuticals, LLC. SB received a Ph D grant from the French Society of Hematology (SFH).
REFERENCES
1. Kirshenbaum AS, Kessler SW, Goff JP, Metcalfe DD. Demonstration of the origin of human mast cells from CD34+ bone marrow progenitor cells. J Immunol. 1991; 146:1410-5.
2. Krishnaswamy G, Ajitawi O, Chi DS. The human mast cell: an overview. Methods Mol Biol. 2006; 315:13-34.
3. Blume-Jensen P, Hunter T. Oncogenic kinase signalling. Nature. 2001; 411:355-65.
4. Galli SJ, Tsai M, Wershil BK, Tam SY, Costa JJ. Regulation of mouse and human mast cell development, survival and function by stem cell factor, the ligand for the c-kit receptor. Int Arch Allergy Immunol. 1995; 107:51-3.
5. Kitamura Y, Oboki K, Ito A. Molecular mechanisms of mast cell development. Immunol Allergy Clin North Am. 2006; 26:387-405; v.
6. Carter MC, Metcalfe DD, Komarow HD. Mastocytosis. Immunol Allergy Clin North Am. 2014; 34:181-96.
7. Horny HP. Mastocytosis: an unusual clonal disorder of bone marrow-derived hematopoietic progenitor cells. Am J Clin Pathol. 2009; 132:438-47.
8. Akin C, Valent P. Diagnostic criteria and classification of mastocytosis in 2014. Immunol Allergy Clin North Am. 2014; 34:207-18.
9. Valent P. Systemic mastocytosis. Cancer Treat Res. 2008; 142:399-419.
10. Arock M, Valent P. Pathogenesis, classification and treatment of mastocytosis: state of the art in 2010 and future perspectives. Expert Rev Hematol. 2010; 3:497-516.
11. Valent P, Horny HP, Escribano L, Longley BJ, Li CY, Schwartz LB, Marone G, Nunez R, Akin C, Sotlar K, Sperr WR, Wolff K, Brunning RD, et al. Diagnostic criteria and classification of mastocytosis: a consensus proposal. Leuk Res. 2001; 25:603-25.
12. Sperr WR, Escribano L, Jordan JH, Schernthaner GH, Kundi M, Horny HP, Valent P. Morphologic properties of neoplastic mast cells: delineation of stages of maturation and implication for cytological grading of mastocytosis. Leuk Res. 2001; 25:529-36.
13. Morgado JM, Sanchez-Munoz L, Teodosio CG, Jara-Acevedo M, Alvarez-Twose I, Matito A, Fernandez-Nunez E, Garcia-Montero A, Orfao A, Escribano L. Immunophenotyping in systemic mastocytosis diagnosis: 'CD25 positive' alone is more informative than the 'CD25 and/or CD2' WHO criterion. Mod Pathol. 2012; 25:516-21.
14. Nagata H, Worobec AS, Oh CK, Chowdhury BA, Tannenbaum S, Suzuki Y, Metcalfe DD. Identification of a point mutation in the catalytic domain of the protooncogene c-kit in peripheral blood mononuclear cells of patients who have mastocytosis with an associated hematologic disorder. Proc Natl Acad Sci U S A. 1995; 92:10560-4.
15. Arock M, Sotlar K, Akin C, Broesby-Olsen S, Hoermann G, Escribano L, Kristensen TK, Kluin-Nelemans HC, Hermine O, Dubreuil P, Sperr WR, Hartmann K, Gotlib J, et al. KIT mutation analysis in mast cell neoplasms: recommendations of the European Competence Network on Mastocytosis. Leukemia. 2015; 29:1223-32.
16. Schwartz LB. Diagnostic value of tryptase in anaphylaxis and mastocytosis. Immunol Allergy Clin North Am. 2006; 26:451-63.
17. Valent P, Sotlar K, Sperr WR, Escribano L, Yavuz S, Reiter A, George TI, Kluin-Nelemans HC, Hermine O, Butterfield JH, Hagglund H, Ustun C, Hornick JL, et al. Refined diagnostic criteria and classification of mast cell leukemia (MCL) and myelomastocytic leukemia (MML): a consensus proposal. Ann Oncol. 2014; 25:1691-700.
18. Bibi S, Langenfeld F, Jeanningros S, Brenet F, Soucie E, Hermine O, Damaj G, Dubreuil P, Arock M. Molecular defects in mastocytosis: KIT and beyond KIT. Immunol Allergy Clin North Am. 2014; 34:239-62.
19. Ma Y, Zeng S, Metcalfe DD, Akin C, Dimitrijevic S, Butterfield JH, McMahon G, Longley BJ. The c-KIT mutation causing human mastocytosis is resistant to STI571 and other KIT kinase inhibitors; kinases with enzymatic site mutations show different inhibitor sensitivity profiles than wild-type kinases and those with regulatory-type mutations. Blood. 2002; 99:1741-4.
20. Gleixner KV, Mayerhofer M, Aichberger KJ, Derdak S, Sonneck K, Bohm A, Gruze A, Samorapoompichit P, Manley PW, Fabbro D, Pickl WF, Sillaber C, Valent P. PKC412 inhibits in vitro growth of neoplastic human mast cells expressing the D816V-mutated variant of KIT: comparison with AMN107, imatinib, and cladribine (2CdA) and evaluation of cooperative drug effects. Blood. 2006; 107:752-9.
21. Gotlib J, Kluin-Nelemans HC, George TI, Akin C, Sotlar K, Hermine O, Awan FT, Hexner E, Mauro MJ, Sternberg DW, Villeneuve M, Huntsman Labed A, Stanek EJ, et al. Efficacy and Safety of Midostaurin in Advanced Systemic Mastocytosis. N Engl J Med. 2016; 374:2530-41.
22. Zappulla JP, Dubreuil P, Desbois S, Letard S, Hamouda NB, Daeron M, Delsol G, Arock M, Liblau RS. Mastocytosis in mice expressing human Kit receptor with the activating Asp816Val mutation. J Exp Med. 2005; 202:1635-41.
23. Gerbaulet A, Wickenhauser C, Scholten J, Peschke K, Drube S, Horny HP, Kamradt T, Naumann R, Muller W, Krieg T, Waskow C, Hartmann K, Roers A. Mast cell hyperplasia, B-cell malignancy, and intestinal inflammation in mice with conditional expression of a constitutively active kit. Blood. 2011; 117:2012-21.
24. Schumacher U, van Damme EJ, Peumans WJ, Butterfield JH, Mitchell BS. Lectin histochemistry of human leukaemic mast cells (HMC-1) transplanted into severe combined immunodeficient (scid) mice. Acta Histochem. 1998; 100:1-9.
25. Demehri S, Corbin A, Loriaux M, Druker BJ, Deininger MW. Establishment of a murine model of aggressive systemic mastocytosis/mast cell leukemia. Exp Hematol. 2006; 34:284-8.
26. Saleh R, Wedeh G, Herrmann H, Bibi S, Cerny-Reiterer S, Sadovnik I, Blatt K, Hadzijusufovic E, Jeanningros S, Blanc C, Legarff-Tavernier M, Chapiro E, Nguyen-Khac F, et al. A new human mast cell line expressing a functional IgE receptor converts to tumorigenic growth by KIT D816V transfection. Blood. 2014; 124:111-20.
27. Wurdinger T, Badr C, Pike L, de Kleine R, Weissleder R, Breakefield XO, Tannous BA. A secreted luciferase for ex vivo monitoring of in vivo processes. Nat Methods. 2008; 5:171-3.
28. Valent P, Arock M, Akin C, Sperr WR, Reiter A, Sotlar K, Hartmann K, George TI, Brockow K, Kluin-Nelemans HC, Gotlib J, Metcalfe DD, Horny HP. The classification of systemic mastocytosis should include mast cell leukemia (MCL) and systemic mastocytosis with a clonal hematologic non-mast cell lineage disease (SM-AHNMD). Blood. 2010; 116:850-1.
29. Li CY, Baek JY. Mastocytosis and fibrosis: role of cytokines. Int Arch Allergy Immunol. 2002; 127:123-6.
30. Savini P, Rondoni M, Poletti G, Lanzi A, Quercia O, Soverini S, De Benedittis C, Musardo G, Martinelli G, Stefanini GF. Serum total tryptase level confirms itself as a more reliable marker of mast cells burden in mast cell leukaemia (aleukaemic variant). Case Rep Hematol. 2015; 2015:737302.
31. Bibi S, Arslanhan MD, Langenfeld F, Jeanningros S, Cerny-Reiterer S, Hadzijusufovic E, Tchertanov L, Moriggl R, Valent P, Arock M. Co-operating STAT5 and AKT signaling pathways in chronic myeloid leukemia and mastocytosis: possible new targets of therapy. Haematologica. 2014; 99:417-29.
32. Frost MJ, Ferrao PT, Hughes TP, Ashman LK. Juxtamembrane mutant V560GKit is more sensitive to Imatinib (STI571) compared with wild-type c-kit whereas the kinase domain mutant D816VKit is resistant. Mol Cancer Ther. 2002; 1:1115-24.
33. Orfao A, Garcia-Montero AC, Sanchez L, Escribano L. Recent advances in the understanding of mastocytosis: the role of KIT mutations. Br J Haematol. 2007; 138:12-30.
34. Jara-Acevedo M, Teodosio C, Sanchez-Munoz L, Alvarez-Twose I, Mayado A, Caldas C, Matito A, Morgado JM, Munoz-Gonzalez JI, Escribano L, Garcia-Montero AC, Orfao A. Detection of the KIT D816V mutation in peripheral blood of systemic mastocytosis: diagnostic implications. Mod Pathol. 2015; 28:1138-49.
35. van Rijn S, Nilsson J, Noske DP, Vandertop WP, Tannous BA, Wurdinger T. Functional multiplex reporter assay using tagged Gaussia luciferase. Sci Rep. 2013; 3:1046.
36. Badr CE, Tannous BA. Bioluminescence imaging: progress and applications. Trends Biotechnol. 2011; 29:624-33.
37. Brockow K, Metcalfe DD. Mastocytosis. Chem Immunol Allergy. 2010; 95:110-24.
38. Brockow K. Epidemiology, prognosis, and risk factors in mastocytosis. Immunol Allergy Clin North Am. 2014; 34:283-95.
39. Meyer LH, Eckhoff SM, Queudeville M, Kraus JM, Giordan M, Stursberg J, Zangrando A, Vendramini E, Moricke A, Zimmermann M et al: Early relapse in ALL is identified by time to leukemia in NOD/SCID mice and is characterized by a gene signature involving survival pathways. Cancer Cell. 2011, 19:206-217
40. Butterfield JH, Weiler D, Dewald G, Gleich GJ. Establishment of an immature mast cell line from a patient with mast cell leukemia. Leuk Res. 1988; 12:345-55.
41. Kristensen T, Vestergaard H, Moller MB. Improved detection of the KIT D816V mutation in patients with systemic mastocytosis using a quantitative and highly sensitive real-time qPCR assay. J Mol Diagn. 2011; 13:180-8.
42. Morse D, Tannous BA. A water-soluble coelenterazine for sensitive in vivo imaging of coelenterate luciferases. Mol Ther. 2012; 20:692-3.
43. Schwartz LB, Bradford TR, Rouse C, Irani AM, Rasp G, Van der Zwan JK, Van der Linden PW. Development of a new, more sensitive immunoassay for human tryptase: use in systemic anaphylaxis. J Clin Immunol. 1994; 14:190-204.
44. Sperr WR, Jordan JH, Baghestanian M, Kiener HP, Samorapoompichit P, Semper H, Hauswirth A, Schernthaner GH, Chott A, Natter S, Kraft D, Valenta R, Schwartz LB, et al. Expression of mast cell tryptase by myeloblasts in a group of patients with acute myeloid leukemia. Blood. 2001; 98:2200-9.