
Introduction
Cutaneous T-cell lymphomas (CTCL), is a non Hodgkin lymphoma that affects skin. The most common type of skin infiltration is mycosis fungoides (MF), with the clinical picture of patches and plaques that may evolve into tumors and erythroderma. Sézary syndrome (SS), is an aggressive CTCL with dissemination of malignant CD4+ T cells with a memory phenotype that can be found in skin, blood, and lymph nodes [1].
During disease progression, a variety of immunologic abnormalities are observed regarding cell-mediated immunity in SS patients [2-3], with decreased production of interferon (IFN)-γ (Th1 cytokine), and down-regulated expression of Th1-specific genes, such as TBX21 (T-bet), NKG7, and SCYA5 (RANTES) [2, 4]. Defects in the production of interleukin 12 (IL-12) [3] and increased Th2 cytokines secretion (IL-4, -5, -10, and -13) by the Sézary cell, which can antagonize with Th1 function, have also been reported [3-5]. Moreover, SS cells exhibit constitutive STAT3 phosphorylation [6-7]. The Jak3/STAT pathway promotes the expression of IL-5, IL-17A/F and IL-10, regulates the production of angiogenetic factors, and confers resistance to treatment with histone deacetylase inhibitors in malignant T cells [8-10].
Considering the altered profile of cytokines secretion in SS, the activation of innate immunity may represent a strategy to potentiate innate immunity and consequently adaptive responses. The enhancement of immunity in SS should be crucial to ameliorate protection against bacterial/viral infections considering that sepsis is common cause of death among SS patients. Moreover, the activation antigen presenting cells by TLRs may lead to an improvement of the adaptive response that is crucial to control tumor burden. The TLRs recognize the pathogen-associated molecular patterns (PAMPs) and have been targeted to treat a variety of skin cancers [11]. Signalling by TLRs involves adaptor proteins known as myeloid differentiation primary-response gene 88 (MyD88), MyD88-adaptor-like (TIRAP/Mal), TIR-domain-containing adaptor protein inducing IFN-β (TRIF) and TRIF-related adaptor molecule (TRAM) [12]. Between the TLRs, TLR2 recognize lipopeptides, TLR4 recognizes lipopolysaccharide (LPS), and TLR5 flagellin. The TLR3, are involved in the recognition of double-stranded RNA, TLR7 and TLR8 recognize single-stranded RNA viruses, and TLR9 is crucial to recognized unmethylated CpG DNA. Human TLR7 and TLR8 recognize guanosine- or uridine-rich ssRNA as well as imidazoquinoline compounds [13-15]. After recognition of PAMPs, TLRs initiate downstream signaling events that culminate in cytokines secretion and initiation of the adaptive immune response.
The modulatory effect of TLR activation in the cytokines response in SS has been showed by the activation of peripheral blood mononuclear cells (PBMC) using the TLR7 agonist (3M-001), which induces increased levels of IFN-α production, and the TLR8 agonist (3M-002), inducing IL-12 and IFN-γ [16]. The combination of IFN-γ or IL-15 with the TLR7/8 agonist 3M-007 was shown to significantly increased Natural Killer (NK) cytotoxic response to CTCL cell lines [17]. Indeed, the in vitro potency of TLR agonists has been translated into clinical benefit. Safe and effective results were observed with subcutaneous CpG (TLR9) [18-19], and topical resiquimod (TLR7/8) in the treatment of CTCL patients [20].
Here, we sought to determine the ability of TLR agonist family members to induce pro-inflammatory and Th2-related cytokine production by PBMC from SS patients. We found that the impaired pro-inflammatory cytokines, Th2/Th1 cytokines and IFN-I production in SS patient cells, were partially restored upon TLR2/TLR4 and TLR7/8 agonist agonists stimulation.
Results
TLR agonists induce cytokine production in SS patient cells
The laboratory characteristics of SS patients are shown in Table S1. All patients were erytrodermic with intense pruritus, and 10/14 patients showed lymphocytosis. Although two patients (patients 5,16) were negative for T cell clones in two compartments (blood, skin or lymph nodes) evaluated, the hospital likely did not employ probes related to the clone present by these patients, whereas the clinical features together with laboratory data were essential to conclude the SS diagnosis.
At baseline levels (unstimulated PBMC), a decreased IL-6 secretion was detected in SS patients than HC subjects (Figure 1). Activation via TLR4 agonists induced high levels of IL-6 in the SS group, although these levels were lower than those noted in HC subjects (Figure 1). TNF secretion was restored by both TLR2 and TLR4 stimulation in the SS group, at the same level as HC subjects. The TLR5 agonist was not able to induce IL-6 and TNF secretion in both the HC and SS groups. For intracellular TLRs, the unique TLR agonist CL097 (TLR7/8) was able to induce IL-6 and TNF secretion in the SS and HC groups. The agonists of TLR5, TLR3, TLR7 and TLR9 did not induce IL-6 and TNF in the SS group.
An important impairment of IFN-γ secretion was noted in PBMC from SS patients in response to TLR ligands, and TLR7/8 agonist was able to induce IFN-γ, although it was lower than the control group. In comparison, for HC, IFN-γ in PBMC was triggered by TLR2, TLR4, TLR7/8 and TLR9 agonists (Figure 1).
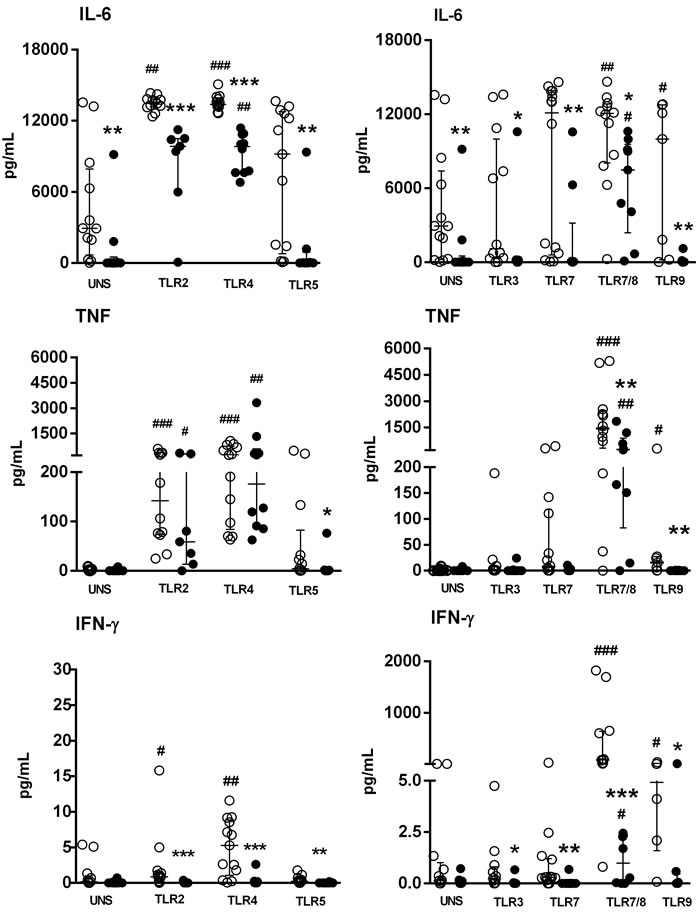
Figure 1: Impaired IL-6, TNF and IFN-γ production by PBMC of SS patients induced by TLR activation. PBMC from SS patients (n = 10, closed circle) and healthy controls (HC, n = 13, open circle) were cultured in medium (unstimulated, UNS) or with TLR agonists (TLR2-TLR9) for 48 h. Supernatants were assessed for IL-6, TNF and IFN-γ secretion with a cytometric bead array. The results are shown as medians and interquartile ranges (IQRs).*p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001 compared with the HC group; #p ≤ 0.05, ##p ≤ 0.01, ###p≤ 0.001 compared with the unstimulated culture.
Moreover, the baseline IL-10 levels were decreased in SS patients. However, these levels were up-regulated upon activation of TLR4 and TLR7/TLR8 in both groups, albeit at lower levels in the SS group (Figure 2). Curiously and in contrast to IL-6 secretion, IL-10 secretion was not induced after TLR2 stimulation in SS patient cells, which was enhanced with a TLR2 agonist.
An enhanced sensitivity flex kit, which can detect fentogram/mL levels by flow cytometry, was used to asses Th2 cytokines (IL-4, IL-5). However, IL-4 and IL-5 were undetectable in the supernatants of PBMC from both HC and SS samples stimulated with TLRs agonists. Figure 2 shows the low-level secretion of IL-13 induced by TLR2, TLR4 and TLR7/8 agonists in the HC group, whereas only activation by TLR4 induced IL-13 in SS samples. These results show that TLR activation induced low levels of Th2 cytokines from PBMC in both analyzed groups.
We next sorted the CD4+CD158k+ T cells (malignant T cells) from SS patients to verify their ability to produce Th2 cytokines upon TLR activation. The sorting procedure was performed with only 3 SS patients (patients 2, 5 and 15) because most patients did not survive. Moreover, only patient 2 showed the presence of CD4+CD158k+ cells, which did not secrete Th2 cytokines upon TLR2/TLR4 or TLR7/8 stimulation.
IL-17A was also undetectable in the supernatants of PBMC activated by TLR agonists in both analyzed groups.
These results revealed an adjuvant effect of TLR2 and TLR4 agonists on IL-6, TNF, IL-10 and IL-13 secretion, while TLR7/8 agonists increased IL-6, TNF, IL-10 and IFN-γ production by PBMCs of SS patients.
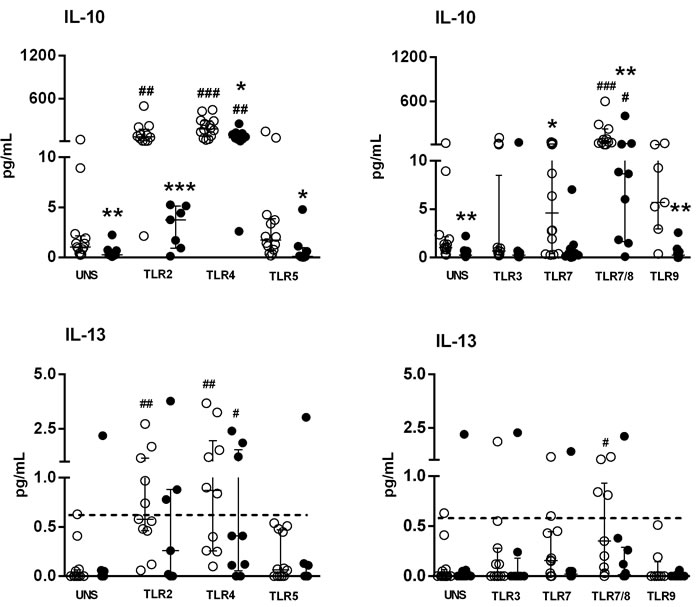
Figure 2: Altered IL-10 and IL-13 secretion by PBMC of SS patients induced by TLR agonists. PBMC from SS patients (n = 10, closed circle) and healthy controls (HC, n = 13, open circle, open circle) were cultured with medium (unstimulated, UNS) or stimulated for 48 h with TLR agonists (TLR2-TLR9). Supernatants were assessed for IL-10 and IL-13 secretion with a cytometric bead array. Horizontal line shows the detection limit for IL-13. The results are shown as medians and IQRs.*p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001 compared with the HC group; #p ≤ 0.05, ##p ≤ 0.01, ###p ≤ 0.001 compared with the unstimulated culture.
Intracellular TLR agonists induce IFN-α production in SS patient cells
We detected IFN-α secretion by PBMC stimulated with TLR9 agonists as well as the TLR7/8 CL097 agonist, but not with TLR3 and TLR7 agonists (Figure 3a). Significant IFN-α secretion was induced following stimulation of PBMC from HC with the TLR9/CpGA agonist, and this level was 29-fold higher compared to that observed in SS patients. In addition, secretion in response to the TLR7/8 agonist was increased 13-fold in HC compared with SS subjects (Figure 3a). Next, we evaluated the profile of TLR mRNA expression in unstimulated PBMC.
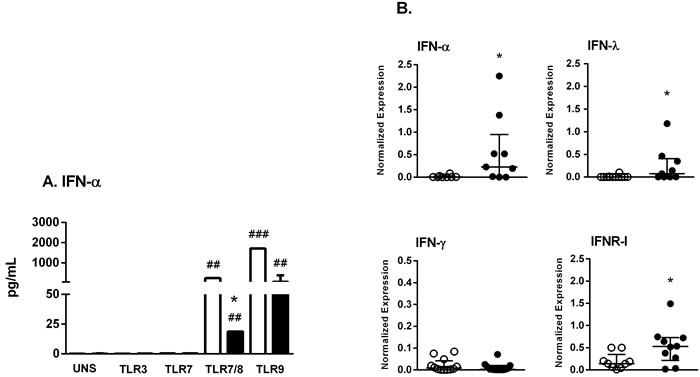
Figure 3: Agonists of TLR7/8 and TLR9 are able to induce IFN-I secretion in SS patient cells. A. PBMC from SS patients (n = 11, closed bar) and healthy controls (HC, n = 13, open bar) were cultured with medium (unstimulated, UNS) or stimulated for 48 h with agonists of TLR3, TLR7, TLR7/8 or TLR9. Supernatants were assessed for IFN-α secretion with a cytometric bead array. B. PBMC from SS patients (closed circle, n = 9) and HC (open circle, n = 12) were assessed for IFN-α, IFNR-I, IFN-λ and IFN-γ mRNA expression by qPCR. The results are presented as medians and IQRs.*p ≤ 0.05 compared with the HC group; #p ≤ 0.05, ##p ≤ 0.01 compared with the unstimulated culture.
Figure 1S shows the similar profile of TLR mRNA expression (TLR2,-3,-4,-7,-8 and -9) in PBMC from HC and SS subjects. Figure 3b presents IFN-I (IFN-α, receptor IFNR-I), II (IFN-γ) and III (IFN-λ) mRNA expression levels. As expected, no alterations in IFN-γ were detected between groups, whereas types I and III IFN were increased in SS subjects. At the time of the experimental analyses, no patient had previously received IFN-α treatment. Moreover, IFN-α was undetectable in the serum of SS patients and HC. The IL-6, TNF-α and IL-10 mRNA levels of unstimulated PBMC were similar in both groups (data not shown).
Next, we assessed the IL-6, TNF, IL-17A, IL-5, IL-2, IL1β, TGF-β1 and IL-10 serum levels. It was detected high levels of IL-5, IL-6, and IL-10 in SS serum compared with HC serum (Figure 4).
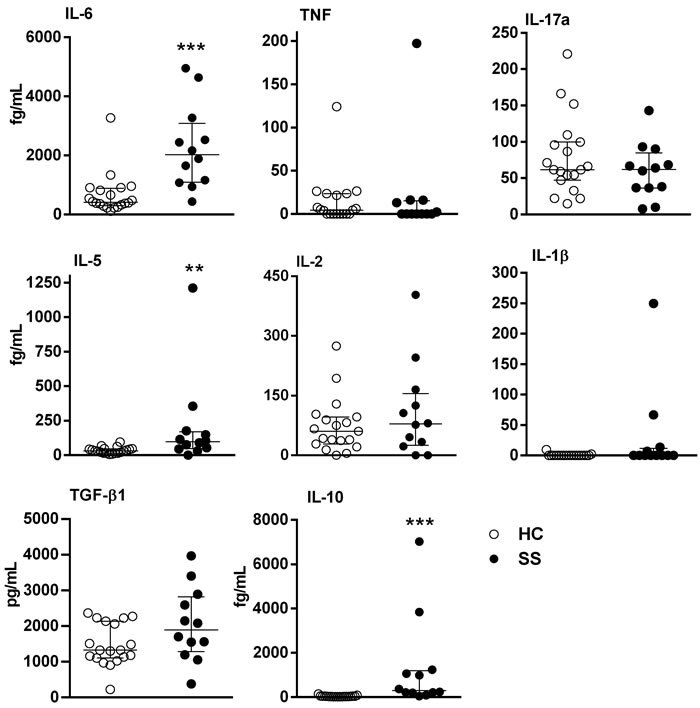
Figure 4: Cytokine serum levels in SS patients. Sera from SS patients (n = 12, closed circle) and healthy controls (HC, n = 19, open circle) were assessed for cytokines using high-sensitivity flex sets for TNF, IL-6, IL-5, IL-10, IL-2 and IL-17A or a flex set for TGF-β1 by flow cytometry. The results are presented as medians and IQRs. **p ≤ 0.01, ***p ≤ 0.001 compared with the HC group.
Discussion
The study evaluated the profile of cytokine production by PBMC induced by TLR agonists in SS patients. Studies focusing on pro-inflammatory, Th2, as well as IFN-I production, in SS patients after stimulation with extracellular and intracellular TLR agonists have not been reported previously. Our findings reveal a dysfunctional cytokine response upon TLR activation in SS patients; indeed, the TLR2/TLR4 agonist was able to partially restore IL-6, TNF, IL-10 and IL-13 production, while TLR7/8 showed an adjuvant effect on IFN I and II, IL-10 and pro-inflammatory cytokine secretion.
Overall, we observed a different profile of cytokine secretion between extracellular and intracellular TLRs agonists, whereby they are more affected following intracellular TLR activation. To determine whether these differences may have been due to different levels of TLRs expression, we observed similar expression of TLRs mRNA levels in PBMC between HC and SS patients, despite the fact that up to 90% of SS lymphocytes represent malignant Sézary cells. This finding showed that the impairment of TLRs activation was not due to altered TLRs expression and may be related to a dysfunctional signaling pathway or epigenetics mechanism, such as hypermethylation of cytokine gene promoters. These mechanisms needs to be further evaluated in SS patients.
We observed that IL-6 and TNF secretion was partially restored by activation with TLR2/4 and TLR7/8 agonists, while the agonists of TLR5, TLR3, TLR7 and TLR9 were unable to induce these cytokines. Although TNF secretion was similar to the control group, IL-6 and IFN-γ production remained low and could possibly explain the susceptibility of SS patients to bacterial infection. In fact, in SS patients, sepsis with Staphylococcus aureus and Pseudomonas aeruginosa can be caused by chronic skin infections and subsequent systemic infections [21].
Differential IL-10 responses were observed by TLRs ligands in SS patients, which were weakly induced by TLR2 and restored by TLR4 agonists, albeit at lower levels than healthy controls. It has been described that TLR2 activation induce a degradation of IL-10 mRNA while TLR4 activation preserves IL-10 mRNA due to the activation of TRIF and p38 signaling, in mouse system [22].
The serum levels of IL-6 in SS patients were increased 4.85-fold compared with those in HC. In SS patients, the levels of neopterin, ß2-MG and sIL-2R are significantly elevated, and IL-6 correlates with tumor burden [23]. CTCL tumor cell lines, but not non-malignant cell lines, produce IL-6 spontaneously [10]. Constitutive STAT5 activation is detected in early stage of CTCL and STAT3 in late stage of disease [24]. Moreover, STAT5 and STAT3 activation could be triggered by constitutively active JAK1 and JAK3 [24]. The high IL-6 serum levels found in our cohort of SS patients together with increased IL-5 levels could reflect the constitutive activation of JAK kinases. However, there was no association between cytokine serum levels and the prominent laboratory findings in SS. Moreover, although increased IL-6 serum levels were detected in SS, the baseline secretion levels of PBMC were decreased. This is probably a consequence of different sources of secreting cells, since several non-immune cells can contribute to this secretion in the serum. A similar effect was detected with IL-10 serum levels and secreted by PBMC in SS.
The etiology of CTCL remains uncertain, and samples have been analyzed for retroviral or other viral causes. However, studies have failed to show associations between viruses and CTCL [25-27]. Notably, we observed an impaired response to agonist treatment via TLR3, TLR7, TLR8 and TLR9 in our SS patients, although activation could be strategically overcome with some TLR agonists. CL097, a derivative of the imidazoquinoline (IMQ) compound R848, recovered IL-6, TNF, and IL-10 as well as IFN-I production by PBMC from SS patients. Given that TLR7 activation does not induce cytokine production, this finding suggests that TLR8 activation overcame the impaired cytokine secretion. In neonates, TLR8 agonist may overcome the inhibitory function of adenosine/cAMP avoiding fully dendritic cells activation [28]. Thus, the adjuvant effect of CL097 must be further evaluated, although our previous reports also demonstrated improvement in cytokine production induced by CL097 in cutaneous diseases, such as lichen planus [29], and during HIV infection [30].
Although IFN-α secretion levels in SS were restored with the TLR9 agonist (CpGA), this positive regulation was not observed for other cytokines, in direct contrast to the broad activity of CL097. These different effects could be due to the distinct signaling pathways triggered by these agonists. IFN-I production stimulated by CpG-A is mediated by PI3K and mTOR [31], whereas TLR7 and TLR8 activate IRF5 and IRF7 [32]. Curiously, upon evaluating type I and type III IFN expression in PBMC, some patients exhibited a constitutive increase in these IFNs (patients 1, 4 and 9) that was not due to seropositivity for viral infections.
Impaired cell-mediated immunity and decreased production of IFN-γ may be due to IL-12 deficiency [33] or a Th2 cytokine profile inhibiting the Th1 response in CTCL patients [9]. Again, the adjuvant effect of CL097 was observed for the IFN-γ response of 50% of SS patients, and very low levels were noted compared with the HC subjects.
The continuous production of IL-4, IL-5, and IL-10 by Sézary cells could represents evasion mechanism to tumoral immune response [18]. Thus, the production of Th2 cytokines might promote disease progression, given that Th2 cytokines may inhibit the antitumor effect. Despite the bias of leukemic Sézary cells toward the Th2 phenotype [34], we did not observe enhanced Th2 secretion by PBMC from SS patients (10 patients) or by malignant Sézary cells derived from one patient. Following TLR stimulation, IL-4 and IL-5 secretion were undetectable in either patients or controls, which indicates that the response to PAMPs is mainly related to Th1 rather than Th2 cytokines. The TLRs activation is crucial to induce stimulation of IL-12p70 and IFN-α secretion from DC and driving TLR-mediated Th1 responses [35]. In fact, very low IL-13 secretion levels by PBMC were detectable upon TLR2/TLR4 and TLR7/8 by healthy control; in contrast, these were almost absent in SS. This finding shows that, despite the known Th2 phenotype of malignant T cells in SS patients upon TLR activation, the mononuclear cells are not able to produce Th2 cytokines.
Together, our data reveal an impaired cytokine response to TLR signaling in SS patients. The decreased TLR response in cytokine production, including pro-inflammatory cytokines and IFN-I, was partially reversed with an agonist of TLR2, TLR4 and TLR7/8, suggesting its adjuvant role in SS.
Material and Methods
Casuistic
Our study included patients with Sézary syndrome (SS; n = 14, 7 males, 7 females), with a median age of 61 years (range 48-75 years), from the Clinic of Cutaneous Lymphomas of the Department of Dermatology, Hospital das Clínicas, University of São Paulo Medical School, Brazil (HC/FMUSP). The SS diagnosis was established based on recognized international clinical, histological, and biological criteria proposed by the World Health Organization (WHO) and European Organization for Research and Treatment of Cancer (EORTC) [36]. Patients were not previously treated with immunomodulating agents or chemotherapy. Healthy controls (HC; n = 19, 8 male, 11 female), with a median age of 58 years (range 33-85 years), were selected from the Laboratory of Medical Investigation, LIM56.. Blood collection was performed before the initiation of treatment. Patients with dermatologic diseases, those using drugs, and those with a history of autoimmune diseases were not included in this evaluation. The exclusion criteria consisted of treatment with immunosuppressant or immune-modifying drugs, pregnancy, and less than 18 years of age. This study was approved by the São Paulo University Institutional Use Committee (CAAE, 07965312.0.0000.0068), and informed consent was obtained from all subjects. All experimental protocols within this study were performed in accordance with the Ethics Committee of this institution.
Cell culture
PBMC were isolated from heparinized venous blood by Ficoll-Hypaque gradient centrifugation (GE Healthcare, Uppsala, Sweden) and diluted in RPMI medium supplemented with 10% AB human serum (Sigma, St Louis, MO,U.S.A.). PBMC cultures (2.0 x 105cells/well) were incubated in 96-well microplates (Costar, Cambridge, MA, U.S.A.) in medium in the presence of ligands for TLR2 (pam3csk4, 0.5 gmL-1); TLR3 [poly(I:C)-RIG, 20 ng mL -1]; TLR4 (LPS, 1.25 µg mL -1); TLR5 (flagellin, 0.5 ng mL -1); TLR7 (imiquimod, 1.25 gmL-1); TLR7/TLR8 (CL097, 2.5 µg mL-1) and TLR9 (CpGA 2206,4 mol L-1) for 48 h at 37°C and 5% CO2. All ligands were obtained from InvivoGen (San Diego, CA, U.S.A.). Cell-free supernatants were harvested and stored at -80°C until cytokine measurements were obtained with a cytometric bead array.
Cytokine measurements
Cytokine levels in the serum were assessed using enhanced sensitivity flex kits for IL-5 (limit of detection 67.8 fg/mL), IL-6 (68.4 fg/mL), IL-10 (13.7 fg/mL),IL-17a (16.1 fg/mL), TNF (67.3 fg/mL), IL-2 (88.9 fg/mL),IL-1β (48.4 fg/mL) and IL-2/TGF-b1 (14.9 pg/mL) from BD Biosciences (CA, USA).Cell culture supernatants were analyzed using cytokine kits to measure IL-6 (2.4 pg/mL), IL-10 (4.5 pg/mL), IL-13 (0.6 pg/mL), TNF (3.8 pg/mL), IFN-γ (3.7 pg/mL) and IFN-α (1.5 pg/mL) or enhanced sensitivity flex kits for IL-4 (144.4 fg/mL), IL-5 (67.8 fg/mL) and IL-17A (26.1 fg/mL).Samples were assessed using flow cytometry (LSR Fortessa, BD, San Jose, CA, U.S.A.).
Real-time PCR
Total RNA was extracted from PBMC using an RNeasy Plus Mini Kit (Qiagen, Valencia, CA, USA), and reverse transcription was performed with a with a iSCRIPT reverse transcriptase kit (Biorad, California, EUA). The primers used in the real-time PCR assay are detailed in Table S2.
GAPDH mRNA levels were used to normalize the mRNA content from PBMC. PCR was performed on an Applied Biosystems 7300 system using specific primers and SYBR Green (Applied Biosystems, Carlsbad, CA, USA) fluorescence detection reagents. The cycling protocol consisted of 10 min at 95°C, followed by 40 cycles of 15 s at 95°C and 60 s at 60°C. The amplification results were visualized and analyzed using Sequence Detection System (SDS) software (Applied Biosystems). Normalized expression was calculated as previously described by Livak [37].
Statistical analysis
The Mann-Whitney U-test was used to compare variables between the groups. Wilcoxon matched pairs test was used to compare baseline level with stimulated sample. p ≤ 0.05 was considered significant.
Acknowledgments
We are grateful to all the individuals who participated in the study.
conflicts of Interest
No conflicts of interest declared.
Grant support
This work was supported by the Fundação de Amparo a Pesquisa do Estado de São Paulo (2014/04865-9), Fundo de Apoio à Dermatologia de São Paulo (018-2015) and the Laboratório de Investigação Médica, Unidade 56 do Hospital das Clínicas da Faculdade de Medicina de São Paulo.
References
1. Imam MH, Shenoy PJ, Flowers CR, Phillips A and Lechowicz MJ. Incidence and survival patterns of cutaneous T-cell lymphomas in the United States. Leukemia & Lymphoma. 2013; 54:752-759.
2. Rook AH, Vowels BR, Jaworsky C, Singh A and Lessin SR. The immunopathogenesis of cutaneous T-cell lymphoma. Abnormal cytokine production by Sezary T cells. Archives of Dermatology. 1993; 129:486-489.
3. Rook AH and Heald P. The immunopathogenesis of cutaneous T-cell lymphoma. Hematology/Oncology Clinics of North America. 1995; 9:997-1010.
4. Vowels BR, Cassin M, Vonderheid EC and Rook AH. Aberrant cytokine production by Sezary syndrome patients: cytokine secretion pattern resembles murine Th2 cells. Journal of Investigative Dermatology. 1992; 99:90-94.
5. Geskin L, Viragova S, Stolz D and Fuschiotti P. Interleukin-13 is overexpressed in cutaneous T-cell lymphoma cells and regulates. Blood. 2015; 125:2798-2805.
6. Zhang Q, Nowak I, Vonderheid EC, Rook AH, Kadin ME, Nowell PC, Shaw LM and Wasik MA. Activation of Jak/STAT proteins involved in signal transduction pathway mediated by receptor for interleukin 2 in malignant T lymphocytes derived from cutaneous anaplastic large T-cell lymphoma and Sezary syndrome. Proceedings of the National Academy of Sciences of the United States of America. 1996; 93:9148-9153.
7. Eriksen KW, Kaltoft K, Mikkelsen G, Nielsen M, Zhang Q, Geisler C, Nissen MH, Ropke C, Wasik MA and Odum N. Constitutive STAT3-activation in Sezary syndrome: tyrphostin AG490 inhibits STAT3-activation, interleukin-2 receptor expression and growth of leukemic Sezary cells. Leukemia. 2001; 15:787-793.
8. Qin JZ, Dummer R, Burg G and Dobbeling U. Constitutive and interleukin-7/interleukin-15 stimulated DNA binding of Myc, Jun, and novel Myc-like proteins in cutaneous T-cell lymphoma cells. Blood. 1999; 93:260-267.
9. Nielsen M, Nissen MH, Gerwien J, Zocca MB, Rasmussen HM, Nakajima K, Ropke C, Geisler C, Kaltoft K and Odum N. Spontaneous interleukin-5 production in cutaneous T-cell lymphoma lines is mediated by constitutively activated Stat3. Blood. 2002; 99:973-977.
10. Krejsgaard T, Ralfkiaer U, Clasen-Linde E, Eriksen KW, Kopp KL, Bonefeld CM, Geisler C, Dabelsteen S, Wasik MA, Ralfkiaer E, Woetmann A and Odum N. Malignant cutaneous T-cell lymphoma cells express IL-17 utilizing the Jak3/Stat3 signaling pathway. Journal of Investigative Dermatology. 2011; 131:1331-1338.
11. Huen AO and Rook AH. Toll receptor agonist therapy of skin cancer and cutaneous T-cell lymphoma. Current Opinion in Oncology. 2014; 26:237-244.
12. Heil F, Hemmi H, Hochrein H, Ampenberger F, Kirschning C, Akira S, Lipford G, Wagner H and Bauer S. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science (New York, NY). 2004; 303:1526-1529.
13. Mijares LA, Wangdi T, Sokol C, Homer R, Medzhitov R and Kazmierczak BI. Airway epithelial MyD88 restores control of Pseudomonas aeruginosa murine infection via an IL-1-dependent pathway. Journal of Immunology. 2011; 186:7080-7088.
14. Uematsu S and Akira S. [Innate immune recognition of viral infection]. Uirusu. 2006; 56:1-8.
15. Lund JM, Alexopoulou L, Sato A, Karow M, Adams NC, Gale NW, Iwasaki A and Flavell RA. Recognition of single-stranded RNA viruses by Toll-like receptor 7. Proceedings of the National Academy of Sciences of the United States of America. 2004; 101:5598-5603.
16. Wysocka M, Newton S, Benoit BM, Introcaso C, Hancock AS, Chehimi J, Richardson SK, Gelfand JM, Montaner LJ and Rook AH. Synthetic imidazoquinolines potently and broadly activate the cellular immune response of patients with cutaneous T-cell lymphoma: synergy with interferon-gamma enhances production of interleukin-12. Clinical Lymphoma & Myeloma. 2007; 7:524-534.
17. Wysocka M, Dawany N, Benoit B, Kossenkov AV, Troxel AB, Gelfand JM, Sell MK, Showe LC and Rook AH. Synergistic enhancement of cellular immune responses by the novel Toll receptor 7/8 agonist 3M-007 and interferon-gamma: implications for therapy of cutaneous T-cell lymphoma. Leukemia & Lymphoma. 2011; 52:1970-1979.
18. Kim EJ, Hess S, Richardson SK, Newton S, Showe LC, Benoit BM, Ubriani R, Vittorio CC, Junkins-Hopkins JM, Wysocka M and Rook AH. Immunopathogenesis and therapy of cutaneous T cell lymphoma. Journal of Clinical Investigation. 2005; 115:798-812.
19. Kim YH, Girardi M, Duvic M, Kuzel T, Link BK, Pinter-Brown L and Rook AH. Phase I trial of a Toll-like receptor 9 agonist, PF-3512676 (CPG 7909), in patients with treatment-refractory, cutaneous T-cell lymphoma. Journal of the American Academy of Dermatology. 2010; 63:975-983.
20. Rook AH, Gelfand JM, Wysocka M, Troxel AB, Benoit B, Surber C, Elenitsas R, Buchanan MA, Leahy DS, Watanabe R, Kirsch IR, Kim EJ and Clark RA. Topical resiquimod can induce disease regression and enhance T-cell effector functions in cutaneous T-cell lymphoma. Blood. 2015; 126:1452-1461.
21. Lorincz AL. Cutaneous T-cell lymphoma (mycosis fungoides). Lancet. 1996; 347:871-876.
22. Teixeira-Coelho M. Guedes J, Ferreirinha P, Howes A, Pedrosa J, Rodrigues F, Lai WS, Blackshear PJ, O´Garra S, Castro AG, Saraiva M. Differential post-transcriptional regulation of IL-10 by TLR2 and TLR4-activated macrophages. EUR J Immunol. 2014 44: 856-66.2014.
23. Hassel JC, Meier R, Joller-Jemelka H, Burg G and Dummer R. Serological immunomarkers in cutaneous T cell lymphoma. Dermatology. 2004; 209:296-300.
24. Netchiporouk E, Litvinov IV, Moreau L, Gilbert M, Sasseville D and Duvic M. Deregulation in STAT signaling is important for cutaneous T-cell lymphoma (CTCL) pathogenesis and cancer progression. Cell Cycle. 2014; 13:3331-3335.
25. Mirvish ED, Pomerantz RG and Geskin LJ. Infectious agents in cutaneous T-cell lymphoma. Journal of the American Academy of Dermatology. 2011; 64:423-431.
26. Dereure O, Cheval J, Du Thanh A, Pariente K, Sauvage V, Manuguerra JC, Caro V, Foulongne V and Eloit M. No evidence for viral sequences in mycosis fungoides and Sezary syndrome skin lesions: a high-throughput sequencing approach. Journal of Investigative Dermatology. 2013; 133:853-855.
27. Dulmage BO, Feng H, Mirvish E and Geskin L. Black cat in a dark room: the absence of a directly oncogenic virus does not eliminate the role of an infectious agent in cutaneous T-cell lymphoma pathogenesis. British Journal of Dermatology. 2015; 172:1449-1451.
28. Philbin VJ, Dowling DJ, Gallington LC, Cortes G, Tan Z, Suter EE, Chi KW, Shuckett A, Stoler-Barak L, Tomai M, Miller RL, Mansfield K and Levy O. Imidazoquinoline Toll-like receptor 8 agonists activate human newborn monocytes and dendritic cells through adenosine-refractory and caspase-1-dependent pathways. Journal of Allergy and Clinical Immunology. 2012; 130:195-204 e199.
29. Domingues R, de Carvalho GC, da Silva Oliveira LM, Futata Taniguchi E, Zimbres JM, Aoki V, da Silva Duarte AJ and Sato MN. The dysfunctional innate immune response triggered by Toll-like receptor activation is restored by TLR7/TLR8 and TLR9 ligands in cutaneous lichen planus. British Journal of Dermatology. 2015; 172:48-55.
30. Cardoso EC, Pereira NZ, Mitsunari GE, Oliveira LM, Ruocco RM, Francisco RP, Zugaib M, da Silva Duarte AJ and Sato MN. TLR7/TLR8 Activation Restores Defective Cytokine Secretion by Myeloid Dendritic Cells but Not by Plasmacytoid Dendritic Cells in HIV-Infected Pregnant Women and Newborns. PLoS One. 2013; 8:e67036.
31. Costa-Mattioli M and Sonenberg N. RAPping production of type I interferon in pDCs through mTOR. Nature Immunology. 2008; 9:1097-1099.
32. Schoenemeyer A, Barnes BJ, Mancl ME, Latz E, Goutagny N, Pitha PM, Fitzgerald KA and Golenbock DT. The interferon regulatory factor, IRF5, is a central mediator of toll-like receptor 7 signaling. Journal of Biological Chemistry. 2005; 280:17005-17012.
33. Wysocka M, Zaki MH, French LE, Chehimi J, Shapiro M, Everetts SE, McGinnis KS, Montaner L and Rook AH. Sezary syndrome patients demonstrate a defect in dendritic cell populations: effects of CD40 ligand and treatment with GM-CSF on dendritic cell numbers and the production of cytokines. Blood. 2002; 100:3287-3294.
34. Guenova E, Watanabe R, Teague JE, Desimone JA, Jiang Y, Dowlatshahi M, Schlapbach C, Schaekel K, Rook AH, Tawa M, Fisher DC, Kupper TS and Clark RA. TH2 cytokines from malignant cells suppress TH1 responses and enforce a global TH2 bias in leukemic cutaneous T-cell lymphoma. Clinical Cancer Research. 2013; 19:3755-3763.
35. Dillon S, Agrawal A, Van Dyke T, Landreth G, McCauley L, Koh A, Maliszewski C, Akira S and Pulendran B. A Toll-like receptor 2 ligand stimulates Th2 responses in vivo, via induction of extracellular signal-regulated kinase mitogen-activated protein kinase and c-Fos in dendritic cells. J Immunol. 2004; 172:4733-4743.
36. Olsen E, Vonderheid E, Pimpinelli N, Willemze R, Kim Y, Knobler R, Zackheim H, Duvic M, Estrach T, Lamberg S, Wood G, Dummer R, Ranki A, Burg G, Heald P, Pittelkow M, et al. Revisions to the staging and classification of mycosis fungoides and Sezary syndrome: a proposal of the International Society for Cutaneous Lymphomas (ISCL) and the cutaneous lymphoma task force of the European Organization of Research and Treatment of Cancer (EORTC). Blood. 2007; 110:1713-1722.
37. Livak KJ and Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001; 25:402-408.