
INTRODUCTION
Childhood acute myeloid leukemia (AML) encompasses a heterogeneous group of malignancies, with great variability in terms of response to therapy. While the majority of patients harbor recurrent chromosomal translocations, almost 20% of childhood AML do not show any recognizable cytogenetic alteration and are defined as cytogenetically-normal (CN-AML) [1]. Recently, an increasing list of molecular markers with prognostics significance has been identified in adult CN-AML [2-8]. However, these alterations are barely detected in pediatric CN-AML [9-11] and there is a considerable proportion of children with CN-AML in whom no genetic abnormality can be unveiled.
In a recent study, we performed whole-transcriptome massively parallel sequencing of 7 cases of pediatric CN-AML; in 3 of them, we identified a novel recurrent fusion transcript involving CBFA2T3 and GLIS2 genes. We then extended the analysis to a larger cohort (N=230) and this novel fusion was detected in 20 out of the 237 pediatric CN-AML cases analyzed (8.4%). The 5-year event-free survival of the 20 positive patients was significantly worse than that of the 217 pediatric CN-AML patients lacking the translocation (27.4%vs 59.6%; P= 0.01), demonstrating that CBFA2T3-GLIS2 fusion transcript is a novel common feature of pediatric CN-AML predicting poorer outcome [12].
Despite this evidence in human AML, expression of CBFA2T3-GLIS2 fusion is not sufficient to foster leukemia development in mice, this suggesting that the fusion protein per se may not promote leukemogenesis [13,14]. Starting from this observation, we reasoned that additional lesions can concur to leukemia development in children harboring CBFA2T3-GLIS2 fusion transcript. Using whole-transcriptome sequencing, we identified a novel fusion transcript that is recurrent in CBFA2T3-GLIS2-positive patients and is instrumental for the identification of a subset of CBFA2T3-GLIS2-rearranged patients characterized by an even poorer outcome.
RESULTS
Identification of DHH-RHEBL1 fusion transcript in pediatric CBFA2T3-GLIS2-positive AML patients by whole-transcriptome sequencing
Blasts from 8 pediatric patients with AML were analyzed by means of whole-transcriptome massively parallel sequencing. Four of them had CN-AML (CN#21, CN#22, CN#23, CN#24), and 4 harbored CBFA2T3-GLIS2 fusion transcript (#1, #3, #13, #17). Among the latter, 2 had already been reported in our previous study [12]. Further analysis on RNA-seq data revealed the presence of a new recurrent fusion transcript in 2 out of 4 CBFA2T3-GLIS2-positive patients. This novel fusion transcript is the result of a read-through that combines at least part of one exon with each of two distinct (parent) genes that are adjacent on the same chromosome in the same orientation [15]. In particular, this transcript involves DHH, a member of the Hedgehog family [16], and RHEBL1, a gene coding for a small GTPase of the Ras family which regulates a wide variety of cellular functions, including cell growth, differentiation, and transformation [17]. Both genes are contiguously localized on the reverse strand of chromosome 12 (Figure 1A) and, although the mechanism that leads to generation of read-through fusion transcripts remains obscure [18], RT-PCR analysis and Sanger sequencing confirmed that all positive cases harbored the in-frame fusion between exon 2 of DHH and exon 2 of RHEBL1 (Figure 1B).
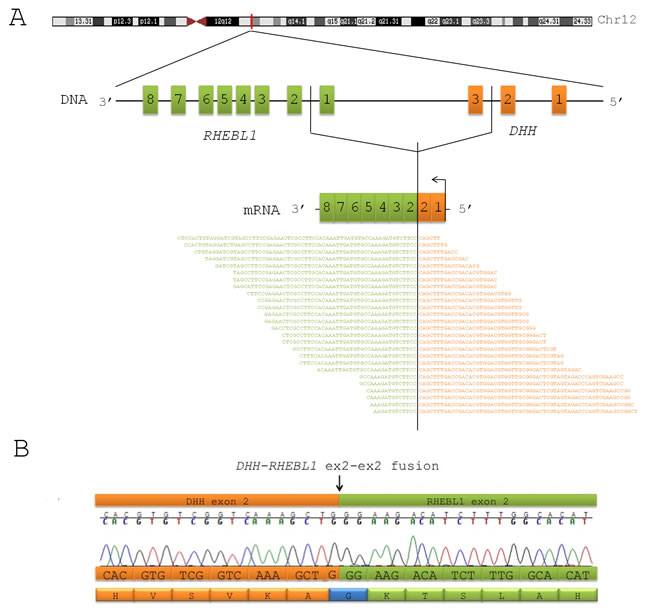
Figure 1: DHH-RHEBL1 is a novel fusion transcript recurrent in pediatric CBFA2T3-GLIS2 positive AML. (A) Schematic representation of the fusion between DHH and RHEBL1 identified by means of whole-transcriptome sequencing. The figure shows the position of DHH and RHEBL1 on chromosome 12 and the fusion transcript detected by RNA-seq. The identification of this novel fusion transcript was supported by an average of 11 span and 21 split reads. (B) Sanger sequencing performed in order to validate the detection of the DHH-RHEBL1 fusion transcript. Electropherogram and predicted sequence of the fusion protein are shown. The black arrow indicates the fusion breakpoint.
DHH-RHEBL1 fusion transcript is recurrent in pediatric CBFA2T3-GLIS2-positive AML
To determine the prevalence of DHH-RHEBL1 fusion in pediatric AML, we then examined a validation cohort of 55 children with AML. The validation cohort included CN-AML patients (N=24), CBFA2T3-GLIS2-positive patients (N=16), patients harboring known cytogenetic/genetic abnormalities (alteration of MLL, NPM1, FLT3, t(8;22)(p11;q13), t(9;11)(p22;q23), inv(16)(p13;q22)) (N=12) and normal CD34+ hematopoietic stem cells (N=3). The DHH-RHEBL1 fusion transcript was detected in 6 out of 16 patients carrying the CBFA2T3-GLIS2 fusion, while it was never found in the other patients with AML, irrespectively of the mutational status, as well as in normal CD34+ cells. Thus, considering also the patients of the sequencing cohort, the DHH-RHEBL1 fusion was globally present in 8 out of 20 (40%) of the CBFA2T3-GLIS2-positive patients, this demonstrating that this novel alteration is a common feature of this peculiar subset of childhood AML.

Figure 2: Implications of DHH-RHEBL1 fusion transcript expression. (A) Expression levels of DHH and RHEBL1 gene obtained from RNA-seq data in DHH-RHEBL1 positive patients (N=2), in CBFA2T3-GLIS2 positive patients (N=2) and in CN-AML patients (N=4). Abbreviations: CPM = count per million, Pos = positive (B) Probability of 8-year overall survival (OS) in CBFA2T3-GLIS2-positive children who did or did not harbor the DHH-RHEBL1 fusion transcript (25%, SE=15 vs 55%, SE=15) (P=0.1).
DHH-RHEBL1–positive patients exhibit a specific gene expression signature and an overexpression of both DHH and RHEBL1
To get insights into the molecular consequences of DHH-RHEBL1 expression, we performed a gene expression analysis on the RNA-seq data for the 8 patients carrying this fusion transcript. Firstly, we analyzed the expression level of the two genes involved in the fusion transcript and we found that the expression of both DHH and RHEBL1is significantly enhanced in the DHH-RHEBL1-positive patients as compared with patients harboring only CBFA2T3-GLIS2 fusion (P=0.007 and P=0.009 respectively) and with the other CN-AML cases (P=0.0005 and P=0.043, respectively) (Figure 2A). Additionally, DHH-RHEBL1-positive patients showed a distinctive gene expression signature both with respect to CBFA2T3-GLIS2-positive patients (518 differentially expressed genes; P<0.05), and CN-AML patients (596 differentially expressed genes; P<0.05). Interestingly, DHH-RHEBL1-positive patients showed higher expression of several genes known to be associated with leukemia occurrence and/or tumor progression, such as FLT3 [19], BEX1 [20], MUC4 [21] and AFAP1L2 [22] (Figure 3). Finally, we also evaluated whether the presence of DHH-RHEBL1 fusion transcript influences the outcome of CBFA2T3-GLIS2-positive patients. The 8-year overall survival of the 8 patients harboring the DHH-RHEBL1 fusion transcript was worse than that of the 12 CBFA2T3-GLIS2-rearranged patients not harboring the DHH-RHEBL1 fusion transcript (25% vs 55%). Likely due to the small number of patients, this difference failed to achieve statistical significance (P=0.1) (Figure 2B).

Figure 3: Analysis of gene expression profile of DHH-RHEBL1-positive patients. Heatmap of the top 30 differentially expressed genes in DHH-RHEBL1-positive patients compared with the patients harboring the CBFA2T3-GLIS2 fusion only and CN-AML patients. (A) Gene expression signature of patients harboring both CBFA2T3-GLIS2 and DHH-RHEBL1 fusion transcript; (B) Gene expression signature of CBFA2T3-GLIS2-positive patients; (C) Gene expression signature of children with CN-AML not harboring any detectable fusion transcript. Abbreviation: Pos = positive.
DISCUSSION
The last decades have witnessed the identification, through chromosomal analysis techniques or RT-PCR, of fusion genes influencing either proliferation/apoptosis or differentiation potential of AML cells [1-5]. Recently, different studies took advantage of next-generation sequencing approach to identify novel mutations or chromosomal aberrations that escape detection by conventional cytogenetic techniques and characterize pediatric AML, as well as other hematopoietic cancers [12,13,23-25]. Next-generation sequencing, as well as gene expression profiling, are also instrumental to refine the biological peculiarities affecting the risk of treatment failure and to detect crucial pathways potentially ‘druggable’ in both adult and childhood AML [2, 26, 27].
In this regards, we recently demonstrated that CBFA2T3-GLIS2 fusion transcript is recurrent in pediatric CN-AML and portends a poor outcome [12]. However, experimental evidence demonstrates that expression of this fusion product in mice is not sufficient to promote leukemogenesis [13,14]. This observation prompted us to investigate by whole-transcriptome sequencing a cohort of CN-AML and CBFA2T3-GLIS2-positive patients. We were able to identify a novel fusion transcript involving DHH and RHEBL1 genes in 2 out of 4 CBFA2T3-GLIS2-positive cases of our sequencing cohort. Extending the analysis to a validation cohort of 55 children, the DHH-RHEBL1 fusion was globally detected (including the 2 cases in the sequencing cohort) in 8 out of 20 (40%) of the CBFA2T3-GLIS2-positive patients, indicating that this novel fusion transcript is recurrent and peculiar to this specific group of pediatric CN-AML. Interestingly, both DHH and RHEBL1 genes have been implicated in a variety of human diseases, including cancer. On the one hand, DHH codes for a member of the Hedgehog (HH) signaling pathway, which, similar to other HH ligands, binds to its receptor Patched and leads to the signaling cascade of repressive interactions, culminating into effects on the transcription of target genes. The HH signaling, during embryogenesis, controls cell proliferation, differentiation and tissue morphogenesis [28]. However, it is also well known to have a role in tumors, and the role that HH signaling plays in the growth of tumors can be classified according to how the pathway is activated [29]. These mechanisms include loss-of-function mutations in inhibitory proteins, such as Patched (PTC1), gain-of-function mutations in positive regulators, such as Smoothened (SMO), and overexpression of the HH ligands (Sonic, Indian and Desert Hedgehog), leading to either autocrine or paracrine activation of the pathway and renewal/propagation of cancer stem cells [28]. Recently, with the identification of CBFA2T3-GLIS2 fusion transcript in pediatric CN-AML, different studies [13,14,23] demonstrated that the presence of this fusion transcript leads to an aberrant activation of the HH signaling due to the ectopic expression of the GLIS2 transcription factor. Notably, in the present work, we demonstrate that patients harboring the DHH-RHEBL1 fusion present an overexpression of DHH compared to both CBFA2T3-GLIS2-positive patients and CN-AML patients (Figure 2A). Considering that overexpression of the HH ligands leads to activation of the HH pathway [28], it is tempting to speculate that overexpression of DHH could contribute to the aberrant activation of the HH pathway. On the other hand, the RHEBL1 protein belongs to the Ras family of small GTPases and, similar to other Ras proteins, is a molecular switch that controls a wide variety of cellular functions including cell growth, differentiation and transformation [17]. Previous studies reported that RHEBL1 could function as an activator of NF-kB [17] and mTOR [30] signaling, both of which are frequently altered in many solid tumors, as well as in leukemias and lymphomas[27, 31-36]. In view of RHEBL1 over-expression in patients harboring the DHH-RHEBL1 fusion transcript compared to those harboring only the CBFA2T3-GLIS2 fusion transcript and to CN-AML children, it will be interesting to investigate more thoroughly its possible role in leukemogenesis.
To define the implications, if any, of DHH-RHEBL1 fusion transcript expression, we performed an analysis of gene expression on RNA-seq data. Firstly, we demonstrated that patients harboring only the CBFA2T3-GLIS2 fusion transcript and patients harboring both CBFA2T3-GLIS2 and DHH-RHEBL1 fusion transcripts exhibited an overexpression of GLIS2 compared to CN-AML patients, this finding being consistent with recently published data [13,14,23]. In addition, and more importantly, gene expression profile revealed that DHH-RHEBL1-positive patients showed a specific gene expression signature, with 518 and 596 genes being significantly overexpressed when compared to patients harboring only the CBFA2T3-GLIS2 fusion transcript (P<0.05) and CN-AML patients (P<0.05), respectively. Interestingly, DHH-RHEBL1-positive patients exhibited an enhanced expression of FLT3, which has been reported to be constitutively activated and over-expressed in a proportion of both pediatric and adult AML [1,19]. Additionally, also BEX1, which is known to be expressed in AML with MLL rearrangements [20], was up-regulated. Furthermore, other genes, such as MUC4 and AFAP1L2, associated with different human cancers [21,22], resulted overexpressed in DHH-RHEBL1-positive patients. Taken together, these findings indicate that DHH-RHEBL1-positive patients exhibit a distinct expression signature compared to both CN-AML patients and to CBFA2T3-GLIS2-positive patients (Figure 3), suggesting that the presence of DHH-RHEBL1 fusion transcript could be important in the definition of a new subgroup among the CBFA2T3-GLIS2-positive patients. To assess whether the presence of DHH-RHEBL1 fusion transcript affects patients’ outcome, we estimated the 8-year overall survival of the 8 patients harboring the DHH-RHEBL1 fusion transcript, finding that it was worse, although not statistically different, than that of the 12 CBFA2T3-GLIS2-rearranged patients not harboring DHH-RHEBL1 fusion (25% vs 55%).
While the discovery of gene fusion resulting from chromosomal aberrations, such as translocations, deletion/insertion, inversion, represents the primary objective of gene fusion analyses, whole-transcriptome sequencing aims at unraveling another category of gene fusion that are referred to as read-through fusion transcripts[15,18]. Several studies indicate that these groups of fusion transcripts are widely observed in virtually all samples analyzed, including samples from non-transformed different tissues [37]. Some of these RNA chimeras, however, appear to be restricted to individual tissue types, and a few of these have been observed to be highly expressed in cancers, this observation underpinning the potential functional relevance with respect to cellular differentiation and disease development [38-42]. To the best of our knowledge, DHH-RHEBL1 is the first read-through fusion transcript reported in a specific subset of pediatric leukemias and its recurrence in CBFA2T3-GLIS2-positive patients suggests it could be important for leukemogenesis. In summary, we discovered a novel DHH-RHEBL1 fusion transcript that is recurrent (40%) in CBFA2T3-GLIS2-positive patients only; it characterizes a subset of patients with an even more dismal outcome among the CBFA2T3-GLIS2-positive patients. The mechanism(s) through which this fusion transcript promotes leukemogenesis and the possibility of targeting it with pathway-specific compounds remain to be thoroughly investigated.
METHODS
Patient samples
After obtaining written informed consent, patient samples analyzed either in the parallel sequencing screening or in the validation cohort were collected from children with newly diagnosed de novo AML other than promyelocytic leukemia, enrolled in the Associazione Italiana Ematologia Oncologia Pediatrica (AIEOP) 2002/01 Protocol [43]. Morphological diagnosis and immunophenotypic analysis was centrally reviewed at the laboratory of Pediatric Hematology of the University Hospital in Padova. Chromosome analysis was performed on bone marrow (BM) aspirates using standard laboratory procedures. Karyotypes were reported according to the International System for Human Cytogenetic Nomenclature (ISCN 2005). For fluorescence in-situ hybridization (FISH), an MLL locus specific (LSI) dual color probe for 11q23 (Abbott-Vysis, Downers Grove, IL) was employed. All the 8 patients of the sequencing cohort were cytogenetically normal, as well as negative for known recurrent genetic abnormalities involving MLL, CBFB, NPM1 and FLT3.
Whole-Transcriptome Sequencing and RNA-seq bioinformatics analyses
RNA library construction and whole-transcriptome sequencing has been described previously [12]. In brief, 250-1000 ng of total RNA were used for the synthesis of cDNA libraries with TruSeq RNA Sample Prep Kit v2 (Illumina, San Diego, CA), and sequenced by synthesis at 75bp in paired-end mode on HiScanSQ sequencer (Illumina). Reads were aligned with TopHat2/BowTie2 [44] to the reference human genome hg19/GRCh37. Defuse [45] and Chimerascan [46] packages were used to detect chimeric transcripts from RNA-seq data. Whole-transcriptome massively parallel sequencing in the 8 children with CN-AML yielded an average of 78.4 million mapped reads/patient, thus reaching an average coverage of 34X.
Gene expression analysis
The mapped reads obtained with TopHat2/BowTie2 pipeline were processed with SAMtools [47] in order to remove the potential optical or PCR duplicate (function “rmdup”) and then the count of the mapped reads for each hg19 gene was performed by applying the Python package “htseq-count” (http://www-huber.embl.de/users/anders/HTSeq/doc/overview.html). Gene annotations were derived from Ensembl Release 70 (January 2013).
The differentially expressed genes were determined with edgeR, a R-bioconductor package suitable for analyzing RNA-seq data [48]. Three different comparisons were performed, corresponding to all the possible couples among the three groups of patients: 1) DHH-RHEBL1-positive and CBFA2T3-GLIS2-positive (N=2); 2) DHH-RHEBL1-negative and CBFA2T3-GLIS2-positive (N=2); 3) CN-AML (N=4).
For each comparison, the complete set of genes, with the corresponding mapped reads count, was firstly reduced in order to consider in our analysis only the genes with count-per-million (CPM) > 3 in more than 2 samples. Then, adopting a statistical method based on the negative binomial distribution, the significance of the differences between the normalized reads count was determined for each gene. Differences with P<0.05 were considered to be statistically significant.
The Multi Experiment Viewer (MeV) tool (http://www.tm4.org/mev.html) was used to visualize the expression data.
Screening forDHH-RHEBL1 fusion transcript in the validation cohort
Total RNA was extracted from BM leukemia cells using TRIzol. DHH-RHEBL1 fusion transcript was detected through RT-PCR and sequenced with the BigDye terminator v3.1 Cycle Sequencing kit (PE Applied Biosystems, Foster City, CA) on an Applied Biosystems 310 analyzer. The RT-PCR was performed at 60°C with the Expand Long Template PCR system (Roche, Mannheim, Germany). Primers used to amplify the full DHH-RHEBL1 fusion transcript (1359bp) were: forward 5’- AGTAGCAGGTCCTAGACACCCCC -3’, reverse 5’- TACCCGTGAAGTCCTGAGGATCT -3’.
ACKNOWLEDGEMENTS
This work was supported in part by grants from Fondazione Umberto Veronesi (Milan) and Fondazione Ginevra Caltagirone to RM, by Cariparo, IRP-Istituto di Ricerca Pediatrica-Città della Speranza Padova to MP, GB and EM, by Special Grant “5x1.000” from AIRC (Associazione Italiana Ricerca sul Cancro) to FL and by MIUR (Ministero dell’Istruzione, dell’Università e della Ricerca Scientifica, Progetti di Rilevante Interesse Nazionale 2009 and 2010) to FL and SR.
References
1. Rubnitz JE, Inaba H. Childhood acute myeloid leukaemia. Br J Haematol. 2012; 159(3):259-76.
2. Pui CH, Carroll WL, Meshinchi S, Arceci RJ. Biology, risk stratification, and therapy of pediatric acute leukemias: an update. J Clin Oncol. 2011; 29(5):551-65.
3. Ho PA, Alonzo TA, Gerbing RB, Pollard J, Stirewalt DL, Hurwitz C, Heerema NA, Hirsch B, Raimondi SC, Lange B, Franklin JL, Radich JP, Meshinchi S. Prevalence and prognostic implications of CEBPA mutations in pediatric acute myeloid leukemia (AML): a report from the Children’s Oncology Group. Blood. 2009; 113(26):6558-66.
4. Cazzaniga G, Dell’Oro MG, Mecucci C, Giarin E, Masetti R, Rossi V, Locatelli F, Martelli MF, Basso G, Pession A, Biondi A, Falini B. Nucleophosmin mutations in childhood acute myelogenous leukemia with normal karyotype. Blood. 2005; 106(4):1419-22.
5. Meshinchi S, Woods WG, Stirewalt DL, Sweetser DA, Buckley JD, Tjoa TK, Bernstein ID, Radich JP. Prevalence and prognostic significance of Flt3 internal tandem duplication in pediatric acute myeloid leukemia. Blood. 2001; 97(1):89-94.
6. Staffas A, Kanduri M, Hovland R, Rosenquist R, Ommen HB, Abrahamsson J, Forestier E, Jahnukainen K, Jónsson ÓG, Zeller B, Palle J, Lönnerholm G, Hasle H, Palmqvist L, Ehrencrona H. Presence of FLT3-ITD and high BAALC expression are independent prognostic markers in childhood acute myeloid leukemia. Blood. 2011; 118(22):5905-13.
7. Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, Chen K, Koboldt DC, Fulton RS, Delehaunty KD, McGrath SD, Fulton LA, Locke DP, Magrini VJ, Abbott RM, Vickery TL, Reed JS, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med; 361(11):1058-66.
8. Ley TJ, Ding L, Walter MJ, McLellan MD, Lamprecht T, Larson DE, Kandoth C, Payton JE, Baty J, Welch J, Harris CC, Lichti CF, Townsend RR, Fulton RS, Dooling DJ, Koboldt DC, et al. DNMT3A mutations in acute myeloid leukemia. N Engl J Med. 2010; 363(25):2424-33.
9. Andersson AK, Miller DW, Lynch JA, Lemoff AS, Cai Z, Pounds SB, Radtke I, Yan B, Schuetz JD, Rubnitz JE, Ribeiro RC, Raimondi SC, Zhang J, Mullighan CG, Shurtleff SA, Schulman BA, et al. IDH1 and IDH2 mutations in pediatric acute leukemia. Leukemia. 2011; 25(10):1570-7.
10. Pigazzi M, Ferrari G, Masetti R, Falini B, Martinolli F, Basso G, Biondi A, Pession A, Cazzaniga G. Low prevalence of IDH1 gene mutation in childhood AML in Italy. Leukemia. 2011; 25(1):173-4.
11. Paganin M, Pigazzi M, Bresolin S, Masetti R, Fagioli F, Chiaretti S, Cazzaniga G, Locatelli F, Pession A, te Kronnie G, Basso G. DNA methyltransferase 3a hot-spot locus is not mutated in pediatric patients affected by acute myeloid or T-cell acute lymphoblastic leukemia: an Italian study. Haematologica. 2011; 96(12):1886-7.
12. Masetti R, Pigazzi M, Togni M, Astolfi A, Indio V, Manara E, Casadio R, Pession A, Basso G, Locatelli F. CBFA2T3-GLIS2 fusion transcript is a novel common feature in pediatric, cytogenetically normal AML, not restricted to FAB M7 subtype. Blood. 2013; 121(17):3469-72.
13. Gruber TA, Larson Gedman A, Zhang J, Koss CS, Marada S, Ta HQ, Chen SC, Su X, Ogden SK, Dang J, Wu G, Gupta V, Andersson AK, Pounds S, Shi L, Easton J, et al. An Inv(16)(p13.3q24.3)-Encoded CBFA2T3-GLIS2 Fusion Protein Defines an Aggressive Subtype of Pediatric Acute Megakaryoblastic Leukemia Cancer Cell. 2012;22(5):683-97.
14. Crispino JD, Le Beau MM. BMP meets AML: induction of BMP signaling by a novel fusion gene promotes pediatric acute leukemia. Cancer Cell. 2012; 22(5):567-8.
15. Maher CA, Kumar-Sinha C, Cao X, Kalyana-Sundaram S, Han B, Jing X, Sam L, Barrette T, Palanisamy N, Chinnaiyan AM. Transcriptome sequencing to detect gene fusions in cancer. Nature. 2009; 458(7234):97-101.
16. Ingham PW, McMahon AP. Hedgehog signaling in animal development: paradigms and principles. Genes Dev. 2001; 15(23):3059-87.
17. Yuan J, Shan Y, Chen X, Tang W, Luo K, Ni J, Wan B, Yu L. Identification and characterization of RHEBL1, a novel member of Ras family, which activates transcriptional activities of NF-kappa B. Mol Biol Rep. 2005; 32(4):205-14.
18. Mitelman F, Johansson B, Mertens F. Fusion genes and rearranged genes as a linear function of chromosome aberrations in cancer. Nat Genet. 2004; 36(4):331-4.
19. Graf M, Hecht K, Reif S, Pelka-Fleischer R, Pfister K, Schmetzer H. Expression and prognostic value of hemopoietic cytokine receptors in acute myeloid leukemia (AML): Implications for future therapeutical strategies. Eur J Haematol 2004; 72(2):89–106.
20. Quentmeier H, Tonelli R, Geffers R, Pession A, Uphoff CC, Drexler HG. Expression of BEX1 in acute myeloid leukemia with MLL rearrangements. Leukemia 2005; 19(8):1488-9.
21. Carraway KL, Perez A, Idris N, Jepson S, Arango M, Komatsu M, Haq B, Price-Schiavi SA, Zhang J, Carraway CA. Prog Nucleic Acid Res Mol Biol. 2002;71:149-85.
22. Emaduddin M, Edelmann MJ, Kessler BM, Feller SM. Odin (ANKS1A) is a Src family kinase target in colorectal cancer cells. Cell Commun Signal. 2008; 9:6:7.
23. Thiollier C, Lopez CK, Gerby B, Ignacimouttou C, Poglio S, Duffourd Y, Guégan J, Rivera-Munoz P, Bluteau O, Mabialah V, Diop M, Wen Q, Petit A, Bauchet AL, Reinhardt D, Bornhauser B et al. Characterization of novel genomic alterations and therapeutic approaches using acute megakaryoblastic leukemia xenograft models. J Exp Med. 2012; 209(11):2017-31.
24. Tschumper RC, Asmann YW, Hossain A, Huddleston PM, Wu X, Dispenzieri A, Eckloff BW, Jelinek DF. Comprehensive assessment of potential multiple myeloma immunoglobulin heavy chain V-D-J intraclonal variation using massively parallel pyrosequencing. Oncotarget. 2012;3(4):502-13.
25. Robins H. Detecting and monitoring lymphoma with high-throughput sequencing. Oncotarget. 2011;2(4):287-8.
26. Bou Samra E, Klein B, Commes T, Moreaux J. Development of gene expression-based risk score in cytogenetically normal acute myeloid leukemia patients. Oncotarget. 2012;3(8):824-32.
27. Martelli AM, Chiarini F, Evangelisti C, Cappellini A, Buontempo F, Bressanin D, Fini M, McCubrey JA. Two hits are better than one: targeting both phosphatidylinositol 3-kinase and mammalian target of rapamycin as a therapeutic strategy for acute leukemia treatment. Oncotarget. 2012;3(4):371-94.
28. Barakat MT, Humke EW, Scott MP. Learning from Jekyll to control Hyde: Hedgehog signaling in development and cancer. Trends Mol Med. 2010; 16(8):337-48.
29. Rubin LL, de Sauvage FJ. Targeting the Hedgehog pathway in cancer. Nat. Rev. Drug Discov. 2006; 5:1026–1033
30. Tee AR, Blenis J, Proud CG. Analysis of mTOR signaling by the small G-proteins, Rheb and RhebL1. FEBS Lett. 2005; 579(21):4763-8.
31. Baud V, Karin M. Is NF-kappaB a good target for cancer therapy? Hopes and pitfalls. Nat Rev Drug Discov. 2009; 8(1):33-40.
32. Kim HJ, Hawke N, Baldwin AS. NF-kappaB and IKK as therapeutic targets in cancer. Cell Death Differ. 2006; 13(5):738-47.
33. Karin M. Nuclear factor-kappaB in cancer development and progression. Nature. 2006; 441(7092):431-6.
34. Fan Y, Dutta J, Gupta N, Fan G, Gélinas C. Regulation of programmed cell death by NF-kappaB and its role in tumorigenesis and therapy. Adv Exp Med Biol. 2008; 615:223-50.
35. Chappell WH, Steelman LS, Long JM, Kempf RC, Abrams SL, Franklin RA, Bäsecke J, Stivala F, Donia M, Fagone P, Malaponte G, Mazzarino MC, Nicoletti F, Libra M, Maksimovic-Ivanic D, Mijatovic S, et al. Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR inhibitors: rationale and importance to inhibiting these pathways in human health. Oncotarget. 2011; 2(3):135-64.
36. Zhou H, Huang S. Role of mTOR signaling in tumor cell motility, invasion and metastasis. Curr Protein Pept Sci. 2011; 12(1):30-42.
37. Prakash T, Sharma VK, Adati N, Ozawa R, Kumar N, Nishida Y, Fujikake T, Takeda T, Taylor TD. Expression of conjoined genes: another mechanism for gene regulation in eukaryotes. PLoS One. 2010;5(10): e13284. doi: 10.1371/journal.pone.0013284.
38. Wang K, Ubriaco G, Sutherland LC. RBM6-RBM5 transcription-induced chimeras are differentially expressed in tumours. BMC Genomics. 2007; 8:348.
39. Kowalski PE, Freeman JD, Mager DL. Intergenic splicing between a HERV-H endogenous retrovirus and two adjacent human genes. Genomics. 1999; 57(3):371-9.
40. Kato M, Khan S, Gonzalez N, O’Neill BP, McDonald KJ, Cooper BJ, Angel NZ, Hart DN. Hodgkin’s lymphoma cell lines express a fusion protein encoded by intergenically spliced mRNA for the multilectin receptor DEC-205 (CD205) and a novel C-type lectin receptor DCL-1. J Biol Chem. 2003; 278(36):34035-41.
41. Zhang Y, Gong M, Yuan H, Park HG, Frierson HF, Li H. Chimeric transcript generated by cis-splicing of adjacent genes regulates prostate cancer cell proliferation. Cancer Discov. 2012; 2(7):598-607.
42. Kumar-Sinha C, Kalyana-Sundaram S, Chinnaiyan AM. SLC45A3-ELK4 chimera in prostate cancer: spotlight on cis-splicing. Cancer Discov. 2012; 2(7):582-5.
43. Pession A, Masetti R, Rizzari C, Putti MC, Casale F, Fagioli F, Luciani M, Lo Nigro L, Menna G, Micalizzi C, Santoro N, Testi AM, Zecca M, Biondi A, Pigazzi M, Rutella S, et al. Results of the AIEOP AML 2002/01 multicenter, prospective trial for treatment of children with acute myeloid leukemia. Blood. 2013; [Epub ahead of print]
44. Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009; 25(9):1105-11.
45. McPherson A, Hormozdiari F, Zayed A, Giuliany R, Ha G, Sun MG, Griffith M, Heravi Moussavi A, Senz J, Melnyk N, Pacheco M, Marra MA, Hirst M, Nielsen TO, Sahinalp SC, et al. deFuse: an algorithm for gene fusion discovery in tumor RNA-Seq data. PLoS Comput Biol. 2011; 7(5):e1001138.
46. Iyer MK, Chinnaiyan AM, Maher CA. ChimeraScan: a tool for identifying chimeric transcription in sequencing data. Bioinformatics. 2011; 27(20):2903-4.
47. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R and 1000 Genome Project Data Processing Subgroup. The Sequence alignment/map (SAM) format and SAMtools. Bioinformatics. 2009; 25:2078-9.
48. Robinson MD, Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biology. 2010; 11, R25.