
INTRODUCTION
Therapeutic monoclonal antibodies (mAb) such as rituximab and ofatumumab have demonstrated the clinical efficacy of targeting the B-cell restricted antigen CD20 for the treatment of B-cell lymphomas and leukemia. Although CD20 is also expressed on healthy B-cells which are depleted in the course of therapy, long-term B-cell aplasia is well manageable [1, 2]. However, refractory disease to CD20-targeted mAb treatment has been reported with various mechanisms of resistance: downregulation of CD20 expression [3–5], internalization of CD20:mAb complex [6], and inhibition of complement-dependent cytotoxicity (CDC) [7–10]. Therefore, additional therapeutic strategies are required.
T-cell receptor (TCR) gene transfer is an attractive strategy to equip T-cells with TCRs of defined antigen-specificity. Due to their high sensitivity for cognate antigen presented in human leukocyte antigen (HLA), TCRs can induce T-cell activation even when antigen abundance is very low [11–13]. Moreover, since HLA molecules sample the entire endogenous protein repertoire, also intracellular antigens can be presented on the cell surface and are accessible for recognition by TCRs. However, the broad application of TCR-based adoptive immunotherapy directed against self-antigens such as CD20 is hampered by lack of an effective immune response against CD20-derived peptides presented in the context of autologous (self) HLA molecules. T-cells carrying high-affinity TCRs reactive to such self-antigens are deleted by negative selection during thymic development to prevent auto-reactivity. An attractive strategy to target self-antigens is to exploit the immunogenicity of such antigens presented in the context of allogeneic (non-self) HLA. Presentation of self-antigens in the context of allogeneic HLA (alloHLA) can induce strong antigen-specific T-cell responses as observed in HLA-mismatched hematopoietic stem cell transplantation [14, 15]. We and others have developed several approaches to generate T-cell responses towards specific antigens from which individual T-cell clones and their TCRs can be isolated [16–20].
We hypothesized that high-affinity TCRs, targeting HLA-presented peptides derived from CD20, would be a valuable addition to current CD20-targeting therapies by providing means to target B-cell malignancies in which CD20 expression is insufficient for mAb-based approaches. To target CD20 in a TCR-based approach, we searched for high-affinity TCRs directed against the CD20-derived peptide SLFLGILSV (CD20SLF) that is endogenously processed and presented in the context of HLA-A*02:01 (HLA-A2) [21]. Furthermore, to broaden the applicability of a CD20-specific TCR-based approach, we searched for additional CD20-derived peptides in the HLA ligandome of B- lymphocytes [22], and were able to identify using a similar high-throughput screening method a high-avidity T-cell clone directed against the CD20-derived peptide RPKSNIVLL (CD20RPK) presented in HLA-B*07:02 (HLA-B7).
RESULTS
Identification of CD20-specific T-cell clones by high-throughput screening
We used peptide-MHC (pMHC)-tetramers composed of CD20-derived peptide SLFLGILSV (CD20SLF) bound to HLA-A2 for the isolation of T-cells reactive to CD20 from PBMCs of 6 healthy HLA-A2neg individuals. Starting with 250 to 1,000 × 106 PBMCs, cells binding to pMHC-tetramers were first enriched by magnetic-associated cell sorting (MACS). From the positive fractions, containing the pMHC-tetramer labelled cells, thousands of pMHC-tetramer+ CD8+ T-cells were single-cell sorted by fluorescent-activated cell sorting (FACS) and clonally expanded for two weeks. In total, 3,632 T-cell clones were isolated and expanded. The number of T-cell clones that could be isolated from an individual varied between 71 and 1,605 (Supplementary Table S1). In an initial high-throughput screen, all 3,632 clones were assessed for their specificity of CD20SLF peptide by stimulation with HLA-A2pos but CD20-negative K562 cells (K562-A2) that were either left untreated or pulsed with 50 nM CD20SLF. The secretion of both IFN-γ and GM-CSF was measured since a substantial number of pMHC-tetramerpos T-cell clones has poor intrinsic IFN-γ production [23]. Therefore, using both cytokines as readout parameters allows for the assessment of more T-cell clones than using IFN-γ alone. Three reactivity patterns could be observed based on the secretion of GM-CSF and IFN-γ. For reasons of brevity and the additional value of GM-CSF over IFN-γ, only data for GM-CSF is shown. More than 55% of T-cell clones did not produce any cytokine upon stimulation with either unloaded or peptide-pulsed K562-A2 cells as exemplified by clone 265 (Supplementary Figure S1A–S1B). These data indicated insufficient avidity to recognize HLA-bound CD20SLF. In contrast, many T-cell clones like 5F9 demonstrated reactivity towards HLA-A2pos K562-A2 cells irrespective of absence or presence of CD20SLF peptide, suggesting a lack of specificity for peptide CD20SLF. Around 38.4% of all tested T-cell clones showed such a reactivity profile in the initial high-throughput screen (Supplementary Figure S1A–S1B). However, 213 T-cell clones, accounting for 5.8% of all tested T-cell clones, were selected that demonstrated peptide specificity by recognizing peptide-pulsed K562-A2 more robustly than unloaded K562-A2 and binding to pMHC-tetramer CD20SLF:A2. Supplementary Table S1 summarizes the distributions of T-cell clones per reactivity profile for each of the 6 HLA-A2neg healthy individuals from which the T-cell clones were isolated.
Next, we continued only with the assessment of the 213 selected T-cell clones that demonstrated specificity for peptide CD20SLF in the initial screening. First, specific binding to pMHC-tetramer CD20SLF:A2 was validated for these T-cell clones. All 213 selected T-cell clones stained with pMHC-tetramer CD20SLF:A2 although the intensity of the staining was variable between clones (Figure 1A and Supplementary Figure S2). Staining with pMHC-tetramer CD20SLF:A2 was specific for all 213 T-cell clones since no binding to a control pMHC-tetramer composed of HLA-A2 and peptide NLVPMVATV derived from the human cytomegalovirus (CMV) protein pp65 (pp65NLV) was observed (Figure 1A and Supplementary Figure S2).
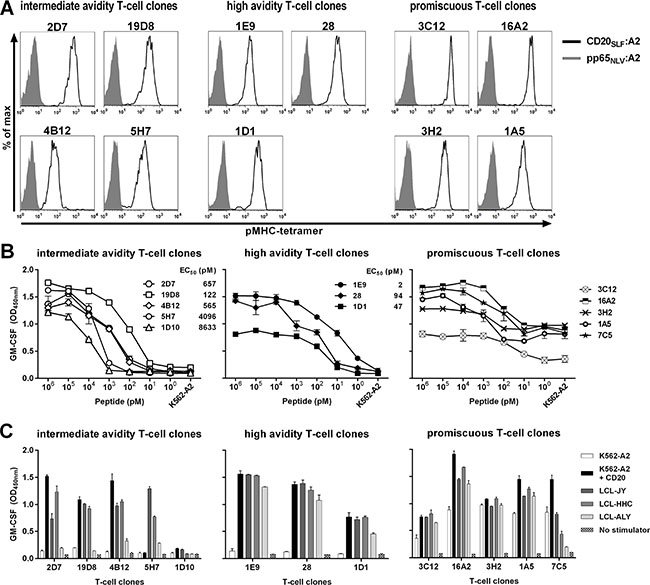
Figure 1: Identification of high-avidity peptide-specific T-cell clones from selected candidate T-cell clones. Shown are data of representative clones among T-cell clones that were selected following high-throughput screening. (A) Shown are histograms of representative T-cell clones that were stained either with pMHC-tetramer composed of CD20SLF:A2 (black line) or a control pMHC-tetramer composed of CMV-derived peptide pp65NLV bound to HLA-A2 (pp65NLV:A2, grey area). See Supplementary Figure S2 for additional T-cell clones and controls. (B) T-cell clones were cocultured with CD20neg K562-A2 cells pulsed with titrated amounts of peptide CD20SLF or left untreated (K562-A2). Values behind T-cell clones indicate peptide concentration at half maximum cytokine production (EC50). (C) T-cell clones were cocultured with K562-A2 cells or K562-A2 cell transduced to express CD20 (K562-A2 + CD20) or 3 HLA-A2pos B-LCLs that endogenously express CD20 (LCL-JY, LCL-HHC, and LCL-ALY). After 18 hours of coculture supernatant was harvested and GM-CSF production was assessed using standard ELISA. Shown are means and standard deviations of one representative experiment carried out in duplicate.
Next, we aimed to identify T-cell clones with high peptide sensitivity and sufficient avidity to recognize endogenously processed peptide. The selected clones demonstrated various degrees of peptide sensitivity when stimulated with titrated amounts of CD20SLF peptide loaded on K562-A2 cells. For many T-cell clones of intermediate avidity, micro- to nanomolar amounts of exogenously loaded CD20SLF were required to induce cytokine production (Figure 1B left). This absence of high peptide sensitivity also translated to poor recognition of endogenously processed peptide, measured by recognition of three HLA-A2pos CD20-expressing Epstein-Barr virus (EBV)-transformed B-lymphoblastic cell-lines (B-LCLs) or K562-A2 cells stably transduced to express CD20 (Figure 1C left). Intermediate avidity T-cell clones either failed to recognize any or reacted only towards a subset of CD20-expressing stimulators. On the other hand, high avidity T-cell clones such as 1E9, 28, and 1D1 demonstrated peptide sensitivities ranging from the nano- to picomolar range (Figure 1B middle). Higher sensitivity to exogenously loaded peptide correlated with the capacity to recognize endogenously processed peptide. High-avidity clones 1E9, 28 and 1D1 readily recognized all three B-LCLs and CD20-transduced K562-A2 cells (Figure 1C middle).
In contrast, many of the T-cell clones lacked exclusive specificity for peptide CD20SLF. This subset of promiscuous T-cell clones recognized peptide-loaded K562-A2 in a peptide concentration-dependent manner (Figure 1B right). However, cytokine production could also be observed towards unloaded K562-A2, albeit to a lesser extent (Figure 1B right and 1C right). These data most likely indicated that besides peptide CD20SLF also other unknown peptides could be recognized in the context of HLA-A2. Similar data was obtained measuring IFN-γ secretion in all experiments (data not shown).
In conclusion, we isolated T-cell clones specific for peptide CD20SLF presented in HLA-A2 from the allorepertoire of HLA-A2neg individuals. Only the most sensitive clones demonstrated sufficient avidity to robustly recognize endogenously processed peptide on all CD20-expressing stimulator cells tested.
Robust recognition of HLA-A2pos ALL cell-lines by high-avidity T-cell clones
Next, we examined the recognition of HLA-A2pos ALL cell-lines by a subset of CD20-specific T-cell clones. High-avidity T-cell clones 1E9 and 28 demonstrated robust recognition of 3 ALL cell-lines ALL-CM, ALL-BV and ALL-RL (Figure 2). Also clone 1D1 recognized all 3 ALL cell-lines, although to a lesser extent. In contrast, intermediate avidity T-cell clones 2D7, 19D8 and 4B12 only weakly recognized or failed to show any reactivity towards the ALL cell-lines. The degree of reactivity closely matched the observed peptide sensitivities. HLA-A2pos but CD20-negative ALL cell-line ALL-GD could not be recognized by any clone.
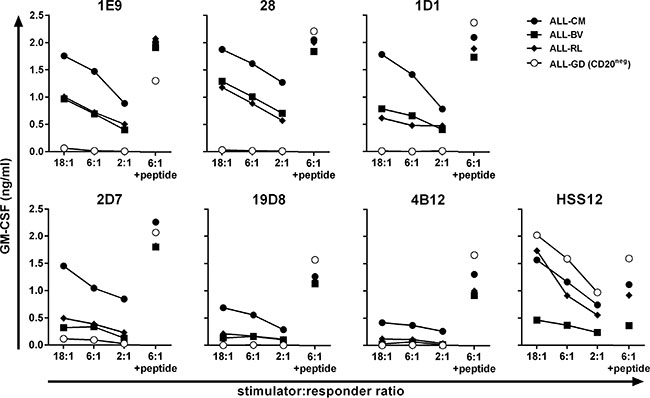
Figure 2: Recognition of malignant cell samples by high-avidity T-cell clones. T-cell clones were cocultured with HLA-A2pos ALL cell-lines at indicated stimulator-to-responder ratios. Stimulator cell-line ALL-GD did not express CD20 as measured by RT-qPCR (Figure 3C). Controls included stimulators pulsed with 50 nM peptide CD20SLF (+ peptide) and T-cell clone HSS12 recognizing peptide FTWEGLYNV from the ubiquitously expressed gene USP11 in the context of HLA-A2 to confirm HLA-A2 expression on the ALL cell-lines. Cytokine production was assessed after 18 hours of coincubation. Representative data from one of three independent experiments.
In summary, T-cell clones demonstrating highest peptide sensitivity also more robustly recognized HLA-A2pos CD20-expressing ALL cell-lines whereas intermediate avidity clones failed to consistently react to these stimulators.
Efficient recognition of CD20low B-cell malignancies
To investigate the clinical potential of high-avidity T-cell clones 1E9 and 28, we assessed their capacity to recognize primary HLA-A2pos B-cell malignancies including chronic lymphocytic leukemia (CLL), acute lymphoblastic leukemia (ALL) and mantle cell lymphoma (MCL). Furthermore, to assess the sensitivity of our T-cell clones, we included primary malignant cell samples with varying degrees of CD20 mRNA expression in our analysis, including CD20low samples with more than 33-fold reduced CD20 mRNA expression compared to healthy B cells. Both clones efficiently recognized all 5 primary CLL samples (Figure 3A and Supplementary Figure S3A). Recognition of all CLL samples was comparable although expression of CD20 mRNA varied greatly between samples (Supplementary Figure S4). Primary CLL sample ACN had a 6-fold reduced CD20 mRNA expression compared to healthy B-cells. In contrast, CLL sample JGN had a 100-fold reduction in CD20 expression but was still sufficiently recognized. Furthermore, 3 out of 4 primary ALL samples were readily recognized by both T-cell clones. Real-time quantitative PCR (RT-qPCR) revealed lack of CD20 mRNA expression in the unrecognized primary ALL sample AGP (Supplementary Figure S4). Strong recognition of CD20low expressing ALL sample MMX was observed, which had a 40-fold reduction in CD20 expression compared to healthy B-cells. Additionally, both T-cell clones readily recognized all 5 tested primary MCL samples (Figure 3B and Supplementary Figure S3B). Of note, clones 1D1, 2D7, 19D8 and 4B12 having demonstrated lower sensitivities for peptide CD20SLF also recognized primary B-cell malignancies, however, to a lower degree than clones 1E9 and 28 (Supplementary Figure S5). High reactivity towards 3 HLA-A2pos ALL cell-lines was observed for clone 1E9 and 28 (Figure 3B and Supplementary Figure S3B). CD20 mRNA expression varied in these cell-lines by orders of magnitude which directly translated to lower CD20 cell surface expression in CD20low expressing cells (Figure 3C). Although CD20 cell surface expression was drastically reduced, clone 1E9 still efficiently lysed CD20low ALL-RL (Figure 3D). In contrast, the CD20-targeting monoclonal antibody ofatumumab did not induce lysis of ALL-RL by complement dependent cytotoxicity (CDC), whereas ALL-CM and ALL-BV were lysed (Figure 3D). Ofatumumab was used over rituximab due its increased induction of CDC [24]. HLA-A2pos ALL-GD, which lacks CD20 mRNA and cell surface expression, could not be lysed by either T-cell clone 1E9 or CDC mediated by mAb ofatumumab.
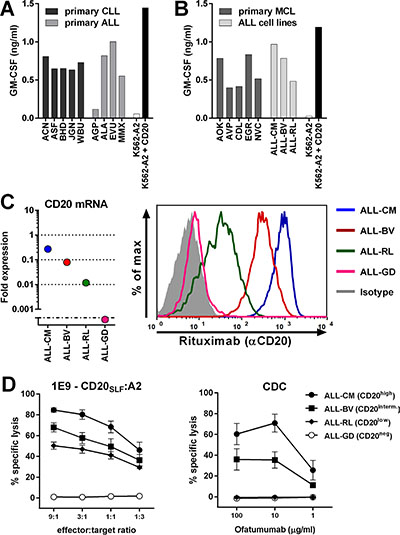
Figure 3: CD20-reactive T-cell clone 1E9 efficiently lyses CD20low B-cell malignancies unsusceptible to CD20-targeting antibodies. T-cell clone 1E9 having shown highest sensitivity for peptide CD20SLF was tested for its recognition of various B-cell malignancies. (A–B) T-cell clone 1E9 was cocultured with HLA-A2pos primary B-cell malignancies or ALL cell-lines. Primary samples included chronic lymphocytic leukemia (CLL) and acute lymphoblastic leukemia (ALL) (A) or primary mantle cell lymphoma (MCL) (B). Primary ALL sample AGP did not demonstrate CD20 expression at the mRNA level (Supplementary Figure S4). Controls included CD20neg K562-A2 and CD20-transduced K562-A2 (K562-A2 + CD20). Supernatant was harvested after 18 hours of coincubation and cytokine production was assessed by standard ELISA. Experiments were carried out in duplicate. One representative experiment of two independent experiments is shown. (C) On the left, expressions of CD20 mRNA in ALL cell-lines was measured by RT-qPCR and is shown as fold expression of average expression in healthy B-cells which was set to 1. The detection limit of the assay was > 0.001 fold. On the right, cell surface CD20 on ALL cell-lines was assessed by FITC-conjugated CD20-specific monoclonal antibody rituximab. Controls included staining with an isotype-matched antibody. (D) Cytotoxicity of clone 1E9 (left panel) or complement dependent cytotoxicity (CDC, right panel) was measured by standard Cr51-release assay. Cr51-labelled targets were incubated with clone 1E9 at different effector-to-target ratios or different concentrations of CD20-targeting monoclonal antibody ofatumumab in the presence of human serum for 5 hours. Targets include HLA-A2pos ALL cell-lines ALL-CM, ALL-BV, ALL-RL and ALL-GD. Controls included incubation with T-cell clone HSS12, recognizing peptide FTWEGLYNV from the ubiquitously expressed gene USP11 and a CMV-reactive T-cell clone CTN (Supplementary Figure S6). Shown are means with standard deviations of one experiment performed in triplicate. CDC shows pooled data using human serum from three different individuals.
These data demonstrated that CD20-reactive T-cell clones 1E9 and 28 potently recognized primary HLA-A2pos B-cell malignancies including CLL, ALL and MCL. Additionally, primary cell samples and cell-lines showing hundred-fold reduced CD20 expression and reduced CD20 cell surface expression could still be efficiently lysed by T-cell clone 1E9 whereas CDC after incubation with CD20-targeting antibodies was absent.
Reactivity of CD20-reactive T-cell clones is restricted to the B-cell compartment
Having established potent reactivity towards B-cell malignancies, we next investigated the safety profile of clones 1E9 and 28 by stimulating them with a panel of HLA-A2pos hematopoietic and nonhematopoietic cell subsets. Clones 1E9 and 28 did not recognize CD20neg primary T-cells, CD14+ monocytes or CD34+ hematopoietic progenitor cells from 2 different individuals (Figure 4A and Supplementary Figure S3C). Moreover, no reactivity against activated T-cells or monocyte-derived immature and mature dendritic cells (DCs) from 2 individuals was observed (Figure 4B and Supplementary Figure S3D). However, CD20-expressing primary and CD40L-stimulated B-cells from both individuals were readily recognized. No reactivity towards 3 HLA-A2pos fibroblasts was observed, even when fibroblasts were pre-treated with 200 IU/ml IFN-γ to simulate inflammation (Figure 4C and Supplementary Figure S3E).
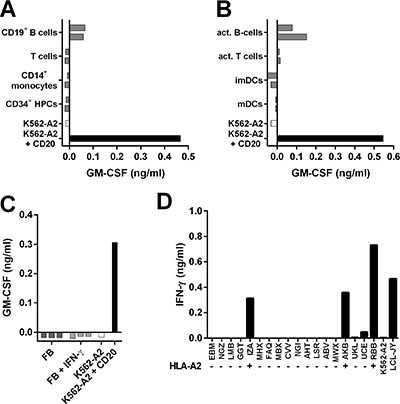
Figure 4: T-cell clone 1E9 demonstrates B-cell-restricted reactivity profile. (A–C) T-cell clone 1E9 was cocultured with different cell subsets of hematopoietic (A–B) or nonhematopoietic (C) origin derived from HLA-A2pos healthy individuals. Clone 1E9 was coincubated with primary (A) or activated (B) hematopoietic cell subsets from two different donors. Activated B-cells were generated by stimulating primary B-cell with CD40 ligand, activated T-cells were generated by stimulation with PHA. Immature and mature dendritic cells (imDCs and mDCs, respectively) were monocyte derived. Clone 1E9 was coincubated with fibroblasts (FB) that had been cultured either in the absence or presence of 200 IU/ml IFN-γ (+IFN-γ) for 4 days prior to the experiment (C). (D) Clone 1E9 was coincubated with a panel of B-LCLs expressing different HLA class I and class II alleles. HLA status regarding presence (+) or absence (−) of HLA-A*0201 is indicated. For a complete list of HLA genotype of all B-LCL see Supplementary Table S1. After 18 hours of coincubation, supernatant was harvested and cytokine production was assessed using standard ELISA. Shown are representative results of two independent experiments.
To test whether clones 1E9 and 28 would be cross-reactive with HLA molecules other than HLA-A2, both clones were stimulated with a panel of B-LCLs expressing 95% of common and rare HLA class I alleles (Supplementary Table S2) [25]. Both clones reacted only towards HLA-A2pos B-LCLs (Figure 4D and Supplementary Figure S3F) indicating no cross-reactivity with other tested HLA class I molecules.
In summary, both T-cell clones followed a CD20-restricted recognition profile correlating with CD20 expression. Reactivity was confined to the B-cell compartment and absent for other hematopoietic and nonhematopoietic cell subsets. No cross-reactivity with other HLA class I alleles was observed for either clone.
TCR gene transfer installs potent CD20-specific reactivity onto recipient T-cells
To test whether CD20-reactivity could be installed onto recipient T-cells by TCR gene transfer, we sequenced and cloned the TCR of clone 1E9 (TCR-1E9). We chose clone 1E9 because of its highest peptide sensitivity and consistent recognition of B-cell malignancies. The V(D) J segments of the TCRα and TCRβ chain were fused to murine TCR α and β constant domains on a modified MP71-TCR-flex retroviral backbone in order to induce preferential pairing and high expression of the introduced TCR chains [26]. The murine constant domain allowed for the isolation of TCR-transduced T-cells by staining with a murine TCR specifc antibody followed by isolation using MACS (Figure 5A). Transduced CD8+ T-cells of HLA-A2pos healthy individuals expressed TCR-1E9 on the cell surface and stained highly with pMHC-tetramer CD20SLF:A2 whereas mock-transduced T-cells did not bind to pMHC-tetramer. TCR-transduced but not mock-transduced T-cells were able to efficiently recognize CD20-expressing HLA-A2pos target cells (Figure 5B). TCR-modified CD8+ T-cells readily recognized primary CLL and MCL samples and autologous activated B-cells, whereas autologous activated T-cells were not recognized. Furthermore, we demonstrate in Figure 5C that CD20-TCR-transduced T-cells efficiently lysed ALL cell-lines, including ALL-RL which demonstrated reduced CD20-expression and insufficient cell surface CD20 to be targeted by monoclonal antibodies. Additionally, TCR-modified but not mock-transduced T-cells efficiently lysed primary ALL samples at very low effector-to-target ratios. Activated B-cells from an autologous source were also lysed by TCR-modified T-cells at levels comparable to parental clone 1E9 (Figure 5D). No lysis of neither primary nor activated autologous T-cells was observed, demonstrating the CD20-specificity and a safe reactivity profile of CD20-TCR-modified T-cells.
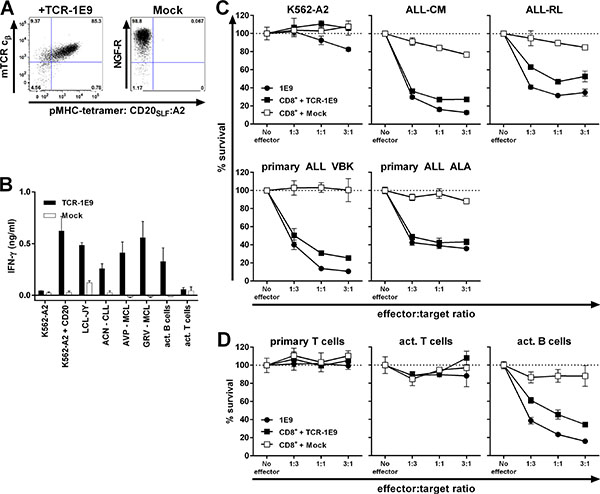
Figure 5: TCR gene transfer installs potent CD20-specific reactivity onto recipient CD8+ T-cells. CD8+ T-cells from an HLA-A2pos healthy individual were retrovirally transduced to express TCR of clone 1E9 (TCR-1E9). V(D)J segments of the TCRα and β chains were fused to the murine constant TCR domains. T-cells transduced with an empty vector (Mock) containing marker gene NGF-R served as control. (A) FACS plots of TCR or mock-transduced CD8+ T-cells following enrichment of transduced cells. TCR-transduced cells were enriched and stained by an antibody binding to the murine constant domain of the TCR β chain (mTCR cβ) of the introduced TCR. Mock-transduced cells were isolated and stained with an antibody specific for marker gene NGF-R. All populations were incubated with pMHC-tetramer CD20SLF:A2. Numbers in corners indicate percentage cells in each quadrant. Dot plots are shown with bi-exponential axes. (B) TCR or mock-transduced T-cells were cocultured with different HLA-A2pos stimulator cells. Primary cell samples include chronic lymphocytic leukemia (ACN) and mantle cell lymphomas (AVP and GRV). Activated B and T-cells were of autologous origin. Controls included CD20neg K562-A2, CD20-transduced K562-A2 (K562-A2 + CD20) and CD20-expressing LCL-JY. IFN-γ production was assessed 18 hours after coculture. Experiment was performed in duplicate. Shown are means with standard deviations. (C–D) Survival of PKH-labelled targets cells was assessed after coculture with either T-cell clone 1E9 or TCR or mock-transduced T-cells at different effector-to-target ratios. Following 18 hours of coincubation, surviving targets cells were counted using FACS analysis and percent surviving cells was calculated. Malignant cell samples include ALL cell-lines ALL-CM and ALL-RL, and primary ALL samples VBK and ALA (C) Primary and activated T-cells and activated B-cells were of same origin as transduced CD8+ T-cells (D). Shown are means with standard deviations of one experiment performed in triplicate.
In conclusion, potent CD20-reactivity and specificity could be installed by TCR gene transfer. TCR-transduced T-cells readily recognized and lysed CD20-expressing B-cell malignancies, including samples with low CD20 expression. No off-target toxicity of CD20-negative targets was observed.
Measured avidity for T-cell clones depends on expression of high-affinity TCR
Before moving to the TCR gene transfer stage, candidate TCRs were primarily selected based on functional data obtained in experiments using the original T-cell clones. Therefore, quality of a TCR was assessed through functional avidity of that particular clone originating from either the naïve or memory compartment. To test whether intrinsic features of T-cell clones could have influenced the selection, we also cloned three additional TCRs for TCR gene transfer. Besides TCR-1E9, we selected the TCRs from clones 1D1, 2D7 and 4B12, since these clones demonstrated varying avidities. All four TCRs were efficiently expressed after transduction into CMV-specific T-cells and could be enriched to great purity (Figure 6A). Sensitivity of the TCR-transduced T-cells for titrated amounts of peptide CD20SLF depended on the introduced TCR (Figure 6B–6C). T-cell transduced with TCR-1E9 were most sensitive. A gradual decrease in sensitivity was observed for T-cells transduced with TCR-1D1, TCR-2D7 and TCR-4B12. Peptide sensitivities of transduced T-cells closely mirrored peptide sensitivities observed for the parental T-cell clones.
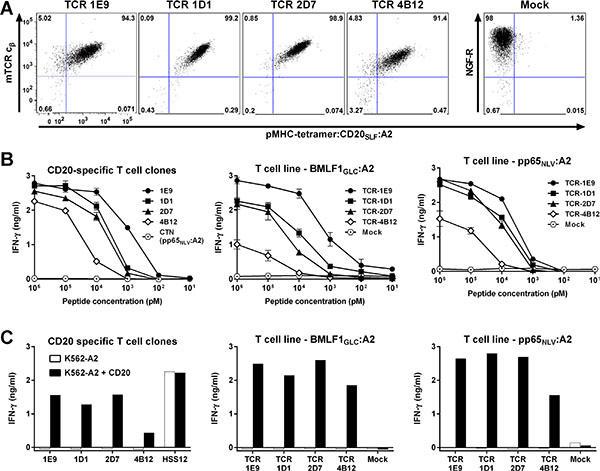
Figure 6: Functional avidity measured of T-cell clones depends on affinity of TCR. Two virus-specific T-cell lines (BMLF1GLC:A2 or pp65NLV:A2) were retrovirally transduced to express the TCRs of clone 1E9, 1D1, 2D7 or 4B12 (TCR-1E9, TCR-1D1, TCR-2D7 or TCR-4B12, respectively). Transduction with an empty backbone (Mock) containing only the marker gene NGF-R served as control. (A) Shown are FACS plots of enriched TCR-transduced T-cells stained with an antibody specific for the murine constant domain of the TCR β chain (mTCR cβ) of the introduced TCR. Cells were also stained with pMHC-tetramer CD20SLF:A2. Mock-transduced T-cells were stained with an antibody specific for marker gene NGF-R. Dot plots are shown with bi-exponential axes. (B) Original T-cell clones (left) or two virus-specific T-cell lines (middle and right) transduced with the different TCR constructs were coincubated with K562-A2 cells pulsed with titrated amounts of peptide CD20SLF. Controls included T-cell clone CTN specific for CMV-derived peptide pp65NLV. Shown are means with standard deviations of one experiment performed in duplicate. (C) Original T-cell clones (left) or two virus-specific T-cell lines (middle and right) transduced with the different TCR constructs were coincubated with K562-A2 cells or CD20-transduced K562-A2 (K562-A2 + CD20). USP11FTW-specific T-cell clone HSS12 served as control. IFN-γ production was assessed after 18 hours of coincubation. Shown are representative results of one of two independent experiments.
These data indicated that screening for candidate TCRs is feasible on clonal populations of T-cells. Observed functional avidities of T-cell clones reflected the affinity of the TCRs and were less likely to be influenced by intrinsic features of each individual clone.
Building a TCR library targeting various CD20-derived peptides and HLA-alleles
An off-the-shelf TCR library targeting different CD20-derived peptides presented in various HLA alleles could broaden the applicability of CD20-specific TCRs. Therefore, we searched for other peptides derived from CD20 in our recently published data describing the HLA ligandome of B-lymphocytes [22]. This study investigated the peptidome of B-lymphocytes by eluting HLA-bound peptides from B-LCLs, and identifying and characterizing these peptides using tandem mass spectrometry. Matching tandem mass spectra of eluted and newly synthesized peptide verified correct identification of the HLA-B7 presented CD20-derived peptide RPKSNIVLL (CD20RPK; data not shown). Using the described high-throughput methodology, three CD20RPK-reactive T-cell clones were isolated from two HLA-B7neg healthy individuals. From these three T-cell clones, clone 3D12 exhibited highest peptide-specific reactivity and strong recognition of three CD20-expressing HLA-B7pos B-LCLs and K562-B7 cells transduced with CD20 (Figure 7A–7B). Furthermore, Clone 3D12 efficiently recognized primary HLA-B7pos B-cell malignancies including ALL, CLL, and MCL despite different CD20 expression levels (Figure 7C and Supplementary Figure S4). Recognition of 5 CLL samples was comparable although CD20 mRNA expression was drastically reduced in CLL sample JGN by more than 100-fold compared to healthy B-cells. Similarly, clone 3D12 highly recognized ALL sample MMX although CD20 mRNA expression was reduced 40-fold compared to healthy B-cells. Recognition of 3 ALL cell-lines was robust (Figure 7D). Clone 3D12 efficiently recognized ALL-KR which had 100-fold reduced CD20 mRNA expression and almost entirely lacked cell surface CD20 (Figure 7E–7F). No reactivity towards healthy hematopoietic or nonhematopoietic cell subsets was observed (Figure 7G–7H).
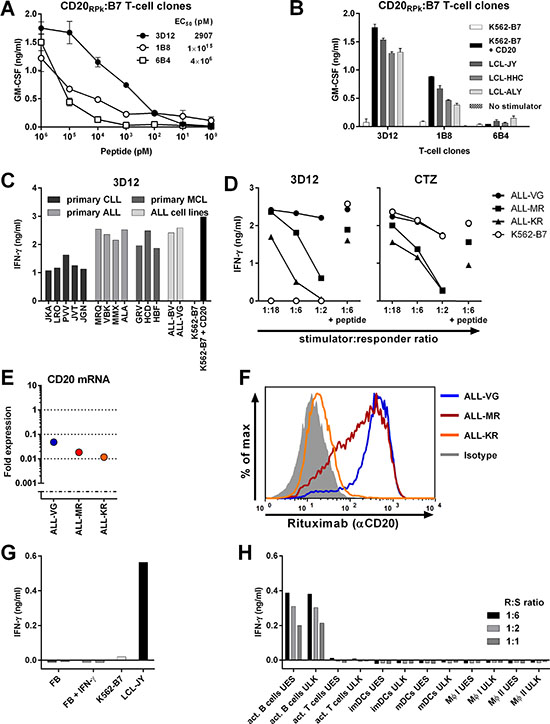
Figure 7: Identification of T-cell clone 3D12 recognizing CD20-derived peptide RPKSNIVLL in HLA-B7. T-cell clones 3D12, 1B8, and 6B4 were isolated from two healthy HLA-B7neg individuals using pMHC-tetramers composed of CD20-derived peptide RPKSNIVLL (CD20RPK) bound to HLA-B7. (A) Clones 3D12, 1B8, and 6B4 were coincubated with CD20-negative K562 cells expressing HLA-B7 (K562-B7) that were exogenously loaded with titrated amounts of peptide CD20RPK. (B) Clones 3D12, 1B8, and 6B4 were cocultured with K562-B7 cells, K562-B7 cells transduced to express CD20 (+ CD20) or three naturally CD20-expressing HLA-B7pos EBV-LCLs. Shown are means and standard deviations of one representative experiment carried out in duplicate. (C) Clone 3D12 was cocultured with primary HLA-B7pos B-cell malignancies and ALL cell lines. Malignant cell samples include chronic lymphocytic leukemia (CLL), acute lymphoblastic leukemia (ALL) and mantle cell lymphoma. (D) Clone 3D12 was cocultured with 3 HLA-B7pos ALL cell lines at indicated responder-to-stimulator ratios. Peptide-pulsed stimulators (1:6 + peptide) served as positive control. T-cell clone CTZ recognizing a ubiquitously expressed antigen in the context of HLA-B7 served as positive control. (E–F). Expressions of CD20 mRNA in ALL cell-lines was measured by RT-qPCR and is shown as fold expression of average expression in healthy B-cells which was set to 1. The detection limit of the assay was > 0.001 fold (E). Cell surface CD20 on ALL cell-lines was assessed by FITC-conjugated CD20-specific monoclonal antibody rituximab (F). Controls included staining with an isotype-matched antibody (gray area). (G–H) Clone 3D12 was cocultured with nonhematopoietic and hematopoietic cell subsets. Fibroblast (FB) of two HLA-B7pos individuals were cultured in the absence or presence of IFN-γ (+ IFN-γ) for 4 days prior to coculture with clone 3D12 (G). Clone 3D12 was cocultured with activated hematopoietic cell subsets of two healthy donors at different responder-to-stimulator (R:S) ratios (H).The stimulatory capacity of stimulator cells was verified by a control clone. Representative data from one of two independent experiments. GM-CSF and IFN-γ secretion was assessed after 18 hours of coculture by standard ELISA. MΦ I and II, Macrophage type I and II, respectively.
These data demonstrated that our high-throughput approach can easily be employed to target additional peptides in different HLA alleles, and that HLA-B7-restricted CD20RPK-specific T-cells can readily recognize CD20low B-cell malignancies.
DISCUSSION
Therapeutic monoclonal antibodies (mAb) targeting the cell surface antigen CD20 have been successfully applied in the clinic. However, relapsed or refractory disease as a result of CD20 cell surface downregulation, internalization of the CD20:mAb complex and other mechanisms has been reported [3–10, 27–29]. Therefore, additional strategies to overcome these mechanisms of resistance are required. Here, we describe the isolation of high-affinity TCRs directed against CD20-derived peptides presented in the context of HLA class I.
To overcome tolerance for self-antigens such as CD20, T-cell clones were isolated from HLA-A2neg or HLA-B7neg individuals, thus exploiting the immunogenicity of allogeneic (non-self) HLA molecules to target self-antigens. Among the T-cell clones isolated using pMHC-tetramers, only a small fraction demonstrated sufficient peptide sensitivity and strong antigen-specificity. These results reflect our previous experiences when employing pMHC-tetramers to isolate T-cell clones [20, 30, 31].
T-cell clones were selected by binding to pMHC-tetramers and subsequently tested for their recognition of peptide-pulsed K562 cells. Although T-cell clones demonstrated CD20SLF-dependent recognition, many T-cell clones reacted to unloaded K562-A2 indicating recognition of other peptides presented in HLA-A2. Of note, reactivity to unloaded CD20-negative K562-A2 cells was an indication for harmful off-target toxicity which could be observed for promiscuous clone 1A5 when stimulated with fibroblasts (Supplementary Figure S7). Peptide promiscuity, i.e. reactivity of one TCR with several peptides, is not uncommon and has been described for several highly peptide-specific TCRs [32–34]. Furthermore, peptide promiscuity can span tens to thousands of different peptides for a given TCR, albeit with strongly varying degrees of peptide sensitivity for each peptide [33]. The ability of TCRs to interact with multiple peptide-HLA molecules is likely to stem from the necessity of the immune systems to cover all possible antigens with a relatively limited TCR repertoire within a single individual [35].
The T-cell clones were isolated from PBMCs of healthy individuals and could therefore stem from the naïve or differentiated effector and memory T-cell pool. To test whether differentiation or activation status of a clone could misguide the selection of candidate TCRs, the TCRs of different CD20-specific clones with varying peptide sensitivities were expressed on the same cellular background in virus-specific T-cells. TCR-transduced T-cells responded more sensitively towards exogenously loaded peptide if the introduced TCR was derived from a T-cell clone also requiring only low amounts of CD20 peptide, indicating that functional data obtained from each T-cell clone is a valid representation of the expressed TCR rather than intrinsic features of the T-cell clone.
T-cell clone 1E9 was selected because of highest peptide-sensitivity and stringent peptide-specificity. Efficient reactivity towards primary B-cell malignancies such as ALL, CLL and MCL was observed. Transfer of TCR-1E9 installed potent CD20-reactivity onto recipient T-cells and led to the lysis of primary B-cell leukemia. Moreover, lysis was also observed for ALL cell-lines that lacked sufficient cell surface CD20 to be susceptible to mAb-mediated CDC. Similarly, HLA-B7-restricted CD20-specific T-cell clone 3D12 recognized ALL-KR lacking almost entirely cell surface CD20. These data indicate a complementary role for CD20-specific TCRs in the treatment of B-cell malignancies. CD20-targeting mAbs are applicable irrespective of HLA genotype but are limited by the degree of cell surface CD20 and presence of secondary cytotoxic mediators such as CDC or immune cells. In contrast, our CD20-specific TCRs showed improved antigen-sensitivity and could be used to administer potent cytotoxic T-cells but are restricted to TCR gene therapy of HLA-A2pos or HLA-B7pos individuals. It could be argued that T-cells expressing CD20-specific chimeric antigen receptors (CARs) will be able to also target malignancies expressing low amounts of extracellular CD20. However, in a patient suffering from primary cutaneous marginal zone lymphoma with spleen involvement only a weak response of the splenomegaly was reported after treatment with CD20-specific CAR-engineered T-cells [36]. It was argued by the authors that very low CD20 expression on malignant cells as has been reported for some indolent splenic lymphoma patients [37] could have caused this weak response among other factors.
Furthermore, TCR-modified T-cells highly sensitive to CD20-derived peptides may prove especially useful in the treatment of CLL. Malignant cells of CLL patients generally express low levels of CD20. In addition, patients suffering from CLL frequently demonstrate complement deficiencies limiting the effectiveness of treatment with monoclonal antibodies [10]. Although CD19-targeting CAR-modified T-cells have been very effective in the treatment of ALL, they have been less successfully applied in the treatment of CLL [38]. Moreover, the emergence of CD19-antigen loss variants after CAR administration urges the development of additional strategies [39]. Therefore, CD20-specific TCRs open an additional avenue to target multiple antigens simultaneously to decrease the risk of tumor-escape variants.
No recognition of a panel of CD20-negative but HLA-A2pos hematopoietic and nonhematopoietic cells was observed indicating a safe reactivity profile of our candidate T-cell clones 1E9 and 3D12. Nonetheless, potential reactivity towards untested cell subsets could exist. Furthermore, the higher sensitivity for CD20 of our TCR-1E9 and clone 3D12 could also increase the risk for on-target off-tumor toxicity that has not been observed for CD20-targeting mAbs. Therefore, to guard against adverse events that may occur in patients due to off-target or on-target off-tumor toxicities, TCR-modified T-cells should be additionally engineered with a suicide switch [40–47].
We demonstrate that a TCR library can be readily built using this approach as demonstrated by the isolation of T-cell clone 3D12 targeting an additional CD20-derived peptide in HLA-B7. This clone also highly recognized CD20low B-cell malignancies in the absence of reactivity towards healthy hematopoietic and nonhematopoietic cell subsets. An off-the-shelf library of various high-affinity CD20-specific TCRs targeting various HLA alleles could significantly broaden the applicability of immunotherapy to a wider patient group.
In summary, we demonstrate that high-affinity CD20-specific TCRs raised from the allorepertoire can be a valuable addition to current treatment options of patients suffering from CD20low B-cell malignancies by administering TCR-engineered T-cells with potent effector function.
MATERIALS AND METHODS
Culture conditions and cells
Peripheral blood was obtained from different individuals after informed consent. Peripheral blood mononuclear cells (PBMCs) were isolated using Ficoll-gradient centrifugation and were cryopreserved. T-cells were cultured in T-cell medium consisting of IMDM (Lonza, Basel, Switzerland) supplemented with 100 IU/ml IL-2 (Proleukine; Novartis Pharma, Arnhem, The Netherlands), 5% fetal bovine serum (FBS; Gibco, Life Technologies, Carlsbad, CA) and 5% human serum. Primary hematopoietic cell subsets were obtained from cryopreserved PBMCs of HLA-A*02:01pos healthy donors that were incubated with either anti-CD4, anti-CD14, anti-CD19 or anti-CD34 magnetic microbeads (Miltenyi Biotec, Bergisch Gladbach, Germany) for 15 min at 4°C. Microbead-labeled cells were isolated on LS column (Miltenyi Biotec) according to manufacturer’s protocol. Purity of isolated cells was assessed using FACS analysis and cells were only used in experiments if purity exceeded 95%. Immature and mature dendritic cells (DCs) were differentiated in vitro from isolated CD14+ cell populations as previously described [15]. Briefly, on day zero 1 × 106 cells/ml were seeded in IMDM supplemented with, 100 ng/ml GM-CSF (Sandoz Novartis Pharma, Almere, The Netherlands), 500 IU/ml IL-4 (Schering-Plough, Kenilworth, NJ), and 10% human serum, and cultured for two days to obtain immature DCs. Mature DCs were generated by culturing immature DCs in IMDM supplemented with 100 ng/ml GM-CSF, 10 ng/ml TNFalpha (CellGenix, Freiburg, Germany), 10 ng/ml IL-1b (Bioscource Invitrogen, Camarillo, CA), 10 ng/ml IL-6 (Sandoz Novartis Pharma), 1 μg/ml PGE-2 (Sigma Aldrich, St. Louis, MO), 500 IU/ml INF-γ (Boehringer Ingelheim, Ingelheim am Rhein, Germany), and 10% human serum for an additional two days. Macrophages type I and II were in vitro differentiated from CD14+ monocytes. CD14+ monocytes were cultured for 8 days in IMDM containing 10% human serum in the presence of 50 ng/ml GM-CSF or 5 IU/ml CSF-1 (R&D Systems, Minneapolis, MN) to obtain Macrophages type I or II, respectively. Activated T-cells were generated by stimulating CD4+ and CD8+ T-cells with irradiated (35 Gy) feeders in a 1:5 ratio in T-cell medium supplemented with 0.8 μg/ml phytohemagglutinin (PHA; Biochrom AG, Berlin, Germany) for 10 days prior to experiment. Activated CD19+ B-cells were generated by coculturing CD19+ cells on CD40L-transduced irradiated (70 Gy) mouse-fibroblasts for 7 days in IMDM supplemented with 2 ng/ml IL-4 and 10% human serum. K562 cells expressing HLA-A2 (K562-A2) or HLA-B7 (K562-B7) were previously described [20, 48]. Acute lymphoblastic leukemia (ALL) cell-lines were derived from primary ALL cells and were previously described [49]. Fibroblasts were cultured from skin biopsies in Dulbecco’s modified Eagle medium (DMEM; Lonza) containing 1g/l glucose and supplemented with 10% FBS as previously described [15]. Fibroblasts treated with IFN-γ were cultured in medium containing 200 IU/ml IFN-γ for four days prior to experiment. All cells were washed twice before use in experiments.
Generation of peptide-MHC complexes
All peptides were synthesized in-house using standard Fmoc chemisty. Recombinant HLA-A2 or HLA-B7 heavy chain and human β2m light chain were in-house produced in Escherichia coli. Major histocompatibility complex (MHC) class I refolding was performed as previously described with minor modifications [50]. Peptide-MHC (pMHC) class I complexes were purified by gel-filtration using HPLC. pMHC-tetramers were generated by labeling biotinylated pMHC-monomers with streptavidine-coupled phycoerythrin (PE; Invitrogen, Carlsbad, CA) or allophycocyanin (APC, Invitrogen). Complexes were stored at 4°C.
Isolation of CD20-reactive T-cell clones
T-cells binding to CD20-specific pMHC-tetramers composed of peptide CD20SLF bound to HLA-A2 or peptide CD20RPK bound to HLA-B7 were isolated from 250 to 1000 × 106 PBMCs of healthy HLA-A2neg or HLA-B7neg individuals, respectively. PBMCs were incubated with PE-labeled pMHC-tetramers for 1 h at 4°C, washed twice, and incubated with anti-PE-microbeads (Miltenyi Biotec) for 15 min at 4°C. PE-labeled cells were isolated on an LS colomn (Miltenyi Biotec) according to manufacturer’s instruction. Positively selected cells were stained with an Alexa700-labelled antibody against CD8 (Invitrogen/Caltag, Buckingham, United Kingdom) in combination with FITC-conjugated antibodies against CD4, CD14, and CD19 (BD Pharmingen, San Jose, CA). pMHC-tetramer+ CD8+ T-cells were single-cell sorted into round-bottom 96-well plates containing 5 × 104 irradiated (35 Gy) feeders in 100 μl T-cell medium supplemented with 0.8 μg/ml PHA.
High-throughput screen
Following 2 weeks of expansion, duplicates of the round-bottom 96-well plates containing the T-cell clones were made by resuspending and distributing T-cell medium containing the T-cell clones over two round-bottom 96-well plates using the Biomek 2000 workstation (Beckman Coulter, Brea, CA) operated through BioWorks software (Version 3.5; Beckman Coulter). Cells in one of the duplicate plates were washed five times before cell pellets were resuspended in 200 μl T-cell medium. Then, using the Biomek 2000 workstation, T-cell clones were distributed over flat-bottom 384-well plates by pipetting 20 μl of cell suspension per well. Subsequently, 20 μl of a cell suspension containing either peptide-pulsed or unloaded K562-A2 cells were added using the Biomek 2000 workstation. K562-A2 cells had been pulsed with peptide CD20SLF by incubating K562-A2 cells in T-cell medium supplemented with 50 nM for 1 h at 37°C. After 18 h coincubation, supernatants were harvested and IFN-γ or GM-CSF production was measured by enzyme-linked immunosorbent assay (ELISA, Sanquin Reagents or R&D Systems, respectively) using the Biomek 2000 workstation.
TCR gene transfer
TRAV and TRBV usage of T-cell clones was determined by reverse transcriptase PCR and sequencing using a previously established protocol [48]. V(D)J segments of the TCRα and TCRβ were codon optimized and cloned into the modified MP71-TCR-flex retroviral backbone. To increase expression and preferential pairing of the introduced TCRαβ chain, the MP71-TCR-flex vector contains codon-optimized and cysteine-modified murine TCRαβ constant domains and porcine teschovirus-derived P2A sequence to link TCR chains [26]. Constructs were ordered from GenScript (Piscataway, NJ).
Virus-specific T-cells were generated by MACS isolation using pMHC-tetramers and stimulated with irradiated feeders and PHA [51]. Purified CD8+ T-cells were activated using irradiated autologous PBMCs and PHA. On day 2 following stimulation of T-cells, retroviral supernatant was loaded on 24-well nontissue culture–treated plates that had been coated with 30 mg/mL retronectine (Takara, Shiga, Japan) and blocked with 2% human serum albumin (Sanquin Reagents, Amsterdam, The Netherlands). Viral supernatant was spun down at 2000 g for 20 minutes at 4°C before activated T-cells were added to retroviral supernatant and incubated at 37°C for 18 hours. Cells were transferred to culture-treated plates containing fresh T-cell medium. Seven days after stimulation, high-purity TCR-transduced T-cells were obtained by MACS isolation based on the expression of the transduced TCR or marker gene NGF-R. Transduced T-cells were incubated with an APC-labelled antibody against the murine constant TCR domain (BD Pharmingen) or nerve growth factor-receptor (NGF-R or CD271, Sanbio, Uden, The Netherlands) for 15 min at 4°C and washed twice. Following incubation with anti-APC microbeads (Miltenyi Biotec) for 15 min at 4°C, TCR-transduced T-cells were isolated on a LS column following manufacturer’s instructions.
FACS analysis
FACS was performed on a LSRII (BD Biosciences, Franklin Lakes, NJ) or a FACS Calibur (BD Biosciences) and analyzed using Diva Software (BD Biosciences) or FlowJo Software (TreeStar, Ashland, OR). 10,000 cells of a T-cell clone were mixed with 10,000 CD4+ T-cells from third party to prevent aggregate formation and stained with 2 μg/ml PE- or APC-labelled pMHC-tetramers for 15 min at 37°C. An Alexa700-conjugated antibody against CD8 (Invitrogen/Caltag) combined with fluorescein isothiocyanate (FITC)-labelled antibodies against CD4, CD14, and CD19 (BD Pharmingen) was added for an additional 15 min at 4°C. Similarly, 25,000 TCR-transduced or mock-transduced T-cells were first incubated with pMHC-tetramers before antibodies against CD8, CD4 and NGF-R were added. PBMCs, purified hematopoietic cell subsets or activated cells were stained with antibodies against CD3, CD4, CD14, CD19, CD34 (BD Pharmingen) for 4°C for 15 min. To analyze the CD20 expression, 50,000 cells of an ALL cell-line were incubated with FITC-conjugated rituximab at a final concentration of 10 μg/ml or an IgG1 isotype control antibody at 4°C for 15 min. To generate FITC-conjugated rituximab, unlabeled rituximab was incubated with FITC isomer I (Sigma-Aldrich) in 0.5 M sodium carbonate buffer (pH 9.5) for 1 hour at room temperature. FITC-conjugated rituximab was purified by filtration using 3kD VivaSpin 20 centrifugal concentrators (Vivaproducts, Littleton, MA) spun at 2750 g. During filtration, sodium carbonate buffer was exchanged for by adding tris buffered saline (TBS, pH 8.2; Sigma Aldrich). FITC-conjugated rituximab was stored at 4°C.
Functional analysis
Stimulator cells were peptide-pulsed at various peptide concentrations for 30 min at 37°C. Responder T-cells and peptide-pulsed or unloaded stimulator cells were coincubated at various responder to stimulator ratios. After 18 h coincubation, supernatants were harvested and IFN-γ or GM-CSF production was measured by ELISA.
Cr51 release assay
Standard Cr51 release assay was performed as previously described [52]. Briefly, target cells were labelled with 100 μCi Na2Cr51O4 for 1 hour at 37°C, washed three times, and added to effector cells in T-cell medium at various effector-to-target ratios to a final volume of 100 μl. Complement dependent cytotoxicity (CDC) was assessed by incubating Cr51-labelled target cells with human serum in the presence of CD20-specific mAb ofatumumab at various concentrations. Spontaneous and maximum release was measured by incubating target cells with medium alone and 1% Triton X-100, respectively. The tests were performed in triplicate. After 5 hours of coincubation, 25 μl of supernatant was harvested and analyzed on a MicroBeta 2450 Microplate Counter (Perkin Elmer, Waltham, MA). The percent specific lysis was calculated as follows: [(experimental release – spontaneous release)/(maximum release – spontaneous release)]×100%.
FACS-based cytotoxicity assay
Adapted from Jedema et al. [53], 10,000 PKH26GL-labelled (Sigma-Aldrich) target cells were coincubated with T-cells at various effector:target ratios in 50 μl T-cell medium for 20 hours. After coincubation, cells were stained with Sytox Blue dead cell stain (Invitrogen/Caltag) in a final concentration of 1 μM for 5 min. 10 μl Flow-count fluorospheres (Beckman Coulter) were added and samples were analyzed using FACS. For each sample, 3,300 Flow-count fluorospheres were acquired and the percent surviving cells was calculated as follows: (PKH26GL-labelled targets in the presence of effector cells)/(PKH26GL-labelled targets in absence of effector cells)×100%.
Real-time quantitative PCR (RT-qPCR) of CD20
Total RNA was isolated using the RNAqueous Micro-Kit and Small Scale Kit (Ambion, Life Technologies) for a maximum of 0.5 × 106 and 10 × 106 cells, respectively, following the manufacturer’s instructions. Total RNA was converted to cDNA using M-MLV reverse transcriptase (Invitrogen). MS4A1 expression (coding gene of CD20) was measured on the Roche Lightcycler 480 (Roche, Basel, Switzerland) using Fast Start TaqDNA Polymerase (Roche) and EvaGreen (Biotium, Hayward, CA) with forward primer 5′-GGGGCTGTCCAGATTATGAA-3′ and reverse primer 5′-GGAGTTTTTCTCCGTTGCTG-3′. mRNA expression of MS4A1 in samples was calculated using the 2-ΔΔCT method [54] with the expression of PBGD serving as endogenous reference gene and the average expression of MS4A1 in 7 healthy CD19+ B-cell samples serving as calibrator.
ACKNOWLEDGMENTS
The authors thank Guido de Roo and Sabrina A.J. Veld (Department of Hematology, Leiden University Medical Center, Leiden, The Netherlands) for providing expert technical assistance in flow cytometric cell sorting; Renate de Boer and Marleen M. van Loenen (Department of Hematology, Leiden University Medical Center, Leiden, The Netherlands) for generating virus-specific T-cells and providing expert technical assistance in TCR transduction experiments; Floris C. Loeff (Department of Hematology, Leiden University Medical Center, Leiden, The Netherlands) for facilitating human serum and providing expert technical assistance in assessing CDC activity of ofatumumab; Carsten Linnemann and Ton N. Schumacher (Division of Immunology, Netherlands Cancer Institute, Amsterdam, The Netherlands) for providing the MP71-TCR-flex retroviral backbone.
CONFLICTS OF INTEREST
The authors disclose no potential conflicts of interest.
Authors’ contributions
L.J. designed, performed, analyzed and interpreted all experiments and wrote the manuscript. D.M.v.d.S. generated pMHC-monomers and performed and analyzed experiments. R.S.H. determined TRAV and TRBV usage and constructed retroviral expression vectors, and designed and performed RT-qPCR. P.H. designed study. M.G.D.K., M.P.S., D.d.R. performed and analyzed experiments. P.A.v.V. designed and cosupervised study. J.H.F.F. designed and supervised study, and wrote the manuscript. M.H.M.H. analyzed and interpreted experiments, designed and supervised study, and wrote the manuscript. All authors revised and edited the manuscript.
GRANT SUPPORT
This research was supported by the financial assistance of the Dutch Cancer Society (2010–4832) and the Landsteiner Foundation for Blood Transfusion Research (LSBR0713).
REFERENCES
1. Shortt J, Spencer A. Adjuvant rituximab causes prolonged hypogammaglobulinaemia following autologous stem cell transplant for non-Hodgkin’s lymphoma. Bone Marrow Transplant. 2006; 38:433–436.
2. Wolverton W, McClenathan B. Prolonged hypogammaglobulinemia after adjuvant rituximab for Hodgkin’s lymphoma presenting with refractory urticaria. Ann Allergy Asthma Immunol. 2012; 109:473–474.
3. Hiraga J, Tomita A, Sugimoto T, Shimada K, Ito M, Nakamura S, Kiyoi H, Kinoshita T, Naoe T. Down-regulation of CD20 expression in B-cell lymphoma cells after treatment with rituximab-containing combination chemotherapies: its prevalence and clinical significance. Blood. 2009; 113:4885–4893.
4. Baig NA, Taylor RP, Lindorfer MA, Church AK, LaPlant BR, Pettinger AM, Shanafelt TD, Nowakowski GS, Zent CS. Induced resistance to ofatumumab-mediated cell clearance mechanisms, including complement-dependent cytotoxicity, in chronic lymphocytic leukemia. J Immunol. 2014; 192:1620–1629.
5. Pozzo F, Bittolo T, Arruga F, Bulian P, Macor P, Tissino E, Gizdic B, Rossi FM, Bomben R, Zucchetto A, Benedetti D, Degan M, D’Arena G, et al. NOTCH1 mutations associate with low CD20 level in chronic lymphocytic leukemia: evidence for a NOTCH1 mutation-driven epigenetic dysregulation. Leukemia. 2016; 30:182–189.
6. Beers SA, French RR, Chan HT, Lim SH, Jarrett TC, Vidal RM, Wijayaweera SS, Dixon SV, Kim H, Cox KL, Kerr JP, Johnston DA, Johnson PW, et al. Antigenic modulation limits the efficacy of anti-CD20 antibodies: implications for antibody selection. Blood. 2010; 115:5191–5201.
7. Golay J, Zaffaroni L, Vaccari T, Lazzari M, Borleri GM, Bernasconi S, Tedesco F, Rambaldi A, Introna M. Biologic response of B lymphoma cells to anti-CD20 monoclonal antibody rituximab in vitro: CD55 and CD59 regulate complement-mediated cell lysis. Blood. 2000; 95:3900–3908.
8. Golay J, Lazzari M, Facchinetti V, Bernasconi S, Borleri G, Barbui T, Rambaldi A, Introna M. CD20 levels determine the in vitro susceptibility to rituximab and complement of B-cell chronic lymphocytic leukemia: further regulation by CD55 and CD59. Blood. 2001; 98:3383–3389.
9. Treon SP, Mitsiades C, Mitsiades N, Young G, Doss D, Schlossman R, Anderson KC. Tumor cell expression of CD59 is associated with resistance to CD20 serotherapy in patients with B-cell malignancies. J Immunother. 2001; 24:263–271.
10. Middleton O, Cosimo E, Dobbin E, McCaig AM, Clarke C, Brant AM, Leach MT, Michie AM, Wheadon H. Complement deficiencies limit CD20 monoclonal antibody treatment efficacy in CLL. Leukemia. 2015; 29:107–114.
11. Sykulev Y, Joo M, Vturina I, Tsomides TJ, Eisen HN. Evidence that a single peptide-MHC complex on a target cell can elicit a cytolytic T cell response. Immunity. 1996; 4:565–571.
12. Irvine DJ, Purbhoo MA, Krogsgaard M, Davis MM. Direct observation of ligand recognition by T cells. Nature. 2002; 419:845–849.
13. Huang J, Brameshuber M, Zeng X, Xie J, Li QJ, Chien YH, Valitutti S, Davis MM. A single peptide-major histocompatibility complex ligand triggers digital cytokine secretion in CD4(+) T cells. Immunity. 2013; 39:846–857.
14. Amir AL, van der Steen DM, Hagedoorn RS, Kester MG, van Bergen CA, Drijfhout JW, de Ru AH, Falkenburg JH, van Veelen PA, Heemskerk MH. Allo-HLA-reactive T cells inducing graft-versus-host disease are single peptide specific. Blood. 2011; 118:6733–6742.
15. Amir AL, van der Steen DM, van Loenen MM, Hagedoorn RS, de Boer R, Kester MD, de Ru AH, Lugthart GJ, van Kooten C, Hiemstra PS, Jedema I, Griffioen M, van Veelen PA, et al. PRAME-specific Allo-HLA-restricted T cells with potent antitumor reactivity useful for therapeutic T-cell receptor gene transfer. Clin Cancer Res. 2011; 17:5615–5625.
16. Savage P, Gao L, Vento K, Cowburn P, Man S, Steven N, Ogg G, McMichael A, Epenetos A, Goulmy E, Stauss HJ. Use of B cell-bound HLA-A2 class I monomers to generate high-avidity, allo-restricted CTLs against the leukemia-associated protein Wilms tumor antigen. Blood. 2004; 103:4613–4615.
17. Abrahamsen IW, Stronen E, Walchli S, Johansen JN, Kjellevoll S, Kumari S, Komada M, Gaudernack G, Tjonnfjord G, Toebes M, Schumacher TN, Lund-Johansen F, Olweus J. Targeting B cell leukemia with highly specific allogeneic T cells with a public recognition motif. Leukemia. 2010; 24:1901–1909.
18. Leisegang M, Wilde S, Spranger S, Milosevic S, Frankenberger B, Uckert W, Schendel DJ. MHC-restricted fratricide of human lymphocytes expressing survivin-specific transgenic T cell receptors. J Clin Invest. 2010; 120:3869–3877.
19. Spranger S, Jeremias I, Wilde S, Leisegang M, Starck L, Mosetter B, Uckert W, Heemskerk MH, Schendel DJ, Frankenberger B. TCR-transgenic lymphocytes specific for HMMR/Rhamm limit tumor outgrowth in vivo. Blood. 2012; 119:3440–3449.
20. Jahn L, Hombrink P, Hassan C, Kester MG, van der Steen DM, Hagedoorn RS, Falkenburg JH, van Veelen PA, Heemskerk MH. Therapeutic targeting of the BCR-associated protein CD79b in a TCR-based approach is hampered by aberrant expression of CD79b. Blood. 2015; 125:949–958.
21. Bae J, Martinson JA, Klingemann HG. Identification of CD19 and CD20 peptides for induction of antigen-specific CTLs against B-cell malignancies. ClinCancer Res. 2005; 11:1629–1638.
22. Hassan C, Kester MG, Ru AH, Hombrink P, Drijfhout JW, Nijveen H, Leunissen JA, Heemskerk MH, Falkenburg JH, Veelen PA. The human leukocyte antigen-presented ligandome of B lymphocytes. MolCell Proteomics. 2013.
23. Hombrink P, Hassan C, Kester MG, Jahn L, Pont MJ, de Ru AH, van Bergen CA, Griffioen M, Falkenburg JH, van Veelen PA, Heemskerk MH. Identification of Biological Relevant Minor Histocompatibility Antigens within the B-lymphocyte-Derived HLA-Ligandome Using a Reverse Immunology Approach. Clin Cancer Res. 2015; 21:2177–2186.
24. Pawluczkowycz AW, Beurskens FJ, Beum PV, Lindorfer MA, van de Winkel JG, Parren PW, Taylor RP. Binding of submaximal C1q promotes complement-dependent cytotoxicity (CDC) of B cells opsonized with anti-CD20 mAbs ofatumumab (OFA) or rituximab (RTX): considerably higher levels of CDC are induced by OFA than by RTX. J Immunol. 2009; 183:749–758.
25. van Loenen MM, de Boer R, Amir AL, Hagedoorn RS, Volbeda GL, Willemze R, van Rood JJ, Falkenburg JH, Heemskerk MH. Mixed T cell receptor dimers harbor potentially harmful neoreactivity. Proc Natl Acad Sci USA. 2010; 107:10972–10977.
26. Linnemann C, Heemskerk B, Kvistborg P, Kluin RJ, Bolotin DA, Chen X, Bresser K, Nieuwland M, Schotte R, Michels S, Gomez-Eerland R, Jahn L, Hombrink P, et al. High-throughput identification of antigen-specific TCRs by TCR gene capture. Nat Med. 2013; 19:1534–1541.
27. Kennedy AD, Beum PV, Solga MD, DiLillo DJ, Lindorfer MA, Hess CE, Densmore JJ, Williams ME, Taylor RP. Rituximab infusion promotes rapid complement depletion and acute CD20 loss in chronic lymphocytic leukemia. J Immunol. 2004; 172:3280–3288.
28. Taylor RP, Lindorfer MA. Immunotherapeutic mechanisms of anti-CD20 monoclonal antibodies. Curr Opin Immunol. 2008; 20:444–449.
29. Lim SH, Vaughan AT, Ashton-Key M, Williams EL, Dixon SV, Chan HT, Beers SA, French RR, Cox KL, Davies AJ, Potter KN, Mockridge CI, Oscier DG, et al. Fc gamma receptor IIb on target B cells promotes rituximab internalization and reduces clinical efficacy. Blood. 2011; 118:2530–2540.
30. Falkenburg WJ, Melenhorst JJ, van de Meent M, Kester MG, Hombrink P, Heemskerk MH, Hagedoorn RS, Gostick E, Price DA, Falkenburg JH, Barrett AJ, Jedema I. Allogeneic HLA-A*02-restricted WT1-specific T cells from mismatched donors are highly reactive but show off-target promiscuity. J Immunol. 2011; 187:2824–2833.
31. Hombrink P, Raz Y, Kester MG, de Boer R, Weissbrich B, von dem Borne PA, Busch DH, Schumacher TN, Falkenburg JH, Heemskerk MH. Mixed functional characteristics correlating with TCR-ligand koff -rate of MHC-tetramer reactive T cells within the naive T-cell repertoire. Eur J Immunol. 2013; 43:3038–3050.
32. Hiemstra HS, van Veelen PA, Willemen SJ, Benckhuijsen WE, Geluk A, de Vries RR, Roep BO, Drijfhout JW. Quantitative determination of TCR cross-reactivity using peptide libraries and protein databases. Eur J Immunol. 1999; 29:2385–2391.
33. Ekeruche-Makinde J, Miles JJ, van den Berg HA, Skowera A, Cole DK, Dolton G, Schauenburg AJ, Tan MP, Pentier JM, Llewellyn-Lacey S, Miles KM, Bulek AM, Clement M, et al. Peptide length determines the outcome of TCR/peptide-MHCI engagement. Blood. 2013; 121:1112–1123.
34. Wucherpfennig KW. T cell receptor crossreactivity as a general property of T cell recognition. Mol Immunol. 2004; 40:1009–1017.
35. Mazza C, Auphan-Anezin N, Gregoire C, Guimezanes A, Kellenberger C, Roussel A, Kearney A, van der Merwe PA, Schmitt-Verhulst AM, Malissen B. How much can a T-cell antigen receptor adapt to structurally distinct antigenic peptides? The EMBO Journal. 2007; 26:1972–1983.
36. Zhang W-y, Wang Y, Guo Y-l, Dai H-r, Yang Q-m, Zhang Y-j, Zhang Y, Chen M-x, Wang C-m, Feng K-c, Li S-x, Liu Y, Shi F-x, et al. Treatment of CD20-directed Chimeric Antigen Receptor-modified T cells in patients with relapsed or refractory B-cell non-Hodgkin lymphoma: an early phase IIa trial report. Signal Transduction And Targeted Therapy. 2016; 1:16002.
37. Tabata R, Iwama H, Tabata C, Yasumizu R, Kojima M. CD5- and CD23-Positive Splenic Diffuse Large B-Cell Lymphoma with Very Low CD20 Expression. Journal of Clinical and Experimental Hematopathology. 2014; 54:155–161.
38. Maude SL, Teachey DT, Porter DL, Grupp SA. CD19-targeted chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Blood. 2015; 125:4017–4023.
39. Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, Teachey DT, Chew A, Hauck B, Wright JF, Milone MC, Levine BL, June CH. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013; 368:1509–1518.
40. Bonini C, Ferrari G, Verzeletti S, Servida P, Zappone E, Ruggieri L, Ponzoni M, Rossini S, Mavilio F, Traversari C, Bordignon C. HSV-TK gene transfer into donor lymphocytes for control of allogeneic graft-versus-leukemia. Science. 1997; 276:1719–1724.
41. Straathof KC, Pule MA, Yotnda P, Dotti G, Vanin EF, Brenner MK, Heslop HE, Spencer DM, Rooney CM. An inducible caspase 9 safety switch for T-cell therapy. Blood. 2005; 105:4247–4254.
42. Ciceri F, Bonini C, Marktel S, Zappone E, Servida P, Bernardi M, Pescarollo A, Bondanza A, Peccatori J, Rossini S, Magnani Z, Salomoni M, Benati C, et al. Antitumor effects of HSV-TK-engineered donor lymphocytes after allogeneic stem-cell transplantation. Blood. 2007; 109:4698–4707.
43. Kieback E, Charo J, Sommermeyer D, Blankenstein T, Uckert W. A safeguard eliminates T cell receptor gene-modified autoreactive T cells after adoptive transfer. Proc Natl Acad Sci USA. 2008; 105:623–628.
44. Griffioen M, van Egmond EH, Kester MG, Willemze R, Falkenburg JH, Heemskerk MH. Retroviral transfer of human CD20 as a suicide gene for adoptive T-cell therapy. Haematologica. 2009; 94:1316–1320.
45. Ciceri F, Bonini C, Stanghellini MT, Bondanza A, Traversari C, Salomoni M, Turchetto L, Colombi S, Bernardi M, Peccatori J, Pescarollo A, Servida P, Magnani Z, et al. Infusion of suicide-gene-engineered donor lymphocytes after family haploidentical haemopoietic stem-cell transplantation for leukaemia (the TK007 trial): a non-randomised phase I-II study. Lancet Oncol. 2009; 10:489–500.
46. Di Stasi A, Tey SK, Dotti G, Fujita Y, Kennedy-Nasser A, Martinez C, Straathof K, Liu E, Durett AG, Grilley B, Liu H, Cruz CR, Savoldo B, et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med. 2011; 365:1673–1683.
47. Wang X, Chang WC, Wong CW, Colcher D, Sherman M, Ostberg JR, Forman SJ, Riddell SR, Jensen MC. A transgene-encoded cell surface polypeptide for selection, in vivo tracking,, ablation of engineered cells. Blood. 2011; 118:1255–1263.
48. Heemskerk MH, de Paus RA, Lurvink EG, Koning F, Mulder A, Willemze R, van Rood JJ, Falkenburg JH. Dual HLA class I and class II restricted recognition of alloreactive T lymphocytes mediated by a single T cell receptor complex. Proc Natl Acad Sci USA. 2001; 98:6806–6811.
49. Nijmeijer BA, Szuhai K, Goselink HM, van Schie ML, van der Burg M, de JD, Marijt EW, Ottmann OG, Willemze R, Falkenburg JH. Long-term culture of primary human lymphoblastic leukemia cells in the absence of serum or hematopoietic growth factors. ExpHematol. 2009; 37:376–385.
50. Garboczi DN, Hung DT, Wiley DC. HLA-A2-peptide complexes: refolding and crystallization of molecules expressed in Escherichia coli and complexed with single antigenic peptides. Proc Natl Acad Sci USA. 1992; 89:3429–3433.
51. van Loenen MM, de Boer R, van Liempt E, Meij P, Jedema I, Falkenburg JH, Heemskerk MH. A Good Manufacturing Practice procedure to engineer donor virus-specific T cells into potent anti-leukemic effector cells. Haematologica. 2014; 99:759–768.
52. van der Veken LT, Hoogeboom M, de Paus RA, Willemze R, Falkenburg JH, Heemskerk MH. HLA class II restricted T-cell receptor gene transfer generates CD4+ T cells with helper activity as well as cytotoxic capacity. Gene Ther. 2005; 12:1686–1695.
53. Jedema I, van der Werff NM, Barge RM, Willemze R, Falkenburg JH. New CFSE-based assay to determine susceptibility to lysis by cytotoxic T cells of leukemic precursor cells within a heterogeneous target cell population. Blood. 2004; 103:2677–2682.
54. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001; 25:402–408.