
INTRODUCTION
Malignant glioma is the most common type (accounting for approximately 80%) of primary malignant brain tumors and associated with exceptionally high morbidity and mortality [1, 2]. The standard therapy for newly diagnosed malignant gliomas involves surgical resection combined with chemotherapy and/or radiotherapy. Although advances in radiotherapy and chemotherapy have brought modest improvements in the survival of patients with malignant glioma, the invasive nature of the disease continue to limit the 5-year survival of glioblastoma (GBM) and its variants to only 4.7% [3-5]. Therefore, there is an urgent need to develop novel therapeutic modalities that specifically target the pathogenesis of malignant gliomas.
Cancer immunotherapy, the idea of boosting the tumor-specific adaptive immune activities instead of directly targeting cancer cells, presents its debut in history more than 100 years ago [6]. After decades of disappointment, it proves its values with recent successes in the treatment of multiple solid and hematological cancers [7]. These successes were built upon incessant efforts to understand the mechanisms underlying cancer immune regulation, and notably, on the discovery of a plethora of immune checkpoints, inhibitory pathways essential for maintaining self-tolerance under physiological conditions and generating the inhibitory microenvironment for tumor to evade immune surveillance during cancer development [8, 9].
These inhibitory pathways are initiated through the ligand-receptor interactions. By far, the best characterized immune checkpoint receptors are programmed cell death protein 1 (PD-1; also known as CD279), cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4; also known as CD152) and indoleamine 2,3-dioxygenase (IDO); agents targeting these molecules are either approved or being extensively tested in clinical trials for multiple solid or hematological cancers [9].
In this review, we will focus on this important strategy of immunotherapy, i.e., targeting immune checkpoints, and discuss its potential in the treatment of malignant gliomas. We will start with a brief overview on the general biology of immune checkpoints, specifically PD-1, CTLA-4, and IDO. Then we will transition to the status of different checkpoint molecules in malignant gliomas, which provides the rationale to targeting these molecules. Finally, we will review the pre-clinical and clinical trials involving the therapies targeting these immune checkpoints.
MALIGNANT GLIOMA
Malignant gliomas are histologically heterogeneous glia-derived tumors that infiltrate the stromal tissues. In 2016, the World Health Organization (WHO) published the new classification of CNS tumors, which, for the first time, combines molecular and histological features to identify many tumor entities [10]. Following this classification system, diffuse gliomas are divided into grade II/III astrocytic tumors, grade II/III oligodendrogliomas, grade IV glioblastomas, and the related diffuse gliomas of childhood. Both grade II diffuse astrocytomas and grade III anaplastic astrocytomas are further divided into isocitrate dehydrogenase (IDH)-wildtype, IDH-mutant and NOS categories. Glioblastomas include: IDH-wildtype glioblastoma; IDH-mutant glioblastoma; and NOS glioblastoma. The NOS designation means that insufficient information is available to assign tumors to the relevant genetic parameter.
The central nervous system (CNS) was once considered immune-privileged, deficit in normal immunological functions, due to its specific anatomical and physiological features: the presence of the blood brain barrier allowing for selective entry of immune cells, the absence of lymphatic vessels or lymph nodes, the critical immune organs in the periphery, the low numbers of traditional antigen-presenting cells (APCs) including dendritic cells (DCs) and macrophages, and the lack of naive T cells in CNS [11, 12]. Nevertheless, recent progresses in neurobiology and neuroimmunology suggest that although challenging, immunotherapy holds extraordinary promises in CNS malignancies. Several recent publications convincingly demonstrated the presence of functional lymphatic vessels within the meningeal compartment [13-15], not only supporting the early descriptions that lymphatic systems exist in the brain [16-18], but also revealing novel routes that enable the communications of glioma antigens and immune cells between the brain and other immune components. Therefore, the glioma antigens may first enter the cerebrospinal fluid (CSF) through perivascular spaces termed Virchow-Robin spaces [19]. Due to the lack of secondary lymphoid tissues in the brain parenchyma, the peripheral lymphoid tissue may be the starting point for initiating tumor-specific immune responses; that is, the antigens may be transported into deep cervical lymph nodes through the newly discovered dural lymphatic, and then be presented by APCs in peripheral lymphoid tissues [13, 20, 21]. The tumor-specific lymphocytes cross the choroid plexus into CSF, are re-stimulated by local APCs, and ultimately traffic through the blood-brain barrier and/or Virchow-Robin spaces into brain parenchyma to mount an efficient immune attack on tumors. Meanwhile, specific chemokines may also play important roles in recruiting glioma-specific tumor-infiltrating lymphocytes (TILs) [21, 22].
Furthermore, the immunosuppressive state observed in patients with malignant gliomas corroborate the significance of immune system in disease development. Locally, malignant gliomas enrich their microenvironment with immunosuppressive factors such as the transforming growth factor beta (TGF-β) and vascular endothelial growth factor (VEGF), both suppressing DC maturation and inhibiting T cell proliferation and cytotoxicity [23, 24]. The reduced absolute counts of CD4+ T cells and increased proportion of immunosuppressive regulatory T cells (Tregs) further nurture the suppressive microenvironment [25]. Systematically, the old age of patient population, cytotoxic chemotherapy and other therapies including corticosteroids all contribute to the deficiency of adaptive immune responses [11]. Hence, the reverse of immunosuppression and the initiation of anti-tumor immunity hold great promise to limit tumor progression and improve patient outcomes for malignant gliomas.
IMMUNE CHECKPOINTS AND MALIGNANT GLIOMA
The accurate execution of each step of immune regulation is fine-tuned by the balance between stimulatory and inhibitory signaling, both of which initiates with membrane receptors, also known as immune checkpoints. Given their accessibility on cell membranes and significance in regulating immune responses, the immune checkpoint receptors and their corresponding ligands become the best druggable targets for immune regulation. Therefore, in contrast to most antibodies currently used for cancer therapy, drugs targeting immune checkpoints do not target tumor cells directly, but instead act on lymphocytes to boost their endogenous anti-tumor activity [26, 27].
Of the many immune checkpoint receptors under intensive investigation, the three best characterized ones in the context of clinical cancer immunotherapy are PD-1, CTLA-4 and IDO, which function by different mechanisms, as detailed below.
PD-1 and its ligands
PD-1 is an inhibitory receptor mainly expressed on activated T cells, including CD4+ and CD8+ T cells [28, 29]. It is also detectable on other lymphocyte subsets, including natural killer (NK) cells and B cells [30, 31]. The elevated expression of PD-1 has been detected in diverse advanced human cancers, such as melanoma, prostate cancer, and renal cell carcinoma [32-34]. The immunosuppressive activities of receptor PD-1 are initiated upon binding to one of its two ligands, PD-L1 or PD-L2, and mediated through multiple mechanisms [28, 35-37]. First, upon ligand engagement, PD-1 recruits the Src homology2 domain-containing tyrosine phosphatase (SHP2), which inhibits the phosphorylation of PI3K, blocks the PI3K-Akt pathway, and suppresses T cell activity [28, 38]. This signaling is activated during antigen presentation to T cells by APCs as well as through T cell-target cell interaction. Some cancer antigens persistently up-regulate PD-1 expression in T cells, which is recognized as a mechanism termed cognate antigen-specific T cells exhaustion (Figure 1A) [39]. Second, PD-1 is highly induced on Tregs upon engagement by PD-L1, which plays a significant role in Treg development and sustaining their suppressive functions (Figure 2A) [40].
The PD-1 ligands, PD-L1 (also known as B7-H1 or CD274) and PD-L2 (also known as B7-DC and CD273) both belong to the B7 family. Through the engagement of PD-1, PD-L1 reduces IFN-γ production by activated T cells and leads to T-cell exhaustion or anergy. Furthermore, PD-L1 drives the differentiation of naive CD4+ T cells into induced Tregs, and maintains their suppressive function [40]. In addition to PD-1, PD-L1 also binds to CD80. The interaction between PD-L1 and CD80 initiates negative signals that inhibit T cell function and cytokine production [41, 42]. PD-L1 was reported to be highly expressed on malignant tumor cells, such as glioma, ovarian, melanoma and lung cancers [43-46]. Two mechanisms have been revealed to regulate PD-L1 expression by tumor cells: innate immune resistance and adaptive immune resistance [47]. The innate immune resistance refers to the finding from certain lymphomas and gliomas that the PD-L1 expression is up-regulated by constitutive oncogenic signaling in the tumor cells, including the PI3K-Akt pathway and constitutive anaplastic lymphoma kinase (ALK) signaling, both independent of inflammatory signals from the tumor microenvironment [48, 49]. Adaptive immune resistance is observed in other tumors including melanoma, where interferons, predominately IFN-γ, up-regulate the expression of PD-L1. The heterogenous expressions of PD-L1 within the tumor tissue are regions of lymphocytes infiltration as part of the immune responses [50, 51]. Other than tumor cells, PD-L1 is also expressed on stromal cells and macrophages within the tumor microenvironment [52-54].
PD-L2, a second ligand for PD-1, is exclusively expressed on APCs including DCs and macrophages under homeostasis, and can be induced in a variety of other immune cells or non-immune cells in response to microenvironmental stimuli [55]. In certain subsets of B cell lymphomas, such as Hodgkin’s lymphoma, follicular cell B cell lymphoma and mediastinal B cell lymphoma, PD-L2 is highly up-regulated [56]. The up-regulation of PD-L2 in lymphomas may result from the gene fusions between the class II major histocompatibility complex (MHC) transactivator (CIITA) and PD-L2 [57]. The downstream signaling or biological functions from PD-L2 interacting with PD-1 are not clear, although studies suggest that PD-L2, through the engagement of PD-1, could inhibit T-cell responses [55, 58]. Besides, it is believed that PD-1 is not the only receptor for PD-L2, since PD-L2 mutant incapable of binding to PD-1 still impact T-cell functions [59].
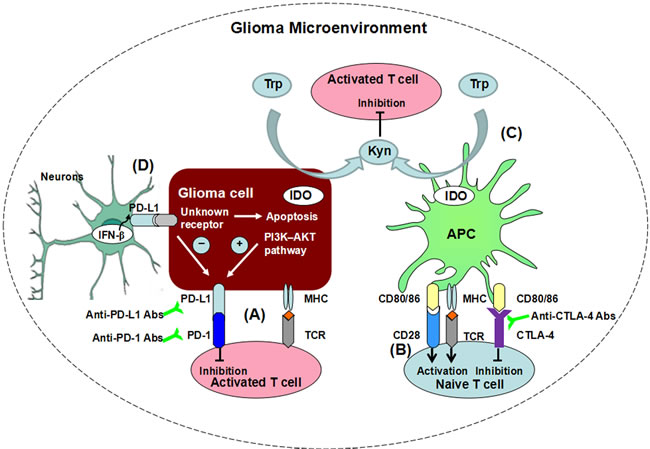
Figure 1: Immune checkpoints suppress T cell function in glioma microenvironment through differential mechanisms. A. The expression of PD-L1 on glioma cells is dependent on the PI3K-AKT pathway. PD-L1, upon engagement to its receptor PD-1 on T cells, inhibits activated T cell functions within the tumor microenvironment. B. CTLA-4 inhibits T cell activation. The co-stimulatory molecules from APCs, CD80 and CD86 bind to both stimulatory receptor CD28 and inhibitory receptor CTLA-4, yet with lower affinity to the former than to the latter. Therefore, CTLA-4 suppresses T cell activation via competitive inhibition. C. The maintenance, and function of T cells require adequate Trp levels, but IDO from tumor cells catabolizes Trp to numerous metabolites, such as Kyn. The decrease of Trp suppressed T cell activation. Meanwhile, the metabolites, such as Kyn can induced T cell apoptosis. D. In neurons surrounding glioma tissue, the expression of PD-L1 is induced by endogenous production of IFN-β. The neurons have the capability to inhibit proliferation of glioma cells and induce its apoptosis. Meanwhile, PD-L1+ neurons reduce the PD-L1 expression on glioma cells.
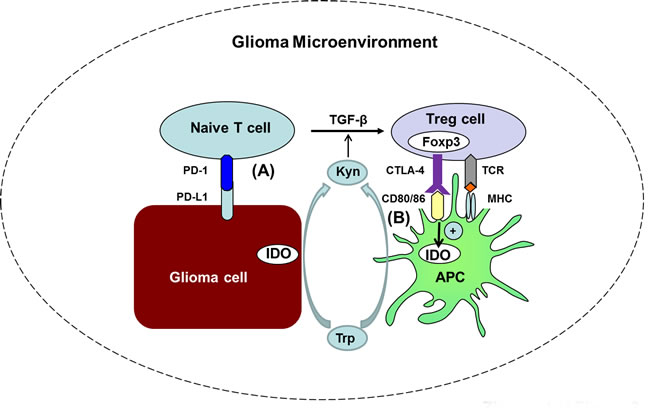
Figure 2: The diversified mechanisms by which various immune checkpoints promote each other, activate Tregs, and contribute to the immunosuppressive microenvironment in glioma. A. IDO from tumor cells catabolizes Trp to Kyn. PD-1/PD-L1 pathway and Kyn could induce FoxP3 expression and promote Treg conversion with the assistance of TGF-β. B. FoxP3 controls CTLA-4 expression by Tregs. The CTLA-4 on Tregs binds to CD80/86, up-regulates the expression of IDO, down-regulates the expression of CD80 and CD86 on APCs, and enhances the suppressive functions of Tregs.
PD-1 and glioma
A recent study showed that the up-regulation of PD-1 on peripheral blood T cells of glioma patients correlates with disease progression. The proportion of PD-1+ cells among peripheral blood CD4+ and CD8+ T cells in glioma patients is higher than that in healthy controls. The CD4+ T cells from astrocytoma patients, the most malignant glioma, express the highest proportion of PD-1 compared with other types [60]. Besides the T cells in peripheral circulation, the frequency and expression level of PD-1 are also significantly higher on glioma-infiltrating CD4+ effector memory T cells, when compared with cells from healthy donors [61]. Functionally, PD-1 expression is closely related to the defects of IFN-γ production from glioma-infiltrating CD4+ effector memory T cells. Further analysis revealed that myeloid-derived suppressor cells (MDSCs), a population of myeloid precursors, are significantly accumulated in patients with glioma and contribute to tumor progression. Glioma-derived MDSCs expressed high levels of PD-L1. When co-cultured with T cells, glioma-derived MDSCs can up-regulated PD-1 expression on CD4+ T effector memory T cells. Consequently, MDSCs in glioma patients contribute to the functional exhaustion of T cells through PD-1/PD-L1 pathway [61].
For patients with GBM, the tumor-infiltrating Tregs significantly correlate with worse prognosis. PD-1 is highly induced on Tregs upon engagement by PD-L1 expressed on glioma, which plays a significant role in Treg development and sustaining their suppressive functions and is responsible for immune escape of glioma [40, 62]. Thus, blockade of the PD-1 signaling may inhibit the production of Treg, and specifically boost anti-tumor immune responses [62].
PD-L1 and glioma
PD-L1 is highly expressed in human malignant gliomas, and its levels significantly correlated with the glioma grade. Recent studies showed that tumor cells from Grade IV gliomas, anaplastic astrocytoma and GBM, present intense positive staining for PD-L1, which was reduced in oligodendroglioma and diffuse astrocytoma (Grade II and III gliomas), and no positive staining of PD-L1 was detected in the tumor-free CNS tissue [46, 63, 64].
The reported positivity of PD-L1 expression in GBM ranges from 61% to 88% [65], potentially due to the differences in sample size, tissue sampling strategies, antibodies and staining protocols, methods for evaluating staining patterns, and assignment of cut-off values. Tissue sampling may critically affect the analysis on PD-L1 expression. Insufficient sampling may not include the PD-L1-positive areas of heterogeneous tumors, generating false negative results and the lower percentage of PD-L1-positive glioblastomas. Therefore, there is an urgent need to standardize staining procedures and evaluation methods to analyze PD-L1 expression in tissue samples [66].
In malignant glioma, PD-L1 expression in tumor cells is controlled through the innate immune resistance mechanism, by the constitutive oncogenic signaling, PI3K-AKT pathway in tumor cells [49]. It is demonstrated that PD-L1 expression is elevated posttranscriptionally in glioma cells after loss of phosphatase and tensin homolog (PTEN) and activation of PI3K pathway (Figure 1A).
Other than glioma cells and tumor-infiltrating lymphocytes [67], PD-L1 expression is also detected on neurons surrounding glioma tissue [68, 69]. There are significant correlations between the clinical outcomes and the level of PD-L1 on neurons from GBM patients. The Up-regulation of PD-L1 on neurons in tumor adjacent brain tissue was associated with prolonged survival, whereas the loss of neuronal PD-L1 was involved in high PD-L1 expression on glioma tissue and poor prognosis. In light of these findings, the expression of PD-L1 is induced by endogenous production of IFN-β in neurons surrounding glioma tissue (Figure 1D). Therefore, the expression of PD-L1 in tumor adjacent brain tissue is important for the survival of patients with malignant glioma [70]. Mutual PD-L1 regulation between tumor-adjacent brain tissue and tumor tissue is a potential prognostic biomarker for GBM.
CTLA-4 and glioma
CTLA-4, also known as CD152, is a member of the immunoglobulin superfamily and the first co-inhibitory molecule identified [71]. CTLA-4 is not expressed by naive T cells, but rapidly induced in both CD4+ and CD8+ T cells upon T-cell activation.
The predominant functions of CTLA-4 are attributed to the effects on the two major subsets of CD4+ T cells: suppression of helper T cell activity and enhancement of Treg immunosuppression [72]. The ligands of CTLA-4 are CD80 and CD86, both of which also signal through the co-stimulatory receptor CD28 and induce T-cell activation. Since CTLA-4 has a much higher affinity for both ligands than CD28, it effectively inhibits the ligand engagement to CD28, transduces inhibitory signals, and suppresses T-cell activation (Figure 1B) [73-75]. Forkhead box P3 (FoxP3), a forkhead transcription factor, controls CTLA-4 expression (Figure 2B). In return, when CTLA-4 is induced in Tregs, CTLA-4 mediates the down-regulating of co-stimulatory molecules, CD80 and CD86 on DCs, and thus reduces the potency of APCs to activate other T cells, critically controlling the immunosuppressive activity of Tregs (Figure 2B) [76]. In addition, CTLA-4 expressed by Tregs could bind to CD80/86 on DCs with high affinity, and then deliver a signal that induces IDO expression, and triggers the IDO activity in DCs (Figure 2B) [77].
A study in glioma patients showed that CTLA-4 is highly expressed on T cells, specifically the effector CD4+T cells and Tregs [78]. More importantly, the expression of CTLA-4 is correlated with the glioma patients’ progression. In patients with malignant glioma, the CD4+T-cell count decreased dramatically in the peripheral blood, spleen, and cervical lymph nodes, while Tregs significantly accumulated in the tumor microenvironment; both collectively contributing to the immunosuppressive milieu of the tumor [25, 79]. Consistent observations were also made in mouse models of gliomas [80]. Consequently, when CTLA-4 inhibitor was applied, CD4+T-cell proliferative capacity was enhanced, and the Treg/CD4+ T ratio was lowered although Treg numbers were not affected, which restored T-cell functional defects and produced stronger anti-tumor response in vitro. Based on these findings, we speculate that the biological activities of CTLA-4 in malignant glioma are carried out preferentially through the inhibition of CD4+ T cell activity.
In response to DC vaccines, an innovative therapeutic method for patients with glioma, the down-regulation of CTLA-4 on peripheral blood lymphocyte closely correlates with good prognosis in glioma patients, further supporting the significance of CTLA-4 in regulating anti-tumor immune responses [78].
IDO and glioma
IDO is expressed by various tumor cells and DCs. Although not an immune checkpoint in the classical sense, IDO mediates several inhibitory pathways in various tumor types and suppresses anti-tumor immunity as a mammalian cytosolic enzyme [81].
IDO is a tryptophan (Trp) catabolic enzyme responsible for Trp degradation through the kynurenine (Kyn) degradation pathway [82-84]. IDO from tumor cells catabolizes Trp to numerous metabolites, including Kyn, 3-hydroxykynurenine (3-HK), and 3-hydroxyanthranilic acid (3-HAA). The maintenance and function of T cells require Trp [85]. Therefore, the Trp degradation mediated by IDO suppresses T cell activation. Meanwhile, the metabolites, such as Kyn, 3-HK, and 3-HAA induce T cell apoptosis (Figure 1C) [86]. In addition, when combined with TGF-β, Kyn could induce FoxP3 expression, and stimulate Treg formation [87-89]. And IDO expressed by tumor cells also induces Treg infiltration into tumor microenvironment (Figure 2B). Together, these mechanisms exacerbate immune suppression associated with cancer development.
In glioma, IDO expressed in glioma cells plays an important role in inhibiting T cell functions and contributes to Treg accumulation [90, 91]. Under normal physiology, the CNS parenchyma does not express IDO [92]. 90% of GBM, however, shows positive expression of IDO [90, 93], which is further boosted in low grade glioma, suggesting that IDO expression level correlates with the severity of glioma [94]. Moreover, GBM patients with higher expression of IDO have a worse prognosis [90]. However, a recent study showed that only 8% of GBM expressed IDO [95]. Considering that the antibodies used in these studies, although different from one another, were all well controlled for the detection specificity, these studies suggest that alternative splice variants and/or posttranslational modifications of IDO protein in GBM samples may result in antigenic variations, and thus the dichotomous findings of IDO in GBM.
Other immune checkpoints and glioma
In addition to PD-1, CTLA-4, and IDO, other immune checkpoints are also involved in the occurrence and development of glioma, including T cell membrane protein-3 (TIM-3; also known as HAVcr2), killer inhibitory receptors (KIR), and V-domain Ig-containing suppressor of T cell activation (VISTA). Similar to PD-1, CTLA-4, and IDO, these immune checkpoints inhibit lymphocyte activity and/or induce lymphocyte anergy, and thus are ideal targets for glioma immunotherapy. Blocking antibodies for these immune checkpoints have shown specific anti-tumor activities in animal models, and some are being tested in clinical trials.
TIM-3 is mainly expressed on activated T cells, NK cells, and monocytes [96-98]. It binds to galectin-9, through which it induces Th1 cell death and suppresses the anti-tumor immune response [96]. The significance of the TIM-3 has been demonstrated in different cancer models, including colon cancer, mammary carcinoma, and melanoma [99, 100]. In glioma patients, the expression of TIM-3 on CD4+T cells and CD8+T cells is significantly elevated than in healthy controls, and the higher expression level of TIM-3 on T cells is associated with tumors of higher grades [101].
KIRs (Killer inhibitory receptors) are a collection of inhibitory receptors that can down-regulate NK cells cytotoxic activity and inhibit cytokine secretion of NK cells [102]. Many KIRs are also expressed on T cells and APCs. Activation of KIRs in T cells suppresses the cytolytic activities of these cells. Consistently, expressions of KIRs are detected in multiple cancer types and correlate with poor prognosis [103, 104]. Among KIRs, CD94/NKG2A is an NK inhibitory receptor expressed by most the astrocytoma-infiltrating T cells [105]. An anti-NKG2A antibody (IPH2201) is undergoing phase I/II trial testing in HNSCC patients (NCT02331875) [104]. This antibody could be applied into glioma clinical trial. Many other anti-KIR mAbs are also being tested in anti-tumor clinical trials. A phase I trial of anti-KIR (IPH2101) in patients with acute myelogenous leukemia has been completed. Several studies involving anti-KIR (lirilumab) combined with anti-PD-1 (clinical trial NCT01714739) or with anti-CTLA4 (clinical trial NCT01750580) are undergoing on patients with hematologic and solid cancers. These trials will investigate the anti-cancer effect by simultaneously boosting both the innate immune activity, via anti-KIR, and the adaptive immune activity via anti-PD-1 or anti-CTLA4.
The VISTA is a member in the CD28 receptor family and is mainly expressed on myeloid and granulocytic cells, including naive T cells, NK cells, macrophages, and DCs, but not on B cells [106]. Through the interaction with an unknown receptor on T cells, VISTA negatively inhibits T cell responses [107]: not only T-cell proliferation, but also the expression of activation markers and the production of cytokines. In vitro, VISTA induces the development of Tregs with the help of TGF-β [108]. In line with its biological activities, VISTA-KO mice are resistant to the development of GL261 glioma [109]. In addition, when combined with a cancer vaccine, VISTA blockade inhibits tumor growth [107]. In a glioma mouse model, GL261 cells were directly injected into the left cerebral hemisphere of WT or VISTA-KO mice and the tumor growth was monitored and recorded using imaging technique system all the time. WT mice tumors died within 35 days after GL261 inoculation, while no tumor developed in approximately 20% of VISTA-KO mice. Therefore, targeting VISTA may present potent therapeutic efficacy on glioma.
Other immune checkpoint molecules, including lymphocyte activation gene-3 (LAG-3; also known as CD223), 2B4 (also known as CD244), B and T lymphocyte attenuator (BTLA; also known as CD272), have not been fully explored for their biological activities and functions in glioma. Given their importance as “next-generation” cancer immunotherapy, further studies should be directed on their significance and therapeutic potentials in glioma.
IMMUNE CHECKPOINTS BLOCKADES IN CLINICAL TRIALS FOR MALIGNANT GLIOMA
The preclinical findings suggest that immune checkpoints are optimal targets for cancer immunotherapy, which stimulated the development of inhibitors targeting these checkpoint receptors and/or their ligands, including antibodies for PD-1 (pembrolizumab, nivolumab, and pidilizumab) [110-112], PD-L1 (MEDI4736, MPDL3280a, and MDX-1105) [113, 114], CTLA-4 (ipilimumab and tremelimumab) [115-117], and IDO (indoximod and INCB024360)[118]. Although some of the antibodies have been approved by US Food and Drug Administration (FDA) to treat melanoma and non-small lung cancer, they are still under intensive investigations for the treatment of malignant gliomas (Table 1).
Table 1: Clinical trials on checkpoint-blocking antibodies in patients with glioma
Target |
Intervention |
Clinical Trials No. |
Phase |
Condition |
PD-1 |
Pidilizumab |
NCT01952769 |
Phase I/II |
Diffuse pontine glioma |
Pembrolizumab + MRI-guided laser ablation |
NCT02311582 |
Phase I/II |
Recurrent malignant glioma |
|
Nivolumab + DC Vaccines |
NCT02529072 |
Phase I |
Recurrent brain tumors |
|
Pembrolizuma + Adenovirus |
NCT02798406 |
Phase II |
Recurrent glioblastoma or gliosarcoma |
|
Nivolumab + FPA008 |
NCT02526017 |
Phase I |
Advanced solid tumors, including glioma |
|
Nivolumab +Galunisertib |
NCT02423343 |
Phase I/II |
Advanced solid tumors, including glioma |
|
Nivolumab + anti-LAG-3 or anti-CD137 |
NCT02658981 |
Phase I |
Recurrent glioblastoma |
|
CTLA-4 & PD-1 |
Ipilimumab/nivolumab, or both + Temozolomide |
NCT02311920 |
Phase I |
Newly diagnosed glioblastoma or gliosarcoma |
Nivolumab ± Ipilimumab vs Bevacizumab |
NCT02017717 |
Phase III |
Recurrent glioblastoma |
|
PD-L1 |
MEDI4736 ± radiotherapy vs MEDI4736 + Bevacizuma |
NCT02336165 |
Phase II |
Glioblastoma |
IDO |
Indoximod + Temozolomide + Bevacizumab + Radiation |
NCT02052648 |
Phase I/II |
Adult patients with primary malignant brain tumors |
Indoximod + Temozolomide + Conformal Radiation |
NCT02502708 |
Phase I |
Pediatric patients with primary malignant brain tumors |
PD-1 blockade
Preclinical studies demonstrated the potential of PD-1 antibodies enhanced the anti-tumor immune responses in a variety of glioma mouse models [60]. At present, at least three PD-1 blocking antibodies are in clinical trials for glioma patients, including pembrolizumab (a humanized IgG4 antibody), nivolumab (a fully human IgG4 antibodies), and pidilizumab (a humanized IgG1 antibody). Pembrolizumab and nivolumab have been approved by the US FDA for melanoma, non-small cell lung cancer, renal cell cancer, Hodgkin’s lymphoma, squamous-cell carcinoma of the head and neck, and bladder cancer [119-124].
For glioma, a Phase I/II clinical trial of pidilizumab (NCT01952769) started in February 2014 in patients with diffuse intrinsic pontine glioma, and is expected to end in November 2018. In this study, treatment-related toxicity and progression-free survival will be recorded to evaluate the safety, toxicities, and efficacy of the antibody. Meanwhile, a phase I/II study of pembrolizumab is ongoing among patients with recurrent malignant gliomas to evaluate the maximal tolerated dose and progression-free survival of pembrolizumab in combination with radiotherapy. This study received the first patient in December 4, 2014, and is expected to be completed in June 2018. (Table 1)
Recently, several studies reported the safety/tolerability of the anti-PD-1 antibody nivolumab, alone or in combination with other treatments in patients with high-grade glioblastoma (GBM) [125, 126]. A randomized trial reported at the ASCO 2015 showed that nivolumab alone or combined with ipilimumab did not generate any serious and specific toxicities in 20 patients with recurrent GBM. Another study reported on the 20th Annual Scientific Meeting of the Society for Neuro tested the concurrent therapy of pembrolizumab or nivolumab with different treatment regimens on 12 patients with recurrent high grade GBM and found no toxicities [127]. Thus, PD-1 inhibitors can be administered safely to patients with other therapies including re-irradiation. An ongoing phase III study (NCT02617589 ) was designed to compare overall survival of nivolumab versus TMZ, each in combination with radiation in newly diagnosed GBM patients with unmethylated MGMT. Therapeutic effects will be judged by tumor progression, safety and tolerability, and survival [128]. There were no serious and specific toxicities associated with PD-1 inhibitors among all clinical trials. Therefore, PD-1 inhibitors can be safely administered to patients with high-grade gliomas in combination with other treatments such as radiation.
PD-L1 blockade
PD-L1 is another promising target for disrupting the PD-1/PD-L1 pathway. Three PD-L1 clinical agents have demonstrated clinical activity in several solid tumor types, including MEDI4736, MPDL3280a, and MDX-1105 antibodies [129, 130]. MEDI4736 and MPDL3280A are two kinds of Fc-modified human IgG1 antibodies. MDX-1105 is a fully human IgG4 antibody. Among these agents, MEDI4736 is being tested in patients with GBM (NCT02336165) (Table 1), where GBM patients are divided into three group: patients with newly diagnosed GBM are given with MEDI4736 every 2 weeks combined with radiotherapy; Bevacizumab-naïve patients with recurrent GBM receive MEDI4736 every 2 weeks as monotherapy or in combination with bevacizumab every 2 weeks; bevacizumab-refractory subjects with recurrent GBM are administered MEDI4736 every 2 weeks in combination with continued bevacizumab. Clinical efficacy and quality of life are being recorded in this phase II study.
CTLA-4 blockade
The anti-CTLA-4 monoclonal antibodies are the first immune checkpoint inhibitors in clinical trial. In 2000, the blockades of CTLA-4, ipilimumab (a fully human IgG1 antibody), and tremelimumab (a fully human IgG2 antibody) entered clinical trials. Ipilimumab is the first agent approved by the US FDA for the treatment of melanoma in 2010 [131].
A phase I and phase III clinical trials of ipilumumab, in combination with another immunomodulatory nivolumab in GBM are currently ongoing (NCT02311920, NCT02017717) (Table1). The phase I clinical trial mainly evaluates the dose limiting toxicities of ipilimumab, nivolumab, and their combination in patients with newly diagnosed GBM who receive temozolomide. The phase III clinical trial mainly evaluates the efficacy and safety of nivolumab, with or without ipilimumab, and compares the efficacy and safety of nivolumab versus bevacizumab in GBM patients at different stages of treatment.
Neuro-Oncology recently published a clinical trial on anti-CTLA-4 monoclonal antibody. In this study, seven patients (age range: 39 to 66 years) with high-grade gliomas (one with recurrent grade 3 and 6 with grade 4 glioma) were treated with ipilimumab between June 2012 and April 2014 [132]. Five patients received other therapies, including bevacizumab, in combination with ipilimumab. Of the 7 patients, 3 died within 5 months of the treatment and 4 have survived for 3.2-23.3 months. Specifically, one patient with anaplastic astrocytoma achieved recurrence-free after 3.2 months of treatment, and has stable disease with improvement in functional status, steroid requirement and other clinical symptoms. The up-regulation of granzyme B expression was also found through immunohistochemistry, indicating a stronger immune response. Accordingly, a subset of patients with recurrent high-grade glioma may benefit from anti-CTLA-4 treatment.
IDO blockade
Several inhibitors targeting IDO, including competitive inhibitor (such as 1-MT), and uncompetitive inhibitor (such as exiguamine A) have been developed. 1-MT has two optical isomers, left-handed form (L-1-MT) and right-handed form (D-1-MT). The phase I clinical trial of D-1-MT (indoximod) combined with chemotherapy has completed [83, 133]. The safety and anti-tumor activity have been demonstrated. Meanwhile, INCB024360, a new-generation IDO inhibitor, has entered clinical tests.
The clinical trials of indoximod on glioma are undergoing (NCT02052648 and NCT02502708), which will lay the foundation for combination therapy of indoximod together with radiation and temozolomide for glioma patients, including adult patients and children. The phase I/II clinical study (NCT02052648) of indoximod are conducted among adult patients with recurrent malignant brain tumors. In this study, the patients will receive the combination of indoximod and temozolomide with either stereotactic radiation or bevacizumab. The safety and the optimal dose of indoximod will be identified. This clinical trial brings IDO-based immunotherapy into glioma therapy. In another phase I clinical trial (NCT02502708) using indoximod in combination with temozolomide plus radiation to treat children with brain tumors, the test dose of indoximod will be from 12.8 mg/kg/dose to 22.4 mg/kg/dose; the safety and tolerability will be also assessed by incidence and severity of adverse events. (Table 1)
THE PRECLINICAL COMBINATION STUDY FOR MALIGNANT GLIOMA
Cancer employs multiple mechanisms to foster its own growth and metastasis [134]. Therefore, the combination therapy targeting distinct mechanisms simultaneously offers advantages over single-drug therapy. In this section, we will discuss the latest combination studies on immune checkpoint inhibitors in mouse model of malignant gliomas.
Combination of different immune checkpoint blockades
The early preclinical studies have shown that the combination of PD-1/PD-L1 and CTLA-4 blockades is more than two times as effective as either alone to eradicate tumor cells. The synergistic anti-tumor activity has been verified in mouse models of melanoma and colon adenocarcinoma [135, 136].
Basic immunologic observations support the notion that PD-1/PD-L1 pathway, CTLA-4, and IDO regulate T-cell responses and promote tumor immunosuppression through overlapping as well as non-redundant mechanisms [40, 75, 76]. In addition, these immune checkpoint molecules regulate each other and further promote the immunosuppression [137]. PD-1/PD-L1 pathway and IDO could induce FoxP3 expression and promote Treg differentiation. FoxP3 controls CTLA-4 expression in Tregs [40, 84]. The CTLA-4 could bind to the CD80/86, and up-regulate the expression of IDO [77]. Consistently with their functions, PD-1/PD-L1, CTLA-4, and IDO are highly expressed in glioma microenvironment. When mAbs for CTLA-4 and PD-L1, together with the IDO inhibitor, 1-MT, were administered to mice bearing glioma, Treg levels in glioma microenvironment decreased significantly, and the combination was more effective than the single agent [138].
To achieve the maximal immune suppression during cancer development, immune checkpoint molecules are frequently co-expressed on lymphocytes. For example, LAG-3 and TIM-3 are commonly co-expressed with PD-1 on tumor-infiltrating T cells and work synergistically to regulate immune functions [139, 140], suppressing the proliferation of T cells and the production of cytokines such as IL-2, TNF, and IFN-γ. Consequently, the combined use of inhibitors for LAG-3/TIM-3 and PD-1 was more effective in stimulating anti-tumor immunity than blocking each individual component in different tumor mode [139, 140]. Similarly, the combination of anti-TIM-3 and anti-PD-1 with stereotactic radiosurgery has shown the potential to enhance anti-tumor immunity and prolong survival in a mouse model of glioma [141].
Immune checkpoint blockades combined with traditional therapy
The traditional therapies for glioma mainly involve surgical removal, chemotherapy, and radiotherapy, which, however, could not eliminate the tumor completely due to the extensive tumor infiltration into the surrounding stroma. In this regard, immunotherapy provides a promising complementary therapy in eradicating glioma.
A recent study in a mouse orthotopic GBM model showed that combining steteotactic radiation with anti-PD-1 blockade increased the tumor-infiltrating cytotoxic T cells while reduced tumor-associated Tregs, resulting in prolonged mouse survival [142]. In this study, the stereotactic radiosurgery delivers high-dose radiation in a single session, which may disrupt the immunosuppressive microenvironment without several immunologic and neurologic side effects associated with conventional radiotherapy. The anti-PD-1 blockade enhances the anti-tumor immunity, maintains the immunologic memory, and synergizes with stereotactic radiosurgery.
In the subcutaneous or intracranial glioma mouse model, oral administration of IDO inhibitors and temozolomide significantly inhibited tumor growth, prolonged survival, and presented synergistic anti-tumor effects. Additional radiation therapy in combination with IDO inhibitors and temozolomide contributes to widespread complement deposition and prolongs mouse survival [143, 144].
Immune checkpoints blockades combined with other immunotherapy
Cancer immunotherapy continues to translate finding from immune molecular mechanisms to novel therapeutic approaches. The current immunotherapeutic approaches mainly include tumor antigen-targeted monoclonal antibodies, immune checkpoint inhibitors, cytokine therapy, and cancer vaccines. Each approach acts through a distinct mechanism, yet all work to boost anti-tumor immunity [7]. Therefore, it is expected that combining different immunotherapeutic approaches would benefit cancer treatment. Agarwalla et al. showed that sequential immunotherapy with vaccination by irradiation glioma cells producing high levels of granulocyte-macrophage colony stimulating factor followed by CTLA-4 blockade significantly improved the survival of mice with intracranial tumors [145]. Berg et al. demonstrated the potency of combining intratumoral IL-12 administration with CTLA-4 blockade in eradicating glioma cells, which was associated with the recruitment of effector T cells and the reduction of Tregs in tumor [146]. In another study, depletion of Tregs by blocking CD25 strongly enhanced the anti-tumor efficacy of DC vaccination in murine glioma model [147]. Moreover, co-administration of anti-CTLA-4 antibody and Treg depletion was used to reverse the tumor immune tolerance, further strengthened CD4+ and CD8+ effector T cells, and resulted in complete glioma eradication without autoimmunity [148]. A triple therapy including the agonist antibodies for 4-1BB (a co-stimulatory molecule expressed on T cells that can enhance the T cell-mediated rejection of tumors), CTLA-4 blocking antibodies, and focal radiation therapy, when compared to component treatment, dramatically improved the long-term tumor-free survival of mice with intracranial gliomas [149].
FUTURE RESEARCH DIRECTION
Although blocking antibodies for immune checkpoints significantly improved the anti-tumor activities of other therapeutic approaches and prove to be a valuable treatment strategy, the high cost associated with generating a decent amount of blocking antibodies for treatment challenges their accessibility to general public. Furthermore, systemic delivery of immune checkpoint blockades may also lead to unwanted side effects, such as dose-dependent autoimmune responses. Therefore, future research should be directed, on one hand, toward improving the delivery vehicles and delivery approaches to boost the concentrations of the blocking antibodies within tumor microenvironment, reducing the therapeutic dosage and minimizing side effects, and on the other hand, toward designing novel approaches to target immune checkpoints.
The blood-brain barrier is the main obstacle for the entrance of therapeutic drugs into the brain. Recently, an advanced self-degradable microneedle patch was invented for the sustained release of anti-PD-1 antibody with minimal invasiveness and least dose of antibody required [150]. The microneedle is made up of hyaluronic acid and pH-sensitive nanoparticles encapsulating anti-PD-1 and glucose oxidase. Glucose oxidase could convert glucose to gluconic acid. The generation of gluconic acid changes the pH, induces the self-dissociation of nanoparticles and promotes substantial release of anti-PD-1 subsequently. This administration strategy has demonstrated its potential for inducing stronger immune responses compared to intratumoral injection of anti-PD-1 with the same dose in a melanoma mouse model [151]. This novel approach allows the compatibility with blocking antibodies for other immune checkpoints (such as anti-CTLA-4) as well as targeting gliomas, since the patch could be directly delivered to the capillary bed within the brain, bypassing the blood-brain barrier. Similar to the microneedle patch, osmotic mini pumps are under intensive investigations in preclinical trial for the treatment of glioma [146].
Unlike blocking antibodies, microRNAs (miRNAs) is a promising strategy to down-regulating gene expression by either inducing mRNA degradation or inhibiting mRNA translation [152]. MiR-138, a miRNA abundant in the brain, is a tumor suppressor, with its inhibition detected in multiple cancers. Both PD-1 and CTLA-4 are downstream targets for miR-138. Upon transfection into CD4+T cells, miR-138 could inhibit the expressions of CTLA-4, PD-1, and FoxP3 in T cells. In vivo, miR-138 treatment potently suppresses glioma growth, decreasing the ratio of intratumoral Treg, reducing the expression of CTLA-4 and PD-1, and leading to a 43% increase in the median survival time [153].
CONCLUSIONS
The area on immune checkpoint blockades has witnessed rapid progression, especially the monoclonal antibodies for PD-1/PD-L1 and CTLA-4, which have been proved by FDA for melanoma and/or lung cancer as sole reagents. Meanwhile, more new reagents are under development, including those targeting LAG-3, TIM-3, and other checkpoint inhibitors. By far, no immune checkpoint blockade has been approved by the FDA for the treatment of malignant glioma. This requires the further understanding on neuroimmunology related to malignant glioma, the translation of mechanistic findings, and the design of rational combinatorial regimens involving immune checkpoint blockades.
Abbreviations
3-HK, 3-hydroxykynurenine; 3-HAA, 3-hydroxyanthranilic acid; AHR, aryl hydrocarbon receptor; ALK, anaplastic lymphoma kinase; APCs, antigen-presenting cells; BTLA, B and T lymphocyte attenuator; CNS, central nervous system; CTLA-4, the cytotoxic T-lymphocyte-associated antigen-4; DCs, dendritic cells; FoxP3, Forkhead box P3; GBM, glioblastoma multiforme; IDO, indoleamine 2,3-dioxygenase; KIR, killer inhibitory receptors; Kyn, kynurenine; LAG-3, Lymphocyte activation gene-3; MDSCs, myeloid-derived suppressor cells; MHC, major histocompatibility complex; NK cells, natural killer cells; PD-1, programmed cell death protein-1; PTEN, phosphatase and tensin homolog; SHP2, Src homology2 domain-containing tyrosine phosphatase; TGF-β, transforming growth factor beta; TILs, tumor infiltrating lymphocytes; TIM-3, T cell membrane protein-3; Tregs, regulatory T cells; Trp, tryptophan; VEGF, vascular endothelial growth factor; VISTA, V-domain Ig-containing suppressor of T cell activation.
ConflictS of Interest
There is no conflict of interest.
GRANT SUPPORT
This work was supported by the National Major Scientific and Technological Special Project for “Significant New Drugs Development” during the Twelfth Five-year Plan Period (No. 2013ZX09102032), the National Natural Science Foundation of China (No. 81402436), the Key Scientific Project of Jilin Province (No. 20140204024YY), the Scientific and Technological Developing Plan of Jilin Province (No. 20150520155JH), the Norman Bethune Program of Jilin University(No. 2015413), and the Foundation for The Excellent Youth Scholars of Jilin University. We sincerely apologize to the authors whose work was not cited in this review due to the space limitations.
REFERENCES
1. Omuro A and DeAngelis LM. Glioblastoma and other malignant gliomas: a clinical review. Jama. 2013; 310:1842-1850.
2. Wen PY and Kesari S. Malignant gliomas in adults. The New England journal of medicine. 2008; 359:492-507.
3. Dolecek TA, Propp JM, Stroup NE and Kruchko C. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2005-2009. Neuro-oncology. 2012; 14 Suppl 5:v1-49.
4. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, Gorlia T, Allgeier A, Lacombe D, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. The New England journal of medicine. 2005; 352:987-996.
5. Woehrer A, Bauchet L and Barnholtz-Sloan JS. Glioblastoma survival: has it improved? Evidence from population-based studies. Current opinion in neurology. 2014; 27:666-674.
6. Coley WB. The treatment of malignant tumors by repeated inoculations of erysipelas. With a report of ten original cases. 1893. Clinical orthopaedics and related research. 1991; :3-11.
7. Mellman I, Coukos G and Dranoff G. Cancer immunotherapy comes of age. Nature. 2011; 480:480-489.
8. Chen L and Han X. Anti-PD-1/PD-L1 therapy of human cancer: past, present, and future. The Journal of clinical investigation. 2015; 125:3384-3391.
9. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nature reviews Cancer. 2012; 12:252-264.
10. Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, Ohgaki H, Wiestler OD, Kleihues P and Ellison DW. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol. 2016; 131:803-820.
11. Heimberger AB and Sampson JH. Immunotherapy coming of age: what will it take to make it standard of care for glioblastoma? Neuro-oncology. 2011; 13:3-13.
12. Sehgal A and Berger MS. Basic concepts of immunology and neuroimmunology. Neurosurgical focus. 2000; 9:e1.
13. Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, Peske JD, Derecki NC, Castle D, Mandell JW, Lee KS, Harris TH and Kipnis J. Structural and functional features of central nervous system lymphatic vessels. Nature. 2015.
14. Aspelund A, Antila S, Proulx ST, Karlsen TV, Karaman S, Detmar M, Wiig H and Alitalo K. A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. The Journal of experimental medicine. 2015; 212:991-999.
15. Ransohoff RM, Schafer D, Vincent A, Blachere NE and Bar-Or A. Neuroinflammation: Ways in Which the Immune System Affects the Brain. Neurotherapeutics. 2015; 12:896-909.
16. Brierley JB and Field EJ. The connexions of the spinal sub-arachnoid space with the lymphatic system. J Anat. 1948; 82:153-166.
17. Foldi M, Csanda E, Simon M, Obal F, Schneider I, Dobranovics I, Zoltan OT, Kozma M and Poberai M. Lymphogenic haemangiopathy. “Prelymphatic” pathways in the wall of cerebral and cervical blood vessels. Angiologica. 1968; 5:250-262.
18. Bradbury M. Lymphatics and the central nervous system. Trends in Neurosciences. 1981; 4:100-101.
19. He G, Lu T, Lu B, Xiao D, Yin J, Liu X, Qiu G, Fang M and Wang Y. Perivascular and perineural extension of formed and soluble blood elements in an intracerebral hemorrhage rat model. Brain Res. 2012; 1451:10-18.
20. Hatterer E, Davoust N, Didier-Bazes M, Vuaillat C, Malcus C, Belin MF and Nataf S. How to drain without lymphatics? Dendritic cells migrate from the cerebrospinal fluid to the B-cell follicles of cervical lymph nodes. Blood. 2006; 107:806-812.
21. Ransohoff RM and Engelhardt B. The anatomical and cellular basis of immune surveillance in the central nervous system. Nature reviews Immunology. 2012; 12:623-635.
22. Dunn GP, Dunn IF and Curry WT. Focus on TILs: Prognostic significance of tumor infiltrating lymphocytes in human glioma. Cancer Immun. 2007; 7:12.
23. Platten M, Wick W and Weller M. Malignant glioma biology: role for TGF-beta in growth, motility, angiogenesis, and immune escape. Microscopy research and technique. 2001; 52:401-410.
24. Roszman T, Elliott L and Brooks W. Modulation of T-cell function by gliomas. Immunology today. 1991; 12:370-374.
25. Fecci PE, Mitchell DA, Whitesides JF, Xie W, Friedman AH, Archer GE, Herndon JE, 2nd, Bigner DD, Dranoff G and Sampson JH. Increased regulatory T-cell fraction amidst a diminished CD4 compartment explains cellular immune defects in patients with malignant glioma. Cancer research. 2006; 66:3294-3302.
26. John LB, Devaud C, Duong CP, Yong CS, Beavis PA, Haynes NM, Chow MT, Smyth MJ, Kershaw MH and Darcy PK. Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells. Clinical cancer research. 2013; 19:5636-5646.
27. Tanaka Y and Okamura H. [Anti-PD-1 antibody: basics and clinical application]. Gan to kagaku ryoho Cancer & chemotherapy. 2013; 40:1145-1149.
28. Kasagi S, Kawano S, Okazaki T, Honjo T, Morinobu A, Hatachi S, Shimatani K, Tanaka Y, Minato N and Kumagai S. Anti-programmed cell death 1 antibody reduces CD4+PD-1+ T cells and relieves the lupus-like nephritis of NZB/W F1 mice. Journal of immunology. 2010; 184:2337-2347.
29. Shu Q, Antalffy B, Su JM, Adesina A, Ou CN, Pietsch T, Blaney SM, Lau CC and Li XN. Valproic Acid prolongs survival time of severe combined immunodeficient mice bearing intracerebellar orthotopic medulloblastoma xenografts. Clinical cancer research. 2006; 12:4687-4694.
30. Fanoni D, Tavecchio S, Recalcati S, Balice Y, Venegoni L, Fiorani R, Crosti C and Berti E. New monoclonal antibodies against B-cell antigens: possible new strategies for diagnosis of primary cutaneous B-cell lymphomas. Immunology letters. 2011; 134:157-160.
31. Terme M, Ullrich E, Aymeric L, Meinhardt K, Desbois M, Delahaye N, Viaud S, Ryffel B, Yagita H, Kaplanski G, Prevost-Blondel A, Kato M, Schultze JL, Tartour E, Kroemer G, Chaput N, et al. IL-18 induces PD-1-dependent immunosuppression in cancer. Cancer research. 2011; 71:5393-5399.
32. Boulis NM, Turner DE, Imperiale MJ and Feldman EL. Neuronal survival following remote adenovirus gene delivery. Journal of neurosurgery. 2002; 96:212-219.
33. Dickinson H, Carico C, Nuno M, Mukherjee D, Ortega A, Black KL and Patil CG. Unplanned readmissions and survival following brain tumor surgery. Journal of neurosurgery. 2015; 122:61-68.
34. Taneja SS. Re: Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. The Journal of urology. 2012; 188:2149.
35. Keir ME, Liang SC, Guleria I, Latchman YE, Qipo A, Albacker LA, Koulmanda M, Freeman GJ, Sayegh MH and Sharpe AH. Tissue expression of PD-L1 mediates peripheral T cell tolerance. The Journal of experimental medicine. 2006; 203:883-895.
36. Philips MF, Muir JK, Saatman KE, Raghupathi R, Lee VM, Trojanowski JQ and McIntosh TK. Survival and integration of transplanted postmitotic human neurons following experimental brain injury in immunocompetent rats. Journal of neurosurgery. 1999; 90:116-124.
37. Pobereskin LH. Influence of premorbid factors on survival following subarachnoid hemorrhage. Journal of neurosurgery. 2001; 95:555-559.
38. Riley JL. PD-1 signaling in primary T cells. Immunological reviews. 2009; 229:114-125.
39. Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, Freeman GJ and Ahmed R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006; 439:682-687.
40. Francisco LM, Salinas VH, Brown KE, Vanguri VK, Freeman GJ, Kuchroo VK and Sharpe AH. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. The Journal of experimental medicine. 2009; 206:3015-3029.
41. Butte MJ, Keir ME, Phamduy TB, Sharpe AH and Freeman GJ. Programmed death-1 ligand 1 interacts specifically with the B7-1 costimulatory molecule to inhibit T cell responses. Immunity. 2007; 27:111-122.
42. Macedo C, Oliveira EH, Almeida RS, Donate PB, Fornari TA, Pezzi N, Sakamoto-Hojo ET, Donadi EA and Passos GA. Aire-dependent peripheral tissue antigen mRNAs in mTEC cells feature networking refractoriness to microRNA interaction. Immunobiology. 2015; 220:93-102.
43. Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, Roche PC, Lu J, Zhu G, Tamada K, Lennon VA, Celis E and Chen L. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nature medicine. 2002; 8:793-800.
44. Ota K, Azuma K, Kawahara A, Hattori S, Iwama E, Tanizaki J, Harada T, Matsumoto K, Takayama K, Takamori S, Kage M, Hoshino T, Nakanishi Y and Okamoto I. Induction of PD-L1 Expression by the EML4-ALK Oncoprotein and Downstream Signaling Pathways in Non-Small Cell Lung Cancer. Clinical cancer research. 2015; 21:4014-4021.
45. Robertson PL, Zeltzer PM, Boyett JM, Rorke LB, Allen JC, Geyer JR, Stanley P, Li H, Albright AL, McGuire-Cullen P, Finlay JL, Stevens KR, Jr., Milstein JM, Packer RJ and Wisoff J. Survival and prognostic factors following radiation therapy and chemotherapy for ependymomas in children: a report of the Children’s Cancer Group. Journal of neurosurgery. 1998; 88:695-703.
46. Wilmotte R, Burkhardt K, Kindler V, Belkouch MC, Dussex G, Tribolet N, Walker PR and Dietrich PY. B7-homolog 1 expression by human glioma: a new mechanism of immune evasion. Neuroreport. 2005; 16:1081-1085.
47. Topalian SL, Drake CG and Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer cell. 2015; 27:450-461.
48. Marzec M, Zhang Q, Goradia A, Raghunath PN, Liu X, Paessler M, Wang HY, Wysocka M, Cheng M, Ruggeri BA and Wasik MA. Oncogenic kinase NPM/ALK induces through STAT3 expression of immunosuppressive protein CD274 (PD-L1, B7-H1). Proceedings of the National Academy of Sciences of the United States of America. 2008; 105:20852-20857.
49. Parsa AT, Waldron JS, Panner A, Crane CA, Parney IF, Barry JJ, Cachola KE, Murray JC, Tihan T, Jensen MC, Mischel PS, Stokoe D and Pieper RO. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nature medicine. 2007; 13:84-88.
50. Chen J, Feng Y, Lu L, Wang H, Dai L, Li Y and Zhang P. Interferon-gamma-induced PD-L1 surface expression on human oral squamous carcinoma via PKD2 signal pathway. Immunobiology. 2012; 217:385-393.
51. Lee SK, Seo SH, Kim BS, Kim CD, Lee JH, Kang JS, Maeng PJ and Lim JS. IFN-gamma regulates the expression of B7-H1 in dermal fibroblast cells. Journal of dermatological science. 2005; 40:95-103.
52. D’Angelo SP, Shoushtari AN, Agaram NP, Kuk D, Qin LX, Carvajal RD, Dickson MA, Gounder M, Keohan ML, Schwartz GK and Tap WD. Prevalence of tumor-infiltrating lymphocytes and PD-L1 expression in the soft tissue sarcoma microenvironment. Human pathology. 2015; 46:357-365.
53. Kuang DM, Zhao Q, Peng C, Xu J, Zhang JP, Wu C and Zheng L. Activated monocytes in peritumoral stroma of hepatocellular carcinoma foster immune privilege and disease progression through PD-L1. The Journal of experimental medicine. 2009; 206:1327-1337.
54. Bloch O, Crane CA, Kaur R, Safaee M, Rutkowski MJ and Parsa AT. Gliomas promote immunosuppression through induction of B7-H1 expression in tumor-associated macrophages. Clinical cancer research. 2013; 19:3165-3175.
55. Shin T, Kennedy G, Gorski K, Tsuchiya H, Koseki H, Azuma M, Yagita H, Chen L, Powell J, Pardoll D and Housseau F. Cooperative B7-1/2 (CD80/CD86) and B7-DC costimulation of CD4+ T cells independent of the PD-1 receptor. The Journal of experimental medicine. 2003; 198:31-38.
56. Rosenwald A, Wright G, Leroy K, Yu X, Gaulard P, Gascoyne RD, Chan WC, Zhao T, Haioun C, Greiner TC, Weisenburger DD, Lynch JC, Vose J, Armitage JO, Smeland EB, Kvaloy S, et al. Molecular diagnosis of primary mediastinal B cell lymphoma identifies a clinically favorable subgroup of diffuse large B cell lymphoma related to Hodgkin lymphoma. The Journal of experimental medicine. 2003; 198:851-862.
57. Steidl C, Shah SP, Woolcock BW, Rui L, Kawahara M, Farinha P, Johnson NA, Zhao Y, Telenius A, Neriah SB, McPherson A, Meissner B, Okoye UC, Diepstra A, van den Berg A, Sun M, et al. MHC class II transactivator CIITA is a recurrent gene fusion partner in lymphoid cancers. Nature. 2011; 471:377-381.
58. Shin T, Yoshimura K, Shin T, Crafton EB, Tsuchiya H, Housseau F, Koseki H, Schulick RD, Chen L and Pardoll DM. In vivo costimulatory role of B7-DC in tuning T helper cell 1 and cytotoxic T lymphocyte responses. The Journal of experimental medicine. 2005; 201:1531-1541.
59. Wang S, Bajorath J, Flies DB, Dong H, Honjo T and Chen L. Molecular modeling and functional mapping of B7-H1 and B7-DC uncouple costimulatory function from PD-1 interaction. The Journal of experimental medicine. 2003; 197:1083-1091.
60. Wei B, Wang L, Zhao X, Du C, Guo Y and Sun Z. The upregulation of programmed death 1 on peripheral blood T cells of glioma is correlated with disease progression. Tumour biology. 2014; 35:2923-2929.
61. Dubinski D, Wolfer J, Hasselblatt M, Schneider-Hohendorf T, Bogdahn U, Stummer W, Wiendl H and Grauer OM. CD4+ T effector memory cell dysfunction is associated with the accumulation of granulocytic myeloid-derived suppressor cells in glioblastoma patients. Neuro-oncology. 2015.
62. El Andaloussi A and Lesniak MS. An increase in CD4+CD25+FOXP3+ regulatory T cells in tumor-infiltrating lymphocytes of human glioblastoma multiforme. Neuro-oncology. 2006; 8:234-243.
63. Avril T, Saikali S, Vauleon E, Jary A, Hamlat A, De Tayrac M, Mosser J and Quillien V. Distinct effects of human glioblastoma immunoregulatory molecules programmed cell death ligand-1 (PDL-1) and indoleamine 2,3-dioxygenase (IDO) on tumour-specific T cell functions. Journal of Neuroimmunology. 2010; 225:22-33.
64. Yao Y, Tao R, Wang X, Wang Y, Mao Y and Zhou LF. B7-H1 is correlated with malignancy-grade gliomas but is not expressed exclusively on tumor stem-like cells. Neuro-oncology. 2009; 11:757-766.
65. Berghoff AS, Ricken G, Widhalm G, Rajky O, Hainfellner JA, Birner P, Raderer M and Preusser M. PD1 (CD279) and PD-L1 (CD274, B7H1) expression in primary central nervous system lymphomas (PCNSL). Clin Neuropathol. 2014; 33:42-49.
66. Preusser M, Berghoff AS, Wick W and Weller M. Clinical Neuropathology mini-review 6-2015: PD-L1: emerging biomarker in glioblastoma? Clin Neuropathol. 2015; 34:313-321.
67. Berghoff AS, Kiesel B, Widhalm G, Rajky O, Ricken G, Wohrer A, Dieckmann K, Filipits M, Brandstetter A, Weller M, Kurscheid S, Hegi ME, Zielinski CC, Marosi C, Hainfellner JA, Preusser M, et al. Programmed death ligand 1 expression and tumor-infiltrating lymphocytes in glioblastoma. Neuro-oncology. 2015; 17:1064-1075.
68. Kingwell K. Neuro-oncology: Glioblastoma prognosis linked to neuronal PD-L1 expression in tumour-adjacent tissue. Nature reviews Neurology. 2013; 9:602-603.
69. Liu Y, Carlsson R, Ambjorn M, Hasan M, Badn W, Darabi A, Siesjo P and Issazadeh-Navikas S. PD-L1 expression by neurons nearby tumors indicates better prognosis in glioblastoma patients. The Journal of neuroscience. 2013; 33:14231-14245.
70. Nduom EK, Wei J, Yaghi NK, Huang N, Kong LY, Gabrusiewicz K, Ling X, Zhou S, Ivan C, Chen JQ, Burks JK, Fuller GN, Calin GA, Conrad CA, Creasy C, Ritthipichai K, et al. PD-L1 expression and prognostic impact in glioblastoma. Neuro-oncology. 2016; 18:195-205.
71. Brunet JF, Denizot F, Luciani MF, Roux-Dosseto M, Suzan M, Mattei MG and Golstein P. A new member of the immunoglobulin superfamily—CTLA-4. Nature. 1987; 328:267-270.
72. Peggs KS, Quezada SA, Chambers CA, Korman AJ and Allison JP. Blockade of CTLA-4 on both effector and regulatory T cell compartments contributes to the antitumor activity of anti-CTLA-4 antibodies. The Journal of experimental medicine. 2009; 206:1717-1725.
73. Azuma M, Ito D, Yagita H, Okumura K, Phillips JH, Lanier LL and Somoza C. B70 antigen is a second ligand for CTLA-4 and CD28. Nature. 1993; 366:76-79.
74. Rudd CE, Taylor A and Schneider H. CD28 and CTLA-4 coreceptor expression and signal transduction. Immunological reviews. 2009; 229:12-26.
75. Walker LS and Sansom DM. Confusing signals: recent progress in CTLA-4 biology. Trends in immunology. 2015; 36:63-70.
76. Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, Nomura T and Sakaguchi S. CTLA-4 control over Foxp3+ regulatory T cell function. Science. 2008; 322:271-275.
77. Munn DH, Sharma MD and Mellor AL. Ligation of B7-1/B7-2 by Human CD4+ T Cells Triggers Indoleamine 2,3-Dioxygenase Activity in Dendritic Cells. The Journal of Immunology. 2004; 172:4100-4110.
78. Fong B, Jin R, Wang X, Safaee M, Lisiero DN, Yang I, Li G, Liau LM and Prins RM. Monitoring of regulatory T cell frequencies and expression of CTLA-4 on T cells, before and after DC vaccination, can predict survival in GBM patients. PloS one. 2012; 7:e32614.
79. Learn CA, Fecci PE, Schmittling RJ, Xie W, Karikari I, Mitchell DA, Archer GE, Wei Z, Dressman H and Sampson JH. Profiling of CD4+, CD8+, and CD4+CD25+CD45RO+FoxP3+ T cells in patients with malignant glioma reveals differential expression of the immunologic transcriptome compared with T cells from healthy volunteers. Clinical cancer research. 2006; 12:7306-7315.
80. Fecci PE, Ochiai H, Mitchell DA, Grossi PM, Sweeney AE, Archer GE, Cummings T, Allison JP, Bigner DD and Sampson JH. Systemic CTLA-4 blockade ameliorates glioma-induced changes to the CD4+ T cell compartment without affecting regulatory T-cell function. Clinical cancer research. 2007; 13:2158-2167.
81. Prendergast GC, Smith C, Thomas S, Mandik-Nayak L, Laury-Kleintop L, Metz R and Muller AJ. Indoleamine 2,3-dioxygenase pathways of pathogenic inflammation and immune escape in cancer. Cancer immunology, immunotherapy. 2014; 63:721-735.
82. Godin-Ethier J, Hanafi LA, Piccirillo CA and Lapointe R. Indoleamine 2,3-dioxygenase expression in human cancers: clinical and immunologic perspectives. Clinical cancer research. 2011; 17:6985-6991.
83. Lob S, Konigsrainer A, Rammensee HG, Opelz G and Terness P. Inhibitors of indoleamine-2,3-dioxygenase for cancer therapy: can we see the wood for the trees? Nature reviews Cancer. 2009; 9:445-452.
84. Mbongue JC, Nicholas DA, Torrez TW, Kim NS, Firek AF and Langridge WH. The Role of Indoleamine 2, 3-Dioxygenase in Immune Suppression and Autoimmunity. Vaccines. 2015; 3:703-729.
85. Fallarino F, Grohmann U, You S, McGrath BC, Cavener DR, Vacca C, Orabona C, Bianchi R, Belladonna ML, Volpi C, Santamaria P, Fioretti MC and Puccetti P. The combined effects of tryptophan starvation and tryptophan catabolites down-regulate T cell receptor zeta-chain and induce a regulatory phenotype in naive T cells. Journal of immunology. 2006; 176:6752-6761.
86. Frumento G, Rotondo R, Tonetti M, Damonte G, Benatti U and Ferrara GB. Tryptophan-derived catabolites are responsible for inhibition of T and natural killer cell proliferation induced by indoleamine 2,3-dioxygenase. The Journal of experimental medicine. 2002; 196:459-468.
87. Chung DJ, Rossi M, Romano E, Ghith J, Yuan J, Munn DH and Young JW. Indoleamine 2,3-dioxygenase-expressing mature human monocyte-derived dendritic cells expand potent autologous regulatory T cells. Blood. 2009; 114:555-563.
88. Mellor AL and Munn DH. Physiologic control of the functional status of Foxp3+ regulatory T cells. Journal of immunology. 2011; 186:4535-4540.
89. Mezrich JD, Fechner JH, Zhang X, Johnson BP, Burlingham WJ and Bradfield CA. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. Journal of immunology. 2010; 185:3190-3198.
90. Wainwright DA, Balyasnikova IV, Chang AL, Ahmed AU, Moon KS, Auffinger B, Tobias AL, Han Y and Lesniak MS. IDO Expression in Brain Tumors Increases the Recruitment of Regulatory T Cells and Negatively Impacts Survival. Clinical Cancer Research. 2012; 18:6110-6121.
91. Zhai L, Lauing KL, Chang AL, Dey M, Qian J, Cheng Y, Lesniak MS and Wainwright DA. The role of IDO in brain tumor immunotherapy. Journal of neuro-oncology. 2015; 123:395-403.
92. O’Connor JC, Lawson MA, Andre C, Briley EM, Szegedi SS, Lestage J, Castanon N, Herkenham M, Dantzer R and Kelley KW. Induction of IDO by bacille Calmette-Guerin is responsible for development of murine depressive-like behavior. Journal of immunology. 2009; 182:3202-3212.
93. Uyttenhove C, Pilotte L, Theate I, Stroobant V, Colau D, Parmentier N, Boon T and Van den Eynde BJ. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nature medicine. 2003; 9:1269-1274.
94. Mitsuka K, Kawataki T, Satoh E, Asahara T, Horikoshi T and Kinouchi H. Expression of indoleamine 2,3-dioxygenase and correlation with pathological malignancy in gliomas. Neurosurgery. 2013; 72:1031-1038; discussion 1038-1039.
95. Theate I, van Baren N, Pilotte L, Moulin P, Larrieu P, Renauld JC, Herve C, Gutierrez-Roelens I, Marbaix E, Sempoux C and Van den Eynde BJ. Extensive profiling of the expression of the indoleamine 2,3-dioxygenase 1 protein in normal and tumoral human tissues. Cancer Immunol Res. 2015; 3:161-172.
96. Hastings WD, Anderson DE, Kassam N, Koguchi K, Greenfield EA, Kent SC, Zheng XX, Strom TB, Hafler DA and Kuchroo VK. TIM-3 is expressed on activated human CD4+ T cells and regulates Th1 and Th17 cytokines. European journal of immunology. 2009; 39:2492-2501.
97. Sun J, Yang M, Ban Y, Gao W, Song B, Wang Y, Zhang Y, Shao Q, Kong B and Qu X. Tim-3 Is Upregulated in NK Cells during Early Pregnancy and Inhibits NK Cytotoxicity toward Trophoblast in Galectin-9 Dependent Pathway. PloS one. 2016; 11:e0147186.
98. Zhang Y, Ma CJ, Wang JM, Ji XJ, Wu XY, Moorman JP and Yao ZQ. Tim-3 regulates pro- and anti-inflammatory cytokine expression in human CD14+ monocytes. Journal of leukocyte biology. 2012; 91:189-196.
99. Fourcade J, Sun Z, Pagliano O, Chauvin JM, Sander C, Janjic B, Tarhini AA, Tawbi HA, Kirkwood JM, Moschos S, Wang H, Guillaume P, Luescher IF, Krieg A, Anderson AC, Kuchroo VK, et al. PD-1 and Tim-3 regulate the expansion of tumor antigen-specific CD8(+) T cells induced by melanoma vaccines. Cancer research. 2014; 74:1045-1055.
100. Kang CW, Dutta A, Chang LY, Mahalingam J, Lin YC, Chiang JM, Hsu CY, Huang CT, Su WT, Chu YY and Lin CY. Apoptosis of tumor infiltrating effector TIM-3+CD8+ T cells in colon cancer. Scientific reports. 2015; 5:15659.
101. Han S, Feng S, Xu L, Shi W, Wang X, Wang H, Yu C, Dong T, Xu M and Liang G. Tim-3 on peripheral CD4(+) and CD8(+) T cells is involved in the development of glioma. DNA and cell biology. 2014; 33:245-250.
102. Pegram HJ, Andrews DM, Smyth MJ, Darcy PK and Kershaw MH. Activating and inhibitory receptors of natural killer cells. Immunology and cell biology. 2011; 89:216-224.
103. Beksac K, Beksac M, Dalva K, Karaagaoglu E and Tirnaksiz MB. Impact of “Killer Immunoglobulin-Like Receptor /Ligand” Genotypes on Outcome following Surgery among Patients with Colorectal Cancer: Activating KIRs Are Associated with Long-Term Disease Free Survival. PloS one. 2015; 10:e0132526.
104. Chester C, Fritsch K and Kohrt HE. Natural Killer Cell Immunomodulation: Targeting Activating, Inhibitory, and Co-stimulatory Receptor Signaling for Cancer Immunotherapy. Front Immunol. 2015; 6:601.
105. Perrin G, Speiser D, Porret A, Quiquerez AL, Walker PR and Dietrich PY. Sister cytotoxic CD8+ T cell clones differing in natural killer inhibitory receptor expression in human astrocytoma. Immunology letters. 2002; 81:125-132.
106. Flies DB, Wang S, Xu H and Chen L. Cutting edge: A monoclonal antibody specific for the programmed death-1 homolog prevents graft-versus-host disease in mouse models. Journal of immunology. 2011; 187:1537-1541.
107. Wang L, Rubinstein R, Lines JL, Wasiuk A, Ahonen C, Guo Y, Lu LF, Gondek D, Wang Y, Fava RA, Fiser A, Almo S and Noelle RJ. VISTA, a novel mouse Ig superfamily ligand that negatively regulates T cell responses. The Journal of experimental medicine. 2011; 208:577-592.
108. Le Mercier I, Chen W, Lines JL, Day M, Li J, Sergent P, Noelle RJ and Wang L. VISTA Regulates the Development of Protective Antitumor Immunity. Cancer research. 2014; 74:1933-1944.
109. Flies DB, Han X, Higuchi T, Zheng L, Sun J, Ye JJ and Chen L. Coinhibitory receptor PD-1H preferentially suppresses CD4+ T cell-mediated immunity. Journal of Clinical Investigation. 2014; 124:1966-1975.
110. Armand P, Nagler A, Weller EA, Devine SM, Avigan DE, Chen YB, Kaminski MS, Holland HK, Winter JN, Mason JR, Fay JW, Rizzieri DA, Hosing CM, Ball ED, Uberti JP, Lazarus HM, et al. Disabling immune tolerance by programmed death-1 blockade with pidilizumab after autologous hematopoietic stem-cell transplantation for diffuse large B-cell lymphoma: results of an international phase II trial. Journal of clinical oncology. 2013; 31:4199-4206.
111. Robert C, Long GV, Brady B, Dutriaux C, Maio M, Mortier L, Hassel JC, Rutkowski P, McNeil C, Kalinka-Warzocha E, Savage KJ, Hernberg MM, Lebbe C, Charles J, Mihalcioiu C, Chiarion-Sileni V, et al. Nivolumab in previously untreated melanoma without BRAF mutation. The New England journal of medicine. 2015; 372:320-330.
112. Robert C, Ribas A, Wolchok JD, Hodi FS, Hamid O, Kefford R, Weber JS, Joshua AM, Hwu WJ, Gangadhar TC, Patnaik A, Dronca R, Zarour H, Joseph RW, Boasberg P, Chmielowski B, et al. Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma: a randomised dose-comparison cohort of a phase 1 trial. Lancet. 2014; 384:1109-1117.
113. Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, Sosman JA, McDermott DF, Powderly JD, Gettinger SN, Kohrt HE, Horn L, Lawrence DP, Rost S, Leabman M, Xiao Y, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014; 515:563-567.
114. Powles T, Eder JP, Fine GD, Braiteh FS, Loriot Y, Cruz C, Bellmunt J, Burris HA, Petrylak DP, Teng SL, Shen X, Boyd Z, Hegde PS, Chen DS and Vogelzang NJ. MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature. 2014; 515:558-562.
115. Hodi FS, Mihm MC, Soiffer RJ, Haluska FG, Butler M, Seiden MV, Davis T, Henry-Spires R, MacRae S, Willman A, Padera R, Jaklitsch MT, Shankar S, Chen TC, Korman A, Allison JP, et al. Biologic activity of cytotoxic T lymphocyte-associated antigen 4 antibody blockade in previously vaccinated metastatic melanoma and ovarian carcinoma patients. Proceedings of the National Academy of Sciences of the United States of America. 2003; 100:4712-4717.
116. Maur M, Tomasello C, Frassoldati A, Dieci MV, Barbieri E and Conte P. Posterior reversible encephalopathy syndrome during ipilimumab therapy for malignant melanoma. Journal of clinical oncology. 2012; 30:e76-78.
117. Ribas A, Kefford R, Marshall MA, Punt CJ, Haanen JB, Marmol M, Garbe C, Gogas H, Schachter J, Linette G, Lorigan P, Kendra KL, Maio M, Trefzer U, Smylie M, McArthur GA, et al. Phase III randomized clinical trial comparing tremelimumab with standard-of-care chemotherapy in patients with advanced melanoma. Journal of clinical oncology. 2013; 31:616-622.
118. Vacchelli E, Aranda F, Eggermont A, Sautes-Fridman C, Tartour E, Kennedy EP, Platten M, Zitvogel L, Kroemer G and Galluzzi L. Trial watch: IDO inhibitors in cancer therapy. Oncoimmunology. 2014; 3:e957994.
119. Hamanishi J and Konishi I. [Targeting the PD-1/PD-L1 immune checkpoint signal - a new treatment strategy for cancer]. [Article in Japanese]. Gan to kagaku ryoho Cancer & chemotherapy. 2014; 41:1071-1076.
120. Rizvi NA, Mazieres J, Planchard D, Stinchcombe TE, Dy GK, Antonia SJ, Horn L, Lena H, Minenza E, Mennecier B, Otterson GA, Campos LT, Gandara DR, Levy BP, Nair SG, Zalcman G, et al. Activity and safety of nivolumab, an anti-PD-1 immune checkpoint inhibitor, for patients with advanced, refractory squamous non-small-cell lung cancer (CheckMate 063): a phase 2, single-arm trial. The Lancet Oncology. 2015; 16:257-265.
121. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, Leming PD, Spigel DR, Antonia SJ, Horn L, Drake CG, Pardoll DM, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. The New England journal of medicine. 2012; 366:2443-2454.
122. Cella D, Grünwald V, Nathan P, Doan J, Dastani H, Taylor F, Bennett B, DeRosa M, Berry S, Broglio K, Berghorn E and Motzer RJ. Quality of life in patients with advanced renal cell carcinoma given nivolumab versus everolimus in CheckMate 025: a randomised, open-label, phase 3 trial. The Lancet Oncology. 2016; 17:994-1003.
123. Ansell SM, Lesokhin AM, Borrello I, Halwani A, Scott EC, Gutierrez M, Schuster SJ, Millenson MM, Cattry D, Freeman GJ, Rodig SJ, Chapuy B, Ligon AH, Zhu L, Grosso JF, Kim SY, et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. The New England journal of medicine. 2015; 372:311-319.
124. Granier C, Karaki S, Roussel H, Badoual C, Tran T, Anson M, Fabre E, Oudard S and Tartour E. [Cancer immunotherapy: Rational and recent breakthroughs]. [Article in French]. La Revue de medecine interne. 2016; 37:694-700.
125. Reardon DA, Sampson JH, Sahebjam S, Lim M, Baehring JM, Vlahovic G, Cloughesy TF, Strauss LC, Latek RR and Paliwal P. Safety and activity of nivolumab (nivo) monotherapy and nivo in combination with ipilimumab (ipi) in recurrent glioblastoma (GBM): Updated results from checkmate-143. Journal of Clinical Oncology. 2016; 34.
126. Sampson JH, Vlahovic G, Sahebjam S, Omuro AMP, Baehring JM, Hafler DA, Voloschin AD, Paliwal P, Grosso J and Coric V. Preliminary safety and activity of nivolumab and its combination with ipilimumab in recurrent glioblastoma (GBM): CHECKMATE-143. Journal of Clinical Oncology. 2015; 33.
127. Schaff L, Donovan L, Lassman AB, Wang T, Carvajal R, Rizvi N, Fox A and Iwamoto FM. Imct-18pd-1 Inhibitors for Recurent High Grade Glioma (Hgg). Neuro-oncology. 2015; 17:v111.113-v111.
128. Sampson JH, Omuro AMP, Preusser M, Lim M, Butowski NA, Cloughesy TF, Strauss LC, Latek RR, Paliwal Pi, Weller M and Reardon DA. A randomized, phase 3, open-label study of nivolumab versus temozolomide (TMZ) in combination with radiotherapy (RT) in adult patients (pts) with newly diagnosed, O-6-methylguanine DNA methyltransferase (MGMT)-unmethylated glioblastoma (GBM): CheckMate-498. Journal of Clinical Oncology. 2016; 34.
129. Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, Pitot HC, Hamid O, Bhatia S, Martins R, Eaton K, Chen S, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. The New England journal of medicine. 2012; 366:2455-2465.
130. Buchwald ZS and Efstathiou JA. Words of wisdom. Re: MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. European urology. 2015; 67:975.
131. Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, Akerley W, van den Eertwegh AJ, Lutzky J, Lorigan P, Vaubel JM, Linette GP, et al. Improved survival with ipilimumab in patients with metastatic melanoma. The New England journal of medicine. 2010; 363:711-723.
132. Hu J, Yu J, Black K and Rudnick J. IT-13IPILIMUMAB FOR RECURRENT HIGH-GRADE GLIOMA: A SINGLE-INSTITUTION CASE SERIES. Neuro-oncology. 2014; 16.
133. Soliman HH, Jackson E, Neuger T, Dees EC, Harvey RD, Han H, Ismail-Khan R, Minton S, Vahanian NN, Link C, Sullivan DM and Antonia S. A first in man phase I trial of the oral immunomodulator, indoximod, combined with docetaxel in patients with metastatic solid tumors. Oncotarget. 2014; 5:8136-8146. doi: 10.18632/oncotarget.2357.
134. Hanahan D and Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011; 144:646-674.
135. Anderson MJ, Shafer-Weaver K, Greenberg NM and Hurwitz AA. Tolerization of tumor-specific T cells despite efficient initial priming in a primary murine model of prostate cancer. Journal of immunology. 2007; 178:1268-1276.
136. Curran MA, Montalvo W, Yagita H and Allison JP. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proceedings of the National Academy of Sciences of the United States of America. 2010; 107:4275-4280.
137. Castro MG, Baker GJ and Lowenstein PR. Blocking immunosuppressive checkpoints for glioma therapy: the more the Merrier! Clinical cancer research. 2014; 20:5147-5149.
138. Wainwright DA, Chang AL, Dey M, Balyasnikova IV, Kim CK, Tobias A, Cheng Y, Kim JW, Qiao J, Zhang L, Han Y and Lesniak MS. Durable therapeutic efficacy utilizing combinatorial blockade against IDO, CTLA-4, and PD-L1 in mice with brain tumors. Clinical cancer research. 2014; 20:5290-5301.
139. Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK and Anderson AC. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. The Journal of experimental medicine. 2010; 207:2187-2194.
140. Woo SR, Turnis ME, Goldberg MV, Bankoti J, Selby M, Nirschl CJ, Bettini ML, Gravano DM, Vogel P, Liu CL, Tangsombatvisit S, Grosso JF, Netto G, Smeltzer MP, Chaux A, Utz PJ, et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer research. 2012; 72:917-927.
141. Kim. JE, Mira A. Patel, Mangraviti. A and Velarde E. the combination of anti-TIM-3 and anti-PD-1 checkpoint inhibitors with focused radiation resulted in a synergistic antitumor immune response in a preclinical glioma model. Neurosurgery. 2014; 62.
142. Zeng J, See AP, Phallen J, Jackson CM, Belcaid Z, Ruzevick J, Durham N, Meyer C, Harris TJ, Albesiano E, Pradilla G, Ford E, Wong J, Hammers HJ, Mathios D, Tyler B, et al. Anti-PD-1 blockade and stereotactic radiation produce long-term survival in mice with intracranial gliomas. International journal of radiation oncology, biology, physics. 2013; 86:343-349.
143. Hanihara M, Kawataki T, Oh-Oka K, Mitsuka K, Nakao A and Kinouchi H. Synergistic antitumor effect with indoleamine 2,3-dioxygenase inhibition and temozolomide in a murine glioma model. Journal of neurosurgery. 2015:1-8.
144. Li M, Bolduc AR, Hoda MN, Gamble DN, Dolisca SB, Bolduc AK, Hoang K, Ashley C, McCall D, Rojiani AM, Maria BL, Rixe O, MacDonald TJ, Heeger PS, Mellor AL, Munn DH, et al. The indoleamine 2,3-dioxygenase pathway controls complement-dependent enhancement of chemo-radiation therapy against murine glioblastoma. Journal for immunotherapy of cancer. 2014; 2:21.
145. Agarwalla P, Barnard Z, Fecci P, Dranoff G and Curry WT, Jr. Sequential immunotherapy by vaccination with GM-CSF-expressing glioma cells and CTLA-4 blockade effectively treats established murine intracranial tumors. Journal of immunotherapy. 2012; 35:385-389.
146. vom Berg J, Vrohlings M, Haller S, Haimovici A, Kulig P, Sledzinska A, Weller M and Becher B. Intratumoral IL-12 combined with CTLA-4 blockade elicits T cell-mediated glioma rejection. Journal of Experimental Medicine. 2013; 210:2803-2811.
147. Maes W, Rosas GG, Verbinnen B, Boon L, De Vleeschouwer S, Ceuppens JL and Van Gool SW. DC vaccination with anti-CD25 treatment leads to long-term immunity against experimental glioma. Neuro-oncology. 2009; 11:529-542.
148. Grauer OM, Nierkens S, Bennink E, Toonen LW, Boon L, Wesseling P, Sutmuller RP and Adema GJ. CD4+FoxP3+ regulatory T cells gradually accumulate in gliomas during tumor growth and efficiently suppress antiglioma immune responses in vivo. International journal of cancer. 2007; 121:95-105.
149. Belcaid Z, Phallen JA, Zeng J, See AP, Mathios D, Gottschalk C, Nicholas S, Kellett M, Ruzevick J, Jackson C, Albesiano E, Durham NM, Ye X, Tran PT, Tyler B, Wong JW, et al. Focal radiation therapy combined with 4-1BB activation and CTLA-4 blockade yields long-term survival and a protective antigen-specific memory response in a murine glioma model. PloS one. 2014; 9:e101764.
150. Koshy ST and Mooney DJ. Biomaterials for enhancing anti-cancer immunity. Current opinion in biotechnology. 2016; 40:1-8.
151. Wang C, Ye Y, Hochu GM, Sadeghifar H and Gu Z. Enhanced Cancer Immunotherapy by Microneedle Patch-Assisted Delivery of Anti-PD1 Antibody. Nano letters. 2016; 16:2334-2340.
152. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004; 116:281-297.
153. Wei J, Nduom EK, Kong LY, Hashimoto Y, Xu S, Gabrusiewicz K, Ling X, Huang N, Qiao W, Zhou S, Ivan C, Fuller GN, Gilbert MR, Overwijk W, Calin GA and Heimberger AB. MiR-138 exerts anti-glioma efficacy by targeting immune checkpoints. Neuro-oncology. 2015.