
Introduction
Hereditary breast and ovarian cancer (HBOC) is characterized by the early onset of breast (BC) and/or ovarian cancer (OC), multiple primaries, bilateral tumors and a family history of cancer of the same spectrum in relatives. About 5-7% of BC and 20-25% of OC are thought to be due to rare variants conferring a hereditary predisposition to cancer [1, 2]. The penetrance of these rare variants may be highly variable depending on the gene considered. The use of multigene panels raises questions about the definition of risk [3]. BRCA1 and BRCA2, the first gene mutations associated with a predisposition for a high risk of breast cancer, confer an estimated risk of 65 and 45% for BC and 39 and 10% for OC, respectively [4]. BRCA1/2 mutations are responsible for 30% of early-onset breast cancers and 90% of family histories of BC and OC [5-8]. Genetic testing and management guidelines are now widely available. Mutations in other highly penetrant genes, such as PTEN, CDH1, STK11 and TP53, can cause cancer susceptibility syndromes [9-12], but remain rare. MLH1, MSH2, MSH6 and PMS2 also contribute to a hereditary risk of OC. The criteria for genetic testing are specific to these predispositions, according to their disease spectrum.
In the past few years, since the arrival of next generation sequencing (NGS), several predisposing genes, mainly of moderate penetrance, have been discovered, thus explaining additional HBOC pedigrees. The majority of these genes are part of the DNA repair/BRCA pathway [13-16]. NGS has made it possible for clinicians to order a single test that evaluates multiple genes simultaneously in a cost-effective and efficient fashion, thus enabling a more complete genetic evaluation [17]. In the last 2 years, the literature assessing the contribution of multigene panel testing in cohorts of patients with HBOC has grown. The results have been consistent even though the inclusion criteria sometimes differ [17-30]. The next challenge is to determine the consequences of these results on clinical management [NCCN clinical practice Guidelines-see URL]. High-penetrance gene mutations have a high well-defined cancer risk by site; actionability for these genes is high, and evidence-based national guidelines exist for risk reduction in at least one organ system; the implications for other family members are thus straightforward. Moderate-penetrance gene mutations carry a more moderate risk, and organ-specific cancer risks are fairly well-defined for at least one cancer site; actionability for these genes is moderate, though there is enough evidence to go beyond empiric risk and to justify enhanced surveillance in the proband for at least one at-risk site. However, the implications for other family members may not be as straightforward. Low-penetrance gene mutations have a lower or uncertain risk and the organ-specific cancer risks are vague; actionability is low, due to the lack of established evidence-based guidelines; screening and management recommendations depend on empiric risk estimates and case reports in the literature. The implications for other family members are not well defined. In 2015, the National Comprehensive Cancer Network (NCCN) in the USA published guidelines for high-penetrance gene mutations as well as a number of moderate penetrance gene mutations, updated in 2016 [see URL]. These included PALB2, CHEK2 and ATM pathogenic or probably pathogenic variants .
The aim of this study was to determine, in a new cohort of 583 patients with HBOC from the French region of Burgundy, the clinical actionability of variants found using a multigene panel for HBOC. Actionability concerned different options, including the modification of cancer surveillance, specific risk-reduction measures, treatment guidance, customized gene-specific treatment options, and the identification of at-risk family members.
Results
Mutations identified in the cohort
Of the 583 subjects tested, a pathogenic or probably pathogenic BRCA1/2 mutation was found in 51 (9%) patients. Besides, thirty-seven pathogenic or probably pathogenic variants were found in 10 other genes of the panel. The distribution of mutations and their clinical presentation are given in Figure 1 and Table 1A and 1B. Six mutations were found in high-penetrance genes, with no diagnostic criteria that could have guided traditional genetic testing in the majority of them. It included two TP53 pedigrees that did not fulfill Chompret criteria (Figure 2A1 and 2C1); four MMR mutations, in two patients with single-affected early-onset BC, one patient with familial BC, and one patient with BC but a family history of two endometrial cancers, which could have led to MMR testing in the relatives (Figure 2B1, B2, B3, C3), with a negative search for microsatellite instability in three breast tumors with available material. Twenty-one mutations were found in moderate-risk HBOC genes. The most frequently mutated genes were CHEK2 (n = 12; 2% of the total cohort, 13% of the positive cohort), ATM (n = 9; 1.5% of the total cohort, 10% of the positive cohort) and PALB2 (n = 4; 0.6% of the total cohort, 4.5% of the positive cohort), mostly in families with BC only. Other results included two BARD1 mutations in families with BC and OC, one BRIP1 and one RAD50 mutation in families with BC only (Figure 1, Table 1). Three patients with a mutation in two different genes were found. One patient had pathogenic mutations in both TP53 and PALB2, one had deleterious mutations in both BRCA2 and CHEK2 and another one in both BRCA1 and PMS2 (Figure 2C1, C2 and C3). One probably pathogenic mutation in SUFU was detected and considered incidental given the absence of a personal or family history of medulloblastoma, and the absence of BC in the spectrum of SUFU-predisposing cancers to date. The index case presented BC at age 49, multiple cancers in her mother at age 69 (OC, endometrial and colorectal cancer) and liver cancer in her maternal grandfather at age 58. The majority of the findings were relevant to the clinical history (34/38, 89%). In addition, seven monoallelic mutations of MUTYH were found, which is the number expected in the general population, and 245 VUS were identified.
Table 1A: Characteristics of patients with pathogenic or probably pathogenic variants in genes other than BRCA
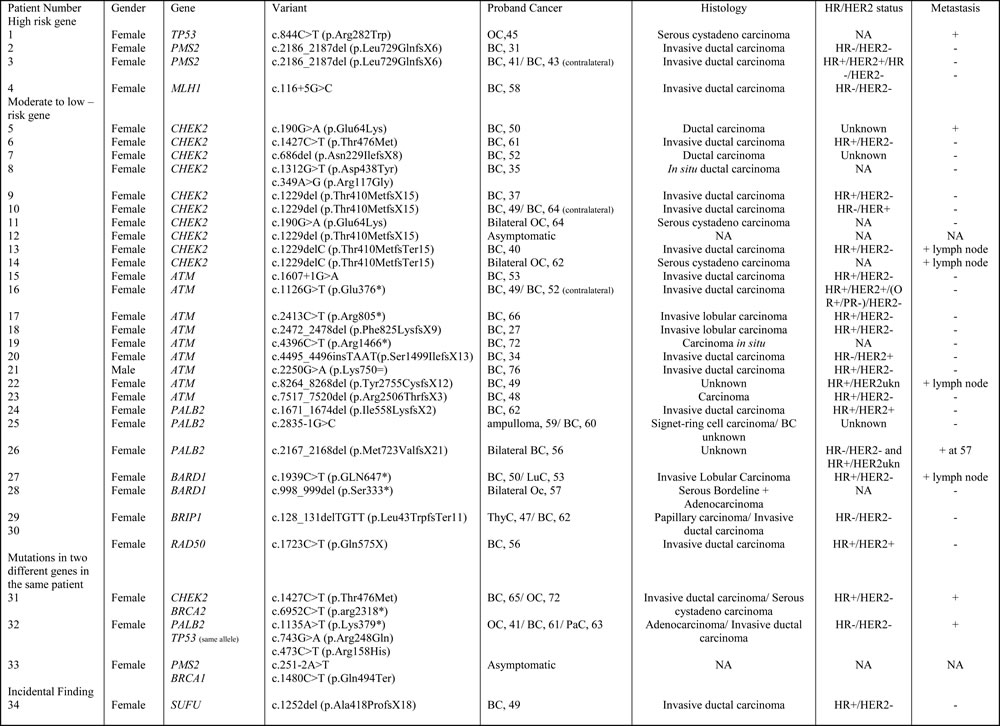
BC: Breast cancer; OC: Ovarian Cancer, PaC: pancreatic cancer, LuC: lung cancer, ThyC: thyroid cancer; ukn: unknown
Table 1B: Family history of cancer of patients with pathogenic or probably pathogenic variants in genes other than BRCA

BC: Breast cancer; OC: Ovarian Cancer, LiC: liver cancer, GyC: gynecological cancer, SyC: systemic cancer, PaC: pancreatic cancer, CeC: cervical cancer, PrC: prostate cancer, CRC: colorectal cancer, TeC: testis cancer, HoD: Hodgkin’s disease, EC: endometrial cancer, Me: melanoma, LuC: lung cancer, ThyC: thyroid cancer
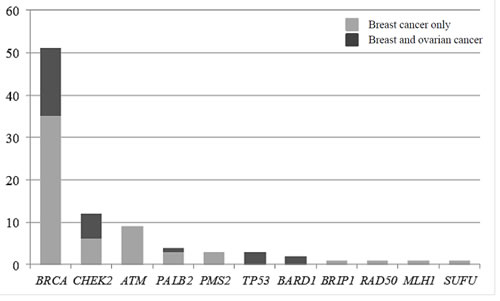
Figure 1: Distribution of pathogenic and likely-pathogenic mutations detected in genes other than BRCA according to cancer presentation in the patient and family. In light gray, families with breast cancer presentation, and in dark grey, families with at least one patient with an ovarian cancer.
Impact of genetic testing on management
The impact of genetic testing on the management of patients is summarized in Table 2, in which patients are classified according to the presence of high-risk, moderate-low-risk, mutations in two different predisposing genes or even incidental findings. All patients were eligible for screening for a genetic predisposition according to our criteria, described in materials and methods. The identification of a mutation in a high-risk gene always led to a modification in genetic counseling and surveillance in patients with good prognosis, but also in some cases to specific risk-reduction measures (prophylactic surgery in females with MMR mutations). No treatment guidance could be counselled (avoidance of radiotherapy in females with Li-Fraumeni syndrome) since our 2 patients with TP53 mutations had metastasis and one of them died. For moderate-low-risk genes, the impact on surveillance (69%) and genetic counseling remained (89%), but there was no impact on specific-risk reduction measures or treatment guidance. These results can be attributed to the recent 2015-2016 NCCN guidelines, which recommend breast magnetic resonance imaging (MRI) screening in patients with ATM, CHEK2 and PALB2 mutations. The question of limiting mammography in patients with ATM heterozygous variants remains open to discussion as there are still discrepancies and few studies regarding the effects of radiation on those patients’ cells [32-34]. The impact on genetic counseling was often limited in this category of patients. Indeed, it is difficult to reassure negative patients, given the possibility that other HBOC predisposing genes unknown to date may be also implicated. The option of proposing breast MRI in positive patients and surveillance in negative patients according to family history was chosen. The eleven patients whose positive results after multigene panel testing had no impact on management were four women with low-penetrance BRIP1, BARD1 and RAD50 mutations, one women with TP53 and PALPB2 mutations who had died at time of the results, four women with a metastasis progression of her disease, one women with an ATM mutation who was 79 at time of the results, and one man with an ATM mutation. None of our patients were given customized gene-specific treatment options at the time of the study as the only example available to date (PARP inhibitors in advanced ovarian cancers) was not available at the time of the decision making. The impact was also major in patients with a mutation in two different genes, since they benefited from surveillance or specific risk-reduction measures, defined according to predispositions due to both genes, and careful genetic counseling, based on the guidelines for the two predisposing genes. Finally, a cautious approach was adopted in the individual with an incidental diagnosis of a SUFU mutation. One should also consider the benefit to all patients of the study in access to future research. The analysis of clinical actionability performed among individuals with mutations other than BRCA revealed that additional disease-specific screening and/or prevention measurers beyond those based on personal and family history alone had been recommended in more than two-third of cases, after determining whether the high-risk surveillance was maintained because of the health status, age (age cut-off for recommending breast MRI set up at 75 years in our center), or unchanged in a context of Boadicea score greater than 20 in the index case.
Table 2: Orientation of clinical management according to genetic information
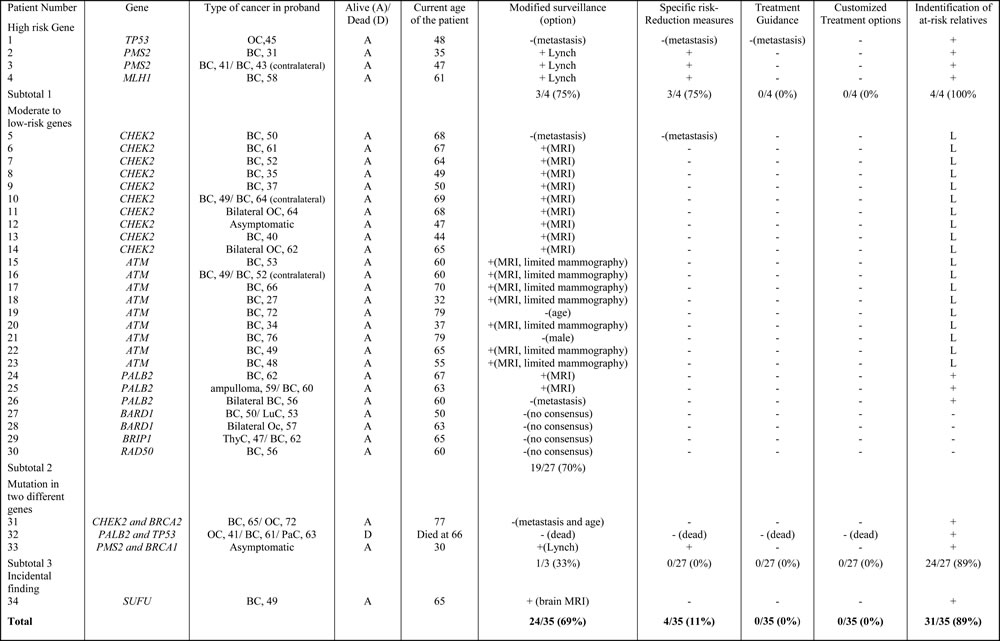
BC: Breast cancer; OC: Ovarian Cancer, PaC: pancreatic cancer, LuC: lung cancer, ThyC: thyroid cancer
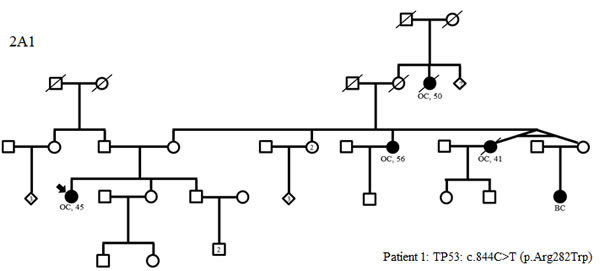
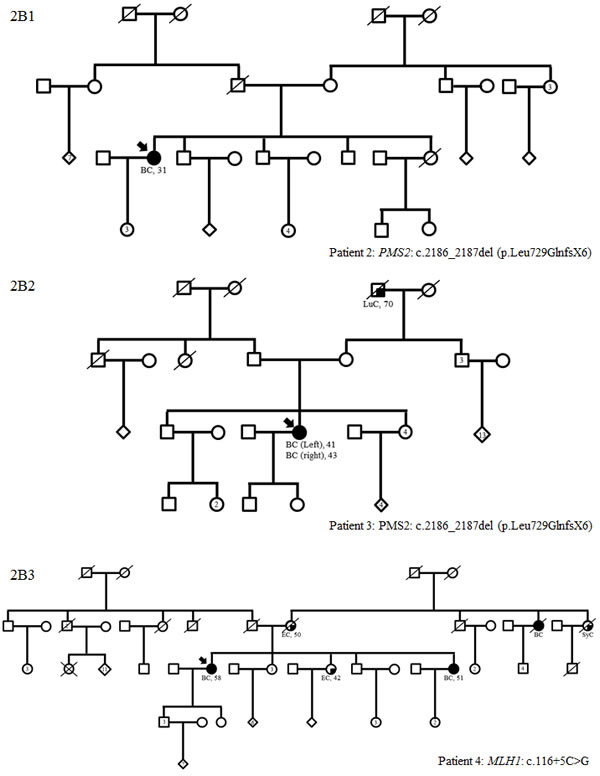
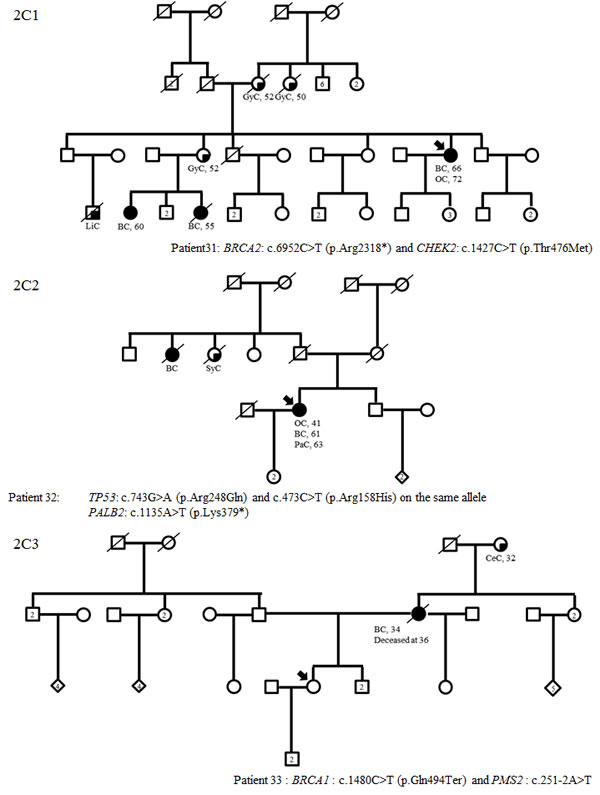
Figure 2: Family trees in patients with TP53 (A1), MMR gene mutations (B1-3) and variants in two predisposing genes (C1-3) (possibly including a TP53 or MMR mutation). BC: breast cancer; OC: ovarian cancer, LiC: liver cancer, GyC: gynecological cancer, SyC: systemic cancer, PaC: pancreatic cancer, CeC: cervical cancer, PrC: prostate cancer, CRC: colorectal cancer, TeC: testis cancer, HoD: Hodgkin’s disease, EC: endometrial cancer, Me: melanoma, LuC: lung cancer
Discussion
Though recent studies have evaluated the positive yield of a multigene panel as compared to BRCA1/2 testing alone, the impact of these results on patient surveillance is rarely reported. In this series, our aim was to answer this question, since it represents a central challenge for the future deployment of genomic medicine [25, 27, 35-36].
Regarding the number of pathogenic variants, our results were quite similar to those in other studies. The frequency of BRCA1/2, PALB2, ATM and CHEK2 mutations were comparable to those in published series with the same inclusion criteria. Table 3A and 3B summarizes the primary aim of these studies, the inclusion criteria, the list of genes included in the selected panels and the results of ten published studies that included more than 500 patients who had undergone diagnostic multigene panel testing for HBOC [18-20, 22-23, 25, 27-28, 30-31]. One study was excluded from this review despite the high number of patients, because the authors chose to study only 36 known pathogenic mutations in the selected HBOC genes [14]. The highest number of patients included was 10030, most of whom had HBOC, although one study was limited to unselected TNBC patient [31] and another one to OC [29]. Two studies included exclusively patients previously tested negatively for BRCA1/2 mutations [25, 27]. The number of genes tested varied between 8 and 27. The most recurrent genes included in the panels were ATM, BARD1, BRIP1, CDH1, CHEK2, MLH1, MSH2, MSH6, PALB2, PSM2, RAD51C, STK11, TP53. The overall detection rate of mutations in genes other than BRCA ranged from 2.9% to 9.3%. Pathogenic mutations in CHEK2 (0-2.1%) and ATM (0.5-2.9%) were by far the most frequently mutated genes in this population of patients, followed by PALB2. NCCN guidelines for those genes have been published very recently, with some differences as compared to those for highly-penetrant genes. Indeed, breast MRI, but not prophylactic surgery, is recommended [NCCN Guideline, see URL], and caution should be exercised regarding genetic counseling in relatives. Indeed, because of the possible co-segregation of several low- or moderate-penetrance genes, surveillance in unaffected relatives not carrying the pathogenic mutation could be recommended depending on the family history. Therefore, a positive result may still be useful, but negative results may not be completely reassuring.
Interestingly, some high-risk genes were detected in our series as in others. We identified two patients carrying three mutations in TP53. Neither of our two patients met the Chompret criteria (Figure 2). Detecting these mutations is extremely important for screening and genetic counseling NCCN guidelines [see URL], including the avoidance of irradiation in the treatment of cancer whenever possible, in order to decrease the risk of a second malignancy. We also identified four mutations in MMR genes (PMS2 and MLH1), but in three of our four families, there were exclusively breast cancer pedigrees (Figure 2). With the experience of gene panels, which included HBOC susceptibility genes among others, some families harboring a mutation in an MMR gene did not meet Amsterdam II or Bethesda criteria for Lynch syndrome testing [37]. To date, it is unclear whether the results in our three families with single-affected early-onset BC should be regarded as incidental or not since an increased risk of BC in Lynch syndrome is still controversial [38-40]. Nevertheless, massively-parallel sequencing targeting multiple candidate genes is changing the paradigm as it gives the opportunity to assess the role of genes that would never have been evaluated in a non-panel approach. Regarding this point, our study suggests that PMS2, which does not contribute greatly to Lynch Syndrome [41], could be considered a good candidate for evaluation. Moreover, as the commonly known technical difficulties of analyzing PMS2 are slowly being overcome by new analysis strategies [41-42], critically needed evidence concerning the role of PMS2 can be expected. For instance, recently, ten Broeke et al. reported a standardized incidence ratio of 3.8 for breast carcinomas, which led them to suggest adding mammography from age 40 years in PMS2 families with evident clusters of BC [43]. Finally, one mutation was found in SUFU, and considered an incidental finding given the absence of a personal or family history of medulloblastoma or Gorlin syndrome.
One of the greatest advantages of gene panels compared with testing single genes is the possibility of diagnosing patients with a mutation in more than one predisposing gene, thus leading to more accurate genetic counseling. Indeed, one of our index cases had two TP53 mutations on the same allele and a PALB2 mutation. Detection of the TP53 mutations alone would have led to inaccurate genetic counseling since negative patients for the TP53 mutations would have been reassured whereas adapted breast screening is justified in PALB2 carriers. The same situation exists for the family with the index case carrying a BRCA and a CHEK2 mutation. Also, the detection of a PMS2 mutation in an index case with a BRCA mutation led to the prescription of digestive and pelvic follow-up.
In this work, we aimed to assess how and whether these advances in technology will be applied in clinical practice, and how best to counsel patients about variants of low or moderate penetrance. Taking advantage of a single clinical genetics team and laboratory in an entire region of France (Burgundy), we had the opportunity to follow the clinical value of multigene testing, especially in the context of recently updated practice guidelines from the USA for moderate-risk HBOC genes. We first identified six individuals with mutations in high-risk genes, associated or not with the clinical presentation of the family, which had been missed by the traditional focused approach to testing. In these cases, the results changed the management of the patients and the at-risk family members, thus justifying the testing of additional family members in all cases. The most challenging results were found in moderate- to low-penetrance HBOC genes. When we applied gene-based consensus practice guidelines when they existed, we found that the sequencing results modified management in the majority of individuals (70%) and prompted testing in the majority of families (89%). In the future, therapeutic options might be evaluated in members of families carrying a mutation in a DNA-repair gene who eventually develop cancer, such as PARP-inhibitor strategies [US FDA breakthrough therapy designation see URL, 44]. Importantly, family members who tested negative for the familial mutation were not reassured, and were managed according to empiric risk depending on the patient’s personal and family history of cancer.
The choice of following the NCCN guidelines as a basis for assessing the impact of panel testing is the main limitation of the work. Indeed, the NCCN has proposed high-risk surveillance including MRI in ATM and CHEK2 mutation carriers, which represent the majority of mutations identified by the panel in predisposing genes other than BRCA. Other authors have warned about the risk of disinformation and the implementation of inappropriate risk-management strategies in cases with limited cancer risk management guidelines, as such guidelines are not sufficiently developed to allow accurate targeted genetic counseling and breast cancer risk management [27]. However, the maintenance of surveillance in non-carrier relatives should limit this risk. Another limitation of the study is the possible overestimation of the percentage of clinically actionable mutations since there is no consensus in France regarding an age cut-off for recommending breast MRI.
In conclusion, multigene panels, which include cancer susceptibility genes that would have escaped clinical investigation because the familial presentation did not meet the adopted criteria for testing, make it possible to identify more individuals with a genetic predisposition for HBOC. Such a strategy, rather than BRCA testing alone, will modify the management of such patients. This strategy also identifies individuals with multiple-gene predispositions, which should lead to caution in genetic counseling. The classification of patients according to their predisposing genes will be of increasing importance with the arrival of customized treatment options. Given the complex issues raised by multi-gene panel analysis, pre- and post-test genetic counseling and informed decision making are of major importance. As increasing data from multigene panel testing become available, we anticipate that international research efforts will lead to more accurate risk estimates and a better classification of cancer spectra and genetic variants. This in turn will drive the development of explicit and specific management guidelines. However, it will also increase the level of uncertainty since massively-parallel sequencing targeting multiple candidate genes will greatly increase the number of patients exhibiting VUS in different candidate genes.
Table 3A: Other publications relating to multigene panel testing for HBOC that included more than 500 patients, including the primary aim of these studies, the inclusion criteria and the overall detection rate of pathogenic or probably pathogenic variant other than BRCA
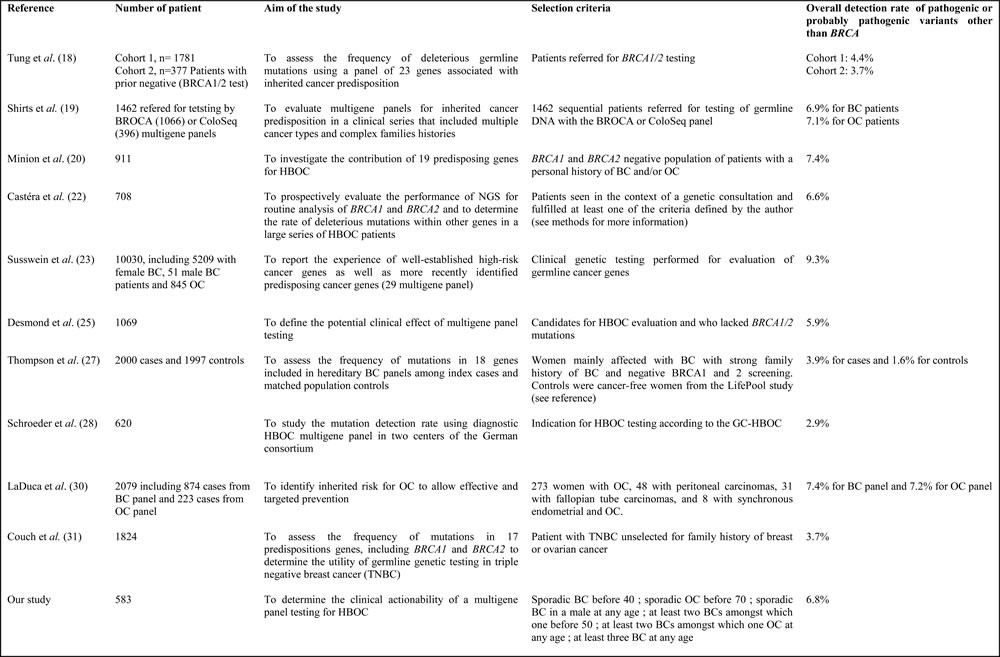
Table 3B: Genes included in the selected panels and their positive yield (number of pathogenic or likely pathogenic variants).

*MUTYH monoallelic pathogenic mutations were not taken into account
Materials and methods
Patients
The study involved the first 583 consecutive patients from Burgundy (France) to benefit from diagnostic panel testing for HBOC, from November 2012 to November 2013 at one of the five genetic clinics of Burgundy. All of the patients, who met the criteria for BRCA1 and BRCA2 testing, had pre- and post-test genetic counseling by a geneticist and/or genetic counselor: i) personal history of single-affected BC before age 40; ii) personal history of single-affected OC before age 70; iii) personal history of single-affected male BC before age 70; iv) personal history of single-affected triple-negative BC before age 50; v) two BC in first or second-degree relatives with at least one cancer before age 50; vi) one BC before age 50 with pancreatic or prostate cancer before age 60 in first or second-degree relatives; vii) three BC in first or second-degree relatives at any age. Testing in first-degree relatives unaffected by cancer of an individual with a very suggestive family history of HBOC was also proposed if it was impossible to test the relatives affected with cancer. Patients had not undergone specific BRCA testing before the multigene panel analysis. For each patient, informed consent for genetic analysis was obtained. General information was entered into the Oncogene database, an in-house database generated via Clinsight, used for the management of Oncogenetic consultations and is registered with the Commission Nationale Information et Liberté (CNIL). This database includes demographic data, self-identified personal and family histories of cancer, and follow-up for patients with a pathogenic or probably pathogenic mutation in a gene predisposing for HBOC.
Sample preparation and next-generation sequencing
DNA was extracted from peripheral blood, using a Maxwell BioRobot system (Promega, Madison, USA). 3 µg of DNA was sonicated using a Covaris S2 (Covaris, Inc, MS, Woburn, MA, USA) and purified. Following capture, samples were barcoded with 16 different indexed primers, pooled per lane and sequenced. The Agilent eArray was used to design the SurSelect solution library (Agilent, Santa Clara, CA, USA) covering a panel of 25 genes including 20 genes with a predisposition for breast and/or ovarian cancer: BRCA1, BRCA2, BARD1, CHEK2, RAD51C, BRIP1, RAD50, MRE11A, PALB2, STK11, MLH1, MSH2, MSH6, EPCAM, PMS2, PTCH1, PTCH2, CDH1, TP53, ATM, PTEN, PIK3CA, SUFU, MUTYH and APC. This design was kindly provided by Nicolas SEVENET’s laboratory (Institut Bergonié, Bordeaux, France) and was developed to meet their diagnostic needs for HBOC, predisposition to digestive cancer or polyposis, and Basal Cell Naevus (Gorlin) syndrome. These genes were not excluded from the bioinformatics analysis since keeping them could have raised the question of incidental findings, which could be important to address for our future practice. The panel did not include all HBOC genes. NBN, NF1, RAD51D and XRCC2 were missing as the panel was designed before the interest of these genes became apparent.
Exon capture was limited to the 451 exonic sequences of the 25 genes with 50 bp surrounding intronic sequences on each side of the exon. The total size of the library was 192 Kb. The library was sequenced on Miseq (Illumina, San Diego, CA, USA) using the paired-end 2x150 bp program.
Large rearrangement analysis for BRCA1 and BRCA2 genes
Large rearrangements were identified by profile comparisons, using the semi-quantitative MLPA method (MRC-Holland, Amsterdam, Netherlands). Briefly, electropherograms from patients were first superposed, the yield of each amplicon in the various samples was evaluated and deletions/duplications of one or more amplicons were revealed by a 2-fold decrease/1.5-fold increase in the corresponding peak, respectively [45]. Large rearrangements were not tested for the other genes of the panel.
Bioinformatics analysis
An average of 215 million high-quality bases were generated for each sample. Raw reads from MiSeq were first aligned to the human genome reference GRCh37/hg19 from UCSC Genome Browser with the Burrows-Wheeler Aligner (BWA, v0.7.6a) [46]. Duplicate paired-end reads were marked with Picard v.1.109. Base quality score recalibration, realignment around indels, and variant discovery were performed with the Genome Analysis Toolkit (GATK) v3.3-0 [47]. Detected variants were annotated with the SeattleSeq annotation portal according to the mutation type, the occurrence in dbSNP and the NHLBI Exome Sequencing Project Exome Variant Server [see URL, 48].
Candidate events were systematically identified by focusing on protein-altering and splice-site variants present at a frequency less than 1% in dbSNP 141, and supported by at least three reads in the subject at base-pair positions covered by at least five reads.
Variant classification
Sequence variants and large insertions and deletions were classified according to the American College of Medical Genetics (ACMG) guidelines for variant interpretation [49]. Variants were classified as pathogenic or probably pathogenic if they conferred a truncating, initiation codon or splice donor/acceptor effect, if functional data demonstrated an effect on protein function relevant to a disease phenotype, or if pathogenicity was otherwise demonstrated in the published literature. If no functional data were available, missense, silent, and intronic variants were classified as variants of uncertain significance (VUS), benign or probably benign based on allele frequency in dbSNP or the Exome Variant Server [see URL]. In the genes other than BRCA, a variant was considered for review when it was classified as pathogenic, probably pathogenic or discordant according to ClinVar. The review was performed by a multidisciplinary team and based on data in the literature. The tools for variant classification included different software and databases (mutation taster, Align GVG Database, BRCA share, ClinVar, NHGRI BIC, ExAc and LOVD BRCA) [see URLs].
Confirmation of the detected variations
For BRCA1 and BRCA2, all the variants detected by NGS within the coding sequence or ± 50bp within the intronic sequences and not recorded as polymorphisms were confirmed by Sanger sequencing, using the BigDye Terminator Cycle Sequencing V1.1 Ready reaction Kit (Life Technologies, Carsbad, CA, USA). For the other genes included in the capture enrichment, all variations inducing a premature stop codon or classified as pathogenic or probably pathogenic were checked by Sanger sequencing.
Data analysis
The incidence of pathogenic and probably pathogenic mutations was calculated for each gene, and the clinical presentations were discussed based on demographic information and clinical history. Genes were grouped in four categories: highly penetrant genes (BRCA1, BRCA2, STK11, MLH1, MSH2, MSH6, EPCAM, PMS2, CDH1, TP53, PTEN), moderately penetrant/HR pathway (BARD1, CHEK2, RAD51C, BRIP1, RAD50, MRE11A, PALB2, ATM), one candidate gene for HBOC (PIK3CA) and other cancer-spectrum predisposing genes (APC, MUTYH, PTCH1, PTCH2, SUFU). Pathogenic mutations in the latter category, in the absence of cancer within the disease spectrum known for these genes in the patient or family members, were considered incidental findings. The impact of having two predisposing genes was particularly studied. Monoallelic MUTYH mutation carriers were not included in the mutation-positive cohort. The incidence of VUS was only given for the cohort overall.
In order to determine whether the discovery of a genetic susceptibility for HBOC guided the clinical management, we stratified the implications for patients according to five options: i) modification of cancer surveillance; ii) suggestion of specific risk-reduction measures, such as prophylactic surgery; iii) offering treatment guidance, such as avoidance of radiotherapy in TP53 patients; iv) providing customized gene-specific treatment options, such as PARP inhibitors in BRCA patients; v) identification of at-risk family members. The possibility of access to research projects was also considered a significant option. Each option was discussed in interdisciplinary meetings, according to the review of the published literature and practice guidelines. Patients with BRCA mutations were excluded from this part of the analysis because such testing is the traditional standard of care, and the aim of this study was to assess the impact on management of testing for any genes beyond the traditional single-gene approach. Since we could expect that, in some cases, the health status of affected patients may not warrant changes to their management (in particular in patients with ovarian cancer), and in other cases, patients would have been candidates for intensive screening anyway because of their very high-risk family history (remaining risk for breast cancer greater than 20%), the effective changes in their medical management guidelines were reviewed case by case.
URL
National Comprehensive Cancer Network (NCCN). Genetic/Familial High-Risk Assessment: Breast and Ovarian. NCCN Clinical Practice Guidelines in Oncology. Fort Washington, PA: NCCN https://www.nccn.org/professionals/physician_gls/f_guidelines_nojava.asp#detection
LynparzaTM (olaparib) granted breakthrough therapy designation by US FDA for treatment of BRCA1/2 or ATM gene mutated metastatic castration resistant prostate cancer http://www.sec.gov/Archives/edgar/data/901832/000119163816001590/azn201601286k.htm
Exome Variant Server, NHLBI GO Exome Sequencing Project (ESP), Seattle, WA http://evs.gs.washington.edu/EVS/
NHS guidelines https://www.nice.org.uk/guidance/cg164/chapter/1-recommendations#surveillance-and-strategies-for-early-detection-of-breast-cancer
dbSNP http://www.ncbi.nlm.nih.gov/projects/SNP/
Exome Variant Server http://evs.gs.washington.edu/EVS/
mutation taster http://www.mutationtaster.org/
Align GVG Database http://agvgd.iarc.fr/
BRCA Share http://www.umd.be/BRCA1/
ClinVar http://www.ncbi.nlm.nih.gov/clinvar/
NHGRI BIC https://research.nhgri.nih.gov/bic/
ExAc http://exac.broadinstitute.org/
LOVD BRCA http://chromium.lovd.nl/LOVD2/cancer/home.php?select_db=BRCA1
Acknowledgments
The authors thank the Institut National du Cancer (INCa) that funds the laboratory for the genetic diagnosis of HBOC patients.
The authors thank Mr. Philip Bastable for the English review
Conflicts of Interest
The authors report no conflict of interest
References
1. Weissman SW, Weiss SM, Newlin AC. Genetic testing by cancer site ovary. Cancer J 2012;18:320-7.
2. Van der Groep P, Van der Wall E, Van Diest PJ. Pathology of hereditary breast cancer. Cell Oncol 2011;34:71-88.
3. Easton DF, Pharoah PDP, Antoniou AC, Tischkowitz M, Tavtigian SV, Nathanson KL, Devilee P, Meindl A, Couch FJ, Southey M, Goldgar DE, Evans GR, Chenevix-Trench G, et al. Gene-Panel Sequencing and the prediction of breast-Cancer Risk. N Engl J Med 2015; 372:2243-57.
4. Antoniou AC, Pharoah PDP, Narod S, Risch HA, Eyfjord JE, Hopper JL, Loman N, Olsson H, Johannsson O, Borg A, Pasini B, Radice P, Manoukian S, et al. Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case series unselected for family history: a combined analysis of 22 studies. Am J Hum Genet 2003; 72: 1117–1130.
5. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin 2013; 63:11-30.
6. Chen S, Parmigiani G. Meta-analysis of BRCA1 and BRCA2 penetrance. J Clin Oncol. 2007;25:1329-33.
7. Mavaddat N, Peock S, Frost D, Ellis S, Platte R, Fineberg E, Evans DG, Izatt L, Eeles RA, Adlard J, Davidson R, Eccles D, Cole T, et al. Cancer risks for BRCA1 and BRCA2 mutation carriers: results from prospective analysis of EMBRACE. J Natl Cancer Inst. 2013;105:812-22.
8. Ford D, Easton DF, Stratton M, Narod S, Goldgar D, Devilee P, Bishop DT, Weber B, Lenoir G, Chang-Claude J, Sobol H, Teare MD, Struewing J, et al. Genetic heterogeneity and penetrance analysis of the BRCA1 and BRCA2 genes in breast cancer families. The Breast Cancer Linkage Consortium. Am J Hum Genet 1998;62:676-689.
9. Walsh T, Casadei S, Coats KH, Swisher E, Stray SM, Higgins J, Roach KC, Mandell J, Lee MK, Ciernikova S, Foretova L, Soucek P, King MC. Spectrum of mutations in BRCA1, BRCA2, CHEK2, and TP53 in families at high risk of breast cancer. JAMA 2006; 295: 1379–1388.
10. Bubien V, Bonnet F, Brouste V, Hoppe S, Barouk-Simonet E, David A, Edery P, Bottani A, Layet V, Caron O, Gilbert-Dussardier B, Delnatte C, Dugast C, et al. High cumulative risks of cancer in patients with PTEN hamartoma tumour syndrome. J Med Genet 2013; 50: 255–263.
11. Giardiello FM, Brensinger JD, Tersmette AC et al: Very high risk of cancer in familial Peutz-Jeghers syndrome. Gastroenterology 2000; 119: 1447–1453.
12. Pharoah PD, Guilford P, Caldas C: Incidence of gastric cancer and breast cancer in CDH1 (E-cadherin) mutation carriers from hereditary diffuse gastric cancer families. Gastroenterology 2001.
13. Daly MB, Pilarski R, Axilbund JE, Berry M, Buys SS, Crawford B, Farmer M, Friedman S, Garber JE, Khan S, Klein C, Kohlmann W, Kurian A, et al. Genetic/Familial High-Risk Assessment: Breast and Ovarian, Version 2.2015. J Natl Compr Canc Netw. 2016 Feb;14:153-62.
14. Domagala P, Jakubowska A, Jaworska-Bieniek K, Kaczmarek K, Durda K, Kurlapska A, Cybulski C, Lubinski J. Prevalence of Germline Mutations in Genes Engaged in DNA Damage Repair by Homologous Recombination in Patients with Triple-Negative and Hereditary Non-Triple-Negative Breast Cancers. PLoS One. 2015;10:e0130393.
15. Wong MW, Nordfors C, Mossman D, Pecenpetelovska G, Avery-Kiejda KA, Talseth-Palmer B, Bowden NA, Scott RJ. BRIP1, PALB2, and RAD51C mutation analysis reveals their relative importance as genetic susceptibility factors for breast cancer. Breast Cancer Res Treat. 2011;127:853-9.
16. Renwick A, Thompson D, Seal S, et al. ATM mutations that cause ataxia-telangiectasia are breast cancer susceptibility alleles. Nat Genet. 2006;38:873-875.
17. Walsh T, Casadei S, Lee MK, Pennil CC, Nord AS, Thornton AM, Roeb W, Agnew KJ, Stray SM, Wickramanayake A, Norquist B, Pennington KP, Garcia RL, et al. Mutations in 12 genes for inherited ovarian, fallopian tube, and peritoneal carcinoma identified by massively parallel sequencing. Proc Natl Acad Sci USA. 2011;108: 18032–18037.
18. Tung N, Battelli C, Allen B, Kaldate R, Bhatnagar S, Bowles K, Timms K, Garber JE, Herold C, Ellisen L, Krejdovsky J, DeLeonardis K, Sedgwick K, Soltis K, Roa B, Wenstrup RJ, Hartman AR. Frequency of mutations in individuals with breast cancer referred for BRCA1 and BRCA2 testing using next-generation sequencing with a 25-gene panel. Cancer. 2015;121:25-33.
19. Shirts BH, Casadei S, Jacobson AL, Lee MK, Gulsuner S, Bennett RL, Miller M, Hall SA, Hampel H, Hisama FM, Naylor LV, Goetsch C, Leppig K, Tait JF, Scroggins SM, Turner EH, Livingston R, Salipante SJ, King MC, Walsh T, Pritchard CC. Improving performance of multigene panels for genomic analysis of cancer predisposition. Genet Med. 2016 Feb 4.
20. Minion LE, Dolinsky JS, Chase DM, Dunlop CL, Chao EC, Monk BJ. Hereditary predisposition to ovarian cancer, looking beyond BRCA1/BRCA2. Gynecol Oncol. 2015;137:86-92.
21. Maxwell KN, Wubbenhorst B, D’Andrea K, Garman B, Long JM, Powers J, Rathbun K, Stopfer JE, Zhu J, Bradbury AR, Simon MS, DeMichele A, Domchek SM, Nathanson KL. Prevalence of mutations in a panel of breast cancer susceptibility genes in BRCA1/2-negative patients with early-onset breast cancer. Genet Med. 2015;17:630-638.
22. Castéra L, Krieger S, Rousselin A, Legros A, Baumann JJ, Bruet O, Brault B, Fouillet R, Goardon N, Letac O, Baert-Desurmont S, Tinat J, Bera O, et al. Next-generation sequencing for the diagnosis of hereditary breast and ovarian cancer using genomic capture targeting multiple candidate genes. European Journal of Human Genetics 2014:22: 1305–1313.
23. Susswein LR, Marshall ML, Nusbaum R, Vogel Postula KJ, Weissman SM, Yackowski L, Vaccari EM, Bissonnette J, Booker JK, Cremona ML, Gibellini F, Murphy PD, Pineda-Alvarez DE, et al. Pathogenic and likely pathogenic variant prevalence among the first 10,000 patients referred for next-generation cancer panel testing. Genet Med. 2015 Dec 17.
24. Slavin TP, Niell-Swiller M, Solomon I, Nehoray B, Rybak C, Blazer K, Weitzel JN. Clinical application of multigene panels : Challenges of next-generation counseling and cancer risk management. Front. Oncol. 2015;5:208.
25. Desmond A, Kurian AW, Gabree M, Mills MA, Anderson MJ, Kobayashi Y, Horick N, Yang S, Shannon KM, Tung N, Ford JM, Lincoln SE, Ellisen LW. Clinical Actionability of Multigene Panel Testing for Hereditary Breast and Ovarian Cancer Risk Assessment. JAMA Oncol. 2015;1:943-951.
26. Kurian AW, Hare EE, Mills MA, Kingham KE, McPherson L, Whittemore AS, McGuire V, Ladabaum U, Kobayashi Y, Lincoln SE, Cargill M, Ford JM. Clinical Evaluation of a Multiple-Gene Sequencing Panel for Hereditary Cancer Risk Assessment. J Clin Oncol 2014:32;2001-2009.
27. Thompson ER, Rowley SM, Li N, McInerny S, Devereux L, Wong-Brown MW, Trainer AH, Mitchell G, Scott RJ, James PA, Campbell IG. Panel Testing for Familial Breast Cancer: Calibrating the Tension Between Research and Clinical Care. J Clin Oncol. 2016 May 1;34:1455-9.
28. Schroeder C, Faust U, Sturm M, Hackmann K, Grundmann K, Harmuth F, Bosse K, Kehrer M, Benkert T, Klink B, Mackenroth L, Betcheva-Krajcir E, Wimberger P, et al. HBOC multi-gene panel testing: comparison of two sequencing centers. Breast Cancer Res Treat 2015;152:129–13.
29. Walsh T, Casadei S, Lee MK, Pennil CC, Nord AS, Thornton AM, Roeb W, Agnew KJ, Stray SM, Wickramanayake A, Norquist B, Pennington KP, Garcia RL, et al. Mutations in 12 genes for inherited ovarian, fallopian tube, and peritoneal carcinoma identified by massively parallel sequencing. Proc Natl Acad Sci USA. 2011;108: 18032–18037.
30. LaDuca H, Stuenkel AJ, Dolinsky JS, Keiles S, Tandy S, Pesaran T, Chen E, Gau CL, Palmaer E, Shoaepour K, Shah D, Speare V, Gandomi S, Chao E. Utilization of multigene panels in hereditary cancer predisposition testing: analysis of more than 2,000 patients. Genet Med. 2014 Nov;16:830-7
31. Couch FJ, Hart SN, Sharma P, Toland AE, Wang X, Miron P, Olson JE, Godwin AK, Pankratz VS, Olswold C, Slettedahl S, Hallberg E, Guidugli L, et al. Inherited mutations in 17 breast cancer susceptibility genes among a large triple-negative breast cancer cohort unselected for family history of breast cancer. J Clin Oncol. 2015 Feb 1;33:304-11.
32. Weeks DE, Paterson MC, Lange K, Andrais B, Davis RC, Yoder F, Gatti RA. Assessment of chronic gamma radiosensitivity as an in vitro assay for heterozygote identification of ataxia-telangiectasia. Radiat Res. 1991; 128: 90-99.
33. Wiencke JK, Wara DW, Little JB, Kelsey KT. Heterogeneity in the clastrogenic response to X-rays in the Lymphocytes from Ataxia-telangiectasia heterozygotes and controls. Cancer Causes Control. 1992. 3: 237-245
34. Kiuru A, Kämäräinen M, Heinävaara S, Pylkäs K, Chapman K, Koivistoinen A, Parviainen T, Winqvist R, Kahim M, Launonen V. Assessment of targeted and Non-targeted responses in cells deficient in ATM function following exposure to low and high dose X-rays. Plos One. 2014. 3: e93211.
35. McGuire AL, McCullough LB, Evans JP: The indispensable role of professional judgment in genomic medicine. JAMA 2013;309:1465-1466.
36. Tuckson RV, Newcomer L, De Sa JM. Accessing genomic medicine: affordability, diffusion, and disparities. JAMA 2013;309:1469-1470.
37. Helder-Woolderink JM, Blok EA, Vasen HF, Hollema H, Mourits MJ, De Bock GH. Ovarian cancer in Lynch syndrome; a systematic review. Eur J Cancer. 2016 Jan 13; 55:65-73
38. Minion LE, Dolinsky JS, Chase DM, Dunlop CL, Chao EC, Monk BJ. Hereditary predisposition to ovarian cancer, looking beyond BRCA1/BRCA2. Gynecol Oncol. 2015;137:86-92.
39. Win AK, Young JP, Lindor NM, Tucker KM, Ahnen DJ, Young GP, Buchanan DD, Clendenning M, Giles GG, Winship I, Macrae FA, Goldblatt J, Southey MC, et al. Colorectal and other cancer risks for carriers and noncarriers from families with a DNA mismatch repair gene mutation: a prospective cohort study, J Clin Oncol 2012;30:958–964.
40. Win AK, Lindor NM, Jenkins MA. Risk of breast cancer in Lynch syndrome: a systematic review. Breast Cancer Res 2013;15:R27.
41. Clendenning M, Hampel H, LaJeunesse J, et al: Long-range PCR facilitates the identification of PMS2-specific mutations. Hum Mutat. 2006. 27:490-495
42. Vaughn CP, Robles J, Swensen JJ, et al: Clinical analysis of PMS2: mutation detection and avoidance of pseudogenes. Hum Mutat. 2010. 31:588-593
43. ten Broeke SW, Brohet RM, Tops CM, van der Klift HM, Velthuizen ME, Bernstein I, Capellá Munar G, Gomez Garcia E, Hoogerbrugge N, Letteboer TG, Menko FH2, Lindblom A, Mensenkamp AR, et al. Lynch syndrome caused by germline PMS2 mutations: delineating the cancer risk. J Clin Oncol. 2015. 33:319-25.
44. Mateo J, Carreira S, Sandhu S, et al. DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. N Engl J Med. 2015 Oct 29;373:1697-708.
45. Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589-95.
46. DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, McKenna A, Fennell TJ, Kernytsky AM, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491-8
47. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL; ACMG Laboratory Quality Assurance Committee. A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405-24.
48. Domchek SM, Bradbury A, Garber JE, et al. Multiplex genetic testing for cancer susceptibility: Out on the high wire without a net? J Clin Oncol 201;31:1267-1270.
49. Chute CG, Kohane IS. Genomic medicine, health information technology, and patient care. JAMA 2013;309:1467-1468.