
INTRODUCTION
Recent epidemiological data identifies prostate cancer (PC) as the most common non-cutaneous cancer and the second-leading cause of cancer-related death among males in the United States following lung cancer [1]. According to the American Cancer Society, approximately 180,000 new cases of PC are diagnosed and 26,000 men, or 1 in 39, die of PC each year [1]. The clinical course of PC is heterogeneous, ranging from indolent to rapidly progressive and fatal. While the five-year survival rate for localized PC is close to 100% due to the availability of curative treatments, some patients experience cancer progression to metastatic castrate-resistant prostate cancer (CRPC), which is currently incurable and carries a poor prognosis (reviewed in [2-4]). Although the recent U.S. Food and Drug Administration (FDA) approval of numerous therapeutic agents for CRPC is promising, an unmet need still exists for the development of rational biomarkers and novel treatment strategies to improve survival.
Prior to 2010, the chemotherapeutic taxane docetaxel (Taxotere®) was the only drug demonstrated to improve survival of CRPC patients in comparison to palliative chemotherapy with mitoxantrone (Novantrone®), increasing median overall survival from 16.3 to 19.2 months [5, 6]. In the last several years, there has been an influx of new therapies mainly due to improved understanding of CRPC biology [4, 7]. These promising drugs have positively altered the therapeutic landscape of CRPC, but emerging resistance mechanisms have already been described for most of these agents (reviewed in [8-11]).
The therapeutic agents receiving FDA approval for treatment of advanced PC in the past five years include 1) abiraterone (Zytiga®; approved 2011), 2) enzalutamide (Xtandi®; approved 2012), 3) cabazitaxel (Jevtana®; approved 2010), 4) sipuleucel-T (Provenge®; approved 2010) and 5) Alpharadin (Xofigo®; approved 2013) (reviewed in [4, 12-14]). Abiraterone is a small-molecule inhibitor of cytochrome P450 17A1 (CYP17A1), an enzyme required for both adrenal and intratumoral de novo biosynthesis of androgens [15]. Enzalutamide is a second-generation antiandrogen and acts as a pure antagonist with no agonist activity [16, 17]. Cabazitaxel is a third-generation chemotherapeutic of the taxane class, which demonstrated superiority to palliative mitoxantrone-based chemotherapy in the post-docetaxel metastatic CRPC setting, although the use of the drug has been hampered by hematological adverse events, most notably febrile neutropenia [18]. Sipuleucel-T is an autologous cellular immunotherapy, also referred to as a therapeutic cancer vaccine, designed to generate an immune response against PC cells expressing prostatic acid phosphatase [19, 20]. Alpharadin is a radioisotope-containing radium-223 dichloride, a nuclide which emits alpha particles, that allows for targeting of PC bone metastases with short-range, high-energy alpha radiation [21]. Clinical trials that investigate the optimal sequence [7, 14] and combinations of these agents in advanced PC to minimize side effects and exploit synergistic mechanisms are needed. Most importantly, novel agents which can be deployed to impose synthetic lethality [22, 23] or applied as second- or third-line treatments [24] in the setting of resistance to current therapies need to be identified and further developed. The clinical limitations of a narrow focus on androgen receptor (AR) as the sole therapeutic target in PC have been increasingly recognized as resistance to any agents targeting AR is inevitable [25-27]. Investigational approaches using combination therapy with pharmacological agents directed against AR and other molecular targets, in addition to AR-negative cells, in advanced PC may prove to be critical to enhance efficacy and delay onset of resistance to agents targeting AR in PC.
THERAPEUTIC TARGETING OF AR IN PROSTATE CANCER GROWTH
Similar to normal prostate, PC cells require androgens for continued growth [28]. The requirement for androgens is effectively exploited through the use of ADT as a first-line therapy for advanced PC (reviewed in [29-31]). AR, a member of the steroid hormone group of nuclear receptors which includes estrogen receptor (ER), progesterone receptor (PR) and mineralocorticoid receptor (MR), functions as a ligand-dependent transcription factor. Binding of androgen ligands, such as testosterone and the more potent dihydrotestosterone (DHT), induces a conformational change in AR that allows for nuclear translocation and induction of androgen-responsive gene expression supporting growth and viability of prostate cells (reviewed in [32-36]). Mechanistically, AR inhibition induces both cell cycle arrest and apoptosis of PC cells [37-41]. Expression of AR is found in primary PC and continues to be detectable throughout progression to CRPC [42, 43].
ADT by means of surgical or pharmacological castration, the latter in the form of luteinizing hormone-releasing hormone (LHRH) / Gonadotropin-releasing hormone (GnRH) agonists (e.g. leuprorelin (Lupron®); goserelin (Zoladex®)) reduces serum testosterone levels by 90-95%. This robust decrease in serum androgens is dampened by the fact that intraprostatic levels of DHT have been reported to decline by only about 50% [44]. The stimulus from the residual intraprostatic DHT and other androgens can be blocked by addition of an antiandrogen drug to produce combined androgen blockade (CAB) [4, 12, 45, 46]. First-generation antiandrogens such as flutamide and bicalutamide have been used clinically for decades and function as AR antagonists by competitive inhibition of androgen binding to AR [4, 12, 45, 46]. In the case of bicalutamide, it also promotes recruitment of AR corepressors which contributes to inhibition of AR transcriptional activity [47]. The second-generation antiandrogen enzalutamide, designed to bind the AR ligand-binding domain (LBD), induces a conformational change in AR that prevents AR nuclear translocation to a greater extent than first-generation agents [16, 17]. In contrast to the use of LHRH/GnRH agonists alone, the addition of antiandrogens should theoretically prevent residual intraprostatic DHT from binding to AR. However, meta-analyses of clinical trials comparing CAB vs. LHRH/GnRH agonist monotherapy for advanced PC have demonstrated only a modest benefit in overall survival, often outweighed by added toxicity and decreased quality of life [48-50].
PROGRESSION OF PROSTATE CANCER TO CASTRATE-RESISTANT DISEASE
PC is considered castrate resistant during ADT with castrate levels of serum testosterone (<50 ng/dL) if it fullfils one or more of the following criteria: 1) a rise of prostate-specific antigen (PSA) serum levels (biochemical progression), 2) development of symptoms in the presence of pre-existing cancer (clinical progression) or 3) detection of new metastatic lesions on imaging (radiographic progression) [51, 52]. CRPC has historically been referred to by several names, including “hormone-refractory” and “androgen-independent” PC [53]; however, the preferred terminology is “castrate-resistant” in recognition of intracrine androgen production which is at least partially responsible for resistance to ADT [54, 55]. CRPC presents as a spectrum of disease, ranging from asymptomatic rising PSA levels without evidence of metastasis to multiple distant metastases with debilitating cancer-related constitutional symptoms.
The mechanisms driving progression from androgen-dependent PC to CRPC are still largely unclear. Continued AR signaling, despite depletion of circulating androgens and AR blockade, is thought to be central to the development of CRPC [56, 57]. Reactivation of AR transcriptional activity has been attributed to various mechanisms, yet no single mechanism has been reliably shown to account completely for progression to CRPC in experimental models or clinical patients. The majority of research efforts have sought to understand how AR signaling is restored in CRPC, but a growing number of studies highlight mechanisms operating outside the AR signaling axis, which may substantially contribute to the development of CRPC. Figure 1 provides a summary of the most widely investigated mechanisms thought to drive CRPC.
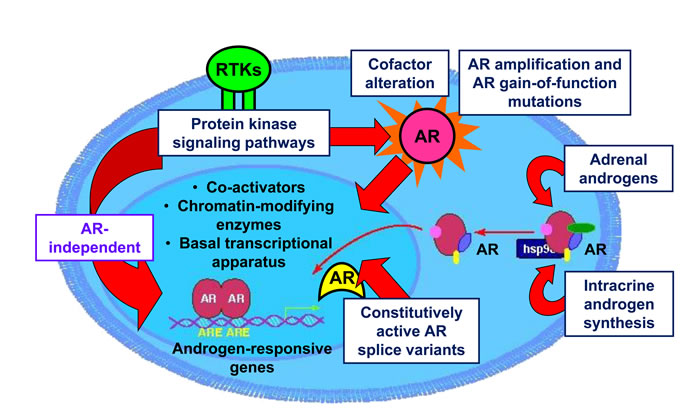
Figure 1: Molecular mechanisms driving CRPC. Development of CRPC has been attributed to numerous potential molecular mechanisms, including: 1) somatic mutations of AR resulting in increased affinity for ligands; 2) amplification of the AR gene locus; 3) intracrine biosynthesis of androgens in prostate cancer cells from adrenal steroids and cholesterol; 4) expression of constitutively active, ligand-independent AR splice variants; 5) non-canonical activation of AR by protein kinase signaling pathways in the absence of ligand by receptor tyrosine kinases (RTK); 6) AR-independent mechanisms operating outside of the AR signaling axis which promote castrate-resistant growth of prostate cancer.
AR-DEPENDENT MOLECULAR MECHANISMS DRIVING CASTRATE-RESISTANT PROSTATE CANCER
AR gene amplification and overexpression
One mechanism of AR reactivation in CRPC is via increased expression of AR, which may be achieved through genomic amplification of the AR locus or upregulation of AR protein levels. Early work using patient-matched clinical specimens indicated that approximately 30% of CRPC tumors harbored high-level AR gene amplification as demonstrated by fluorescence in situ hybridization (FISH), in comparison to none of the androgen-dependent primary tumors [58, 59]. Increased AR expression can sensitize PC cells to sub-physiological levels of androgen [60] and overcome inhibition by antiandrogens such as bicalutamide [61]. PC cells which overexpress AR are capable of accessing a greater number of binding sites on chromatin, generating an altered AR transcriptome [62]. In one report, cDNA microarray analysis of CRPC xenograft tumors found that the only change consistently associated with antiandrogen resistance was a modest increase in AR mRNA levels [63].
Somatic AR mutations
Somatic AR mutations may occur selectively in response to androgen deprivation, although the exact frequency of AR mutations in PC remains unknown. A review of 27 clinical studies found that AR mutations in androgen-dependent tumors ranged from 2-25%, while the incidence in CRPC tumors was slightly higher at 10-40% [64]. Additional work has identified the AR LBD as a mutational hotspot, with more recent studies placing the incidence of AR LBD point mutations in CRPC at ~15-20% [65-68]. Several gain-of-function AR LBD mutations which confer hypersensitivity to androgens or broaden ligand specificity have been characterized. Two well-known examples are AR-T877A and AR-W741C, originally described in LNCaP cells, which convert the antiandrogens flutamide and bicalutamide to partial agonists, respectively [69, 70].
Resistance to the second-generation antiandrogens enzalutamide (formerly MDV3100) and apalutamide (formerly ARN-509) was shown to be conferred by the novel AR LBD mutation F876L (alternatively reported as F877L in a different genomic build) in in vitro and in vivo models of PC [25-27]. Moreover, AR-F876L was also detected circulating as cell-free tumor DNA in the serum of patients who had received apalutamide as part of a phase I clinical trial [26]. AR-F876L repositions the co-activator docking helix 12 of AR, promoting an antagonist-to-agonist switch of enzalutamide and apalutamide [25-27]. However, the overall contribution of AR-F876L to clinical resistance to second-generation antiandrogens remains to be determined; a genomic landscape study of 150 men with metastatic CRPC using next-generation sequencing did not detect the mutation, despite some patients previously receiving enzalutamide [68].
Analysis of plasma from patients with metastatic CRPC for circulating cell-free tumor DNA detected several AR LBD mutations associated with abiraterone resistance, including AR-T878A and AR-L702H [71, 72]. In one such study, emergence of AR-T878A and AR-L702H was observed in 13% of CRPC tumors progressing on abiraterone, while total AR copy number remained unchanged pre- and post-treatment with abiraterone [71]. Mechanistically, these AR LBD mutants were demonstrated to sensitize AR to activation by progesterone (T878A) [73] and glucocorticoids (L702H) [74], circumventing the effects of CYP17A1 inhibition on AR signaling. Interestingly, it is necessary for abiraterone to be administered concurrently with the glucocorticoid prednisone to prevent the common adverse reaction of mineralocorticoid excess syndrome resulting from prolonged CYP17A1 blockade [74].
Prostate intracrine androgen biosynthesis
In the absence of circulating androgens produced by the testes, an alternative mechanism to activate AR signaling is through conversion of cholesterol and adrenal androgen precursors into testosterone and DHT in PC cells (reviewed in [75, 76]). It has been demonstrated that PC cells contain all components required for androgen biosynthesis, with many of the key enzymes being elevated in recurrent or metastatic tumors [77, 78]. Enzymes responsible for the conversion of cholesterol to androgen precursors (CYP17A1, HSDD3B2) and conversion of androgen precursors to testosterone and DHT (AKR1C3, SRD5A1/2) were shown to be elevated approximately 8-10 fold in metastatic PC [79]. Therefore, upregulation of the androgen production machinery may increase local androgen concentrations in CRPC, further facilitating reactivation of the AR signaling pathway. Production of androgen precursors, such as dehydroepiandrosterone (DHEA) and androstenedione, is blocked by abiraterone, which inhibits the enzymatic activity of the CYP17A1 enzyme [80, 81]. Other androgen biosynthesis inhibitors in clinical development are Ortonel (TAK-700), Galeterone (TOK-001/VN1/124-1) and VT-464 [46]. However, several mechanisms of resistance to abiraterone were documented shortly after FDA approval, including elevated intratumoral levels of CYP17A1 [82] and gain-of-function mutations which sensitize AR to non-androgen ligands [73, 74]; it remains to be seen whether these represent the primary means by which resistance develops to abiraterone.
AR splice variants (AR-Vs)
Constitutively active AR splice variants (AR-Vs), generated through alternative gene splicing or rearrangement of the AR gene, have been increasingly studied as a contributor to castrate-resistant growth over the past several years. The expression of AR-Vs has now been well-documented in PC cell lines, xenograft tumors, and clinical PC samples [83-85]. The majority of AR-Vs have extensive truncation or exon skipping of the complete C-terminus, leading to loss of the AR LBD. AR-Vs, in which the bipartite nuclear localization signal (NLS) contained in exons 3 and 4 is disrupted, were shown to be predominantly cytoplasmic in AR transactivation reporter assays [86]. However, other AR-Vs contain both exons 3 and 4 and are thus capable of nuclear translocation and activation of AR target genes in a ligand-independent manner. A notable exception is AR-V7, the best characterized AR-V to date, which is constitutively active despite lacking a full NLS, through a mechanism yet to be fully determined [84, 86, 87].
Whether AR-Vs promote castrate-resistant growth of PC primarily by reactivating the normal AR transcriptome or generating an altered AR gene signature remains disputed. Although AR-Vs have been shown to be capable of activating canonical AR target genes such as KLK3 (encoding PSA), several studies have investigated whether AR-Vs can generate alternative gene signatures in CRPC cells [85-87]. One report found that the AR-V gene signature was largely independent of the transcriptional profile generated by full-length AR [88]. In contrast, the Dehm group found that AR-Vs, including AR-V7, preferentially bind the same canonical high-affinity androgen response elements (AREs) engaged by full-length AR on a genome-wide scale [89]. Of note, the Dehm group developed a unique model, the R1-D567 cell line, engineered to exclusively express constitutively active AR-Vs in the absence of full-length AR [89], while other investigators have used PC cell lines with heterogeneous expression of both full-length AR and AR-Vs [85-87]. These findings suggest that AR-V genomic binding specificity and transcriptional output may be highly dependent on the experimental model selected and its particular cellular context.
The constitutive, ligand-independent activity of AR-Vs has substantial implications for treatment of CRPC. AR-V7 transcript and protein levels were found to be upregulated in clinical CRPC bone metastases and correlated with decreased survival rate after surgery [90]. In a more recent study, detection of AR-V7 in circulating tumor cells from enzalutamide- or abiraterone-treated patients was shown to be associated with therapeutic resistance to both agents [91]. AR-V7-positive patients treated with either enzalutamide or abiraterone were found to have lower PSA response rates, shorter PSA progression-free survival, shorter clinical or radiographic progression-free survival and shorter overall survival, compared to AR-V7-negative patients [91]. Based on the noted association, disruption of AR-V activity may be a means to prevent or reverse resistance to enzalutamide and abiraterone. Indeed, short-interfering RNA (siRNA)-mediated knockdown of AR-V7 was shown to confer sensitivity to enzalutamide in CWR22Rv1 cells, which intrinsically express high levels of AR-V7 [92]. On the other hand, overexpression of AR-V7 in LNCaP cells negative for AR-Vs failed to confer resistance to enzalutamide in vitro or in vivo [86]. Further mechanistic work is therefore needed to determine the contribution of AR-V7 and other AR-Vs to development of resistance to enzalutamide and abiraterone.
A novel therapeutic strategy capable of inhibiting both full-length AR and AR-Vs relies on targeting of the AR N-terminus, which is conserved across all AR isoforms. EPI-001, a bisphenol A-derived small molecule antagonist, which covalently binds the AR N-terminal domain, was shown to block transcriptional activity of AR and several AR-Vs, including AR-V7 [93-96]. Based on the preclinical data, a prodrug formulation of the compound, EPI-506, is currently undergoing a Phase I/II clinical trial (NCT02606123) in CRPC patients previously treated with enzalutamide and/or abiraterone. Targeting of the AR N-terminus may prove to be a viable therapeutic approach for circumventing AR LBD-based deletions, which confer resistance to conventional antiandrogens and ligand-depleting agents such as abiraterone.
Non-canonical AR transactivation
Numerous growth factors, cytokines, and hormones have been implicated in the activation of AR when androgens are absent or present at sub-physiological concentrations [97]. Non-canonical induction of AR signaling has been linked to mechanisms which promote AR phosphorylation [98]. Insulin-like growth factor-1 (IGF-1) was shown to induce AR transcriptional activity under androgen-deprived conditions in vitro, an effect which could be inhibited by bicalutamide [99]. Separately, it was reported that IGF-1-mediated AR activation required expression of β(1A) integrins, which facilitated functional interaction between the activated IGF-1 receptor and AR, subsequently leading to AR-mediated anchorage-independent growth of PC [100]. Interleukin-6 (IL-6) was shown to activate AR reporter gene constructs in DU145 PC cells and upregulate PSA secretion in LNCaP cells [101]. AR activation by IL-6 has been proposed to be mediated by both the Stat3 and MAPK signaling pathways in PC cells [102, 103]. Specifically, in the absence of androgens, IL-6 was shown to induce association of Stat3 and AR, followed by increased expression of AR-regulated genes [102].
Promotion of AR signaling in PC may also occur through loss of negative regulatory signals, which suppress AR function. This concept is supported by findings demonstrating that the cyclin D1b isoform of the widely studied cell cycle regulator does not possess AR inhibitory function attributed to the more common cyclin D1a isoform [104], and that cyclin D1b was found to be elevated in clinical PC specimens [105]. Additionally, a recent paper demonstrated that cyclin D1b cooperates with AR signaling to promote tumor cell growth and metastatic behavior of PC cells, partially by inducing expression of the pro-oncogenic transcription factor SNAI2 (Slug) [106]. In another example, loss or inactivation of the retinoblastoma (Rb) tumor suppressor gene induced an E2F1-mediated increase in AR mRNA and protein levels, which promoted castrate-resistant growth and resistance to bicalutamide in PC cells [107].
Modulation of AR activity by receptor tyrosine kinases may contribute to progression of PC to castrate-resistant cancer. Overexpression of Her2/neu (ErbB2) has been shown to enhance AR transcriptional activity in both the presence and absence of androgens, stimulating proliferation of LNCaP cells under both settings [108, 109]. Moreover, Src kinase promoted AR transcriptional activity in castrate-resistant C4-2 cells [110]. In line with these findings, several other studies demonstrated that AR transcriptional activity was attenuated by protein kinase inhibitors [101, 111].
AR-INDEPENDENT MOLECULAR MECHANISMS DRIVING CRPC
AR-independent bypass pathways
While most research to date has focused on the continued importance of AR in CRPC, alternative signaling pathways supporting proliferation and survival of CRPC cells have been shown to be capable of completely bypassing AR [112-114]. Consequently, AR bypass pathways are not dependent on AR for their downstream effects and can theoretically remain active even in the absence of AR expression [112-114]. Sustained blockade of AR signaling may contribute to the selection of PC cell clones able to upregulate AR bypass pathways, conferring a castrate-resistant phenotype.
Several studies have investigated candidate genes which may support CRPC growth independently of AR. Whitworth and colleagues used an RNA interference (RNAi) phenotypic screen to profile 673 kinases and identify those which contributed most to castrate-resistant growth of LNCaP cells under androgen-depleted conditions [112]. Moreover, expression of the anti-apoptotic protein Bcl-2 was reported to be induced in PC xenograft tumors initially negative for Bcl-2 expression following castration of mice [115]. At the same time, inhibition of Bcl-2 using antisense oligonucleotides delayed onset of castrate resistance in an LNCaP xenograft tumor model [116]. Bcl-2 has been reported to be overexpressed in both mouse models of CRPC as well as clinical samples from CRPC patients [117, 118], although increased Bcl-2 expression is not strictly a prerequisite for progression to CRPC [118, 119].
A recent body of work supports the concept that the glucocorticoid receptor (GR) may be able to substitute for AR in binding to androgen response elements (AREs) and driving PC cell survival under certain circumstances [113, 120, 121]. Chromatin immunoprecipitation followed by sequencing (ChIP-seq) and mRNA expression analysis indicated that these two steroid hormone receptors have highly overlapping cistrome and transcriptome profiles in PC [113, 120]. Furthermore, acquired resistance to enzalutamide in LNCaP cells and xenograft tumors correlated with upregulation of GR expression and was demonstrated to be dependent on GR for enzalutamide-resistant growth [113]. Glucocorticoid-induced activation of GR in VCaP cells was also found to confer resistance to enzalutamide [113]. In the same study, PC xenograft tumors which developed resistance to the second-generation antiandrogens enzalutamide and apalutamide showed a 27-fold higher expression of GR mRNA levels compared to controls [113]. Although research is ongoing, evidence thus far suggests that proliferation and survival signals may be able to bypass AR through GR in PC cells treated with antiandrogens, including second-generation agents. Whether other steroid hormone receptors, such as progesterone receptor (PR), contribute to AR bypass signaling remains to be determined.
AR-negative cell populations: prostate cancer stem-like cells
The cancer stem cell theory states that only a rare subpopulation of tumor cells possessing stem cell-like properties, most notably unlimited self-renewal capacity, and multilineage differentiation, can initiate tumor formation [122]. The more general term “tumor-initiating cells (TICs)” is also used interchangeably [123] with cancer stem-like cells, and refers to a population of cells within the original clinical cancer sample that has the ability and is critical for forming a tumor when xenotransplanted to immunodeficient mice [124]. In other words, the presence of TICs in patient-derived cancer samples and supportive growth environment for their proliferation in vivo determines the success rates of the tumor formation in mice [124]. Cancer stem cells are believed to be resistant to most therapies used to debulk tumors, harboring the ability to repopulate a tumor with therapy-resistant progeny more prone to metastasis (reviewed in [125, 126]). The gold standard for assessment of putative cancer stem-like cell populations is the capability of the cancer stem-like cells to generate serially transplantable xenograft tumors in immunodeficient mice that histopathologically resemble the parental tumor. Other surrogate assays/markers of cancer stem-like properties include cell surface markers, transcriptional profiles and tumor sphere-forming ability (reviewed in [127]).
Several putative populations of PC stem-like cells (PCSCs) have been reported in the literature, most commonly identified through fluorescence-activated cell sorting (FACS) of bulk tumor cell lines, xenograft tumors or clinical samples for stem-like cell surface markers. Reported marker profiles include CD44+/α2β1+/CD133+ [128], TRA-1-60/CD151/CD166 [129], ALDH [130] and PSA−/lo [131], among others. While PC stem-like cells are positive for CD44 marker, the expression of the standard isoform of CD44 is typically lost in PC due to aberrant splicing[132]. Importantly, a number of reports have suggested that PCSCs are AR-negative or express very low levels of AR, predicting lack of responsiveness to antiandrogens and other therapies targeting the AR signaling axis. Patrawala and colleagues demonstrated that CD44+ PCSCs were negative for AR expression, as well as approximately 10-100x more tumorigenic in immunodeficient mice than CD44- non-PCSCs [133]. Moreover, putative CD44+/α2β1+/CD133+ PCSCs isolated from clinical PCs following prostatectomy were found to be AR-negative and exhibited a prostate basal cell phenotype [128]. More recent work supports enrichment of a basal cell gene expression profile in metastatic CRPC, establishing a link between expansion of a stem-like cell population and castrate resistance [134]. Collectively, this body of work suggests that residual AR-negative/low PCSCs may remain after androgen deprivation, potentially repopulating tumors with castrate-resistant tumor cells.
AR-negative cell populations: neuroendocrine prostate cancer cells
Interspersed within prostate epithelium are neuroendocrine (NE) cells, which can be identified by morphology under electron microscopy or through positive staining for chromogranin A, synaptophysin, neuron-specific enolase or other markers of NE differentiation [135]. These specialized cells secrete a variety of neuropeptides, such as bombesin, neurotensin, parathyroid hormone-related protein (PTHrP), serotonin and calcitonin, which are known to promote proliferation and survival of prostate adenocarcinoma cells [135]. In the normal prostate, NE cells constitute <1% of the total cell population and typically remain quiescent [135].
Neuroendocrine prostate cancer (NEPC) describes a heterogeneous group of tumors, including prostate adenocarcinoma with NE differentiation, adenocarcinoma with Paneth cell NE differentiation, carcinoid tumors, pure small cell carcinomas, pure large cell NE cancer and adenocarcinomas with mixed histologies [136]. Approximately 5-10% of primary prostate adenocarcinomas contain NE tumor cells, with greatest abundance in high-grade PCs [135, 137]. NEPC can be classified as de novo or treatment-related. NEPC arising de novo is a rare entity, typically constituting less than 2% of all primary PCs and displays a high propensity for metastasis, limited treatment options and dismal prognosis, with most patients succumbing to the cancer within a year of diagnosis [138, 139]. Much more commonly, NEPC emerges in CR recurrent tumors in patients who have previously had androgen deprivation therapy for prostate adenocarcinoma (reviewed in [140-154]), suggesting that epithelial cells are able to transdifferentiate into NE cells and/or NE cells possess a proliferative advantage under androgen deprivation.
NEPC cells are negative for AR and PSA expression and highly proliferative [155]. Recent work by Beltran and colleagues [156] indicates divergent evolution of NEPC cells from one or more CRPC cells rather than linear clonal evolution. Moreover, NE differentiation in PC has been shown to be a predictive factor of unfavorable clinical outcome [147, 157-160]. Most importantly, emergence of NE differentiation during PC progression to CRPC has therapeutic implications. Specifically, molecular mechanisms driving AR-independent/negative NE cells in CRPC need to be identified for development of strategies to therapeutically target NE cell populations in CRPC.
The lack of AR expression in NEPC cells [135, 137] has led to efforts to determine the key signaling pathways that are dysregulated in these cells. Molecular profiling of NEPC revealed that deletion of RB1 and PTEN, mutations in TP53 and amplification of MYCN and Aurora kinase A (AURKA) are common in NE cells [161, 162]. Work from the Huang group demonstrated that mutant p53 inactivates the IL-8/CXCR2/p53 signaling pathway which maintains NE cells in a quiescent state [163] and simultaneously results in AURKA overexpression, leading to hyperproliferation of NE cells [164]. Overexpression of N-Myc in LNCaP cells induced NE differentiation, downregulated AR levels and decreased AR target gene expression [162]. Moreover, the Witte group showed that aberrant N-Myc expression combined with activated AKT1 was sufficient to transform human prostate epithelial cells into NEPC with phenotypic features characteristic of metastatic CRPC in patients [165]. With the growing clinical use of abiraterone and enzalutamide to suppress AR signaling, NE differentiation may become an increasingly common resistance mechanism underlying castrate resistance.
JAK2-STAT5 SIGNALING PROMOTES GROWTH OF PROSTATE CANCER THROUGH AR-DEPENDENT AND AR-INDEPENDENT MOLECULAR MECHANISMS
Jak2-Stat5a/b signaling pathway in prostate cancer
In order to target AR-negative cell populations in CRPC and to impose an additional survival strain for AR-positive PC cells during ADT, identification of non-AR therapeutic targets may provide novel treatment strategies for CRPC. Stat5a/b are latent cytoplasmic proteins that function as both signaling molecules and nuclear transcription factors, which are critical for PC cell viability in vitro, in vivo and in patient-derived PCs ex vivo [166-174]. Two highly homologous isoforms, Stat5a and Stat5b (hereafter referred to as Stat5a/b), display over 90% amino acid identity and are encoded by genes juxtaposed on chromosome 17q21.2 [175].
In PC, Stat5a/b is predominantly activated by binding of prolactin (Prl) to the membrane-bound prolactin receptor (PrlR), which transmits signals through Jak2 kinase [173, 176, 177]. In addition, Stat5a/b may also be activated by Src kinase or members of the EGF receptor family [178-180]. Prl is a polypeptide hormone whose actions are mediated by the PrlR, a non-kinase single-pass transmembrane receptor part of the class 1 hematopoietic cytokine receptor superfamily (reviewed in [181, 182]). Prl is primarily secreted by lactotroph cells of the anterior pituitary gland and carries out a wide variety of functions, including the well-known effect of stimulating lactation in females; circulating Prl is present in males, though at lower levels than in females (reviewed in [181, 182]). A large body of work provides evidence that Prl is expressed within the prostate epithelium and functions as a local growth factor in an autocrine/paracrine manner distinct from its endocrine mechanism [183-187]; (reviewed in [188]).
Signaling through the Jak2-Stat5a/b pathway is initiated upon Prl binding to PrlR and subsequent receptor-associated Jak2 kinase activation by autophosphorylation. This leads to phosphorylation of cytoplasmic tyrosine residues of the cytokine receptor which serves as docking sites for Stat5a/b via binding of the Stat5a/b SH2 domain [189]. Stat5a/b monomers recruited to the receptor-tyrosine kinase complex are activated by tyrosine kinase-mediated phosphorylation at conserved C-terminal tyrosine residues (Stat5a: Y694, Stat5b: Y699) (reviewed in [190]). Phosphorylated Stat5a/b (pY694/699) monomers undergo homo- or heterodimerization, followed by nuclear localization of functional Stat5a/b dimers in an active, energy-dependent process directed by Ran-dependent nuclear import machinery [191, 192]. Specifically, a nuclear shuttling complex is formed by the chaperone protein MgcRacGAP, the small G protein Rac1 and the carrier protein importin/karyopherin β1, which cooperate to translocate the Stat5a/b dimer from cytosol to nucleus [193]. Within the nucleus, Stat5a/b dimers bind to palindromic DNA consensus sequences (TTC(C/T)N(G/A)GAA), referred to as gamma-interferon activation (GAS) elements, to initiate transcription of Stat5a/b regulated genes [194-196]. Homodimers of Stat5a/b have equivalent specificity for binding palindromes spaced three base pairs apart [196, 197], while tetramers of Stat5a/b can bind tandem consensus and non-consensus GAS motifs spaced six base pairs apart [196]. The sequence of molecular events in the canonical Jak2-Stat5a/b signaling cascade leading to activation of Stat5a/b-regulated genes is shown in Figure 2.
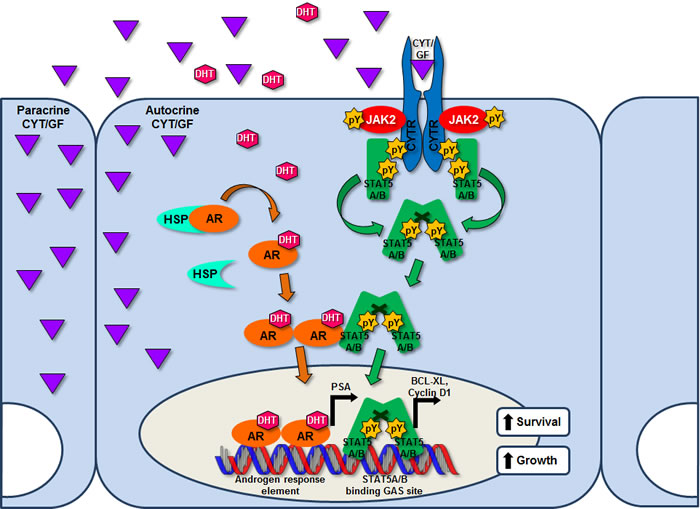
Figure 2: Canonical Jak2-Stat5a/b signaling in prostate cancer cells and functional interaction with AR signaling. Various levels of inhibition are possible in the Stat5a/b signaling cascade to block Stat5a/b target gene expression, including targeting of cytokine receptor activation, Jak2 kinase-mediated phosphorylation, and Stat5a/b dimerization, DNA binding and transcriptional activity. Physical interaction between Stat5a/b and AR has been reported, providing a mechanistic basis for crosstalk between these two pathways, which results in reciprocal synergistic effects on nuclear localization and transcriptional activity. Depicted are known Stat5a/b target genes Bcl-xL and cyclin D1 and AR target gene PSA. Abbreviations: AR, androgen receptor; ARE, androgen response element; CYT, cytokine; CYTR, cytokine receptor; DHT, dihydrotestosterone; GAS, gamma-interferon activation sequence; GF, growth factor; HSP, heat shock protein; pY, phosphotyrosine.
Stat5a/b is a predictive biomarker and therapeutic target in prostate cancer
Over the last decade, numerous reports have supported the role of Stat5a/b in clinical progression of PC. Stat5a/b is constitutively active in human PC, but not in normal prostate epithelium [174]. Examination of the distribution of active Stat5a/b across 114 paraffin-embedded clinical PC specimens of various histological grades revealed that active Stat5a/b was strongly associated with high-grade PC [173]. In a separate study, analysis of tissue microarrays derived from 357 PC patients accompanied by 30-year clinical follow-up data found that high expression levels of active Stat5a/b were correlated with poorly differentiated, high-grade PC [172]. Importantly, active Stat5a/b status was predictive of early PC recurrence after initial treatment by radical prostatectomy or transurethral resection [172]. Active Stat5a/b was found to be an independent prognostic marker of early cancer recurrence not only in high-grade PC but also in intermediate grade PC [172]. Patients who had received androgen deprivation therapy prior to radical prostatectomy were more likely to display active Stat5a/b when compared to patients not receiving neoadjuvant therapy [172]. More recent work analyzing nuclear Stat5a/b expression levels in two cohorts of PC patients treated by radical prostatectomy or deferred palliative therapy showed that nuclear Stat5a/b was an independent prognostic marker for both cohorts [166]. Corroborating earlier work, high levels of nuclear Stat5a/b predicted increased risk of early cancer recurrence following radical prostatectomy in PCs of intermediate grade [166]. At the same time, active Stat5a/b predicted early PC-specific death in the deferred palliative therapy cohort [166]. In the cohort of PC patients treated by radical prostatectomy, each patient was represented by 10 tissue cores taken from different areas of the tumor. Interestingly, the presence of active Stat5a/b even in one of the 10 tissue cores already predicted likely cancer recurrence after radical prostatectomy [166], suggesting that those PCs with elevated nuclear Stat5a/b anywhere in the tumor may already have micrometastasized at the time of surgery. Further work demonstrated that the Stat5a/b gene locus is amplified in approximately 30% of CRPC metastases [198]. Similar to the presence of nuclear Stat5a/b protein in specific areas of prostate tumor, Stat5a/b locus amplification was dispersed into groups of PC cells within the tumor tissue, as demonstrated by FISH of paraffin-embedded tissue sections. It is important to note, however, that presence of specific areas with Stat5a/b gene locus amplification shown by FISH may not be detectable in analyses of RNA/DNA extracted from ground whole tissue specimens which include a mix of both epithelial and stromal components. Collectively, these findings indicate that Stat5a/b may provide a robust biomarker for recurrence of lethal PC after failure of first-line therapy for early stage cancer, warranting further investigation.
Stat5a/b promotes growth of prostate cancer cells and xenograft tumors
A substantial body of work supports the concept that Stat5a/b is highly critical for the viability of human PC cells in vitro and xenograft tumor growth in vivo. Inhibition of Stat5a/b by varied methods induced massive apoptotic death of PC cells in culture and suppressed the growth of xenograft tumors in nude mice [170, 171, 174, 199]. Ahonen and colleagues provided the first evidence of Stat5a/b in promotion of PC growth [174]. Transduction of PC cells with adenovirus expressing dominant-negative Stat5a/b lacking the transactivation domain, induced extensive apoptosis of PC cells, as determined by cell morphology, DNA fragmentation, caspase-3 and -9 activation, and cell viability assays [174]. Stat5a/b regulation of PC cell viability was further validated in the transgenic adenocarcinoma mouse prostate (TRAMP) model. In TRAMP-derived PC cell lines, use of an inducible carboxy terminal-truncated Stat5a/b construct to inhibit wild-type Stat5a/b activity led to decreased PC cell growth in soft agar and tumor formation in nude mice [200]. More recent studies further indicated a critical role of Stat5a/b in maintenance of PC viability, as inhibition of Stat5a/b by various methodological approaches (antisense oligonucleotides, RNA interference or adenoviral expression of dominant-negative Stat5a/b) triggered extensive apoptosis across multiple Stat5a/b-positive PC cell lines. Mechanistically, Stat5a/b was found to sustain viability and promote cell cycle progression of PC cells through activation of Bcl-xL and cyclin D1, respectively [171]. Based on gene array analyses, target genes regulated by Stat5a/b include genes regulating proliferation, apoptosis and metastatic processes [169, 170]. These results also translated to the in vivo setting, with inhibition of Stat5a/b decreasing the incidence and growth of both subcutaneous and orthotopic PC xenograft tumors in nude mice [171]. Most importantly, inhibition of Stat5a/b blocked not only primary growth of PC xenograft tumors, but also recurrent growth of castrate-resistant tumors [177] demonstrated by pharmacological inhibition of Stat5a/b signaling by a potent Jak2 kinase inhibitor, AZD1480, in the CWR22Pc model of CRPC growth. In fact, Stat5a/b inhibition was more efficacious than docetaxel chemotherapy at both preventing recurrence of castrate-resistant CWR22Pc tumors following androgen deprivation as well as suppressing growth of established castrate-resistant CWR22Pc tumors [177].
Stat5a/b induces metastatic behavior and stem-like cell properties of prostate cancer cells
The first evidence of Stat5a/b promotion of metastatic behavior of PC cells in vitro and in vivo [169] was provided using an experimental lung metastasis model; inoculation of nude mice with PC cells expressing an active Stat5 construct (Stat5aS710F) increased in vivo formation of lung metastases by 11-fold compared to β-galactosidase (LacZ)-expressing control cells [169]. Active Stat5a/b upregulated hallmarks of the epithelial-to-mesenchymal transition (EMT) that precedes metastasis, including enhanced migration, decreased expression of cell surface E-cadherin, increased expression of mesenchymal markers and heterotypic adhesion of both AR-positive and AR-negative PC cells to endothelial cells [169, 201]. In conjunction with promotion of EMT, Stat5a/b induced stem-like properties in PC cells [201], such as tumor sphere formation and expression of the cancer stem cell marker Bmi1, a core component of Polycomb-Repressive Complex 1 (PRC1), which overrides cellular senescence and promotes self-renewal [202-204]. Mechanistically, Stat5a/b regulation of both EMT and cancer stem-like cell properties were found to be mediated by Twist1, a well-characterized EMT transcription factor [205-207]. Indeed, genetic knockdown of Twist1 suppressed Stat5a/b-induced functional endpoints of EMT, and Stat5a/b-induction of stem-like features in PC cells. These findings establish Stat5a/b in the promotion of EMT processes and a cancer stem-like cell phenotype, which enable the formation of distant PC metastases. Pharmacological targeting of Stat5a/b may prevent metastatic dissemination of PC.
Stat5a/b induces AR activity in prostate cancer cells, but also promotes PC cell growth independently of AR
Stat5a/b has been shown to promote AR signaling in PC cells (Figure 2). The first paper to report functional interaction between Stat5a/b and AR demonstrated that active Stat5a/b increased nuclear localization and transcriptional activity of AR in PC cells [208]. Moreover, Stat5a/b and AR were reported to physically interact in PC cells [208]. Follow-up studies implicated Stat5a/b in regulating proteasomal degradation of androgen-liganded AR in PC cells [209]. Specifically, Stat5a/b was shown to physically interact with and sequester AR liganded by antiandrogens, including enzalutamide, from trafficking to the proteasome [209]. Furthermore, antiandrogen therapy induced proteasomal degradation of AR in PC cells, a degradative mechanism which can be accelerated by disruption of Stat5a/b expression or activation [209]. A high-profile study using ChIP-Seq to map genome-wide occupancy of AR in clinical CRPC specimens revealed that AR binding sites in clinical PCs differed markedly from those in human PC cell lines. Importantly, novel AR binding sites identified in CRPC patients were strongly enriched for Stat, Myc and E2F binding motifs, suggesting that genomic repositioning of AR in CRPC may be induced by cooperating transcription factors in clinical CRPC [210]. Notably, this work also demonstrated that physical interaction of Stat5a/b and AR occurs in clinical CRPC specimens [210].
Importantly, Stat5a/b blockade, by either genetic or pharmacological knockdown, induces extensive apoptotic death of PC cells that do not express AR. Specifically, inhibition of Stat5a/b was found to induce rapid apoptotic death of DU145 PC cells, which are negative for AR expression, providing evidence that Stat5a/b sustains viability of PC cells independently of AR [170, 211]. At the same time, Stat5a/b overexpression in DU145 cells increased colony formation in vitro and accelerated xenograft tumor growth in vivo [198]. These findings indicate that Stat5a/b acts through mechanisms operating independently of AR to promote growth of PC, in addition to mechanisms directly impacting AR function. Future challenges include identification of the specific mechanisms underlying the capability of Stat5a/b to maintain and promote viability of PC cells independently of AR. Moreover, it will be important to determine if Stat5 inhibition will be able to induce death of AR-negative cell populations in castrate-resistant prostate tumors.
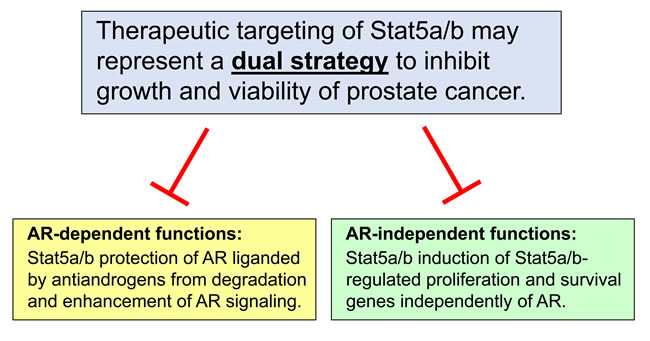
Figure 3: Therapeutic targeting of Stat5a/b inhibits growth of prostate cancer through AR-dependent and AR-independent mechanisms. Disruption of Stat5a/b signaling inhibits growth and viability of prostate cancer through two distinct mechanisms, serving as a potential dual strategy to eliminate prostate cancer cells. First, targeting of Stat5a/b inhibits the abilities of Stat5a/b to protect antiandrogen-liganded AR from proteasomal degradation and enhance AR signaling, including downstream molecular events such as AR nuclear localization, chromatin binding, and activation of target gene expression in prostate cancer cells. Second, targeting of Stat5a/b prevents the induction of AR-independent, Stat5a/b-regulated genes involved in proliferation and survival of prostate cancer cells.
Pharmacological targeting of Stat5a/b as a treatment strategy for prostate cancer
Recent studies demonstrate that enzalutamide provides a modest improvement in survival in patients with PC due to rapid development of resistance [8, 9]. Moreover, in a preclinical model, enzalutamide-resistant xenograft tumors treated with an experimental third-generation antiandrogen responded for only eight weeks before resistance developed again [212]. A treatment strategy that targets both AR and Stat5a/b concurrently potentially provides an additive or even synthetic lethal approach to kill PC cells as well as decrease the likelihood of selection for resistance mechanisms to the new-generation anti-androgens.
Pharmacological inhibition of the Stat5a/b signaling pathway can be accomplished through blockade at various levels, with inhibitors directed against the cytokine/growth factor receptor, tyrosine kinase or Stat5a/b itself. Of note, highest specificity with least clinical side-effects is likely achieved through direct targeting of Stat5 instead of its upstream activators. Indeed, past work has shown that disruption of Stat5a/b dimerization, transactivation and/or DNA binding is possible by targeting of the appropriate domain(s). Initial approaches utilized a Stat5a/b dominant-negative mutant lacking the transactivation domain [171, 174] and a Stat3 decoy phosphotyrosyl peptide [213, 214] to demonstrate proof-of-principle of direct Stat transcription factor inhibition. More recent investigations have shown that natural or synthetic small molecule compounds are capable of inhibiting Stat5a/b activity and downstream signaling (Table 1).
Table 1: Comparison of selected Stat5a/b inhibitors in efficacy of inhibition of Stat5a/b tyrosine phosphorylation (pYStat5a/b), as detected by Western blot analysis.
Publication (Fig. of Interest): |
Inhibitor Name: |
pYStat5a/b Inhibition (µM): |
Nam et al. 2012 (Fig. 2A) |
E804 |
10-20 |
Nelson et al. 2011 (Fig. 2A) |
Pimozide |
>10 |
Elumalai et al. 2015 (Fig. 4C) |
Stafib-1 (Comp. #17) |
10 |
Müller et al. 2008 (Fig. 3A) |
Comp. #6 |
400 |
Page et al. 2012 (Fig. 2A) |
BP-1-107 BP-1-108 SF-1-087 SF-1-088 |
40 |
Cumaraswamy et al. 2014 (Fig. 4A) |
AC-3-019 |
15 |
Liao et al. 2015 (Fig. 2F) |
IST5-002 |
5 |
Compounds which are currently in clinical use for a variety of oncologic and non-oncologic indications have been found to inhibit Stat5a/b in several preclinical studies (Table 1). Nam and colleagues screened a number of indirubicin derivatives, demonstrating that the most potent compound (E804) blocked Stat5a/b phosphorylation at 10-20 µmol/L in K562 human chronic myelogenous leukemia (CML) cells positive for Bcr-Abl, which drives constitutive activation of Stat5a/b [215]. Pimozide (Orap®), an antipsychotic drug, was shown to inhibit Stat5 phosphorylation in K562 cells by 35-55% at 10 µmol/L [216]. Other investigators have pursued rational drug design strategies in development of Stat5a/b inhibitors (Table 1). Elumalai and colleagues used fosfosal, a salicylic acid derivative and previously identified inhibitor of Stat5b [217], as a lead compound for optimization studies targeting the Stat5b SH2 domain [218]. The most promising compound, known as #17 or Stafib-1 (Stat five b inhibitor-1), specifically inhibited Stat5b phosphorylation in K562 cells at an IC50 of approximately 10 μmol/L [218]. In another study, a lead compound identified from a 17,000 molecule library was modified with chemical substitutions to yield a family of Stat5a/b small molecule inhibitors, of which compound #6 blocked IFN-α-stimulated Stat5a/b phosphorylation in human Burkitt’s lymphoma B cells at approximately 400 µmol/L [219]. A different investigator group generated a series of SH2-domain binding, salicylic acid-derived Stat5a/b inhibitors (BP-1-107, BP-1-108, SF-1-087, SF-1-088), which were all capable of fully blocking Stat5a/b phosphorylation at 40 µmol/L [220]. Building on this work, the investigators used one of their previously identified Stat5a/b inhibitors as a scaffold for further optimization [221]. A new library of 24 compounds was generated, of which AC-3-019 emerged as the most promising candidate, inhibiting Stat5a/b phosphorylation at 15 µmol/L in K562 cells [221].
Recently, the Nevalainen group identified and validated a small molecule inhibitor which targets the Stat5a/b SH2 domain in both PC and CML, malignancies known to be driven by Stat5a/b signaling [211]. In silico screening of chemical structure databases and medicinal chemistry modeling yielded 30 top-ranked candidate molecules, of which the lead compound IST5-002 proved most potent in biological assays [211]. Specifically, IST5-002 blocked transient docking of the SH2-domain of the Stat5 monomer to the phosphotyrosyl moiety of a tyrosine kinase-receptor complex, resulting in inhibition of both Jak2 and Bcr-Abl-mediated phosphorylation of Stat5. Secondly, dimerization of Stat5 was inhibited by IST5-002, which may be caused by decreased Stat5 phosphorylation or additional suppression of binding of the phosphorylated Stat5 monomer to the pY694/699 residues of the partner Stat5 monomer. Nuclear translocation, DNA binding and transcriptional activity of Stat5 were all inhibited at considerably low IC50 values, while IL-6-stimulated transcriptional activation of Stat3 was unaffected [211]. IST5-002 triggered extensive apoptosis of PC cells in culture, suppressed growth of PC xenograft tumors in nude mice and induced death of patient-derived PCs in ex vivo organ cultures [211]. IST5-002 also triggered apoptosis of both imatinib-sensitive and imatinib-resistant CML cell lines and induced death of primary CML cells [211]. Importantly, IST5-002 inhibited Bcr-Abl-driven Stat5a/b phosphorylation in K562 cells at an IC50 of less than 5 µM and, thus, IST5-002 is more potent in cell-based assays than any existing published Stat5a/b inhibitors (Table 1). Moreover, IST5-002 is the first small molecule inhibitor of Stat5a/b to demonstrate efficacy in experimental models of solid and hematological malignancies at low micromolar potency, providing a highly attractive lead structure for further optimization and clinical development.
CONCLUSIONS
PC remains a leading cause of cancer-related death for American men, with a subset of patients progressing to metastatic CRPC despite receiving aggressive treatment for localized cancer. An improved understanding of the molecular biology driving cancer progression has allowed substantial expansion of the therapeutic landscape for CRPC. Over the past five years, a diverse array of novel agents has been approved by the FDA for use in CRPC, with many other experimental therapies now in late-stage clinical trials. Most notable in the newly expanded treatment options for CRPC are the androgen synthesis inhibitor abiraterone and second-generation antiandrogen enzalutamide, both of which have been widely adopted in clinical practice by medical oncologists. However, additional studies are required to optimally integrate these new and emerging therapies with existing standard-of-care treatment paradigms. In addition, drug sequencing strategies need to be designed to anticipate and overcome newly described resistance mechanisms. Despite the impressive progress to date, the lack of a curative therapy for CRPC underscores a remaining unmet need, which stems from the still incompletely understood transition from androgen-dependence to castrate-resistance.
Identification of therapeutic targets which regulate non-AR-mediated proliferation and survival pathways represent an opportunity to improve treatment for advanced PC. Stat5a/b has been previously validated as a biomarker and therapeutic target in PC. Active Stat5a/b expression levels carry prognostic value by identifying patients at risk for cancer recurrence and poor clinical outcomes. Levels of active Stat5a/b carry predictive value as well, serving to predict treatment response to radical prostatectomy in patients with early stage PC. There is a strong rationale for pharmacological targeting of Stat5a/b based on its pleiotropic effects on sustaining PC growth and viability, enhancement of AR transcriptional activity and promotion of metastatic behavior. Stat5a/b serves not only as a critical survival factor for AR-positive but also AR-negative PC cells and xenograft tumors, providing a means for targeting PC outside of the AR signaling axis. The collective findings described in this review indicate that Stat5a/b acts through both AR-dependent and AR-independent molecular mechanisms, which are distinct but not mutually exclusive and therefore hold the potential for imposing synthetic lethal effects if blocked simultaneously. Pharmacological inhibition of Stat5a/b may thus represent a dual strategy to target growth of PC and delay onset of resistance to agents targeting AR in PC.
ACKNOWLEDGEMENTS
This work is supported in part by grants from the National Institutes of Health/National Cancer Institute to M.T.N. (7R01CA113580-10, 5R21CA178755-02) and Advancing a Healthier Wisconsin (#5520368) and D.T.H. (5F31CA180626-03).
CONFLICTs OF INTERESTS
The authors declare no conflict of interests.
REFERENCES
1. Siegel RL, Miller KD and Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016; 66(1):7-30.
2. Kirby M, Hirst C and Crawford ED. Characterising the castration-resistant prostate cancer population: a systematic review. Int J Clin Pract. 2011; 65(11):1180-1192.
3. Chang AJ, Autio KA, Roach M, 3rd and Scher HI. High-risk prostate cancer-classification and therapy. Nat Rev Clin Oncol. 2014; 11(6):308-323.
4. Attard G, Parker C, Eeles RA, Schroder F, Tomlins SA, Tannock I, Drake CG and de Bono JS. Prostate cancer. Lancet. 2016; 387(10013):70-82.
5. Petrylak DP, Tangen CM, Hussain MH, Lara PN, Jr., Jones JA, Taplin ME, Burch PA, Berry D, Moinpour C, Kohli M, Benson MC, Small EJ, Raghavan D and Crawford ED. Docetaxel and estramustine compared with mitoxantrone and prednisone for advanced refractory prostate cancer. N Engl J Med. 2004; 351(15):1513-1520.
6. Tannock IF, de Wit R, Berry WR, Horti J, Pluzanska A, Chi KN, Oudard S, Theodore C, James ND, Turesson I, Rosenthal MA and Eisenberger MA. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N Engl J Med. 2004; 351(15):1502-1512.
7. Lorente D, Mateo J, Perez-Lopez R, de Bono JS and Attard G. Sequencing of agents in castration-resistant prostate cancer. Lancet Oncol. 2015; 16(6):e279-292.
8. Nelson WG and Yegnasubramanian S. Resistance emerges to second-generation antiandrogens in prostate cancer. Cancer Discov. 2013; 3(9):971-974.
9. Ning YM, Pierce W, Maher VE, Karuri S, Tang SH, Chiu HJ, Palmby T, Zirkelbach JF, Marathe D, Mehrotra N, Liu Q, Ghosh D, Cottrell CL, Leighton J, Sridhara R, Ibrahim A, et al. Enzalutamide for treatment of patients with metastatic castration-resistant prostate cancer who have previously received docetaxel: U.S. Food and Drug Administration drug approval summary. Clin Cancer Res. 2013; 19(22):6067-6073.
10. Claessens F, Helsen C, Prekovic S, Van den Broeck T, Spans L, Van Poppel H and Joniau S. Emerging mechanisms of enzalutamide resistance in prostate cancer. Nat Rev Urol. 2014; 11(12):712-716.
11. Watson PA, Arora VK and Sawyers CL. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat Rev Cancer. 2015; 15(12):701-711.
12. Yap TA, Zivi A, Omlin A and de Bono JS. The changing therapeutic landscape of castration-resistant prostate cancer. Nat Rev Clin Oncol. 2011; 8(10):597-610.
13. Trewartha D and Carter K. Advances in prostate cancer treatment. Nat Rev Drug Discov. 2013; 12(11):823-824.
14. Drake CG, Sharma P and Gerritsen W. Metastatic castration-resistant prostate cancer: new therapies, novel combination strategies and implications for immunotherapy. Oncogene. 2014; 33(43):5053-5064.
15. Bianchini D, Zivi A, Sandhu S and de Bono JS. Horizon scanning for novel therapeutics for the treatment of prostate cancer. Expert Opin Investig Drugs. 2010; 19(12):1487-1502.
16. Scher HI, Beer TM, Higano CS, Anand A, Taplin ME, Efstathiou E, Rathkopf D, Shelkey J, Yu EY, Alumkal J, Hung D, Hirmand M, Seely L, Morris MJ, Danila DC, Humm J, et al. Antitumour activity of MDV3100 in castration-resistant prostate cancer: a phase 1-2 study. Lancet. 2010; 375(9724):1437-1446.
17. Tran C, Ouk S, Clegg NJ, Chen Y, Watson PA, Arora V, Wongvipat J, Smith-Jones PM, Yoo D, Kwon A, Wasielewska T, Welsbie D, Chen CD, Higano CS, Beer TM, Hung DT, et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science. 2009; 324(5928):787-790.
18. de Bono JS, Oudard S, Ozguroglu M, Hansen S, Machiels JP, Kocak I, Gravis G, Bodrogi I, Mackenzie MJ, Shen L, Roessner M, Gupta S and Sartor AO. Prednisone plus cabazitaxel or mitoxantrone for metastatic castration-resistant prostate cancer progressing after docetaxel treatment: a randomised open-label trial. Lancet. 2010; 376(9747):1147-1154.
19. Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, Redfern CH, Ferrari AC, Dreicer R, Sims RB, Xu Y, Frohlich MW and Schellhammer PF. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010; 363(5):411-422.
20. Di Lorenzo G, Buonerba C and Kantoff PW. Immunotherapy for the treatment of prostate cancer. Nat Rev Clin Oncol. 2011; 8(9):551-561.
21. Parker C, Nilsson S, Heinrich D, Helle SI, O’Sullivan JM, Fossa SD, Chodacki A, Wiechno P, Logue J, Seke M, Widmark A, Johannessen DC, Hoskin P, Bottomley D, James ND, Solberg A, et al. Alpha emitter radium-223 and survival in metastatic prostate cancer. N Engl J Med. 2013; 369(3):213-223.
22. Kaelin WG, Jr. The concept of synthetic lethality in the context of anticancer therapy. Nat Rev Cancer. 2005; 5(9):689-698.
23. Nijman SM. Synthetic lethality: general principles, utility and detection using genetic screens in human cells. FEBS Lett. 2011; 585(1):1-6.
24. Mateo J, Carreira S, Sandhu S, Miranda S, Mossop H, Perez-Lopez R, Nava Rodrigues D, Robinson D, Omlin A, Tunariu N, Boysen G, Porta N, Flohr P, Gillman A, Figueiredo I, Paulding C, et al. DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. N Engl J Med. 2015; 373(18):1697-1708.
25. Balbas MD, Evans MJ, Hosfield DJ, Wongvipat J, Arora VK, Watson PA, Chen Y, Greene GL, Shen Y and Sawyers CL. Overcoming mutation-based resistance to antiandrogens with rational drug design. Elife. 2013; 2:e00499.
26. Joseph JD, Lu N, Qian J, Sensintaffar J, Shao G, Brigham D, Moon M, Maneval EC, Chen I, Darimont B and Hager JH. A clinically relevant androgen receptor mutation confers resistance to second-generation antiandrogens enzalutamide and ARN-509. Cancer Discov. 2013; 3(9):1020-1029.
27. Korpal M, Korn JM, Gao X, Rakiec DP, Ruddy DA, Doshi S, Yuan J, Kovats SG, Kim S, Cooke VG, Monahan JE, Stegmeier F, Roberts TM, Sellers WR, Zhou W and Zhu P. An F876L mutation in androgen receptor confers genetic and phenotypic resistance to MDV3100 (enzalutamide). Cancer Discov. 2013; 3(9):1030-1043.
28. Huggins C and Hodges CV. Studies on prostatic cancer: The effects of castration, of estrogen and of androgen injection on serum phosphatases in metastatic carcinoma of the prostate. Cancer Res. 1941; 1:293-297.
29. Chen Y, Sawyers CL and Scher HI. Targeting the androgen receptor pathway in prostate cancer. Curr Opin Pharmacol. 2008; 8(4):440-448.
30. Mohler JL. A role for the androgen-receptor in clinically localized and advanced prostate cancer. Best Pract Res Clin Endocrinol Metab. 2008; 22(2):357-372.
31. Knudsen KE and Penning TM. Partners in crime: deregulation of AR activity and androgen synthesis in prostate cancer. Trends Endocrinol Metab. 2010; 21(5):315-324.
32. Gelmann EP. Molecular biology of the androgen receptor. J Clin Oncol. 2002; 20(13):3001-3015.
33. Huang H and Tindall DJ. The role of the androgen receptor in prostate cancer. Crit Rev Eukaryot Gene Expr. 2002; 12(3):193-207.
34. Bennett NC, Gardiner RA, Hooper JD, Johnson DW and Gobe GC. Molecular cell biology of androgen receptor signalling. Int J Biochem Cell Biol. 2010; 42(6):813-827.
35. Heinlein CA and Chang C. Androgen receptor in prostate cancer. Endocr Rev. 2004; 25(2):276-308.
36. Brinkmann AO, Blok LJ, de Ruiter PE, Doesburg P, Steketee K, Berrevoets CA and Trapman J. Mechanisms of androgen receptor activation and function. J Steroid Biochem Mol Biol. 1999; 69(1-6):307-313.
37. Agus DB, Cordon-Cardo C, Fox W, Drobnjak M, Koff A, Golde DW and Scher HI. Prostate cancer cell cycle regulators: response to androgen withdrawal and development of androgen independence. J Natl Cancer Inst. 1999; 91:1869-1876.
38. Denmeade SR, Lin XS and Isaacs JT. Role of programmed (apoptotic) cell death during the progression and therapy for prostate cancer. Prostate. 1996; 28(4):251-265.
39. Kyprianou N and Isaacs JT. Activation of programmed cell death in the rat ventral prostate after castration. Endocrinology. 1988; 122(2):552-562.
40. Knudsen KE, Cavenee WK and Arden KC. D-type cyclins complex with the androgen receptor and inhibit its transcriptional transactivation ability. Cancer Res. 1999; 59:2297-2301.
41. Balk SP and Knudsen KE. AR, the cell cycle, and prostate cancer. Nucl Recept Signal. 2008; 6:e001.
42. Sadi MV, Walsh PC and Barrack ER. Immunohistochemical study of androgen receptors in metastatic prostate cancer. Comparison of receptor content and response to hormonal therapy. Cancer. 1991; 67(12):3057-3064.
43. Ruizeveld de Winter JA, Janssen PJ, Sleddens HM, Verleun-Mooijman MC, Trapman J, Brinkmann AO, Santerse AB, Schroder FH and van der Kwast TH. Androgen receptor status in localized and locally progressive hormone refractory human prostate cancer. Am J Pathol. 1994; 144(4):735-746.
44. Labrie F. Mechanism of action and pure antiandrogenic properties of flutamide. Cancer. 1993; 72(12 Suppl):3816-3827.
45. Labrie F, Belanger A, Simard J, Labrie C and Dupont A. Combination therapy for prostate cancer. Endocrine and biologic basis of its choice as new standard first-line therapy. Cancer. 1993; 71:1059-1067.
46. Wong YN, Ferraldeschi R, Attard G and de Bono J. Evolution of androgen receptor targeted therapy for advanced prostate cancer. Nat Rev Clin Oncol. 2014; 11(6):365-376.
47. Masiello D, Cheng S, Bubley GJ, Lu ML and Balk SP. Bicalutamide functions as an androgen receptor antagonist by assembly of a transcriptionally inactive receptor. J Biol Chem. 2002; 277:26321-26326.
48. Lukka H, Waldron T, Klotz L, Winquist E, Trachtenberg J, Genitourinary Cancer Disease Site G and Cancer Care Ontario Program in Evidence-based C. Maximal androgen blockade for the treatment of metastatic prostate cancer--a systematic review. Curr Oncol. 2006; 13(3):81-93.
49. Samson DJ, Seidenfeld J, Schmitt B, Hasselblad V, Albertsen PC, Bennett CL, Wilt TJ and Aronson N. Systematic review and meta-analysis of monotherapy compared with combined androgen blockade for patients with advanced prostate carcinoma. Cancer. 2002; 95(2):361-376.
50. Group PCTC. Maximum androgen blockade in advanced prostate cancer: an overview of the randomised trials. Prostate Cancer Trialists’ Collaborative Group. Lancet. 2000; 355(9214):1491-1498.
51. Shaffer DR and Scher HI. Prostate cancer: a dynamic illness with shifting targets. Lancet Oncol. 2003; 4(7):407-414.
52. Hotte SJ and Saad F. Current management of castrate-resistant prostate cancer. Curr Oncol. 2010; 17 Suppl 2:S72-79.
53. Feldman BJ and Feldman D. The development of androgen-independent prostate cancer. Nat Rev Cancer. 2001; 1:34-45.
54. Mostaghel EA, Page ST, Lin DW, Fazli L, Coleman IM, True LD, Knudsen B, Hess DL, Nelson CC, Matsumoto AM, Bremner WJ, Gleave ME and Nelson PS. Intraprostatic androgens and androgen-regulated gene expression persist after testosterone suppression: therapeutic implications for castration-resistant prostate cancer. Cancer Res. 2007; 67(10):5033-5041.
55. Scher HI, Halabi S, Tannock I, Morris M, Sternberg CN, Carducci MA, Eisenberger MA, Higano C, Bubley GJ, Dreicer R, Petrylak D, Kantoff P, Basch E, Kelly WK, Figg WD, Small EJ, et al. Design and end points of clinical trials for patients with progressive prostate cancer and castrate levels of testosterone: recommendations of the Prostate Cancer Clinical Trials Working Group. J Clin Oncol. 2008; 26(7):1148-1159.
56. Isaacs JT and Isaacs WB. Androgen receptor outwits prostate cancer drugs. Nat Med. 2004; 10(1):26-27.
57. Scher HI and Sawyers CL. Biology of progressive, castration-resistant prostate cancer: directed therapies targeting the androgen-receptor signaling axis. J Clin Oncol. 2005; 23(32):8253-8261.
58. Visakorpi T, Hyytinen E, Koivisto P, Tanner M, Keinanen R, Palmberg C, Palotie A, Tammela T, Isola J and Kallioniemi OP. In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nat Genet. 1995; 9(4):401-406.
59. Koivisto P, Kononen J, Palmberg C, Tammela T, Hyytinen E, Isola J, Trapman J, Cleutjens K, Noordzij A, Visakorpi T and Kallioniemi OP. Androgen receptor gene amplification: a possible molecular mechanism for androgen deprivation therapy failure in prostate cancer. Cancer Res. 1997; 57(2):314-319.
60. Waltering KK, Helenius MA, Sahu B, Manni V, Linja MJ, Janne OA and Visakorpi T. Increased expression of androgen receptor sensitizes prostate cancer cells to low levels of androgens. Cancer Res. 2009; 69(20):8141-8149.
61. Kawata H, Ishikura N, Watanabe M, Nishimoto A, Tsunenari T and Aoki Y. Prolonged treatment with bicalutamide induces androgen receptor overexpression and androgen hypersensitivity. Prostate. 2010; 70(7):745-754.
62. Linja MJ, Savinainen KJ, Saramaki OR, Tammela TL, Vessella RL and Visakorpi T. Amplification and overexpression of androgen receptor gene in hormone-refractory prostate cancer. Cancer Res. 2001; 61:3550-3555.
63. Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, Vess R, Rosenfeld MG and Sawyers CL. Molecular determinants of resistance to antiandrogen therapy. Nat Med. 2004; 10:33-39.
64. Koochekpour S. Androgen receptor signaling and mutations in prostate cancer. Asian J Androl. 2010; 12(5):639-657.
65. Beltran H, Yelensky R, Frampton GM, Park K, Downing SR, MacDonald TY, Jarosz M, Lipson D, Tagawa ST, Nanus DM, Stephens PJ, Mosquera JM, Cronin MT and Rubin MA. Targeted next-generation sequencing of advanced prostate cancer identifies potential therapeutic targets and disease heterogeneity. Eur Urol. 2013; 63(5):920-926.
66. Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, Quist MJ, Jing X, Lonigro RJ, Brenner JC, Asangani IA, Ateeq B, Chun SY, Siddiqui J, Sam L, Anstett M, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012; 487(7406):239-243.
67. Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, Arora VK, Kaushik P, Cerami E, Reva B, Antipin Y, Mitsiades N, Landers T, Dolgalev I, Major JE, Wilson M, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010; 18(1):11-22.
68. Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, Montgomery B, Taplin ME, Pritchard CC, Attard G, Beltran H, Abida W, Bradley RK, Vinson J, Cao X, Vats P, et al. Integrative clinical genomics of advanced prostate cancer. Cell. 2015; 161(5):1215-1228.
69. Hara T, Miyazaki J, Araki H, Yamaoka M, Kanzaki N, Kusaka M and Miyamoto M. Novel mutations of androgen receptor: a possible mechanism of bicalutamide withdrawal syndrome. Cancer Res. 2003; 63(1):149-153.
70. Sun C, Shi Y, Xu LL, Nageswararao C, Davis LD, Segawa T, Dobi A, McLeod DG and Srivastava S. Androgen receptor mutation (T877A) promotes prostate cancer cell growth and cell survival. Oncogene. 2006; 25(28):3905-3913.
71. Romanel A, Gasi Tandefelt D, Conteduca V, Jayaram A, Casiraghi N, Wetterskog D, Salvi S, Amadori D, Zafeiriou Z, Rescigno P, Bianchini D, Gurioli G, Casadio V, Carreira S, Goodall J, Wingate A, et al. Plasma AR and abiraterone-resistant prostate cancer. Sci Transl Med. 2015; 7(312):312re310.
72. Azad AA, Volik SV, Wyatt AW, Haegert A, Le Bihan S, Bell RH, Anderson SA, McConeghy B, Shukin R, Bazov J, Youngren J, Paris P, Thomas G, Small EJ, Wang Y, Gleave ME, et al. Androgen Receptor Gene Aberrations in Circulating Cell-Free DNA: Biomarkers of Therapeutic Resistance in Castration-Resistant Prostate Cancer. Clin Cancer Res. 2015; 21(10):2315-2324.
73. Chen EJ, Sowalsky AG, Gao S, Cai C, Voznesensky O, Schaefer R, Loda M, True LD, Ye H, Troncoso P, Lis RL, Kantoff PW, Montgomery RB, Nelson PS, Bubley GJ, Balk SP, et al. Abiraterone treatment in castration-resistant prostate cancer selects for progesterone responsive mutant androgen receptors. Clin Cancer Res. 2015; 21(6):1273-1280.
74. Attard G, Reid AH, Auchus RJ, Hughes BA, Cassidy AM, Thompson E, Oommen NB, Folkerd E, Dowsett M, Arlt W and de Bono JS. Clinical and biochemical consequences of CYP17A1 inhibition with abiraterone given with and without exogenous glucocorticoids in castrate men with advanced prostate cancer. J Clin Endocrinol Metab. 2012; 97(2):507-516.
75. Mostaghel EA and Nelson PS. Intracrine androgen metabolism in prostate cancer progression: mechanisms of castration resistance and therapeutic implications. Best Pract Res Clin Endocrinol Metab. 2008; 22(2):243-258.
76. Cai C and Balk SP. Intratumoral androgen biosynthesis in prostate cancer pathogenesis and response to therapy. Endocr Relat Cancer. 2011; 18(5):R175-182.
77. Titus MA, Gregory CW, Ford OH, 3rd, Schell MJ, Maygarden SJ and Mohler JL. Steroid 5alpha-reductase isozymes I and II in recurrent prostate cancer. Clin Cancer Res. 2005; 11(12):4365-4371.
78. Penning TM and Byrns MC. Steroid hormone transforming aldo-keto reductases and cancer. Ann N Y Acad Sci. 2009; 1155:33-42.
79. Montgomery RB, Mostaghel EA, Vessella R, Hess DL, Kalhorn TF, Higano CS, True LD and Nelson PS. Maintenance of intratumoral androgens in metastatic prostate cancer: a mechanism for castration-resistant tumor growth. Cancer Res. 2008; 68(11):4447-4454.
80. de Bono JS, Logothetis CJ, Molina A, Fizazi K, North S, Chu L, Chi KN, Jones RJ, Goodman OB, Jr., Saad F, Staffurth JN, Mainwaring P, Harland S, Flaig TW, Hutson TE, Cheng T, et al. Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med. 2011; 364(21):1995-2005.
81. Efstathiou E, Titus M, Tsavachidou D, Tzelepi V, Wen S, Hoang A, Molina A, Chieffo N, Smith LA, Karlou M, Troncoso P and Logothetis CJ. Effects of abiraterone acetate on androgen signaling in castrate-resistant prostate cancer in bone. J Clin Oncol. 2012; 30(6):637-643.
82. Cai C, Chen S, Ng P, Bubley GJ, Nelson PS, Mostaghel EA, Marck B, Matsumoto AM, Simon NI, Wang H, Chen S and Balk SP. Intratumoral de novo steroid synthesis activates androgen receptor in castration-resistant prostate cancer and is upregulated by treatment with CYP17A1 inhibitors. Cancer Res. 2011; 71(20):6503-6513.
83. Dehm SM, Schmidt LJ, Heemers HV, Vessella RL and Tindall DJ. Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer Res. 2008; 68(13):5469-5477.
84. Hu R, Dunn TA, Wei S, Isharwal S, Veltri RW, Humphreys E, Han M, Partin AW, Vessella RL, Isaacs WB, Bova GS and Luo J. Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res. 2009; 69(1):16-22.
85. Sun S, Sprenger CC, Vessella RL, Haugk K, Soriano K, Mostaghel EA, Page ST, Coleman IM, Nguyen HM, Sun H, Nelson PS and Plymate SR. Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. J Clin Invest. 2010; 120(8):2715-2730.
86. Watson PA, Chen YF, Balbas MD, Wongvipat J, Socci ND, Viale A, Kim K and Sawyers CL. Constitutively active androgen receptor splice variants expressed in castration-resistant prostate cancer require full-length androgen receptor. Proc Natl Acad Sci U S A. 2010; 107(39):16759-16765.
87. Guo Z, Yang X, Sun F, Jiang R, Linn DE, Chen H, Chen H, Kong X, Melamed J, Tepper CG, Kung HJ, Brodie AM, Edwards J and Qiu Y. A novel androgen receptor splice variant is up-regulated during prostate cancer progression and promotes androgen depletion-resistant growth. Cancer Res. 2009; 69(6):2305-2313.
88. Hu R, Lu C, Mostaghel EA, Yegnasubramanian S, Gurel M, Tannahill C, Edwards J, Isaacs WB, Nelson PS, Bluemn E, Plymate SR and Luo J. Distinct transcriptional programs mediated by the ligand-dependent full-length androgen receptor and its splice variants in castration-resistant prostate cancer. Cancer Res. 2012; 72(14):3457-3462.
89. Chan SC, Selth LA, Li Y, Nyquist MD, Miao L, Bradner JE, Raj GV, Tilley WD and Dehm SM. Targeting chromatin binding regulation of constitutively active AR variants to overcome prostate cancer resistance to endocrine-based therapies. Nucleic Acids Res. 2015; 43(12):5880-5897.
90. Hornberg E, Ylitalo EB, Crnalic S, Antti H, Stattin P, Widmark A, Bergh A and Wikstrom P. Expression of androgen receptor splice variants in prostate cancer bone metastases is associated with castration-resistance and short survival. PLoS One. 2011; 6(4):e19059.
91. Antonarakis ES, Lu C, Wang H, Luber B, Nakazawa M, Roeser JC, Chen Y, Mohammad TA, Chen Y, Fedor HL, Lotan TL, Zheng Q, De Marzo AM, Isaacs JT, Isaacs WB, Nadal R, et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N Engl J Med. 2014; 371(11):1028-1038.
92. Li Y, Chan SC, Brand LJ, Hwang TH, Silverstein KA and Dehm SM. Androgen receptor splice variants mediate enzalutamide resistance in castration-resistant prostate cancer cell lines. Cancer Res. 2013; 73(2):483-489.
93. Banuelos CA, Tavakoli I, Tien AH, Caley DP, Mawji NR, Li Z, Wang J, Yang YC, Imamura Y, Yan L, Wen JG, Andersen RJ and Sadar MD. Sintokamide A is a Novel Antagonist of Androgen Receptor that Uniquely Binds Activation Function-1 in its Amino-Terminal Domain. J Biol Chem. 2016.
94. Kato M, Banuelos CA, Imamura Y, Leung JK, Caley DP, Wang J, Mawji NR and Sadar MD. Cotargeting Androgen Receptor Splice Variants and mTOR Signaling Pathway for the Treatment of Castration-Resistant Prostate Cancer. Clin Cancer Res. 2016; 22(11):2744-2754.
95. Yang YC, Banuelos CA, Mawji NR, Wang J, Kato M, Haile S, McEwan IJ, Plymate S and Sadar MD. Targeting Androgen Receptor Activation Function-1 with EPI to Overcome Resistance Mechanisms in Castration-Resistant Prostate Cancer. Clin Cancer Res. 2016; 22(17):4466-4477.
96. Myung JK, Banuelos CA, Fernandez JG, Mawji NR, Wang J, Tien AH, Yang YC, Tavakoli I, Haile S, Watt K, McEwan IJ, Plymate S, Andersen RJ and Sadar MD. An androgen receptor N-terminal domain antagonist for treating prostate cancer. J Clin Invest. 2013; 123(7):2948-2960.
97. Zhu ML and Kyprianou N. Androgen receptor and growth factor signaling cross-talk in prostate cancer cells. Endocr Relat Cancer. 2008; 15(4):841-849.
98. Wang LG, Liu XM, Kreis W and Budman DR. Phosphorylation/dephosphorylation of androgen receptor as a determinant of androgen agonistic or antagonistic activity. Biochem Biophys Res Commun. 1999; 259:21-28.
99. Culig Z, Hobisch A, Cronauer MV, Radmayr C, Trapman J, Hittmair A, Bartsch G and Klocker H. Androgen receptor activation in prostatic tumor cell lines by insulin- like growth factor-I, keratinocyte growth factor, and epidermal growth factor. Cancer Res. 1994; 54(20):5474-5478.
100. Sayeed A, Alam N, Trerotola M and Languino LR. Insulin-like growth factor 1 stimulation of androgen receptor activity requires beta(1A) integrins. J Cell Physiol. 2012; 227(2):751-758.
101. Hobisch A, Eder IE, Putz T, Horninger W, Bartsch G, Klocker H and Culig Z. Interleukin-6 regulates prostate-specific protein expression in prostate carcinoma cells by activation of the androgen receptor. Cancer Res. 1998; 58(20):4640-4645.
102. Chen T, Wang LH and Farrar WL. Interleukin 6 activates androgen receptor-mediated gene expression through a signal transducer and activator of transcription 3-dependent pathway in LNCaP prostate cancer cells. Cancer Res. 2000; 60(8):2132-2135.
103. Yang L, Wang L, Lin HK, Kan PY, Xie S, Tsai MY, Wang PH, Chen YT and Chang C. Interleukin-6 differentially regulates androgen receptor transactivation via PI3K-Akt, STAT3, and MAPK, three distinct signal pathways in prostate cancer cells. Biochem Biophys Res Commun. 2003; 305(3):462-469.
104. Burd CJ, Petre CE, Morey LM, Wang Y, Revelo MP, Haiman CA, Lu S, Fenoglio-Preiser CM, Li J, Knudsen ES, Wong J and Knudsen KE. Cyclin D1b variant influences prostate cancer growth through aberrant androgen receptor regulation. Proc Natl Acad Sci U S A. 2006; 103(7):2190-2195.
105. Comstock CE, Augello MA, Benito RP, Karch J, Tran TH, Utama FE, Tindall EA, Wang Y, Burd CJ, Groh EM, Hoang HN, Giles GG, Severi G, Hayes VM, Henderson BE, Le Marchand L, et al. Cyclin D1 splice variants: polymorphism, risk, and isoform-specific regulation in prostate cancer. Clin Cancer Res. 2009; 15(17):5338-5349.
106. Augello MA, Burd CJ, Birbe R, McNair C, Ertel A, Magee MS, Frigo DE, Wilder-Romans K, Shilkrut M, Han S, Jernigan DL, Dean JL, Fatatis A, McDonnell DP, Visakorpi T, Feng FY, et al. Convergence of oncogenic and hormone receptor pathways promotes metastatic phenotypes. J Clin Invest. 2013; 123(1):493-508.
107. Sharma A, Yeow WS, Ertel A, Coleman I, Clegg N, Thangavel C, Morrissey C, Zhang X, Comstock CE, Witkiewicz AK, Gomella L, Knudsen ES, Nelson PS and Knudsen KE. The retinoblastoma tumor suppressor controls androgen signaling and human prostate cancer progression. J Clin Invest. 2010; 120(12):4478-4492.
108. Craft N, Shostak Y, Carey M and Sawyers CL. A mechanism for hormone-independent prostate cancer through modulation of androgen receptor signaling by the HER-2/neu tyrosine kinase. Nat Med. 1999; 5(3):280-285.
109. Yeh S, Lin HK, Kang HY, Thin TH, Lin MF and Chang C. From HER2/Neu signal cascade to androgen receptor and its coactivators: a novel pathway by induction of androgen target genes through MAP kinase in prostate cancer cells. Proc Natl Acad Sci U S A. 1999; 96(10):5458-5463.
110. Asim M, Siddiqui IA, Hafeez BB, Baniahmad A and Mukhtar H. Src kinase potentiates androgen receptor transactivation function and invasion of androgen-independent prostate cancer C4-2 cells. Oncogene. 2008; 27(25):3596-3604.
111. Nazareth LV and Weigel NL. Activation of the human androgen receptor through a protein kinase A signaling pathway. J Biol Chem. 1996; 271(33):19900-19907.
112. Whitworth H, Bhadel S, Ivey M, Conaway M, Spencer A, Hernan R, Holemon H and Gioeli D. Identification of kinases regulating prostate cancer cell growth using an RNAi phenotypic screen. PLoS One. 2012; 7(6):e38950.
113. Arora VK, Schenkein E, Murali R, Subudhi SK, Wongvipat J, Balbas MD, Shah N, Cai L, Efstathiou E, Logothetis C, Zheng D and Sawyers CL. Glucocorticoid receptor confers resistance to antiandrogens by bypassing androgen receptor blockade. Cell. 2013; 155(6):1309-1322.
114. Saraon P, Jarvi K and Diamandis EP. Molecular alterations during progression of prostate cancer to androgen independence. Clin Chem. 2011; 57(10):1366-1375.
115. Liu AY, Corey E, Bladou F, Lange PH and Vessella RL. Prostatic cell lineage markers: emergence of BCL2+ cells of human prostate cancer xenograft LuCaP 23 following castration. Int J Cancer. 1996; 65(1):85-89.
116. Gleave M, Tolcher A, Miyake H, Nelson C, Brown B, Beraldi E and Goldie J. Progression to androgen independence is delayed by adjuvant treatment with antisense Bcl-2 oligodeoxynucleotides after castration in the LNCaP prostate tumor model. Clin Cancer Res. 1999; 5(10):2891-2898.
117. Colombel M, Symmans F, Gil S, O’Toole KM, Chopin D, Benson M, Olsson CA, Korsmeyer S and Buttyan R. Detection of the apoptosis-suppressing oncoprotein bc1-2 in hormone-refractory human prostate cancers. Am J Pathol. 1993; 143(2):390-400.
118. Furuya Y, Krajewski S, Epstein JI, Reed JC and Isaacs JT. Expression of bcl-2 and the progression of human and rodent prostatic cancers. Clin Cancer Res. 1996; 2(2):389-398.
119. Sternberg CN, Dumez H, Van Poppel H, Skoneczna I, Sella A, Daugaard G, Gil T, Graham J, Carpentier P, Calabro F, Collette L, Lacombe D and Group EGTC. Docetaxel plus oblimersen sodium (Bcl-2 antisense oligonucleotide): an EORTC multicenter, randomized phase II study in patients with castration-resistant prostate cancer. Ann Oncol. 2009; 20(7):1264-1269.
120. Sahu B, Laakso M, Pihlajamaa P, Ovaska K, Sinielnikov I, Hautaniemi S and Janne OA. FoxA1 specifies unique androgen and glucocorticoid receptor binding events in prostate cancer cells. Cancer Res. 2013; 73(5):1570-1580.
121. Isikbay M, Otto K, Kregel S, Kach J, Cai Y, Vander Griend DJ, Conzen SD and Szmulewitz RZ. Glucocorticoid receptor activity contributes to resistance to androgen-targeted therapy in prostate cancer. Horm Cancer. 2014; 5(2):72-89.
122. Reya T, Morrison SJ, Clarke MF and Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001; 414(6859):105-111.
123. Chen X, Rycaj K, Liu X and Tang DG. New insights into prostate cancer stem cells. Cell Cycle. 2013; 12(4):579-586.
124. Clarke MF, Dick JE, Dirks PB, Eaves CJ, Jamieson CH, Jones DL, Visvader J, Weissman IL and Wahl GM. Cancer stem cells--perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res. 2006; 66(19):9339-9344.
125. Dean M, Fojo T and Bates S. Tumour stem cells and drug resistance. Nat Rev Cancer. 2005; 5(4):275-284.
126. Chaffer CL and Weinberg RA. A perspective on cancer cell metastasis. Science. 2011; 331(6024):1559-1564.
127. Visvader JE and Lindeman GJ. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer. 2008; 8(10):755-768.
128. Collins AT, Berry PA, Hyde C, Stower MJ and Maitland NJ. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res. 2005; 65(23):10946-10951.
129. Rajasekhar VK, Studer L, Gerald W, Socci ND and Scher HI. Tumour-initiating stem-like cells in human prostate cancer exhibit increased NF-kappaB signalling. Nat Commun. 2011; 2:162.
130. van den Hoogen C, van der Horst G, Cheung H, Buijs JT, Lippitt JM, Guzman-Ramirez N, Hamdy FC, Eaton CL, Thalmann GN, Cecchini MG, Pelger RC and van der Pluijm G. High aldehyde dehydrogenase activity identifies tumor-initiating and metastasis-initiating cells in human prostate cancer. Cancer Res. 2010; 70(12):5163-5173.
131. Qin J, Liu X, Laffin B, Chen X, Choy G, Jeter CR, Calhoun-Davis T, Li H, Palapattu GS, Pang S, Lin K, Huang J, Ivanov I, Li W, Suraneni MV and Tang DG. The PSA(-/lo) prostate cancer cell population harbors self-renewing long-term tumor-propagating cells that resist castration. Cell Stem Cell. 2012; 10(5):556-569.
132. Iczkowski KA, Bai S and Pantazis CG. Prostate cancer overexpresses CD44 variants 7-9 at the messenger RNA and protein level. Anticancer Res. 2003; 23(4):3129-3140.
133. Patrawala L, Calhoun T, Schneider-Broussard R, Li H, Bhatia B, Tang S, Reilly JG, Chandra D, Zhou J, Claypool K, Coghlan L and Tang DG. Highly purified CD44+ prostate cancer cells from xenograft human tumors are enriched in tumorigenic and metastatic progenitor cells. Oncogene. 2006; 25(12):1696-1708.
134. Zhang D, Park D, Zhong Y, Lu Y, Rycaj K, Gong S, Chen X, Liu X, Chao HP, Whitney P, Calhoun-Davis T, Takata Y, Shen J, Iyer VR and Tang DG. Stem cell and neurogenic gene-expression profiles link prostate basal cells to aggressive prostate cancer. Nat Commun. 2016; 7:10798.
135. Sun Y, Niu J and Huang J. Neuroendocrine differentiation in prostate cancer. Am J Transl Res. 2009; 1(2):148-162.
136. Epstein JI, Amin MB, Beltran H, Lotan TL, Mosquera JM, Reuter VE, Robinson BD, Troncoso P and Rubin MA. Proposed morphologic classification of prostate cancer with neuroendocrine differentiation. Am J Surg Pathol. 2014; 38(6):756-767.
137. Abrahamsson PA. Neuroendocrine differentiation in prostatic carcinoma. Prostate. 1999; 39(2):135-148.
138. Schwartz LH, LaTrenta LR, Bonaccio E, Kelly WK, Scher HI and Panicek DM. Small cell and anaplastic prostate cancer: correlation between CT findings and prostate-specific antigen level. Radiology. 1998; 208(3):735-738.
139. Humphrey PA. Histological variants of prostatic carcinoma and their significance. Histopathology. 2012; 60(1):59-74.
140. Li Z, Chen CJ, Wang JK, Hsia E, Li W, Squires J, Sun Y and Huang J. Neuroendocrine differentiation of prostate cancer. Asian J Androl. 2013; 15(3):328-332.
141. Lipianskaya J, Cohen A, Chen CJ, Hsia E, Squires J, Li Z, Zhang Y, Li W, Chen X, Xu H and Huang J. Androgen-deprivation therapy-induced aggressive prostate cancer with neuroendocrine differentiation. Asian J Androl. 2014; 16(4):541-544.
142. Hu CD, Choo R and Huang J. Neuroendocrine differentiation in prostate cancer: a mechanism of radioresistance and treatment failure. Front Oncol. 2015; 5:90.
143. Beltran H, Tagawa ST, Park K, MacDonald T, Milowsky MI, Mosquera JM, Rubin MA and Nanus DM. Challenges in recognizing treatment-related neuroendocrine prostate cancer. J Clin Oncol. 2012; 30(36):e386-389.
144. Abrahamsson PA, Falkmer S, Falt K and Grimelius L. The course of neuroendocrine differentiation in prostatic carcinomas. An immunohistochemical study testing chromogranin A as an “endocrine marker”. Pathol Res Pract. 1989; 185(3):373-380.
145. Terry S and Beltran H. The many faces of neuroendocrine differentiation in prostate cancer progression. Front Oncol. 2014; 4:60.
146. Hirano D, Okada Y, Minei S, Takimoto Y and Nemoto N. Neuroendocrine differentiation in hormone refractory prostate cancer following androgen deprivation therapy. Eur Urol. 2004; 45(5):586-592; discussion 592.
147. Berruti A, Mosca A, Porpiglia F, Bollito E, Tucci M, Vana F, Cracco C, Torta M, Russo L, Cappia S, Saini A, Angeli A, Papotti M, Scarpa RM and Dogliotti L. Chromogranin A expression in patients with hormone naive prostate cancer predicts the development of hormone refractory disease. J Urol. 2007; 178(3 Pt 1):838-843; quiz 1129.
148. Ito T, Yamamoto S, Ohno Y, Namiki K, Aizawa T, Akiyama A and Tachibana M. Up-regulation of neuroendocrine differentiation in prostate cancer after androgen deprivation therapy, degree and androgen independence. Oncol Rep. 2001; 8(6):1221-1224.
149. Shen R, Dorai T, Szaboles M, Katz AE, Olsson CA and Buttyan R. Transdifferentiation of cultured human prostate cancer cells to a neuroendocrine cell phenotype in a hormone-depleted medium. Urol Oncol. 1997; 3(2):67-75.
150. Jiborn T, Bjartell A and Abrahamsson PA. Neuroendocrine differentiation in prostatic carcinoma during hormonal treatment. Urology. 1998; 51(4):585-589.
151. di Sant’Agnese PA. Neuroendocrine differentiation in carcinoma of the prostate. Diagnostic, prognostic, and therapeutic implications. Cancer. 1992; 70(1 Suppl):254-268.
152. Lin D, Wyatt AW, Xue H, Wang Y, Dong X, Haegert A, Wu R, Brahmbhatt S, Mo F, Jong L, Bell RH, Anderson S, Hurtado-Coll A, Fazli L, Sharma M, Beltran H, et al. High fidelity patient-derived xenografts for accelerating prostate cancer discovery and drug development. Cancer Res. 2014; 74(4):1272-1283.
153. Wafa LA, Palmer J, Fazli L, Hurtado-Coll A, Bell RH, Nelson CC, Gleave ME, Cox ME and Rennie PS. Comprehensive expression analysis of L-dopa decarboxylase and established neuroendocrine markers in neoadjuvant hormone-treated versus varying Gleason grade prostate tumors. Hum Pathol. 2007; 38(1):161-170.
154. Beltran H, Tomlins S, Aparicio A, Arora V, Rickman D, Ayala G, Huang J, True L, Gleave ME, Soule H, Logothetis C and Rubin MA. Aggressive variants of castration-resistant prostate cancer. Clin Cancer Res. 2014; 20(11):2846-2850.
155. Wang W and Epstein JI. Small cell carcinoma of the prostate. A morphologic and immunohistochemical study of 95 cases. Am J Surg Pathol. 2008; 32(1):65-71.
156. Beltran H, Prandi D, Mosquera JM, Benelli M, Puca L, Cyrta J, Marotz C, Giannopoulou E, Chakravarthi BV, Varambally S, Tomlins SA, Nanus DM, Tagawa ST, Van Allen EM, Elemento O, Sboner A, et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat Med. 2016; 22(3):298-305.
157. Berruti A, Vignani F, Russo L, Bertaglia V, Tullio M, Tucci M, Poggio M and Dogliotti L. Prognostic role of neuroendocrine differentiation in prostate cancer, putting together the pieces of the puzzle. Open Access J Urol. 2010; 2:109-124.
158. Conteduca V, Burgio SL, Menna C, Carretta E, Rossi L, Bianchi E, Masini C, Amadori D and De Giorgi U. Chromogranin A is a potential prognostic marker in prostate cancer patients treated with enzalutamide. Prostate. 2014; 74(16):1691-1696.
159. Krijnen JL, Bogdanowicz JF, Seldenrijk CA, Mulder PG and van der Kwast TH. The prognostic value of neuroendocrine differentiation in adenocarcinoma of the prostate in relation to progression of disease after endocrine therapy. J Urol. 1997; 158(1):171-174.
160. Berruti A, Mosca A, Tucci M, Terrone C, Torta M, Tarabuzzi R, Russo L, Cracco C, Bollito E, Scarpa RM, Angeli A and Dogliotti L. Independent prognostic role of circulating chromogranin A in prostate cancer patients with hormone-refractory disease. Endocr Relat Cancer. 2005; 12(1):109-117.
161. Tan HL, Sood A, Rahimi HA, Wang W, Gupta N, Hicks J, Mosier S, Gocke CD, Epstein JI, Netto GJ, Liu W, Isaacs WB, De Marzo AM and Lotan TL. Rb loss is characteristic of prostatic small cell neuroendocrine carcinoma. Clin Cancer Res. 2014; 20(4):890-903.
162. Beltran H, Rickman DS, Park K, Chae SS, Sboner A, MacDonald TY, Wang Y, Sheikh KL, Terry S, Tagawa ST, Dhir R, Nelson JB, de la Taille A, Allory Y, Gerstein MB, Perner S, et al. Molecular characterization of neuroendocrine prostate cancer and identification of new drug targets. Cancer Discov. 2011; 1(6):487-495.
163. Chen H, Sun Y, Wu C, Magyar CE, Li X, Cheng L, Yao JL, Shen S, Osunkoya AO, Liang C and Huang J. Pathogenesis of prostatic small cell carcinoma involves the inactivation of the P53 pathway. Endocr Relat Cancer. 2012; 19(3):321-331.
164. Li Z, Sun Y, Chen X, Squires J, Nowroozizadeh B, Liang C and Huang J. p53 Mutation Directs AURKA Overexpression via miR-25 and FBXW7 in Prostatic Small Cell Neuroendocrine Carcinoma. Mol Cancer Res. 2015; 13(3):584-591.
165. Lee JK, Phillips JW, Smith BA, Park JW, Stoyanova T, McCaffrey EF, Baertsch R, Sokolov A, Meyerowitz JG, Mathis C, Cheng D, Stuart JM, Shokat KM, Gustafson WC, Huang J and Witte ON. N-Myc Drives Neuroendocrine Prostate Cancer Initiated from Human Prostate Epithelial Cells. Cancer Cell. 2016; 29(4):536-547.
166. Mirtti T, Leiby BE, Abdulghani J, Aaltonen E, Pavela M, Mamtani A, Alanen K, Egevad L, Granfors T, Josefsson A, Stattin P, Bergh A and Nevalainen MT. Nuclear Stat5a/b predicts early recurrence and prostate cancer-specific death in patients treated by radical prostatectomy. Hum Pathol. 2013; 44(3):310-319.
167. Dagvadorj A, Tan SH, Liao Z, Xie J, Nurmi M, Alanen K, Rui H, Mirtti T and Nevalainen MT. N-terminal truncation of Stat5a/b circumvents PIAS3-mediated transcriptional inhibition of Stat5 in prostate cancer cells. Int J Biochem Cell Biol. 2011; 42(12):2037-2046.
168. Thomas C, Zoubeidi A, Kuruma H, Fazli L, Lamoureux F, Beraldi E, Monia BP, MacLeod AR, Thuroff JW and Gleave ME. Transcription factor Stat5 knockdown enhances androgen receptor degradation and delays castration-resistant prostate cancer progression in vivo. Mol Cancer Ther. 2010; 10(2):347-359.
169. Gu L, Vogiatzi P, Puhr M, Dagvadorj A, Lutz J, Ryder A, Addya S, Fortina P, Cooper C, Leiby B, Dasgupta A, Hyslop T, Bubendorf L, Alanen K, Mirtti T and Nevalainen MT. Stat5 promotes metastatic behavior of human prostate cancer cells in vitro and in vivo. Endocr Relat Cancer. 2010; 17(2):481-493.
170. Gu L, Dagvadorj A, Lutz J, Leiby B, Bonuccelli G, Lisanti MP, Addya S, Fortina P, Dasgupta A, Hyslop T, Bubendorf L and Nevalainen MT. Transcription factor Stat3 stimulates metastatic behavior of human prostate cancer cells in vivo, whereas Stat5b has a preferential role in the promotion of prostate cancer cell viability and tumor growth. Am J Pathol. 2010; 176(4):1959-1972.
171. Dagvadorj A, Kirken RA, Leiby B, Karras J and Nevalainen MT. Transcription factor signal transducer and activator of transcription 5 promotes growth of human prostate cancer cells in vivo. Clin Cancer Res. 2008; 14(5):1317-1324.
172. Li H, Zhang Y, Glass A, Zellweger T, Gehan E, Bubendorf L, Gelmann EP and Nevalainen MT. Activation of signal transducer and activator of transcription-5 in prostate cancer predicts early recurrence. Clin Cancer Res. 2005; 11(16):5863-5868.
173. Li H, Ahonen TJ, Alanen K, Xie J, LeBaron MJ, Pretlow TG, Ealley EL, Zhang Y, Nurmi M, Singh B, Martikainen PM and Nevalainen MT. Activation of signal transducer and activator of transcription 5 in human prostate cancer is associated with high histological grade. Cancer Res. 2004; 64(14):4774-4782.
174. Ahonen TJ, Xie J, LeBaron MJ, Zhu J, Nurmi M, Alanen K, Rui H and Nevalainen MT. Inhibition of transcription factor Stat5 induces cell death of human prostate cancer cells. J Biol Chem. 2003; 278(29):27287-27292.
175. Levy DE and Darnell JE, Jr. Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol. 2002; 3(9):651-662.
176. Dagvadorj A, Collins S, Jomain JB, Abdulghani J, Karras J, Zellweger T, Li H, Nurmi M, Alanen K, Mirtti T, Visakorpi T, Bubendorf L, Goffin V and Nevalainen MT. Autocrine prolactin promotes prostate cancer cell growth via Janus kinase-2-signal transducer and activator of transcription-5a/b signaling pathway. Endocrinology. 2007; 148(7):3089-3101.
177. Gu L, Liao Z, Hoang DT, Dagvadorj A, Gupta S, Blackmon S, Ellsworth E, Talati P, Leiby B, Zinda M, Lallas CD, Trabulsi EJ, McCue P, Gomella L, Huszar D and Nevalainen MT. Pharmacologic inhibition of Jak2-Stat5 signaling By Jak2 inhibitor AZD1480 potently suppresses growth of both primary and castrate-resistant prostate cancer. Clin Cancer Res. 2013; 19(20):5658-5674.
178. Bidosee M, Karry R, Weiss-Messer E and Barkey RJ. Regulation of growth hormone receptors in human prostate cancer cell lines. Mol Cell Endocrinol. 2009; 309(1-2):82-92.
179. Feldman L, Wang Y, Rhim JS, Bhattacharya N, Loda M and Sytkowski AJ. Erythropoietin stimulates growth and STAT5 phosphorylation in human prostate epithelial and prostate cancer cells. Prostate. 2006; 66(2):135-145.
180. Kazansky AV, Kabotyanski EB, Wyszomierski SL, Mancini MA and Rosen JM. Differential effects of prolactin and src/abl kinases on the nuclear translocation of STAT5B and STAT5A. J Biol Chem. 1999; 274(32):22484-22492.
181. Kelly PA, Djiane J, Postel-Vinay MC and Edery M. The prolactin/growth hormone receptor family. Endocr Rev. 1991; 12(3):235-251.
182. Bole-Feysot C, Goffin V, Edery M, Binart N and Kelly PA. Prolactin (PRL) and its receptor: actions, signal transduction pathways and phenotypes observed in PRL receptor knockout mice. Endocr Rev. 1998; 19(3):225-268.
183. Nevalainen MT, Valve EM, Ingleton PM, Nurmi M, Martikainen PM and Harkonen PL. Prolactin and prolactin receptors are expressed and functioning in human prostate. J Clin Invest. 1997; 99(4):618-627.
184. Nevalainen MT, Valve EM, Ahonen T, Yagi A, Paranko J and Harkonen PL. Androgen-dependent expression of prolactin in rat prostate epithelium in vivo and in organ culture. Faseb J. 1997; 11(14):1297-1307.
185. Nevalainen MT, Harkonen PL, Valve EM, Ping W, Nurmi M and Martikainen PM. Hormone regulation of human prostate in organ culture. Cancer Res. 1993; 53(21):5199-5207.
186. Nevalainen MT, Valve EM, Ingleton PM and Harkonen PL. Expression and hormone regulation of prolactin receptors in rat dorsal and lateral prostate. Endocrinology. 1996; 137(7):3078-3088.
187. Ahonen TJ, Harkonen PL, Laine J, Rui H, Martikainen PM and Nevalainen MT. Prolactin is a survival factor for androgen-deprived rat dorsal and lateral prostate epithelium in organ culture. Endocrinology. 1999; 140(11):5412-5421.
188. Goffin V, Hoang DT, Bogorad RL and Nevalainen MT. Prolactin regulation of the prostate gland: a female player in a male game. Nat Rev Urol. 2011; 8(11):597-607.
189. Pezet A, Ferrag F, Kelly PA and Edery M. Tyrosine docking sites of the rat prolactin receptor required for association and activation of stat5. J Biol Chem. 1997; 272(40):25043-25050.
190. Tan SH and Nevalainen MT. Signal transducer and activator of transcription 5A/B in prostate and breast cancers. Endocr Relat Cancer. 2008; 15(2):367-390.
191. Sekimoto T, Nakajima K, Tachibana T, Hirano T and Yoneda Y. Interferon-gamma-dependent nuclear import of Stat1 is mediated by the GTPase activity of Ran/TC4. J Biol Chem. 1996; 271(49):31017-31020.
192. Sekimoto T, Imamoto N, Nakajima K, Hirano T and Yoneda Y. Extracellular signal-dependent nuclear import of Stat1 is mediated by nuclear pore-targeting complex formation with NPI-1, but not Rch1. Embo J. 1997; 16(23):7067-7077.
193. Kawashima T, Bao YC, Minoshima Y, Nomura Y, Hatori T, Hori T, Fukagawa T, Fukada T, Takahashi N, Nosaka T, Inoue M, Sato T, Kukimoto-Niino M, Shirouzu M, Yokoyama S and Kitamura T. A Rac GTPase-activating protein, MgcRacGAP, is a nuclear localizing signal-containing nuclear chaperone in the activation of STAT transcription factors. Mol Cell Biol. 2009; 29(7):1796-1813.
194. Decker T, Lew DJ, Mirkovitch J and Darnell JE, Jr. Cytoplasmic activation of GAF, an IFN-gamma-regulated DNA-binding factor. Embo J. 1991; 10(4):927-932.
195. Horvath CM, Wen Z and Darnell JE, Jr. A STAT protein domain that determines DNA sequence recognition suggests a novel DNA-binding domain. Genes Dev. 1995; 9(8):984-994.
196. Soldaini E, John S, Moro S, Bollenbacher J, Schindler U and Leonard WJ. DNA binding site selection of dimeric and tetrameric Stat5 proteins reveals a large repertoire of divergent tetrameric Stat5a binding sites. Mol Cell Biol. 2000; 20(1):389-401.
197. Ehret GB, Reichenbach P, Schindler U, Horvath CM, Fritz S, Nabholz M and Bucher P. DNA binding specificity of different STAT proteins. Comparison of in vitro specificity with natural target sites. J Biol Chem. 2001; 276(9):6675-6688.
198. Haddad BR, Gu L, Mirtti T, Dagvadorj A, Vogiatzi P, Hoang DT, Bajaj R, Leiby B, Ellsworth E, Blackmon S, Ruiz C, Curtis M, Fortina P, Ertel A, Liu C, Rui H, et al. STAT5A/B gene locus undergoes amplification during human prostate cancer progression. Am J Pathol. 2013; 182(6):2264-2275.
199. Thomas C, Zoubeidi A, Kuruma H, Fazli L, Lamoureux F, Beraldi E, Monia BP, MacLeod AR, Thuroff JW and Gleave ME. Transcription factor Stat5 knockdown enhances androgen receptor degradation and delays castration-resistant prostate cancer progression in vivo. Mol Cancer Ther. 2011; 10(2):347-359.
200. Kazansky AV, Spencer DM and Greenberg NM. Activation of signal transducer and activator of transcription 5 is required for progression of autochthonous prostate cancer: evidence from the transgenic adenocarcinoma of the mouse prostate system. Cancer Res. 2003; 63(24):8757-8762.
201. Talati PG, Gu L, Ellsworth EM, Girondo MA, Trerotola M, Hoang DT, Leiby B, Dagvadorj A, McCue PA, Lallas CD, Trabulsi EJ, Gomella L, Aplin AE, Languino L, Fatatis A, Rui H, et al. Jak2-Stat5a/b Signaling Induces Epithelial-to-Mesenchymal Transition and Stem-Like Cell Properties in Prostate Cancer. Am J Pathol. 2015; 185(9):2505-2522.
202. Jacobs JJ, Kieboom K, Marino S, DePinho RA and van Lohuizen M. The oncogene and Polycomb-group gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature. 1999; 397(6715):164-168.
203. Sharpless NE and DePinho RA. The INK4A/ARF locus and its two gene products. Curr Opin Genet Dev. 1999; 9(1):22-30.
204. Park IK, Morrison SJ and Clarke MF. Bmi1, stem cells, and senescence regulation. J Clin Invest. 2004; 113(2):175-179.
205. Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C, Savagner P, Gitelman I, Richardson A and Weinberg RA. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell. 2004; 117(7):927-939.
206. Qin Q, Xu Y, He T, Qin C and Xu J. Normal and disease-related biological functions of Twist1 and underlying molecular mechanisms. Cell Res. 2012; 22(1):90-106.
207. Alexander NR, Tran NL, Rekapally H, Summers CE, Glackin C and Heimark RL. N-cadherin gene expression in prostate carcinoma is modulated by integrin-dependent nuclear translocation of Twist1. Cancer Res. 2006; 66(7):3365-3369.
208. Tan SH, Dagvadorj A, Shen F, Gu L, Liao Z, Abdulghani J, Zhang Y, Gelmann EP, Zellweger T, Culig Z, Visakorpi T, Bubendorf L, Kirken RA, Karras J and Nevalainen MT. Transcription factor Stat5 synergizes with androgen receptor in prostate cancer cells. Cancer Res. 2008; 68(1):236-248.
209. Hoang DT, Gu L, Liao Z, Shen F, Talati PG, Koptyra M, Tan SH, Ellsworth E, Gupta S, Montie H, Dagvadorj A, Savolainen S, Leiby B, Mirtti T, Merry DE and Nevalainen MT. Inhibition of Stat5a/b Enhances Proteasomal Degradation of Androgen Receptor Liganded by Antiandrogens in Prostate Cancer. Mol Cancer Ther. 2015; 14(3):713-726.
210. Sharma NL, Massie CE, Ramos-Montoya A, Zecchini V, Scott HE, Lamb AD, MacArthur S, Stark R, Warren AY, Mills IG and Neal DE. The androgen receptor induces a distinct transcriptional program in castration-resistant prostate cancer in man. Cancer Cell. 2013; 23(1):35-47.
211. Liao Z, Gu L, Vergalli J, Mariani SA, De Dominici M, Lokareddy RK, Dagvadorj A, Purushottamachar P, McCue PA, Trabulsi E, Lallas CD, Gupta S, Ellsworth E, Blackmon S, Ertel A, Fortina P, et al. Structure-Based Screen Identifies a Potent Small Molecule Inhibitor of Stat5a/b with Therapeutic Potential for Prostate Cancer and Chronic Myeloid Leukemia. Mol Cancer Ther. 2015; 14(8):1777-1793.
212. Kuruma H, Matsumoto H, Shiota M, Bishop J, Lamoureux F, Thomas C, Briere D, Los G, Gleave M, Fanjul A and Zoubeidi A. A novel antiandrogen, Compound 30, suppresses castration-resistant and MDV3100-resistant prostate cancer growth in vitro and in vivo. Mol Cancer Ther. 2013; 12(5):567-576.
213. Turkson J, Ryan D, Kim JS, Zhang Y, Chen Z, Haura E, Laudano A, Sebti S, Hamilton AD and Jove R. Phosphotyrosyl peptides block Stat3-mediated DNA binding activity, gene regulation, and cell transformation. J Biol Chem. 2001; 276(48):45443-45455.
214. Turkson J, Kim JS, Zhang S, Yuan J, Huang M, Glenn M, Haura E, Sebti S, Hamilton AD and Jove R. Novel peptidomimetic inhibitors of signal transducer and activator of transcription 3 dimerization and biological activity. Mol Cancer Ther. 2004; 3(3):261-269.
215. Nam S, Scuto A, Yang F, Chen W, Park S, Yoo HS, Konig H, Bhatia R, Cheng X, Merz KH, Eisenbrand G and Jove R. Indirubin derivatives induce apoptosis of chronic myelogenous leukemia cells involving inhibition of Stat5 signaling. Mol Oncol. 2012; 6(3):276-283.
216. Nelson EA, Walker SR, Weisberg E, Bar-Natan M, Barrett R, Gashin LB, Terrell S, Klitgaard JL, Santo L, Addorio MR, Ebert BL, Griffin JD and Frank DA. The STAT5 inhibitor pimozide decreases survival of chronic myelogenous leukemia cells resistant to kinase inhibitors. Blood. 2011; 117(12):3421-3429.
217. Graber M, Janczyk W, Sperl B, Elumalai N, Kozany C, Hausch F, Holak TA and Berg T. Selective targeting of disease-relevant protein binding domains by O-phosphorylated natural product derivatives. ACS Chem Biol. 2011; 6(10):1008-1014.
218. Elumalai N, Berg A, Natarajan K, Scharow A and Berg T. Nanomolar inhibitors of the transcription factor STAT5b with high selectivity over STAT5a. Angew Chem Int Ed Engl. 2015; 54(16):4758-4763.
219. Muller J, Sperl B, Reindl W, Kiessling A and Berg T. Discovery of chromone-based inhibitors of the transcription factor STAT5. Chembiochem. 2008; 9(5):723-727.
220. Page BD, Khoury H, Laister RC, Fletcher S, Vellozo M, Manzoli A, Yue P, Turkson J, Minden MD and Gunning PT. Small molecule STAT5-SH2 domain inhibitors exhibit potent antileukemia activity. J Med Chem. 2012; 55(3):1047-1055.
221. Cumaraswamy AA, Lewis AM, Geletu M, Todic A, Diaz DB, Cheng XR, Brown CE, Laister RC, Muench D, Kerman K, Grimes HL, Minden MD and Gunning PT. Nanomolar-Potency Small Molecule Inhibitor of STAT5 Protein. ACS Med Chem Lett. 2014; 5(11):1202-1206.