
Introduction
The 70 KDa ribosomal S6 kinases (S6K) RPS6KB1 (S6K1) and RPS6KB2 (S6K2) are key effectors of PI3K/mTOR-regulated signalling, and have been implicated in a variety of human diseases including diabetes and cancer [1-5]. They belong to the family of AGC kinases and are highly homologous with a sequence identity of 83% in their catalytic domains [6]. S6K1, which is the most extensively studied of the two, has been shown to phosphorylate a number of substrates that regulate protein synthesis, including the 40S ribosomal protein S6 (RPS6), and proteins involved in translation, such as the eukaryotic initiation factor 4B (EIF4B) and eukaryotic elongation factor 2 kinase [7].
In turn, S6K1 is activated by phosphorylation of the activation loop residue Thr252 (S6K1 numbering) by PDK1, and by phosphorylation of Thr412, located in the kinase extension region. Phosphorylation of Thr412 is achieved by mTORC1, which is a heterotrimeric complex comprising mTOR, raptor and mLST-8 [1]. However, for full activation of S6K1, these phosphorylation events have to be preceded by phosphorylation of a series of serine and threonine residues in the C-terminal autoinhibitory domain and by phosphorylation of Ser394 in the turn-motif [8].
The identification of S6K1 as a potential therapeutic target in oncology is supported by the fact that the S6K1 gene (RPS6KB1) is located at human chromosome 17q23, a region that is amplified in 20% of primary breast cancers as measured by comparative genomic hybridisation [9]. In addition, increased expression of S6K1 has been associated with cell transformation and elevated proliferation rates in tumours [10, 11]. Moreover, amplification and overexpression of S6K1 have been linked with poor prognosis and an increased risk of local recurrence [12]. Studies in cultured cells have confirmed that S6K activity is enhanced by mechanisms activating the PI3-kinase/Akt/mTOR pathway, such as loss of the tumour suppressor PTEN, and have shown a positive correlation between S6K activity and tumour growth [13]. In addition, loss of either of the tumour suppressive TSC1/2 complex proteins can result in activation of mTORC1 and S6K. The TSC2 protein acts as a GTPase-activating protein (GAP) towards Rheb (Ras homologue enriched in brain) and promotes conversion of active Rheb-GTP into the inactive form, Rheb-GDP, that can no longer stimulate mTORC1 [14]. Furthermore, S6K1 regulates the turnover of the oncogenic protein MDM2 in the ovarian cancer cell line OVCAR-3 [15]. In DU-145 prostate cancer cells, S6K1 increased survivin expression produced by upstream activation of the pathway, whereas acute ablation of endogenous S6K1 by small interfering RNA down-regulated survivin levels [16]. Finally, increased phosphorylation of S6K and corresponding phosphorylation of RPS6 was observed in HPV16-infected cervical cancer tissue samples [17], and the constitutive activation of S6K1 is associated with cisplatin resistance in human H69 small cell lung cancer cells [18].
Ribosomal S6 kinases are thus interesting therapeutic targets for cancer treatment. In this study we describe a high-throughput screening (HTS) campaign against S6K1 using an amplified luminescent proximity homogeneous assay, and the subsequent hit evaluation for the three hit series we identified. In addition, we report a novel PKA-S6K1 chimeric protein, which was used to elucidate the binding mode of two of the HTS hit series including the improved C-6-substituted azabenzimidazole S6K inhibitors. Finally, for the most potent compound from the azabenzimidazole series we demonstrate inhibition of S6K in intact human bladder tumour cells exhibiting loss of expression of the TSC1/2 complex.
Results and Discussion
A PKA-S6K1 Chimera for Structure-Based Drug Design.
To date, the available structural data for S6 kinases are limited to three crystal structures of the S6K1 kinase domain. Two structures are of unphosporylated S6K1 and consistent with an inactive state of the kinase. The third is of S6K1 phosphorylated on Thr252 in the activation loop and consistent with a partially activated state of the kinase [19]. The three crystal structures of S6K1 were solved as protein-ligand complexes with the pan-kinase inhibitor staurosporine, but the authors were not able to generate an apo-S6K1 structure, which they attributed to a susceptibility of the apo-enzyme to aggregation upon concentration and upon storage. Whilst the published S6K1 crystal structures provided an exciting first structural insight in S6 kinases, the crystal system might be less applicable to iterative protein-ligand structural studies informing the structure-based design of potent S6K inhibitors.
In order to generate a more robust crystal system suitable for the generation of high-resolution protein-ligand structural data required to facilitate S6K inhibitor design, we decided to develop a S6K1 chimeric protein based on the catalytic subunit of the closely related AGC kinase PKAα (PKA). Chimeric proteins based on PKA have been successfully used in the discovery of PKB inhibitors [20, 21], the analysis of Aurora kinase inhibitors [22], and to study the selectivity determinants of Rho kinase inhibitors [23]. S6K1 and PKA share a sequence identity of approximately 33% in their kinase domains and their ATP-binding sites are nearly identical. Our PKA-S6K1 chimera was created by mutation of five residues in or near the ATP-binding site (F54Y, M120L, V123L, L173M and Q181K) that differ between the two kinases. Analogous to PKA and other PKA-based chimeric proteins, the PKA-S6K1 chimera could be expressed in E. coli and the tetra-phosphorylated enzyme purified using a protocol described previously (see Materials and Methods). Co-crystals of purified PKA-S6K1 chimera with PKA inhibitor peptide (PKI, residues 5-24) were successfully grown, routinely diffracted to between 1.5 and 2.0 Å resolution, and ternary complexes with inhibitors could easily be obtained using soaking experiments.
To validate the PKA-S6K1 chimera as a structural surrogate for S6K1, we solved the structure of staurosporine bound to the PKA-S6K1 chimera and compared it with the publicly available staurosporine-bound crystal structures of PKA (PDB code: 1STC) and the phosphorylated and partially activated S6K1 (PDB code: 3A62). As expected, the overall conformations of the staurosporine-bound PKA and PKA-S6K1 structures are nearly identical (rmsd 0.51 Å for 330 equivalent Cα atoms, Figure 1), except for residues 316 to 320 in the C-terminal tail, which adopt a different conformation in the PKA-S6K1 chimera compared to PKA (Figure 1A). In both structures, the PDK1 phosphorylation site in the activation loop (Thr197) is in its phosphorylated state, the activation loop is in an optimal conformation for substrate binding and both structures are ternary complexes with the peptide inhibitor PKI. However, it is important to note that the binding of staurosporine induces substantial conformational changes in the structure of PKA [24] and therefore also in the structure of the PKA-S6K1 chimera. During its catalytic cycle, the conformation of the PKA kinase domain shuttles between an open unliganded conformation, and a closed ATP- and substrate-bound conformation, which together with several intermediate conformations, have been captured in different PKA crystal structures [25]. Both the staurosporine-bound PKA and PKA-S6K1 structures are most similar to an intermediate conformation represented by the adenosine-bound PKA structure (PDB code: 1BKX), in which the P-loop adopts a conformation halfway between the fully open and closed forms of the kinase (Figure 1C). In both staurosporine-bound crystal structures, changes in the side chain conformations of residues lining the respective ATP-binding sites help to accommodate the binding of the bulky staurosporine molecule. The biggest differences include the conformation of the respective Phe54 or Tyr54 residue at the tip of the P-loop, which in both structures is tucked underneath the loop and points towards staurosporine, and the conformation of Phe327 in the C-terminal tail, whose side chain is not only shifted, but also rotated by approximately 90 degrees to allow binding of the inhibitor (Figure 1C).
The construct used to elucidate the native S6K1 structure lacks 75 N-terminal residues and 126 C-terminal residues of the S6K1 sequence. In addition, the S6K1 crystallographic coordinates lack a further 10 N-terminal residues and 27 C-terminal residues due to disorder of these residues within the crystals used for structure elucidation. Therefore, the native S6K1 crystal structures do not contain the C-terminal auto-inhibitory domain and the majority of the characteristic AGC kinase C-terminal tail [26], which folds back from the C-terminal lobe across the ATP-binding site to the N-terminal lobe, is not present. Although the S6K1 construct is considerably shorter than the PKA and PKA-S6K1 chimera sequences, the overall tertiary SK61 structure is very similar to the PKA-S6K1 chimera (rmsd 1.33 Å for 247 equivalent Cα atoms, Figure 1B). The most notable differences with the PKA-S6K1 chimera include the disordered αB-helix and partially disordered activation loop and αC-helix, which are consistent with the low activity of the phospho-Thr252 form of the enzyme. The binding-mode of staurosporine to native S6K1 and the PKA-S6K1 chimera is nearly identical, and the amino acid residues lining the respective ATP-binding sites and contacting staurosporine have very similar side chain conformations. The exception is the conformation of the tyrosine residue (Tyr102 in S6K1 and Tyr54 in PKA-S6K1) at the tip of the P-loop, which extends outward in S6K1, but is folded back underneath the P-loop in the PKA-S6K1 chimera (Figure 1D). This could potentially be a cause of differences in inhibitor-binding between native S6K1 and the PKA-S6K1 chimera, but because of the high similarity of the overall kinase domain structures and the very minor differences in side chain conformations within the respective staurosporine-occupied ATP-binding sites, we concluded that the PKA-S6K1 chimera was a suitable, robust and high-resolution surrogate system to guide our structure-based S6K inhibitor design.
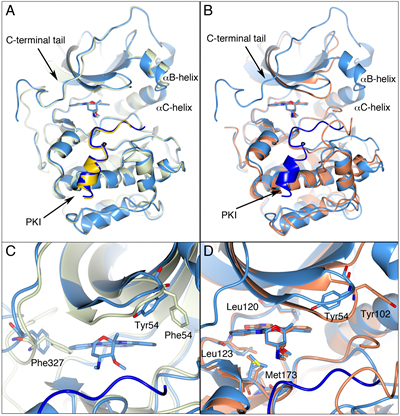
Figure 1: PKA, the PKA-S6K1 chimera and native S6K1. A) Superposition of the staurosporine-bound structures of PKA (PDB code: 1STC) in light green and the PKA-S6K1 chimera in light blue showing the close similarity of the structures. The inhibitor peptide PKI is shown in yellow and blue in the respective structures. The staurosporine molecules are shown in cylinder representation. Relevant secondary structure elements are labelled. B) Superposition of the staurosporine-bound structures of the PKA-S6K1 chimera in light blue and native S6K1 (PDB code: 3A62) in orange showing the similarity in their tertiary structures and the nearly identical binding mode of staurosporine. The inhibitor peptide PKI present in the PKA-S6K1 chimera structure is shown in blue. C) Close-up of the superposition of the staurosporine bound PKA-S6K1 chimera and the adenosine-bound PKA structure (PDB code: 1BKX) which represents an intermediate conformation in the PKA catalytic cycle. The colour scheme is the same as in panel A, but the adenosine molecule bound in 1BKX is not displayed for clarity. Phe327 in the C-terminal tail and the aromatic residue at the tip of the P-loop (Tyr54/Phe54) adopt different conformations in the respective structures. D) Close-up of the S6K1 and PKA-S6K1 chimera superposition. The positions of four of the five mutations in the PKA-S6K1 chimera ATP-binding site (Tyr54, Leu120, Leu123, Met173) are highlighted using the PKA-S6K1 sequence numbering. The fifth mutation near the ATP-binding site (Lys181) could not be shown in this orientation. Also shown is Tyr102 in S6K1. All structural figures were made using CCP4MG [48].
High-throughput screening of S6K1.
To identify inhibitors of S6K1, an HTS using an AlphaScreen™ kinase assay was carried out which measured the inhibition of the catalytic domain of S6K1 by the reduction in phosphorylation of an RPS6 substrate peptide. A compound library of approximately 67,000 compounds with lead-like properties was screened at a final concentration of 30 μM. With a mean Z’ factor of 0.69 ± 0.07 (CV% = 9.3) the overall performance of the HTS was considered to be good (Figure 2). A total of 414 hits were identified yielding an initial hit rate of 0.6%. To exclude compounds active through aggregation, all initial hits were reanalysed using the same assay conditions in the presence of Triton™ X100 [27]. The 67 resulting hits (confirmed hit rate 0.1%) were inspected for chemical tractability, toxicophores and undesired compounds, which left 19 progressible hits of which 12 were commercially available and repurchased for further analysis. The IC50 values of 11 of the 12 hits ranged from 0.25 to 22.6 μM, but the remaining compound did not reconfirm in a secondary DELFIA phosphorylation assay and was therefore discarded.
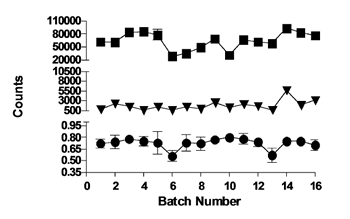
Figure 2: Overall performance of the S6K1 high-throughput screen. Alphascreen™ counts (mean ± SD) for total activity wells (■); no enzyme blanks (▼) and Z’ factor (●) for 16 batches of 14 plates containing 320 compounds each.
Hit evaluation.
The resulting confirmed HTS hits could be grouped into three chemical series and three singletons. The three chemical series were prioritised for investigation.
The first series comprised three carboxamidobenzimidazoles (1 – 3) with respective IC50 values of 0.56 μM, 1.04 μM and 10.14 μM (Figure 3 and Table 1). Their favourable physicochemical properties, including molecular weights ≤ 283 Da, ClogP ≤ 2.1, total polar surface areas (tPSA) ≤ 97 Å2, and their good ligand efficiencies made them an attractive series for further investigation.
Figure 3: Carboxamidobenzimidazole hits
The carboxamidobenzimidazoles are known inhibitors of poly(ADP-ribose) polymerase (PARP) with activities in the low nanomolar range [28] and therefore our first aim was to attempt to divorce their S6K1 and PARP activities. It is known that the 4-carboxamide and its intramolecular hydrogen bond to the N-3 of the benzimidazole are crucial for PARP activity as this mimics the natural cofactor nicotinamide adenine dinucleotide (NAD+). Replacement of the amide by a carboxylic acid group gave compound 4, which is known to be inactive against PARP but also led to loss of activity against S6K1. Similarly, the N-methyl amide 5 was inactive against S6K1, as was the regioisomeric 5-carboxamide 6, which is another carboxamidobenzimidazole lacking PARP activity [28]. Initial docking studies and subsequent determination of the crystal structure of compound 1 bound to the PKA-S6K1 chimera showed that the 4-carboxamide group is involved in crucial hydrogen interactions with the hinge region (Figure 4A), which explains the loss of potency upon modifications in this area of the molecule. The phenol group of 1 extends past the medium-sized Leu120 gatekeeper into the selectivity pocket and interacts with the conserved Glu91 in the αC-helix. A superposition of the compound 1-bound PKA-S6K1 chimera structure with the S6K1 structure shows that 1 would fit in the ATP-binding site (Figure 4B). However, because the key features of these 2-arylbenzimidazole-4-carboxamides that lead to potent inhibition of S6K1 are also crucial to inhibition of PARP, we did not pursue this series any further.
Table 1: IC50 values and ligand efficiencies of carboxamidobenzimidazole hits 1 – 3.
Compound |
R1 |
R2 |
R3 |
R4 |
S6K1 IC50 (μM)a |
LEc |
||
1 |
OH |
H |
- |
- |
0.56 ± 0.05 |
0.45 |
||
2 |
H |
OH |
- |
- |
1.04 ± 0.16 |
0.44 |
||
3 |
OH |
OMe |
- |
- |
10.1 ± 2.9 |
0.33 |
||
4 |
- |
- |
CO2H |
H |
>100b |
n/a |
||
5 |
- |
- |
CONHMe |
H |
>100b |
n/a |
||
6 |
- |
- |
H |
CONH2 |
>100b |
n/a |
||
aThe IC50 values were taken from the confirmation DELFIA assays on repurchased samples and are expressed as the mean ± standard error for duplicate measurements. bSingle measurement. cLigand efficiencies were calculated using the mean IC50 values [49].
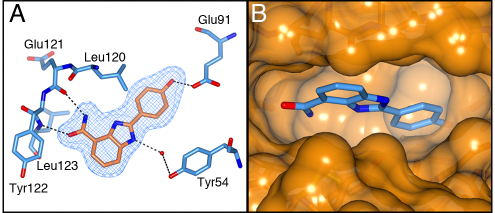
Figure 4: The binding mode of compound 1. A) Crystal structure of the PKA-S6K1 chimera in complex with compound 1. The compound is shown in cylinder representation with orange carbon atoms. PKA-S6K1 amino acids interacting with 1 are displayed in cylinder representation with blue carbon atoms. Solvent atoms are displayed as red spheres and hydrogen bonds are shown as black dashed lines. The electron density shown as a blue mesh is a Fo-Fc omit map contoured at 3σ. B) Superposition of the structure of 1 bound to PKA-S6K1 with the S6K1 structure (PDB code: 3A62) showing that 1 fits in the S6K1 ATP-binding site. Compound 1 is shown in cylinder representation with blue carbon atoms. The S6K1 solvent accessible surface is shown in orange.
The second hit series consisted of three 2,3-diaryl(3H)-quinazolin-4-ones (Figure 5). Although the compounds in this series had slightly higher ClogP values (2.8–4.0) than the benzimidazoles from series 1, the potencies and ligand efficiencies (Table 2) were still attractive. However, upon resynthesis of these compounds, it became clear that their physicochemical properties are far from ideal. Obtaining reliable assay results was difficult owing to limited aqueous solubility, and despite developing a new synthetic route to this series [29], it proved impossible to develop any directional structure-activity relationships (SAR), which led us to discard this series.
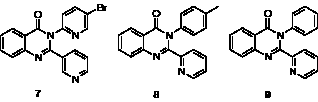
Figure 5: Diarylquinazolinone hits
Table 2: IC50 values and ligand efficiencies of diarylquinazoline hits.
Compound |
S6K1 IC50 (μM)a |
LEb |
7 |
0.26 ± 0.15 |
0.38 |
8 |
1.55 ± 0.92 |
0.33 |
9 |
2.97 ± 1.53 |
0.33 |
aThe IC50 values were taken from the confirmation DELFIA assays on repurchased samples and are expressed as the mean ± standard error for duplicate measurements. bLigand efficiencies were calculated using the mean IC50 values [49].
The third hit series consisted of the two N-alkylbenzimidazoles compound 10 and compound 11 which both have an amino-oxadiazole substituent at the C-2 position (Figure 6). Their respective IC50 values of 340 nM and 7.0 μM translate to good ligand efficiencies (Table 3). At the start of our programme, compounds containing this ring system had been reported as inhibitors of the AGC kinases mitogen and stress-activated protein kinase (MSK1), Rho kinase (ROCK1), and AKT [30-32], and the excellent properties of these hits (ClogP = 2.8, tPSA = 76 Å2, Mw = 241/243 Da), led us to select the benzimidazole oxadiazole scaffold as a starting point for our SAR studies. During the course of our work our findings were reinforced by Bandarage et al. [33], who also reported benzimidazole oxadiazoles as attractive S6K1 inhibitors.

Figure 6: Benzimidazole oxadiazole hits
Table 3: IC50 values and ligand efficiencies of benzimidazole oxadiazole hits.
Compound |
S6K1 IC50 (μM)a |
LEb |
10 |
0.34 ± 0.14 |
0.50 |
11 |
6.98 ± 2.07 |
0.40 |
aThe IC50 values were taken from the confirmation DELFIA assays on repurchased samples and are expressed as the mean ± standard error for duplicate measurements. bLigand efficiencies were calculated using the mean IC50 values.
Exploration of the benzimidazole N-1 position.
It was initially decided to investigate simple alkyl modifications at the N-1 position of the benzimidazole scaffold. Several analogues were prepared according to well-established literature methods (Scheme 1) [33].
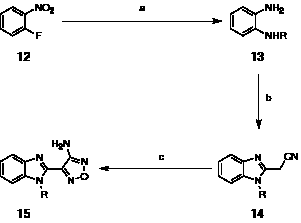
Scheme 1: Preparation of 2-aminooxadiazole benzimidazole derivatives. Reagents and conditions: (a) i. RNH2, KF, K2CO3, 50-100 ºC, ii. Pd(10%)/C, EtOH, rt or SnCl2·2H2O, reflux (77-86%, 2 steps); (b) ethyl cyanoacetate, 195 ºC, 2 h (43-70%); (c) NaNO2, 6 M aq. HCl, MeOH then 6 M aq. NaOH, 50% aq. NH2OH, reflux (10-45%).
In agreement with the results of Bandarage et al., compounds 15b, 15c and 15e with the respective ethyl, cyclopropyl and cyclopropylmethyl substituents were most potent, but the N-methyl analogue compound 15a was about six-fold less active (Table 4). In addition, incorporation of the larger benzyl substituent (15d) resulted in complete loss of biochemical activity. The crystal structure of the PKA-S6K1 chimera bound to 15e (Figure 7a) shows that its cyclopropylmethyl group fits snugly in a hydrophobic pocket formed by Gly50, Tyr54, Val57, and Phe327, as proposed by Banderage et al. [33], and suggests that this pocket is too small to accommodate the benzyl group of 15d. Superposition of 15e on the staurosporine-bound S6K1 structure shows that the compound also fits in the native S6K1 ATP binding site, but owing to the disorder in the S6K1 C-terminal tail and the different conformation of the P-loop, the presence of the hydrophobic pocket binding the cyclopropyl group could not be verified (Figure 7b).
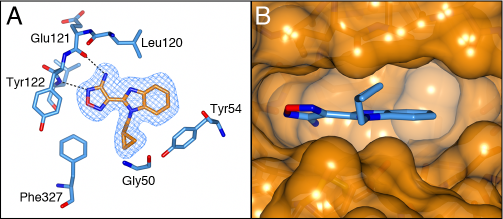
Figure 7: Binding mode of compound 15e. A) Crystal structure of the PKA-S6K1 chimera in complex with compound 15e. The compound is shown in cylinder representation with orange carbon atoms. PKA-S6K1 amino acids interacting with 15e are displayed in cylinder representation with blue carbon atoms. Hydrogen bonds are shown as black dashed lines. The electron density shown as a blue mesh is a Fo-Fc omit map contoured at 3σ. B) Superposition of the structure of 15e bound to PKA-S6K1 with the S6K1 structure (PDB code: 3A62) showing that 15e fits well in the S6K1 ATP-binding site. However, due to the flexibility in the S6K1 P-loop and disorder in the C-terminal tail it cannot be confirmed if the hydrophobic pocket binding the cyclopropyl group of 15e in the PKA-S6K1 chimera is present in native S6K1. Compound 15e is shown in cylinder representation with blue carbon atoms. The S6K1 solvent accessible surface is shown in orange.
Acetylation on the exocyclic amino group of the aminooxadiazole moiety renders this series inactive against S6K, as shown by compound 16, with an IC50 value of greater than 100 μM (Scheme 2). This is consistent with the binding mode observed in the 15e-bound PKA-S6K1 structure, in which the oxadiazole group binds to the hinge by the formation of a hydrogen bond between its N-2 atom and the backbone amide group of Leu123 and through a hydrogen bond between the exocyclic amino group with the backbone carbonyl of Glu121. Although the closely related acetylated compound 16 could in principle still form these hydrogen bonds, in this binding mode the acetyl group would clash with the gatekeeper residue Leu120 and Val104, situated at the bottom of the ATP-binding site.
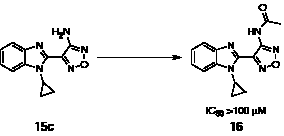
Scheme 2: Preparation of 2-acetamide-oxadiazole derivative 16. Reagents and conditions: AcCl, rt (70%).
Table 4: IC50 values and ligand efficiencies for N-1 substituted compounds 15 (Scheme 1).
Compound |
R |
S6K1 IC50 (nM)a |
LEe |
15a |
Methyl |
194d |
0.58 |
15b |
Ethyl |
59.8 ± 32.4b |
0.59 |
15c |
Cyclopropyl |
36.6 ± 7.0b |
0.57 |
15d |
Benzyl |
>100000c |
n/a |
15e |
Cyclopropyl methyl |
19.8d |
0.56 |
aThe IC50 values were determined with a mobility shift assay. bMean ± standard deviation from triplicate measurements. cDuplicate measurement. dSingle measurement. eLigand efficiencies were calculated using the mean IC50 values.
Exploration of the benzimidazole C-5 and C-7 positions.
Modifications on the benzene ring at the C-5 and C-7 positions were investigated as shown in Scheme 3. Preparation of the requisite bromo-precursors 20a and 20f (Scheme 3) allowed the synthesis of analogues incorporating phenyl (20b), cyclopropyl (20c), methoxy (20d) and methylamino (20e) on the R1 position and 3-pyridyl on the R2 position (20f, Scheme 3). However, unfortunately none of these modifications yielded an improvement in potency (Table 5).
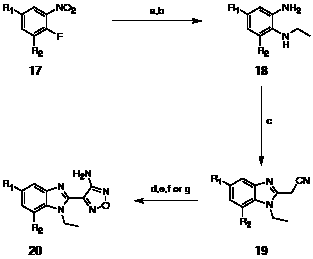
Scheme 3: Preparation of C-5 and C-7 substituted benzimidazoles; Reagents and conditions: (a) EtNH2, THF (quant.); (b) Na2S2O4, EtOH, reflux (90%); (c) i. CDI, cyanoacetic acid, THF, reflux; ii. acetic acid (81%, 2 steps); (d) NaNO2, 6 M aq. HCl, MeOH then 6 M aq. NaOH, 50% aq. NH2OH, reflux (54%); (e) i. n-Buli, B(OMe)3, THF then aq. H2O2 (41%); ii. MeI, K2CO3, acetone (80%); (f) i. trifluoroacetamide, CuI, N,N’-dimethylethylenediamine, K2CO3, dioxane (25%); ii. MeI, KOH, acetone, reflux then 5% aq. NaOH, EtOH, reflux (70%); (g) PhB(OH)2 or cyclopropylboronate ester or 3-pyridine boronic acid, Pd(PPh3)4, 1 M aq. NaOH, DME, 150 ºC (71%,15% and 25% respectively).
Table 5: IC50 values and ligand efficiencies of C-5 and C-7 substituted benzimidazoles (Scheme 3).
Compound |
R1 |
R2 |
S6K1 IC50 (nM) a |
LEc |
20a |
Br |
H |
66.4 ± 5.6 |
0.55 |
20b |
Ph |
H |
1640 ± 530 |
0.35 |
20c |
cyclopropyl |
H |
334 ± 25.1 |
0.45 |
20d |
OMe |
H |
63.4b |
0.52 |
20e |
NHMe |
H |
148b |
0.50 |
20f |
H |
Br |
303 ± 21 |
0.50 |
20g |
H |
|
352 ± 173 |
0.39 |
aThe IC50 values were determined with a mobility shift assay and expressed as the mean ± standard deviation for triplicate measurements. bSingle measurement. cLigand efficiencies were calculated using the mean IC50 values.
Azabenzimidazoles.
In order to diversify this scaffold, we chose to replace the benzimidazole ring with an azabenzimidazole. Azabenzimidazole aminooxadiazoles have been reported as potent Rho kinase inhibitors and upon analysis of their selectivity profile were also found to have a strong affinity for S6K [34]. Therefore we synthesised a limited set of azabenzimidazole analogues from the bromo-precursor 21a using the synthetic route reported (see Scheme 4) [34]. Interestingly, 21a (Table 6) was equipotent with the N-1 cyclopropyl-substituted benzimidazole oxadiazole 15c. Introduction of a phenoxy group in the C-6 position (21b) conferred some activity as already suggested by the selectivity studies on Rho kinase. Substitution with a phenyl group at C-6 (21c) turned out to be detrimental and abolished all affinity for S6K1. An attempt to improve the physicochemical properties by introducing amino-side chains such as a morpholine group α-to the N-atom (21d) also proved unsuccessful. However, the methyl-substituted compound 21e represented the most potent inhibitor within this series, and with its ligand efficiency of 0.6 could prove to be an attractive tool compound for further S6K studies (Table 6).
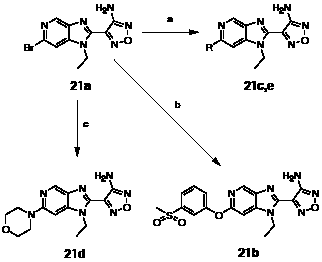
Scheme 4: Preparation of azabenzimidazole derivatives. Reagents and conditions: (a) RB(OH)2, Pd(PPh3)4, 1 M aq. NaOH, DME, 150 ºC microwave [R = Me (25%), R = Ph (59%)]; (b) methyl-(3-hydroxyphenyl)sulfone, Cs2CO3, Cu (10 mol%), Fe(acac)3 (30 mol%), DMF, 150 ºC, microwave (17%); (c) morpholine, NMP, 160 ºC microwave (51%).
Table 6: IC50 values and ligand efficiencies of azabenzimidazoles (Scheme 4).
Compound |
R |
S6K1 IC50 (nM) a |
LEd |
21a |
Br |
19.7 ± 2.6 |
0.59 |
21b |
|
103 ± 23b |
0.34 |
21c |
Ph |
>100000 |
n/a |
21d |
morpholine |
110c |
0.42 |
21e |
Me |
17.4 ± 6.0 |
0.60 |
aThe IC50 values were determined with a mobility shift assay and expressed as the mean ± standard deviation for triplicate measurements. bMean value from duplicate measurement ± standard error. cSingle measurement. dLigand efficiencies were calculated using the mean IC50 values.
The binding mode of both the synthetic precursor 21a and 21e was determined by solving the crystal structures of these inhibitors in complex with the PKA-S6K1 chimera (Figure 8). Both compounds bind to PKA-S6K1 in the same orientation as the benzimidazole oxadiazoles with the azabenzimidazole nitrogen forming an additional hydrogen bond interaction with the catalytic Lys72, which in turn forms the conserved salt bridge with Glu91 located in the αC-helix. In both structures the respective bromo and methyl C-6 substituents pack against the side chain ring of Tyr54, located at the tip of the P-loop and are found to be involved in protein-inhibitor interactions in other kinases such as PKB [21].
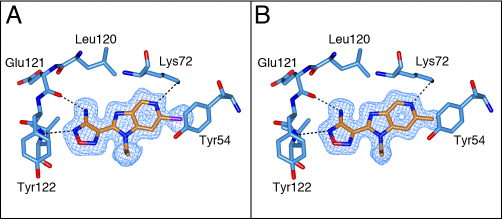
Figure 8: The binding mode of azabenzimidazoles 21a and 21e. A) Crystal structure of the PKA-S6K1 chimera in complex with the synthetic precursor compound 21a. B) Crystal structure of the PKA-S6K1 chimera in complex with compound 21e. The compounds are shown in cylinder representation with orange carbon atoms. PKA-S6K1 amino acids interacting with the respective compounds are displayed in cylinder representation with blue carbon atoms. Hydrogen bonds are shown as black dashed lines. The electron density shown as a blue mesh are Fo-Fc omit maps contoured at 3σ.
Comparison of the binding modes of identified S6K1 inhibitors.
The PKA-S6K1 inhibitor structures described in this paper are most similar to the intermediate adenosine-bound PKA conformation (PDB code: 1BKX). In all structures, Phe327 adopts a conformation similar to the one observed in PKA and consistent with shielding the bound ligand from the solvent.
The carboxamidobenzimidazole 1 is the only inhibitor which binds in the selectivity pocket, whereas the benzimidazole-oxadiazole 15e and azabenzimidazole-oxadiazoles 21a and 21e bind in a very similar manner exploiting the small hydrophobic pocket formed by Phe327 from the C-terminal tail, Gly50 and Tyr54 from the P-loop (Figure 9A). Interestingly, in the benzimidazole and azabenzimidazole inhibitors the aromatic Tyr54 at the tip of the P-loop closes in over the inhibitor and packs against the hydrophobic side of the inhibitors. In compounds 21a and 21e, Tyr 54 interacts with the respective bromine and methyl groups, but the effect is most pronounced for compound 15e, which is unsubstituted in this position allowing Tyr54 to interact directly with the core scaffold of the inhibitor (Figure 9B). The closing of the P-loop over these inhibitors effectively shields them from the solvent, which together with the hydrophobic stacking interactions is likely to contribute to the potency of these inhibitors.
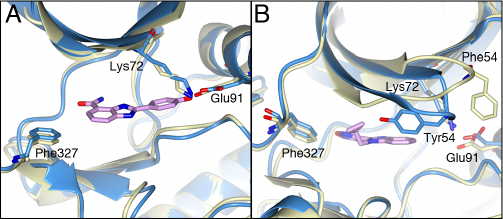
Figure 9: Comparision of 4-carboxamidobenzimidazole and benzimidazole-oxadiazole with PKA. A). Superposition of the compound 1 bound PKAS6K1 structure (light-blue) with the intermediate adenosine-bound PKA structure (PDB code: 1BKX, lemon yellow). Compound 1 is shown in pink. The phenol group of compound 1 extends into the selectivity pocket. B) Superposition of the compound 15e bound PKA-S6K1 structure (light blue) with 1BKX (lemon yellow). Compound 15e is shown in pink. Tyr54 at the tip of the P-loop of the PKA-S6K1 chimera swings towards the benzimidazole core scaffold of the inhibitor. In both inhibitor-bound structures Phe327 is in a similar conformation as observed in the adenosine-bound structure.
Cellular activity of azabenzimidazole 21e (CCT239066).
We examined the activity of the substituted-azabenzimidazole 21e (CCT239066) in a human bladder carcinoma line (HCV29) that has activated mTOR signalling as a result of losing expression of the TSC complex which allows activation of mTORC1 [35]. CCT239066 (21e) inhibits S6K1 with IC50 values of 17.4 ± 6.0 nM and S6K2 at 300 ± 102 nM respectively and demonstrated 50% tumour growth inhibition (GI50) at a concentration of 3.5 μM following 96 hours continuous exposure. CCT239066 (21e) also inhibited phosphorylation of a biomarker of S6K1 activity, phosphorylation of RPS6 at Ser235/236 (Figure 10) in intact cells. The EC50 for 50% inhibition of Ser235/236 phosphorylation following 2 hours exposure was 9.1 ± 1.5 μM (n=3) compared to IC50 values > 15 μM for AKT-Thr308, AKT-Ser473, GSK3b-Ser9 and S6K-Ser421/424 as determined by electrochemoluminescent ELISA in intact cells.
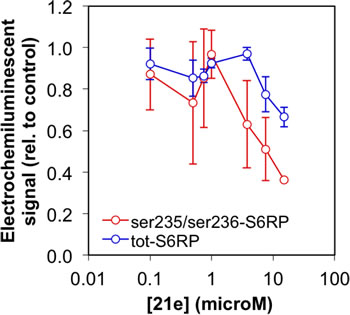
Figure 10: Inhibition of RPS6 phosphorylation. Results from an electrochemiluminscent ELISA assay showing the dose-dependent inhibition of phosphorylation of RPS6 on serine 235 and serine 236 in HCV29 human bladder cancer cells following a two hour exposure to compound 21e.
In conclusion, we have identified three different chemical series of S6 kinase inhibitors. Optimisation of the benzimidazole oxadiazole series resulted in a potent, ligand efficient azabenzimidazole S6K1/S6K2 inhibitor with good physicochemical properties and cellular activity. In addition, we developed a novel robust crystallography system based on a PKA-S6K1 chimera and experimentally determined the binding mode of two of the three inhibitor series, including the improved and cellularly active azabenzimidazole inhibitor. The availability of both a robust crystallography system and cellularly active tool compounds will aid the future design of potent and selective S6K inhibitors.
Methods
High-throughput screening of S6K1.
The catalytic domain of S6K1 was screened against our in-house HTS library of lead-like compounds using an AlphaScreen™ kinase assay in which inhibition of S6K1 was measured by a reduction in the phosphorylation of a S6 peptide. Compounds were screened at a final concentration of 30 μM and 0.3% (v/v) DMSO by dispensing 3 μL compound solution from a source plate containing the compounds at a concentration of 200 μM in 2% (v/v) DMSO, into the central 320 wells of a polypropylene 384-well assay plate (#781287; Greiner Bio-One, Frickenhausen, Germany). 0.3% (v/v) DMSO was used as a vehicle control. 17 μL of the reaction mixture containing 1 nM S6K1 (#14-486; Millipore, Billerica, MA, USA), 30 μM ATP and 250 nM S6 peptide (Biotin-KRRRLSSLRASTSKSESSQKI, synthesised by J. Metcalfe, ICR) in assay buffer consisting of 40 mM HEPES pH 7.5, 10 mM MgCl2, 1 mM DTT and 0.02% (v/v) Tween 20, was added to the compound plate and incubated for an hour at room temperature. Each 384 well plate contained 32 control wells for total enzyme activity, no enzyme blanks and the positive control, staurosporine at a final concentration of 20 nM. The reaction was stopped by the addition of 80 μL of 50 mM EDTA pH 7.5. An aliquot containing 10 μL of the assay solution was transferred to a white OptiPlate™ (#6007299; PerkinElmer Life Sciences, Waltham, MA, USA). AlphaScreen™ streptavidin-coated donor beads, protein A-conjugated acceptor beads (#6760617M; PerkinElmer Life Sciences) and anti-phospho S6 antibody (#2211; Cell Signalling Technology Inc., Danvers, MA, USA) were diluted in bead buffer comprising 40 mM HEPES pH 7.5, 40 mM NaCl, 5% (v/v) glycerol and 0.125% BSA and added to the OptiPlate™ resulting in a final concentration of 0.1 nM antibody and 10 μg/mL beads. Plates were sealed and incubated overnight at room temperature in the dark before being read on the Fusion™ Multilabel Reader (PerkinElmer Life Sciences). Using a 300 ms excitation at 680 nm and 700 ms per well reading time at 520-620 nm the signal was expressed in counts per second. A MiniTrak™ V (PerkinElmer Life Sciences) was used for compound and reagent addition. Primary screening data were analysed in ActivityBase (IDBS, Guildford, UK). Compounds with a percentage inhibition of 50% or better were classed as initial hits. All hits were cherry-picked and re-assayed in triplicate to confirm activity. In addition, the hits were tested for aggregation by assaying them in triplicate with 0.01% (v/v) Triton™ X-100 added to the assay buffer.
General synthetic chemistry.
Reactions were carried out under nitrogen or argon when necessary. Organic solutions were dried over MgSO4. Starting materials and solvents were purchased from commercial suppliers and were used without further purification. Microwave reactions were carried out using a Biotage Initiator 60 microwave reactor. Flash silica chromatography was performed using Merck silica gel 60 (0.025-0.04 mm) and ethylacetate/petroleum spirit (40-60). Ion exchange chromatography was performed using Isolute Flash SCX-II (acidic). 1H NMR spectra were recorded on a Bruker AMX500 instrument using internal deuterium lock. 13C NMR spectra were recorded at 125 MHz on a Bruker AMX500 instrument. Chemical shifts (δ) are reported relative to TMS (δ=0) and/or referenced to the solvent in which they were measured. Combined HPLC-MS analyses were recorded using a Waters Alliance 2795 separations module and Waters/Micromass LCT mass detector with electro spray ionization (+ve or -ve ion mode as indicated) and with HPLC performed using Supelco DISCOVERY C18, 50 mm x 4.6 mm or 30 mm x 4.6 mm i.d. columns, at a temperature of 22 °C with gradient elution of 10-90% MeOH/0.1% aqueous formic acid at a flow rate of 1 mL/min and a run time of 6 min. Compounds were detected at 254 nm using a Waters 2487 dual λ absorbance detector. All tested compounds gave 95% purity as determined by this method. All purified synthetic intermediates gave 95% purity as determined by this method except where indicated in the text. High-resolution mass spectra were measured on an Agilent 6210 ToF HPLC-MS with a Phenomenex Gemini 3 μm C18 (3 cm x 4.6 mm i.d.) column.
The synthesis of the key oxadiazole-substituted benzimidazoles and azabenzimidazoles 15a-e, 16, and 21b-e from their respective precursors is described below. For details of the synthesis of all precursors and compounds 20a-g see the supplementary information.
4-(1-Methyl-1H-benzoimidazol-2-yl)-1,2,5-oxadiazole-3-ylamine (15a).
Benzimidazole 14a (342 mg, 2.00 mmol) was diluted in MeOH (2 mL) and aqueous HCl (6 M, 1.5 mL). Solid NaNO2 (166 mg, 2.40 mmol, 1.2 equiv.) was added portionwise over 10 min. After the addition, the reaction mixture was made basic with the addition of aqueous NaOH (6 M, 1.5 mL) and hydroxylamine (50% aqueous solution, 0.68 mL, 10.27 mmol, 5.1 equiv.) was added. The reaction mixture was heated to reflux for 4 h, cooled and filtered (H2O and MeOH). Purification of the resulting solid by chromatography (SiO2, petroleum spirit/EtOAc: 70/30) afforded the title compound as a light yellow solid (254 mg, 45%). 1H NMR (500 MHz, DMSO-d6): δ=7.82 (d, J=8.1 Hz, 1H), 7.76 (d, J=8.1 Hz, 1H), 7.44 (ddd, J=1.0, 7.1, 8.1 Hz, 1H), 7.35 (ddd, J=1.0, 7.1, 8.1 Hz, 1H), 6.99 (br s, 2H), 4.16 ppm (s, 3H); 13C NMR (125 MHz, DMSO-d6): δ=156.1 (s), 141.5 (s), 140.9 (s), 138.4 (s), 135.8 (s), 124.4 (d), 123.0 (d), 119.8 (d), 111.1 (d), 32.2 ppm (q); LC-MS: m/z: 216 [M+H]+, Rt = 4.39 min; HR LC-MS: m/z calcd for C10H10N5O: 216.0879 [M+H]+; found: 216.0878.
4-(1-Ethyl-1H-benzoimidazol-2-yl)-1,2,5-oxadiazole-3-ylamine (15b).
The benzimidazole 14b (574 mg, 3.10 mmol) was dissolved in MeOH (2.5 mL) and aqueous HCl (6 M, 2.3 mL). Solid NaNO2 (256 mg, 3.72 mmol, 1.2 equiv.) was added portionwise over 10 min. After the addition, the reaction mixture was made basic with aqueous NaOH (6 M, 2.3 mL) and hydroxylamine hydrochloride (226 mg, 3.25 mmol, 1.05 equiv.) was added. The reaction mixture was heated to reflux for 2 h, cooled and filtered (H2O and MeOH). Purification of the resulting solid by chromatography (SiO2, petroleum spirit/EtOAc: 70/30) afforded the title compound as a yellow solid (62 mg, 9%). 1H NMR (500 MHz, DMSO-d6): δ=7.82 (d, J=7.8 Hz, 1H), 7.79 (d, J=7.8 Hz, 1H), 7.43 (m, 1H), 7.36 (m, 1H), 6.97 (br s, 2H), 4.70 (m, 2H), 1.39 ppm (t, J=6.3 Hz, 3H); 13C NMR (125 MHz, DMSO-d6): δ=156.1 (s), 141.7 (s), 140.1 (s), 138.1 (s), 134.7 (s), 124.5 (d), 123.1 (d), 120.0 (d), 111.0 (d), 40.1 (t), 14.8 ppm (q); LC-MS: m/z: 230 [M+H]+, Rt = 4.65 min; HR LC-MS: m/z calcd for C11H12N5O: 230.1036 [M+H]+; found: 230.1036.
4-(1-Cyclopropyl-1H-benzoimidazol-2-yl)-1,2,5-oxadiazole-3-ylamine (15c).
The benzimidazole 14c (318 mg, 1.61 mmol) was diluted in MeOH (1 mL) and aqueous HCl (6 M, 1.5 mL). Solid NaNO2 (135 mg, 1.93 mmol, 1.2 equiv.) was added portionwise over 10 min. After the addition, the reaction mixture was made basic with aqueous NaOH (6 M, 1.5 mL) and hydroxylamine (50% aqueous solution, 0.55 mL, 8.30 mmol, 5.1 equiv.) was added. The reaction mixture was heated to reflux for 3 h, cooled and filtered (H2O and MeOH). Purification of the resulting solid by chromatography (SiO2, petroleum spirit/EtOAc: 70/30) afforded the title compound as a green solid (154 mg, 40%). 1H NMR (500 MHz, DMSO-d6): δ=7.81 (d, J=8.1 Hz, 1H), 7.76 (d, J=8.1 Hz, 1H), 7.43 (apparent td, J=1.0, 8.1 Hz,1H), 7.35 (apparent td, J=1.0, 8.1 Hz, 1H), 6.91 (br s, 2H), 3.66 (m, 1H), 1.31-1.27 (m, 2H), 1.04-1.01 ppm (m, 2H); 13C NMR (125 MHz, DMSO-d6): δ=156.1 (s), 142.3 (s), 141.4 (s), 138.1 (s), 135.9 (s), 124.4 (d), 123.1 (d), 120.1 (d), 112.0 (d), 26.7 (d), 8.3 ppm (t); LC-MS: m/z: 242 [M+H]+, Rt = 4.67 min; HR LC-MS: m/z calcd for C12H12N5O: 242.1036 [M+H]+; found: 242.1038.
4-(1-Benzyl-1H-benzoimidazol-2-yl)-1,2,5-oxadiazole-3-ylamine (15d).
The benzimidazole (14d) (249 mg, 1.00 mmol) was diluted in MeOH (3 mL) and aqueous HCl (6 M, 1 mL). Solid NaNO2 (83 mg, 1.2 mmol, 1.2 equiv.) was added portionwise over 10 min. After the addition, the reaction mixture was made basic with aqueous NaOH (6 M, 1 mL) and hydroxylamine (50% aqueous solution, 0.34 mL, 5.1 mmol, 5.10 equiv.) was added. The reaction mixture was heated to reflux for 5 h, cooled and filtered (H2O and MeOH). Purification of the resulting solid by chromatography (SiO2, petroleum spirit/EtOAc: 70/30) afforded the title compound as a white solid (124 mg, 43%). 1H NMR (500 MHz, DMSO-d6): δ=7.87 (d, J=7.0 Hz, 1H), 7.72 (d, J=7.0 Hz, 1H), 7.41 (ddd, J=1.2, 7.1, 8.3 Hz, 1H), 7.37 (ddd, J=1.2, 7.1, 8.3 Hz, 1H), 7.30 (m, 2H), 7.24 (m, 1H), 7.15 (m, 2H), 7.03 (br s, 2H), 5.97 ppm (s, 2H); 13C NMR (125 MHz, DMSO-d6): δ=156.1 (s), 141.7 (s), 140.6 (s), 138.1 (s), 136.4 (s), 135.3 (s), 128.7 (2d), 127.5 (d), 126.5 (2d), 124.8 (d), 123.4 (d), 120.1 (d), 111.5 (d), 48.0 ppm (t); LC-MS: m/z: 292 [M+H]+, Rt = 5.07 min; HR LC-MS: m/z calcd for C16H14N5O: 292.1193 [M+H]+; found: 292.1199.
4-(1-Cyclopropylmethyl-1H-benzoimidazol-2-yl)-1,2,5-oxadiazole-3-ylamine (15e).
NaH (30 mg, 0.765 mmol) was added to a stirred solution of 2-aminoxadiazole benzimidazole (0.140 g, 0.696 mmol), in DMF (10 mL) at room temperature. The mixture was stirred at room temperature for 45 min, followed by the drop-wise addition of (bromomethyl)cyclopropane (0.188 g, 1.392 mmol) dissolved in DMF (1 mL). This mixture was heated at 65 °C for 18 h. After cooling to room temperature, the excess DMF was removed under reduced pressure and the resulting mixture was diluted with diethyl ether (50 mL). The organic mixture was washed with water and brine before drying over MgSO4. The excess solvent was removed under reduced pressure and the residue purified by flash chromatography (SiO2; EtOAc:hexane; 1:4) to afford title compound 15e (86 mg, 46%) as a white solid. 1H NMR (500 MHz, DMSO-d6): δ=7.82 (m, 2H), 7.43 (m, 1H), 7.36 (m, 1H), 6.99 (s, 2H), 4.59 (d, J=7.0 Hz, 2H), 1.36 (m, 1H), 0.48 ppm (m, 4H); 13C NMR (125 MHz, DMSO-d6): δ=156.2, 141.7, 140.2, 138.2, 135.4, 124.6, 123.1, 119.9, 111.5, 48.8, 11.3, 3.4 ppm; LC-MS: m/z: 256.11 [M+H]+, Rt = 2.90 min; HR LC-MS: m/z calcd for C13H13N5O: 256.2752 [M+H]+; found: 256.1194.
N-[4-(1-Cyclopropyl-1H-benzoimidazol->2-yl)-1,2,5-oxadiazole-3-yl]-acetamide (16).
A mixture of amino-oxadiazole (15c) (50 mg, 0.207 mmol) and acetyl chloride (0.5 mL, 7.03 mmol) was stirred at room temperature for 2 h. The reaction mixture was partitioned between EtOAc and saturated aqueous sodium bicarbonate solution (200 mL). After separation, the organic phase was washed with brine, dried over MgSO4 and evaporated under vacuum. The crude product was purified by flash chromatography (SiO2, petroleum spirit/EtOAc, gradient: 80/20 to 70/30) to give 41 mg (70%) of the title compound as a white solid. 1H NMR (500 MHz, CDCl3): δ=11.07 (s, 1H), 7.84 (d, J=8.1 Hz, 1H), 7.72 (d, J=8.1 Hz, 1H), 7.60 (td, J=7.2, 1.0 Hz, 1H), 7.42 (td, J=7.2, 1.0 Hz, 1H), 3.77–3.45 (m, 1H), 2.46 (s, 3H), 1.45 (q, J=6.7 Hz, 2H), 1.20–1.04 ppm (m, 2H); 13C NMR (125 MHz, DMSO-d6): δ=168.3 (s), 150.1 (s), 141.6 (s), 141.2 (s), 135.9 (s), 124.4 (d), 123.1 (d), 120.2 (d), 111.9 (d), 99.5 (s), 26.0 (d), 23.3 (t), 7.7 ppm (t); LC-MS: m/z: 242 [M–(CH3C=O)+H]+, Rt = 2.41 min; HR LC-MS: m/z calcd for C14H14N5O2: 284.1142 [M+H]+; found: 284.1143.
4-[1-Ethyl-6-(3-methanesulfonyl-phenoxy)-1H-imidazo[4,5-c]pyridin-2-yl]-1,2,5-oxadiazole-3-ylamine (21b).
A glass tube (evacuated and back-filled with Ar) was charged with Fe(acac)3 (16.5 mg, 0.047 mmol), Cu powder (1.0 mg, 0.016 mmol), methyl-(3-hydroxyphenyl)sulfone (40 mg, 0.23 mmol), and Cs2CO3 (102 mg, 0.31 mmol). Bromoazabenzimidazole 21a (50 mg, 0.156 mmol) was added under Ar followed by anhydrous DMF (1.5 mL). The tube was sealed under Ar, and the mixture was heated to 150 ºC under microwave irradiation for 30 min. After cooling to room temperature, the mixture was diluted with dichloromethane and filtered. The filtrate was concentrated in vacuo and partitioned between water and EtOAc. After separation, the water phase was extracted with EtOAc and the organics were combined, washed twice with water, dried over MgSO4 and concentrated. Purification of the residue by flash chromatography on silica gel (EtOAc/petroleum spirit, gradient: 40/60 to 70/30) afforded 21b (12 mg, 25%) as a white solid (13 mg, 20%). 1H NMR (500 MHz, DMSO-d6): δ=8.79 (s, 1H), 7.75 (apparent dt, J=1.2, 7.9 Hz, 1H), 7.70 (apparent t, J=7.9 Hz, 1H), 7.67 (s, 1H), 7.63 (apparent t, J=2.0 Hz, 1H), 7.50 (ddd, J=1.0, 2.3, 8.0 Hz, 1H), 6.92 (s, 2H), 4.70 (q, J=7.1 Hz, 2H), 3.25 (s, 3H), 1.41 ppm (t, J=7.1 Hz, 3H); 13C NMR (125 MHz, DMSO-d6): δ=158.2 (s), 156.1 (s), 155.6 (s), 143.1 (s), 143.0 (s), 142.2 (s), 140.0 (d), 137.8 (s), 137.1 (s), 130.9 (d), 125.3 (d), 122.1 (d), 118.2 (d), 93.7 (d), 43.3 (q), 40.6 (t), 14.6 ppm (q); LC-MS: m/z: 401 [M+H]+, Rt = 2.27 min; HR LC-MS: m/z calcd for C17H17N6O4S: 401.1026 [M+H]+; found: 401.1026.
4-(1-Ethyl-6-phenyl-1H-imidazo[4,5-c]pyridin-2-yl)-1,2,5-oxadiazole-3-ylamine (21c).
PhB(OH)2 (30 mg, 0.24 mmol), 1 M aqueous NaOH (0.48 mL, 0.48 mmol) and Pd(PPh3)4 (9.3 mg, 0.008 mmol) were added to a solution of 21a (50 mg, 0.16 mmol) in dimethoxyethane (DME, 2 mL). The reaction mixture was heated in a microwave reactor at 140 ºC for 1.5 h. The reaction mixture was then partitioned between saturated aqueous NH4Cl solution and EtOAc. After separation, the organic phase was washed with brine, dried over MgSO4 and evaporated under vacuum. The residue was first purified by ion exchange chromatography on SCX-II acidic resin (2 g) eluting with MeOH then a 2 M solution of NH3 in MeOH. After concentration, the resulting solid was purified on a small pad of silica gel (EtOAc/petroleum spirit, gradient: 30/70 to 50/50) to afford 21c (29 mg, 59%) as a white solid. 1H NMR (500 MHz, DMSO-d6): δ=9.20 (s, 1H), 8.46 (s, 1H), 8.25 (d, J=7.3 Hz, 2H), 7.53 (dd, app t, J=7.7 Hz, 2H), 7.44 (dd, app t, J=7.3 Hz, 1H), 6.96 (s, 2H), 4.79 (q, J=7.1 Hz, 2H), 1.45 ppm (t, J=7.1 Hz, 3H); 13C NMR (125 MHz, DMSO-d6): δ=156.2 (s), 150.5 (s), 142.4 (s), 142.3 (d), 140.8 (s), 139.1 (s), 138.5 (s), 137.9 (s), 128.8 (d), 128.6 (d), 128.5 (d), 126.8 (d), 102.4 (d), 40.5 (t), 14.9 ppm (q); LC-MS: m/z: 307 [M+H]+, Rt = 2.23 min; HR LC-MS: m/z calcd for C16H15N6O: 308.1329 [M+H]+; found: 308.1337.
4-(1-Ethyl-6-morpholin-4-yl-1H-imidazo[4,5-c]pyridin-2-yl)-1,2,5-oxadiazole-3-ylamine (21d).
Morpholine (0.07 mL, 0.81 mmol) was added to a solution of 21a (25 mg, 0.081 mmol) in N-methylpyrrolidinone (0.5 mL). The reaction mixture was heated in a microwave reactor at 160 ºC for 3 h. The mixture was cooled and concentrated. Purification of the crude by preparative TLC, eluting with EtOAc afforded 21d (13 mg, 51%) as a yellow solid. 1H NMR (500 MHz, DMSO-d6): δ=8.73 (s, 1H), 7.04 (s, 1H), 6.90 (s, 2H), 4.61 (q, J=7.1 Hz, 2H), 3.87-3.67 (m, 4H), 3.61-3.42 (m, 4H), 1.36 ppm (t, J=7.1 Hz, 3H); 13C NMR (125 MHz, DMSO-d6): δ=156.7 (s), 156.0 (s), 142.5 (s), 140.6 (s), 140.3 (d), 137.9 (s), 133.7 (s), 86.1 (d), 66.0 (t), 46.2 (t), 40.0 (t), 14.6 ppm (q); LC-MS: m/z: 316 [M+H]+, Rt = 2.50 min; HR LC-MS: m/z calcd for C14H18N7O2: 316.1516 [M+H]+; found: 316.1515.
4-(1-Ethyl-6-methyl-1H-imidazo[4,5-c]pyridin-2-yl)-1,2,5-oxadiazole-3-ylamine (21e).
MeB(OH)2 (29 mg, 0.48 mmol), 1 M aqueous NaOH (0.65 mL, 0.65 mmol) and Pd(PPh3)4 (9.3 mg, 0.008 mmol) were added to a solution of 21a (50 mg, 0.16 mmol) in DME (2 mL). The reaction mixture was heated in a microwave reactor at 140 ºC for 2 h. The reaction mixture was then partitioned between saturated aqueous NH4Cl solution and EtOAc. After separation, the organic phase was washed with brine, dried over MgSO4 and evaporated under vacuum. Purification of the residue by flash chromatography (SiO2, EtOAc) afforded 21e (12 mg, 25%) as a white solid. 1H NMR (500 MHz, DMSO-d6): δ=8.99 (s, 1H), 7.70 (s, 1H), 6.93 (s, 2H), 4.66 (q, J=7.2 Hz, 2H), 2.63 (s, 3H), 1.39 ppm (t, J=7.2 Hz, 3H); 13C NMR (125 MHz, DMSO-d6): δ=156.1 (s), 151.7 (s), 141.8 (d), 141.5 (s), 140.2 (s), 137.9 (s), 137.3 (s), 104.5 (d), 40.3 (t), 24.3 (t), 14.7 ppm (q); LC-MS: m/z: 245 [M+H]+, Rt = 1.73 min; HR LC-MS: m/z calcd for C11H13N6O: 245.1145 [M+H]+; found: 245.1151.
Crystallisation and structure determination.
Crystallisation of the PKA-S6K1 chimera was performed using the same conditions as previously reported for the PKA/PKB chimera [21]. Prior to crystallisation, the purified protein was concentrated to 18 mg/mL in crystallisation buffer consisting of 25 mM MES-Bis/Tris pH 6.5, 75 mM LiCl, 0.1 mM EDTA, 1 mM octanoyl-N-methylglucamide and 1 mM DTT and containing 1 mM PKA-inhibitor peptide PKI (residues 5-24, Sigma-Aldrich) or an in-house synthesised peptide corresponding to residues 5-22 of the PKA-inhibitor peptide, but terminating with an amide at the C-terminus. Crystals of the PKA-S6K1/PKI staurosporine ternary complex were obtained by streak-seeding into fresh hanging drops containing the same protein and reservoir solutions as used for the crystallisation of the binary PKA-S6K1/PKI complex, with the addition of 0.5 mM staurosporine. All other protein-inhibitor complexes were obtained by soaking experiments of selected inhibitors at a final concentration of 1 mM as previously described for PKA [21]. Crystals were briefly transferred to a cryoprotectant solution containing 22.5% (v/v) 2-methyl-2,4-pentanediol, 25 mM MES-Bis/Tris pH 6.5, 0.1 mM EDTA and 1 mM DTT, prior to flash-cooling in liquid nitrogen. The data for the binary PKA-S6K1/PKI complex were collected at beamline ID14EH2 at the ESRF (Grenoble, France). Data for the ternary PKA-S6K1/PKI/inhibitor complexes were collected at the following beamlines: staurosporine- and compound 1-bound complexes were collected at beamline I02 of the Diamond Light Source (Oxfordshire, UK) and the data for the protein-inhibitor complexes with compounds 15e, 21a, and 21e were collected at beamline ID14EH4 at the ESRF. All data sets were integrated, merged and scaled using the programs iMosflm [36], Pointless and Aimless [37, 38] from the CCP4 suite [39] (see Supplementary Table S1 for data collection and refinement statistics). The PKA-S6K1/PKI structure was solved by molecular replacement using PHASER [40] with a PKA structure (PDB code: 2GFC) as a search model, after removal of all water molecules and truncation of the 5 mutated amino acids to alanine. All protein-inhibitor structures were solved by molecular replacement with the PKA-S6K1/PKI structure as a search model after removal of all water molecules. All structures were manually rebuilt in COOT [41] and refined with BUSTER [42] in iterative cycles. Ligand restraints were generated with Grade [43] and Mogul [44]. The quality of the structures was assessed with MOLPROBITY [45].
Cellular assays.
The human bladder carcinoma cell line HCV29 was obtained from Dr Richard Lamb (University of Liverpool, UK). This cell line has elevated mTORC1 and S6K activity through loss of TSC1 [46] as a result of a missense mutation and TSC2 loss that was confirmed by immunoblotting (data not shown). Cells were cultured in RPMI (Invitrogen, Life Technologies Ltd, Paisley, UK) containing 2 mmol/L glutamine (Invitrogen), and supplemented with non-essential amino acids (Invitrogen) 10% fetal bovine serum (PAA Laboratories Ltd, Yeovil, UK) in 5% CO2 in air at 37 °C. GI50 values (concentrations) causing 50% inhibition of proliferation for tumour cells were determined using a sulforhodamine B assay. Electrochemiluminescent immunoassays (Meso Scale Discovery, Rockville, MD, USA) measuring the inhibition of phosphorylation of RPS6-ser235/236, AKT-Thr308, AKT-Ser473, GSK3b-Ser9 and S6K-Ser421/424 were carried out as described in Raynaud et al. [47].
Disclosure of Potential Conflicts of Interest.
The authors are current or former employees of The Institute of Cancer Research, which has a commercial interest in the development of kinase inhibitors. Rob van Montfort is a former employee of Astex Pharmaceuticals, Spiros Linardopoulos a former employee of Onyx pharmaceuticals and Paul Workman a former employee of Astra Zeneca.
Accession Codes.
The coordinates of the PKA-S6K1/PKI chimera and of the PKA-S6K1 chimera complexed with staurosporine and compounds 1, 15e, 21a and 21e and their associated structure factors have been deposited in the Protein Data Bank with accession codes: 4C33, 4C34, 4C35, 4C36, 4C37 and 4C38, respectively.
Acknowledgements
This work was supported by Cancer Research UK program grant number C309/A8274 and The Institute of Cancer Research. We acknowledge National Health Service funding to the National Institute for Health Biomedical Research Centre. Chiau Ling Chow is supported by a Wellcome Trust Ph.D. program in Mechanism-Based Drug Discovery at The Institute of Cancer Research, grant number 090174/Z/09/Z. Paul Workman is a Cancer Research UK Life Fellow. We thank the staff of the DIAMOND Light Source beamline I02 and the ESRF beamlines ID14EH2 and ID14EH4 for their support during data collection, and Dr Nora Cronin for support during data collection.
References
1. Fenton TR and Gout IT. Functions and regulation of the 70kDa ribosomal S6 kinases. Int J Biochem Cell Biol. 2011; 43(1): 47-59.
2. Bjornsti MA and Houghton PJ. The TOR pathway: a target for cancer therapy. Nat Rev Cancer. 2004; 4(5): 335-348.
3. Shamji AF, Nghiem P and Schreiber SL. Integration of growth factor and nutrient signaling: implications for cancer biology. Mol Cell. 2003; 12(2): 271-280.
4. Peralba JM, DeGraffenried L, Friedrichs W, Fulcher L, Grunwald V, Weiss G and Hidalgo M. Pharmacodynamic Evaluation of CCI-779, an Inhibitor of mTOR, in Cancer Patients. Clin Cancer Res. 2003; 9(8): 2887-2892.
5. Jaeschke A, Hartkamp J, Saitoh M, Roworth W, Nobukuni T, Hodges A, Sampson J, Thomas G and Lamb R. Tuberous sclerosis complex tumor suppressor-mediated S6 kinase inhibition by phosphatidylinositide-3-OH kinase is mTOR independent. J Cell Biol. 2002; 159(2): 217-224.
6. Gout I, Minami T, Hara K, Tsujishita Y, Filonenko V, Waterfield MD and Yonezawa K. Molecular cloning and characterization of a novel p70 S6 kinase, p70 S6 kinase beta containing a proline-rich region. J Biol Chem. 1998; 273(46): 30061-30064.
7. Pearce LR, Komander D and Alessi DR. The nuts and bolts of AGC protein kinases. Nat Rev Mol Cell Biol. 2010; 11(1): 9-22.
8. Dennis PB, Pullen N, Pearson RB, Kozma SC and Thomas G. Phosphorylation sites in the autoinhibitory domain participate in p70(s6k) activation loop phosphorylation. J Biol Chem. 1998; 273(24): 14845-14852.
9. Courjal F and Theillet C. Comparative genomic hybridization analysis of breast tumors with predetermined profiles of DNA amplification. Cancer Res. 1997; 57(19): 4368-4377.
10. Kwon HK, Bae GU, Yoon JW, Kim YK, Lee HY, Lee HW and Han JW. Constitutive activation of p70S6k in cancer cells. Arch Pharm Res. 2002; 25(5): 685-690.
11. Miyakawa M, Tsushima T, Murakami H, Wakai K, Isozaki O and Takano K. Increased expression of phosphorylated p70S6 kinase and Akt in papillary thyroid cancer tissues. Endocr J. 2003; 50(1): 77-83.
12. van der Hage JA, van den Broek LJ, Legrand C, Clahsen PC, Bosch CJ, Robanus-Maandag EC, van de Velde CJ and van de Vijver MJ. Overexpression of P70 S6 kinase protein is associated with increased risk of locoregional recurrence in node-negative premenopausal early breast cancer patients. Br J Cancer. 2004; 90(8): 1543-1550.
13. Neshat MS, Mellinghoff IK, Tran C, Stiles B, Thomas G, Petersen R, Frost P, Gibbons JJ, Wu H and Sawyers CL. Enhanced sensitivity of PTEN-deficient tumors to inhibition of FRAP/mTOR. Proc Natl Acad Sci USA. 2001; 98(18): 10314-10319.
14. Tomasoni R and Mondino A. The tuberous sclerosis complex: balancing proliferation and survival. Biochem Soc Trans. 2011; 39(2): 466-471.
15. Fang J, Meng Q, Vogt PK, Zhang R and Jiang BH. A downstream kinase of the mammalian target of rapamycin, p70S6K1, regulates human double minute 2 protein phosphorylation and stability. J Cell Physiol. 2006; 209(2): 261-265.
16. Vaira V, Lee CW, Goel HL, Bosari S, Languino LR and Altieri DC. Regulation of survivin expression by IGF-1/mTOR signaling. Oncogene. 2007; 26(19): 2678-2684.
17. Zhou Y, Pan Y, Zhang S, Shi X, Ning T and Ke Y. Increased phosphorylation of p70 S6 kinase is associated with HPV16 infection in cervical cancer and esophageal cancer. Br J Cancer. 2007; 97(2): 218-222.
18. Dhar R and Basu A. Constitutive activation of p70 S6 kinase is associated with intrinsic resistance to cisplatin. Int J Oncol. 2008; 32(5): 1133-1137.
19. Sunami T, Byrne N, Diehl RE, Funabashi K, Hall DL, Ikuta M, Patel SB, Shipman JM, Smith RF, Takahashi I, Zugay-Murphy J, Iwasawa Y, Lumb KJ, Munshi SK and Sharma S. Structural Basis of Human p70 Ribosomal S6 Kinase-1 Regulation by Activation Loop Phosphorylation. J Biol Chem. 2010; 285(7): 4587-4594.
20. Gassel M, Breitenlechner CB, Ruger P, Jucknischke U, Schneider T, Huber R, Bossemeyer D and Engh RA. Mutants of protein kinase A that mimic the ATP-binding site of protein kinase B (AKT). J Mol Biol. 2003; 329(5): 1021-1034.
21. Davies TG, Verdonk ML, Graham B, Saalau-Bethell S, Hamlett CC, McHardy T, Collins I, Garrett MD, Workman P, Woodhead SJ, Jhoti H and Barford D. A structural comparison of inhibitor binding to PKB, PKA and PKA-PKB chimera. J Mol Biol. 2007; 367(3): 882-894.
22. Pflug A, de Oliveira TM, Bossemeyer D and Engh RA. Mutants of protein kinase A that mimic the ATP-binding site of Aurora kinase. Biochem J. 2011; 440(1): 85-93.
23. Bonn S, Herrero S, Breitenlechner CB, Erlbruch A, Lehmann W, Engh RA, Gassel M and Bossemeyer D. Structural analysis of protein kinase A mutants with Rho-kinase inhibitor specificity. J Biol Chem. 2006; 281(34): 24818-24830.
24. Prade L, Engh RA, Girod A, Kinzel V, Huber R and Bossemeyer D. Staurosporine-induced conformational changes of cAMP-dependent protein kinase catalytic subunit explain inhibitory potential. Structure. 1997; 5(12): 1627-1637.
25. Akamine P, Madhusudan, Wu J, Xuong NH, Ten Eyck LF and Taylor SS. Dynamic features of cAMP-dependent protein kinase revealed by apoenzyme crystal structure. J Mol Biol. 2003; 327(1): 159-171.
26. Kannan N, Haste N, Taylor SS and Neuwald AF. The hallmark of AGC kinase functional divergence is its C-terminal tail, a cis-acting regulatory module. Proc Natl Acad Sci USA. 2007; 104(4): 1272-1277.
27. Ryan AJ, Gray NM, Lowe PN and Chung CW. Effect of detergent on “promiscuous” inhibitors. J Med Chem. 2003; 46(16): 3448-3451.
28. White AW, Almassy R, Calvert AH, Curtin NJ, Griffin RJ, Hostomsky Z, Maegley K, Newell DR, Srinivasan S and Golding BT. Resistance-modifying agents. 9. Synthesis and biological properties of benzimidazole inhibitors of the DNA repair enzyme poly(ADP-ribose) polymerase. J Med Chem. 2000; 43(22): 4084-4097.
29. Kalusa A, Chessum N and Jones K. An efficient synthesis of 2,3-diaryl (3H)-quinazolin-4-ones via imidoyl chlorides. Tetrahedron Lett. 2008; 49(41): 5840-5842.
30. Bamford MJ, Alberti MJ, Bailey N, Davies S, Dean DK, Gaiba A, Garland S, Harling JD, Jung DK, Panchal TA, Parr CA, Steadman JG, Takle AK, Townsend JT, Wilson DM and Witherington J. (1H-Imidazo[4,5-c]pyridin-2-yl)-1,2,5-oxadiazol-3-ylamine derivatives: A novel class of potent MSK-1-inhibitors. Bioorg Med Chem Lett. 2005; 15(14): 3402-3406.
31. Bamford MJ, Bailey N, Davies S, Dean DK, Francis L, Panchal TA, Parr CA, Sehmi S, Steadman JG, Takle AK, Townsend JT and Wilson DM. (1H-imidazo[4,5-c]pyridin-2-yl)-1,2,5-oxadiazol-3-ylamine derivatives: Further optimisation as highly potent and selective MSK-1-inhibitors. Bioorg Med Chem Lett. 2005; 15(14): 3407-3411.
32. Heerding DA, Rhodes N, Leber JD, Clark TJ, Keenan RM, Lafrance LV, Li M, Safonov IG, Takata DT, Venslavsky JW, Yamashita DS, Choudhry AE, Copeland RA, Lai Z, Schaber MD, Tummino PJ, et al. Identification of 4-(2-(4-amino-1,2,5-oxadiazol-3-yl)-1-ethyl-7-{[(3S)-3-piperidinylmethyl]oxy}-1H-imidazo[4,5-c]pyridin-4-yl)-2-methyl-3-butyn-2-ol (GSK690693), a novel inhibitor of AKT kinase. J Med Chem. 2008; 51(18): 5663-5679.
33. Bandarage U, Hare B, Parsons J, Pham L, Marhefka C, Bemis G, Tang Q, Moody CS, Rodems S, Shah S, Adams C, Bravo J, Charonnet E, Savic V, Come JH and Green J. 4-(Benzimidazol-2-yl)-1,2,5-oxadiazol-3-ylamine derivatives: Potent and selective p70S6 kinase inhibitors. Bioorg Med Chem Lett. 2009; 19(17): 5191-5194.
34. Stavenger RA, Cui H, Dowdell SE, Franz RG, Gaitanopoulos DE, Goodman KB, Hilfiker MA, Ivy RL, Leber JD, Marino JP, Jr., Oh H-J, Viet AQ, Xu W, Ye G, Zhang D, Zhao Y, et al. Discovery of aminofurazan-azabenzimidazoles as inhibitors of Rho-kinase with high kinase selectivity and antihypertensive activity. J Med Chem. 2007; 50(1): 2-5.
35. Zhang HB, Cicchetti G, Onda H, Koon HB, Asrican K, Bajraszewski N, Vazquez F, Carpenter CL and Kwiatkowski DJ. Loss of Tsc1/Tsc2 activates mTOR and disrupts PI3K-Akt signaling through downregulation of PDGFR. J Clin Invest. 2003; 112(8): 1223-1233.
36. Battye TG, Kontogiannis L, Johnson O, Powell HR and Leslie AG. iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr Sect D 2011; 67(Pt 4): 271-281.
37. Evans P. Scaling and assessment of data quality. Acta Crystallogr Sect D 2006; 62(Pt 1): 72-82.
38. Evans PR. An introduction to data reduction: space-group determination, scaling and intensity statistics. Acta Crystallogr Sect D 2011; 67(Pt 4): 282-292.
39. Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, et al. Overview of the CCP4 suite and current developments. Acta Crystallogr Sect D 2011; 67(Pt 4): 235-242.
40. McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC and Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007; 40: 658-674.
41. Emsley P, Lohkamp B, Scott WG and Cowtan K. Features and development of Coot. Acta Crystallogr Sect D. 2010; 66: 486-501.
42. Bricogne G, Blanc E, Brandl M, Flensburg C, Keller P, Paciorek W, Roversi P, Sharff A, Smart OS, Vonrhein C and Womack TO. (2011). BUSTER version 2.11.2. Cambridge, United Kingdom: Global Phasing Ltd.
43. Smart OS, Womack TO, Sharff A, Flensburg C, Keller P, Paciorek W, Vonrhein C and Bricogne G. (2011). Grade, version 1.1.1. Cambridge, United Kingdom: Global Phasing Ltd., http://www.globalphasing.com.
44. Bruno IJ, Cole JC, Kessler M, Luo J, Motherwell WD, Purkis LH, Smith BR, Taylor R, Cooper RI, Harris SE and Orpen AG. Retrieval of crystallographically-derived molecular geometry information. J Chem Inf Comput Sci. 2004; 44(6): 2133-2144.
45. Chen VB, Arendall WB, 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS and Richardson DC. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr Sect D. 2010; 66(Pt 1): 12-21.
46. Pymar LS, Platt FM, Askham JM, Morrison EE and Knowles MA. Bladder tumour-derived somatic TSC1 missense mutations cause loss of function via distinct mechanisms. Hum Mol Genet. 2008; 17(13): 2006-2017.
47. Raynaud FI, Eccles SA, Patel S, Alix S, Box G, Chuckowree I, Folkes A, Gowan S, De Haven Brandon A, Di Stefano F, Hayes A, Henley AT, Lensun L, Pergl-Wilson G, Robson A, Saghir N, et al. Biological properties of potent inhibitors of class I phosphatidylinositide 3-kinases: from PI-103 through PI-540, PI-620 to the oral agent GDC-0941. Mol Cancer Ther. 2009; 8(7): 1725-1738.
48. Potterton E, McNicholas S, Krissinel E, Cowtan K and Noble M. The CCP4 molecular-graphics project. Acta Crystallogr Sect D. 2002; 58(Pt 11): 1955-1957.
49. Hopkins AL, Groom CR and Alex A. Ligand efficiency: a useful metric for lead selection. Drug Discov Today. 2004; 9(10): 430-431.