
INTRODUCTION
Prostate cancer remain the most common malignancy and the second most frequent cause of cancer-related mortality in men in developed countries [1, 2]. Metastatic prostate cancer represents a yet unsolved clinical problem due to the high frequency of relapse and treatment resistance. Understanding the pathways that lead to prostate cancer progression in primary prostate tumors is an important task to prevent this deadly disease. Several studies have provided evidence of the presence of tumor-initiating stem-like cancer cells with high self-renewing properties in human cancers, including prostate cancer [3–5]. Cancer stem cells (CSCs) with acquired stem-like properties can originate from transformation of normal tissue/adult stem cells or from more differentiated progenitor cells [6, 7]. CSCs within the primary tumors are likely a major source of tumor heterogeneity, disease progression and treatment failure. The cancer stem cell model postulates a hierarchical organization of cells such that only a small subset is responsible for tumorigenesis in primary tumors. On the other hand, several factors contribute to the solid tumor heterogeneity including genetic mutations and epigenetic changes and the presence or absence of a cellular hierarchy. Thus, understanding the mechanisms controlling the expansion and maintenance of prostate CSCs could be an important step toward development of more effective CSC-directed strategies for treatment of prostate cancer.
ETS transcription factors are main elements in differentiation and developmental programs in many tissues. Expression of ETS factors is tightly regulated according to tissue-specific and time-dependent programs [8, 9]. Deregulated expression of ETS factors has oncogenic consequences on tissue developmental programs and is one of the most frequent findings in human tumors. About 50% of prostate cancers exhibit gene rearrangements and ectopic expression of ETS genes, like ERG and ETV1 [10–13]. ESE3/EHF is an ETS factor expressed in normal epithelial cells, including prostate epithelial cells [9]. Notably, ESE3/EHF ranks among most highly expressed transcription factors [14]. Previously we reported that ESE3/EHF is frequently downregulated in prostate tumors and that its loss is associated with robust inflammatory gene signatures [15, 16]. Furthermore, we showed that ESE3/EHF controls the differentiation program of prostate epithelial cells and its loss alters cell differentiation and conferred a CSC-like phenotype along with tumor-initiating and metastatic capability [17]. We found that ESE3/EHF controls a large network of targets transcriptionally, inducing genes related to epithelial cell differentiation and repressing genes connected with self-renewal and CSC phenotype [17]. Relevantly, we have recently shown that ESE3/EHF directly controls the level and activity of distinct components of the Lin28/let-7 axis, a key pathway involved in stem cell biology and expansion of cancer stem cell compartment [18].
IL-6 is a cytokine involved in many physiologic and pathophysiologic processes. IL-6 signaling leads to activation of JAK/STAT pathway and recent reports demonstrated that STAT3 activation occurrs frequently in metastatic prostate cancer [19]. While the oncogenic role of IL-6 has been widely investigated, little is known about factors regulating IL-6, particularly at the transcriptional level. Understanding the factors regulating IL-6 expression might be relevant for novel approach targeting its activation. In these studies we identify IL-6 as a novel direct target of ESE3/EHF. These data establish that repression of IL-6 is an important mechanism by which ESE3/EHF restrains stemness and tumor progression. This opens new perspective to target aggressive tumors with IL-6 elevation and loss of ESE3/EHF.
RESULTS
IL-6 is inversely correlated to ESE3/EHF in prostate cells and tumors
In an effort to understand the role of ESE3/EHF in restraining stem-like phenotypes, we observed that in a panel of human prostate cancer cells IL-6 level was inversely correlated to ESE3/EHF and increased gradually from less aggressive, androgen-dependent and ESE3/EHF positive LNCaP to the more aggressive androgen-independent and ESE3/EHF negative DU145 cells (Figure 1A upper and lower panels). Moreover, IL-6 was significantly elevated in prostate epithelial cells with stable ESE3/EHF knockdown at the mRNA and protein level suggesting that ESE3/EHF could maintain IL-6 repressed in normal epithelial cells (Figure 1B and 1C). In keeping with this hypothesis, IL-6 level was significantly elevated in ESE3low tumors (p < 0.01) compared to normal prostate [16]. To determine whether the link between ESE3/EHF and IL-6 observed in our cell line models was seen also in clinical samples, we analyzed gene expression data from two large (n = 545 and n = 131, respectively) human prostate cancer patients datasets [20, 21]. We found a significant inverse correlation between ESE3/EHF and IL-6 expression in human tumors (Figure 1D–1E). Notably, the inverse correlation between ESE3/EHF and IL-6 was also observed in metastatic prostate tumors (Figure 1F). Collectively, these data suggested that IL-6 could be a transcriptional target of ESE3/EHF.
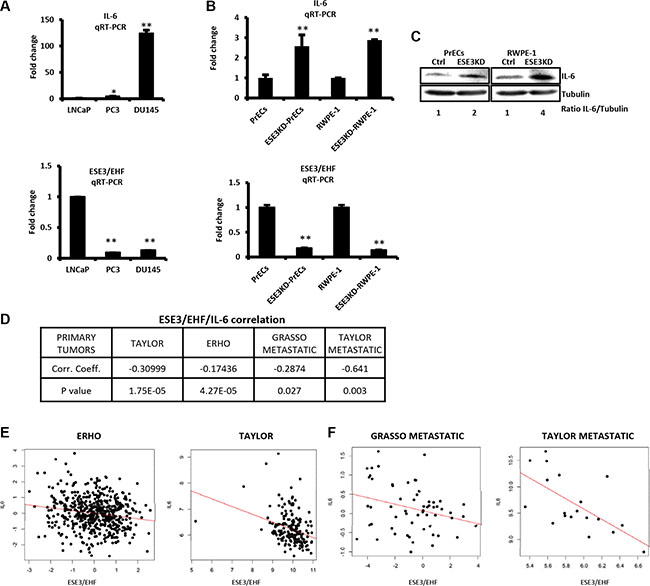
Figure 1: ESE3/EHF and IL-6 expression are inversely correlated. (A) IL-6 (top) and ESE3/EHF (bottom) mRNA levels evaluated by qRT-PCR in indicated PCa cell lines. β-actin was used as reference for loading control. Data are presented as fold change relative to LNCaP cells. (B) IL-6 (top) and ESE3/EHF (bottom) mRNA levels evaluated by qRT-PCR in ESE3KD cell line models. β-actin was used as reference for loading control. Data are presented as fold change relative to PrECs and RWPE-1 cells. (C) Immunoblots of IL-6 in indicated cell lines. IL-6/Tubulin ratio determined by band intensity is reported. (D) ESE3/IL-6 correlation analysis. Table shows correlation coefficient and p value in indicated datasets. (E–F). Pearson distribution plots showing significant inverse correlation between IL-6 and ESE3/EHF in human primary (E) and metastatic prostate tumors (F). P values were determined using t-test. *P < 0.05; **P < 0.01. Data are representative of three independent experiments with at least three replicates per experiment.
ESE3/EHF transcriptionally represses IL-6
To better define the relationship between ESE3/EHF and IL-6, we scanned the IL-6 gene promoter for ETS binding sites (EBS). Computational analysis showed multiple highly scored EBS corresponding to the consensus EHF motif in the promoter region (Figure 2A). To test whether ESE3/EHF bound to the promoter and controlled IL-6 transcription, we selected a high-confidence EBS which was also nearest (-550/-557 bp) to the transcription starting site (TSS) and performed chromatin immunoprecipitation (ChIP). We found that ESE3/EHF bound to the IL-6 promoter in LNCaP cells that express high level of ESE3/EHF (Figure 2B left panel). Consistent with a repressive function on the IL-6 promoter, we found enrichment of repressive (H3K9me and H3K27me) histone marks in LNCaP cells (Figure 2B right panel). The transcriptional effect of ESE3/EHF on IL-6 was further assessed by measuring IL-6 promoter activity in DU145 cells, which do not express endogenous ESE3/EHF. Activity of the IL-6 promoter reporter was significantly reduced by transient expression of ESE3/EHF in DU145 cells, consistent with transcriptional repression of the gene by ESE3/EHF (Figure 2C). Collectively these data support that ESE3/EHF directly control IL-6 transcription and maintains the gene under a repressive status.
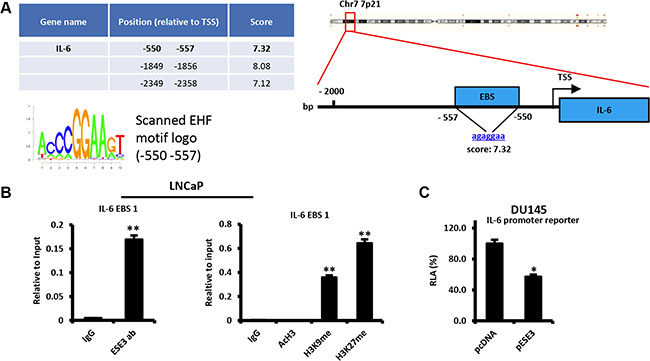
Figure 2: ESE3/EHF transcriptionally represses IL-6. (A) Predicted ETS binding sites (EBS) in the human IL-6 promoter (left). Position relative to the transcription start site (TSS) of the gene, sequence, corresponding scores (right) and EHF scanned motif logo (lower left). (B) Binding of ESE3/EHF to the IL-6 promoter (left panel) and chromatin marks (right panel) determined by chromatin immunoprecipitation (ChIP) in LNCaP cells. (C) Transcriptional activity of IL-6 promoter reporters in control (pcDNA) and stably ESE3/EHF expressing (pESE3) DU145 cells evaluated by dual luciferase assay. P values were determined using t-test. *P < 0.05; **P < 0.01. Data are representative of three independent experiments.
IL-6 is a mediator of prostate epithelial cell transformation and stem cell properties upon loss of ESE3/EHF
To understand the contribution of IL-6 to the transformed phenotype observed in prostate epithelial cells upon loss of ESE3/EHF, we transiently knockdown IL-6 by siRNA in ESE3KD-PrECs and ESE3KD RWPE- 1 cells for 48 h and assessed the consequences on the cell phenotype. IL-6 knockdown was evaluated at mRNA level by qRT-PCR (Figure 3A). We observed a significant decrease in the colony number in soft agar in ESE3KD cells transfected with siRNA targeting IL-6 compared to control (siGL3) transfected cells (Figure 3B). Moreover, tumor sphere formation efficiency (SFE) was also significantly reduced in siIL-6 treated cells compared to control cells suggesting an impact on the cancer stem-like compartment (Figure 3C). To determine whether IL-6 ablation also reversed target gene activation observed in ESE3KD cells, we evaluated the expression of selected gene markers. We observed a significant reduction of the level of STAT3, BMI-1, NANOG and POU5F1 indicating that IL-6 activation contributes to the activation of these target genes upon loss of ESE3/EHF (Figure 3D). Collectively, these data indicate that knockdown of IL-6 in ESE3KD cells is sufficient to reverse the transforming and the stem-like phenotype acquired upon loss of ESE3/EHF. Thus, activation of the IL-6 pathway is an important mediator of the effects observed in ESE3KD cells and targeting IL-6 could be a useful strategy in the context of low ESE3/EHF expressing prostate tumors.
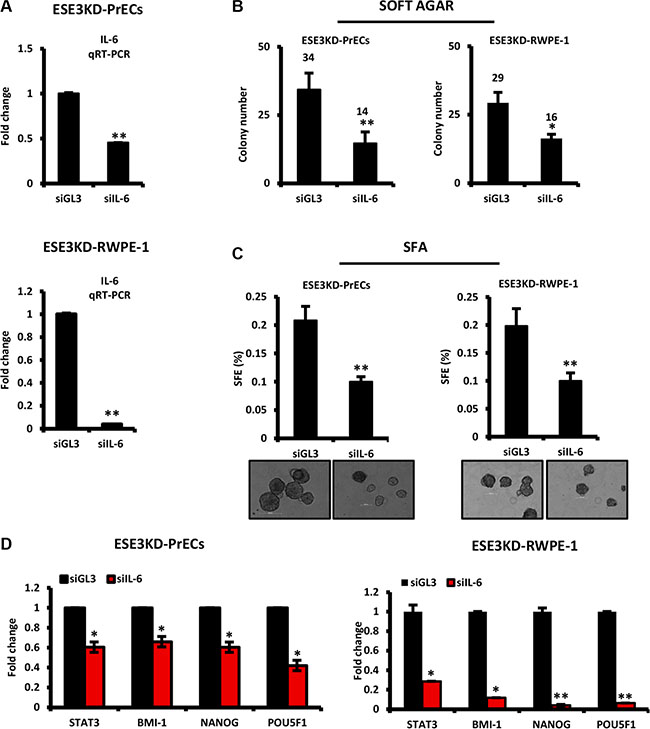
Figure 3: IL-6 is a mediator of prostate epithelial cell transformation and stem cell properties upon loss of ESE3/EHF. (A) Knockdown efficiency of IL-6 evaluated by qRT-PCR following transfection with siRNAs targeting IL-6 in ESE3KD-PrECs (top) and RWPE-1 cells (bottom). (B–C) Colony formation (B) and sphere forming efficiency (C) in ESE3KD-PrECs and ESE3KD-RWPE-1 following transfection of IL-6 siRNA. (D) STAT3 and canonical CSC marker mRNA level evaluated by qRT-PCR in ESE3KD-PrECs (left) and ESE3KD-RWPE-1 (right) following transfection with siRNAs targeting IL-6. β-actin was used as reference gene. Data are presented as fold change relative to control siGL3. P values were determined using t-test. *P < 0.05; **P < 0.01. Data are representative of three independent experiments.
IL-6 induces transformation and cancer stem-like phenotypes
To further understand the consequences of IL-6 upregulation, we exposed normal PrECs and RWPE-1 and corresponding ESE3KD cells to IL-6 stimulation. We observed that IL-6 treatment significantly increased tumor sphere formation in normal (PrECs) and ESE3KD prostate epithelial cells compared to vehicle treated cells (Figure 4A–4B). Consistently, canonical CSC markers like Lin28A, Lin28B, BMI-1, NANOG and POU5F1 were also significantly induced in both normal and ESE3KD cells compared to vehicle treated cells (Figure 4C–4D). IL-6 and STAT3 mRNA level were also induced after IL-6 treatment (Figure 4C–4D). Overall, the response to IL-6 was stronger in ESE3KD compared to the normal prostate epithelial cells. Flow cytometry analysis revealed that IL-6 stimulation induced a robust elevation of pSTATyr705 in ESE3KD-PrECs and ESE3KD-RWPE-1 in comparison to cells treated with vehicle (Figure 4E). Collectively these data support a role of IL-6 in expanding the prostate cancer stem-like phenotype and suggest that ESE3/EHF controls both basal and IL-6 induced STAT3 response. Low level of ESE3/EHF in the cell context and human tumors might render cells hypersensitive to IL-6 stimulation.
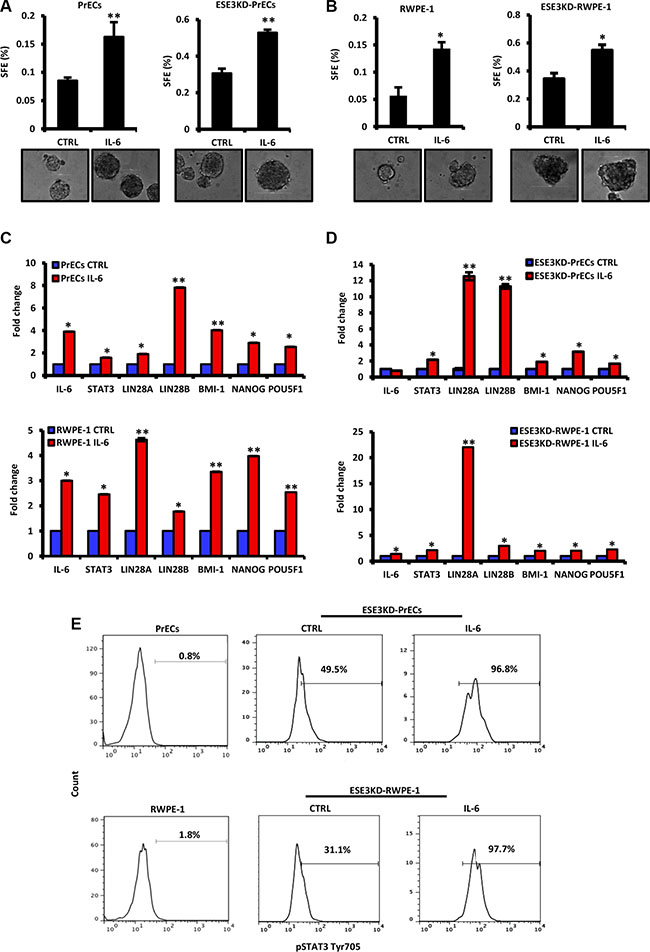
Figure 4: IL-6 induces transformation and cancer stem-like phenotypes in normal and more significantly in ESE3KD prostate epithelial cells. (A–B) Sphere forming efficiency (SFE) in PrECs and ESE3KD-PrECs (A) and in RWPE-1 and ESE3KD-RWPE-1 (B) cells after IL-6 treatment in vitro. (C–D) mRNA levels of STAT3, IL-6 and indicated genes evaluated by qRT-PCR in PrECs and ESE3KD-PrECs (left) (C) and in RWPE-1 and ESE3KD-RWPE-1 cells (right) (D) following 4 hours of treatments in vitro with IL-6 (50 ng/ml) or DMSO as control (CTRL). Data are presented as fold change relative to control (CTRL) cells. (E) Flow cytometry analysis of STAT3-Tyr705 staining following IL-6 treatment of control PrECs and ESE3KD-PrECs (upper) and RWPE-1 and ESE3KD-RWPE-1 (lower) cells. Percentages of positive cells are shown. P values were determined using t-test. *P < 0.05; **P < 0.01. Data are representative of three independent experiments.
Targeting IL-6/STAT3 activation by JAK2 inhibitors inhibits stemness and self–renewal properties in ESE3KD-PrECs in vivo
IL-6 could play a role in transforming prostate epithelial cells by activating the JAK/STAT3 pathway. Consistent with this hypothesis, the level of IL-6 was significantly enriched in the CSC compartment in ESE3KD-PrECs in comparison to the adherent counterpart (Figure 5A). Intriguingly, we also found that pSTAT3 Tyr705 was increased in ESE3KD prostatospheres compared to adherent counterpart cells by ICC staining (Figure 5B) and by flow cytometry (Figure 5C) in ESE3KD-PrECs and RWPE-1 cells.
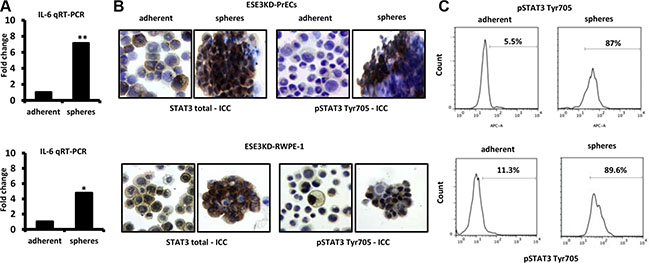
Figure 5: IL-6 elevation and STAT3 induction in cancer stem-like compartment of ESE3KD prostate epithelial cells. (A) IL-6 mRNA level evaluated by qRT-PCR in adherent and prostatosphere cells derived from ESE3KD-PrECs (top) and ESE3KD-RWPE-1 (bottom) cells. Data are presented as fold change relative to the corresponding adherent cells. (B) ICC staining for STAT3 total and pSTAT3-Tyr705 for adherent cells and prostatospheres derived from ESE3KD-PrECs (upper) and ESE3KD-RWPE-1 (lower) cultures. (C) Flow cytometry analysis of STAT3-Tyr705 staining for indicated cells. Percentages of positive cells are shown.
Thus, targeting the JAK/STAT3 pathway could antagonize stemness and self-renewal phenotypes in these cells. To test this, we used NVP-BSK805, a potent and selective JAK2 inhibitor, to inhibit STAT3 phosphorylation at Tyr705. NVP-BSK805 treatment significantly reduced prostatosphere formation in adherent ESE3KD-PrECs and ESE3KD-RWPE-1 cells (Figure 6A and 6B). Next, we tested the ability of NVP-BSK805 to inhibit tumorigenicity and self-renewal of ESE3KD-PrECs prostatosphere-derived tumor xenografts (ESE3KD-PrECs-xeno) in vivo. ESE3KD-PrECs xenografts were grown for the first generation in vivo. Then, the tumors were explanted, dissociated and re-engrafted subcutaneously. Treatment was initiated 21 days after the engraftment and continued for 2 weeks (Figure 6C). Notably, in vivo treatments significantly reduced the growth rate and size of the prostatosphere-derived tumor xenografts (Figure 6D– 6E). Notably, the level of IL-6 was elevated in G1-G3 prostatosphere-derived tumor xenografts being higher than those observed in ESE3KD adherent cells in culture (Figure 6F). Importantly,IL-6 was significantly reduced by the treatment with the JAK2 inhibitor (Figure 6G). Furthermore, when we explanted the tumors and evaluated ex vivo the SFE we found that the prostatosphere forming ability was significantly compromised in cells derived from NVP-BSK805 treated xenografts compared to control xenografts, suggesting that the CSC-like compartment was affected by the treatment (Figure 6H). Consistent with a specific inhibition of the JAK-STAT3 pathway, flow cytometry analysis revealed significant reduction of pSTAT3 in tumor xenografts treated with NVP-BSK805 (Figure 6I). Collectively, these data suggest that the expansion of stem-like cancer cells in tumor xenografts from ESE3KD-PrECs was associated with elevation of IL-6 and consequent activation of JAK/STAT3 and that it could be efficiently targeted by JAK/STAT3 inhibitors.
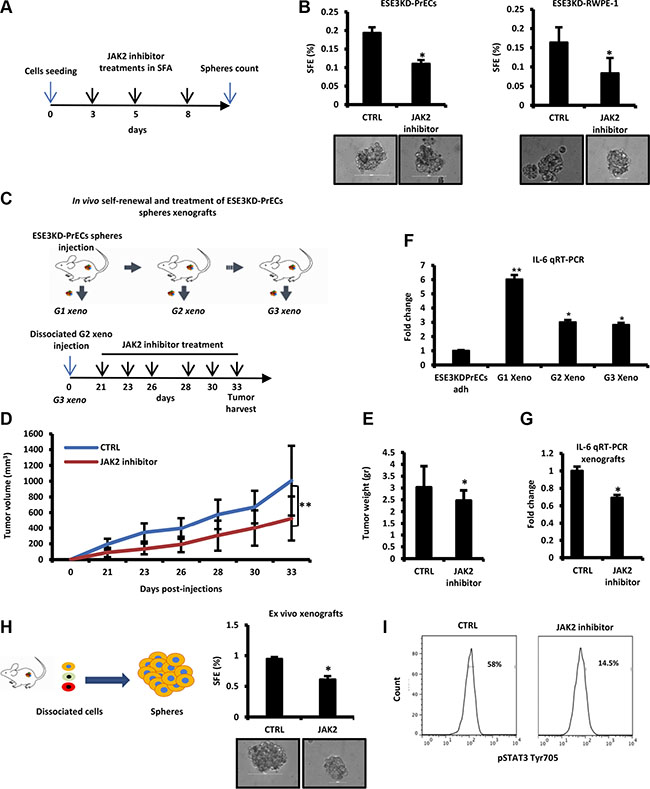
Figure 6: Targeting IL-6/STAT3 activation by JAK2 inhibitors reverses stemness and self–renewal properties in vivo. (A) Experimental plan of JAK2 inhibitor (NVP) treatment of prostatospheres derived from ESE3KD-PrECs cells during SFA. (B) SFE of ESE3KD-PrECs and ESE3KD-RWPE-1 cells upon treatment described in A. (C) Experimental plan of the treatment with JAK2 inhibitor in vivo. Prostatospheres obtained from SFA were dissociated and injected subcutaneously in NSG mice (n = 4/group). Tumors formed by ESE3KD-PrECs prostatospheres cells (G1 xeno) were dissociated and re-implanted (2 × 105 cells/site) in NSG mice (n = 4/group) for two consecutive generations (G2 xeno and G3 xeno). Treatment was initiated 21 days post-engraftment of the G3 xeno and continued as indicated (lower panel). (D–E) Tumor growth determined by caliper (D) and tumor weight (E) of G3 xeno following treatment as described in C. (F) IL-6 evaluation by qRT-PCR in ESE3KD prostatospheres and G1-G3 xeno. (G) IL-6 evaluation by qRT-PCR in control (CTRL) and following treatment with JAK2 inhibitor. (H–I). SFE ex vivo (H) and flow cytometry analysis of STAT3-Tyr705 staining (I) following JAK2 treatment described in C. P values were determined using t-test. *P < 0.05; **P < 0.01. Data are representative of three independent experiments with at least three replicates per experiment. Percentages of positive cells are shown.
IL-6 elevation and loss of ESE3/EHF are associated with STAT3 activation and enrichment of aggressive features in human prostate tumors
To further understand the clinical relevance of our data and verify the association between loss of ESE3/EHF, IL-6 upregulation and STAT3 activation with aggressive features in prostate tumors, we selected a group of tumors characterized by high level of IL-6 and low level of ESE3/EHF (IL-6high/ESE3low) (Figure 7A) in a large primary prostate tumor dataset [21]. By applying differential gene expression analysis we extracted a gene signature comparing the expression profile of the IL-6high/ESE3low (n = 51) with the remaining tumors (n = 495). The IL-6high/ESE3low tumors displayed a robust signature with several genes up and downregulated. We analyzed the top overexpressed genes (≥ 2 fold) in the IL-6high/ESE3low signature by ChIP Enrichment Analysis (CHEA) and found a significant enrichment of STAT3 targets among other transcription factors (Figure 7B). Relevantly, the EBS sites were also significantly over-represented among the genes in the IL-6high/ESE3low signature (Figure 7C) suggesting that the genes extracted could be direct targets of EHF. Functional annotation analysis of the genes activated in IL-6high/ESE3low signature by Metacore supported enrichment of inflammatory genes and genes related to cell adhesion and metastatic spread (Figure 7D).To further understand the functional relevance of this signature we performed gene set enrichment analysis (GSEA) (Figure 7E). This analysis revealed that the IL-6high/ESE3low tumors were significantly enriched of genes attenuated in IL-6 deprivation (Croonquist_IL-6_DN), underlying the accuracy of our approach in extracting relevant IL-6 targets in prostate tumors. Moreover, the IL- 6high/ESE3low tumors were also enriched of an ensemble of genes encoding extracellular matrix and extracellular matrix-associated proteins (Naba_Matrisome), genes conferring migratory and metastatic properties. Intriguingly, there was also enrichment of genes acting as a barrier for senescence, a pathway opposing the induction of a cancer stem cell phenotype. Collectively, these findings supported the activation of the IL-6/STAT3 pathway and aggressive cancer stem-like phenotype in IL-6high/ESE3low tumors and suggested that these tumors could be targeted by therapy opposing this pathway.
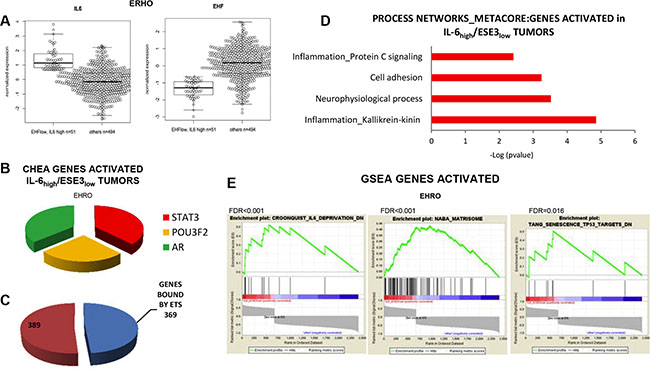
Figure 7: IL-6 elevation and loss of ESE3/EHF are associated with STAT3 activation and inflammatory features in human prostate tumors. (A) Box–plots showing the level of IL-6 (left) and ESE3/EHF (right) in the selected tumor populations. (B) Transcription factor analysis of the top genes activated in IL-6high/ESE3low compared to all the other tumors using CHEA by Enrich. (C) Number of ESE3/EHF putative binding sites among the top activated genes in the IL-6high/ESE3low signature. (D) Functional annotation analysis by Metacore using the upregulated genes. (E) GSEA using indicated list of genes and comparing IL-6high/ESE3low with all the other tumors in the EHRO dataset.
DISCUSSION
In this study we report a link between the tumor suppressor ESE3/EHF and IL-6. Specifically, loss of ESE3/EHF leads to upregulation of IL-6 and activation of the JAK/STAT3 pathway with consequent induction of EMT and expansion of the cancer stem cell compartment (Figure 8).
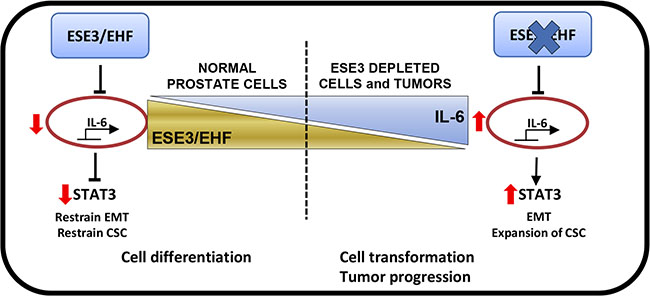
Figure 8: ESE3/EHF controls the activation status of IL-6 and JAK/STAT pathway in normal prostate. Loss of ESE3/EHF leads to upregulation of IL-6 and activation of the JAK/STAT3 pathway with consequent induction of EMT and expansion of the cancer stem cell compartment.
The ETS factor ESE3/EHF is an endogenously expressed ETS factor whose level is higher in normal prostate and decreased in aggressive and metastatic tumors [15]. We recently reported a tumor suppressor role of ESE3/EHF in controlling differentiation and cancer stem-like phenotype in human prostate epithelial cells [17]. ESE3/EHF loss in prostate epithelial cells leads to cell transformation, EMT, acquisition of stem-like properties and broad reprogramming of the cell transcriptome. More recently, we have reported that ESE3/EHF controls the Lin28/let-7 axis acting as a critical barrier to malignant transformation and preventing cancer stem cell expansion [18]. In this study, in an effort to further understand the mechanisms by which ESE3/EHF controls cancer stem-like cells we uncovered that IL-6 is a key target repressed by ESE3/EHF. IL-6 is a cytokine involved in many pathophysiologic processes. IL-6 signals through a cell-surface type I cytokine receptor complex. IL-6 interaction with the receptor triggers its activation initiating a signal transduction cascade leading to activation of the JAK/STAT pathway. Several pieces of evidences have associated IL-6 with an aggressive prostate cancer phenotype and metastatic processes through the regulation of EMT and the homing of cancer cells to the bone [22]. Recent evidence indicates that IL-6 plays a major role in the transition from hormone-dependent to CRPC [22]. Moreover, IL-6 is upregulated in many epithelial cancers such as breast and prostate [23–25]. Collectively, the evidence reported so far points to an important role of IL-6 in transformation and a stem-like phenotype in tumors. Additionally, IL-6 is a key molecule in the dynamic equilibrium between CSCs and non cancer stem cells (NCSCs) via IL-6 secretion, and IL-6 can convert NCSCs to CSCs in breast and prostate cell lines as well as from cells derived from human breast tumors [26]. Importantly, IL-6 has been reported as a key mediator of an inflammatory positive feedback loop occurring from non-transformed to transformed breast epithelial cells and involving other key oncogenic proteins such as NF-kB, Lin28 and STAT3 [27].
Thus, a deep understanding of factors regulating IL-6 expression might be relevant for the development of novel therapeutic approaches targeting its activity [28, 29]. We have previously reported that IL-6 is induced in prostate cancer cells and that the ETS factor ESE1 was an important mediator enhancing the NFKb response [30].
In this study we identify IL-6 as a novel direct target of ESE3/EHF in prostate epithelial cells. Intriguingly, to our knowledge this is the first reported inhibitory transcription factor controlling IL-6.
IL-6 expression has been reported to be inhibited by wild type p53 and the retinoblastoma (RB) tumor suppressor gene [31]. However, it is not known if the effect is direct or mediated by the modulation of other transcription factors. Future studies should address the possibility of a crosstalk between ESE3, Rb and p53 in the control of IL-6 transcription.
We have recently reported that ESE3/EHF restrains stemness by repressing the LIN28/let7 axis [18]. The data presented here establish an additional mechanism by which ESE3/EHF restrains stemness and tumor progression through repression of IL-6. On the other hand, elevation of IL-6 by loss of ESE3/EHF might also impact the activation of Lin28/let7b axis. Accordingly, we found that IL-6 treatment promoted transformation and cancer stem-like features in normal prostate epithelial cells. In contrast, IL-6 inhibition by siRNA reverted these malignant phenotypes in cancer cells. We have previously reported that tumors with low levels of ESE3/EHF have aggressive clinical characteristics and are phenotypically associated with EMT and cancer stem cell features.The data reported here support the notion that IL-6 could contribute to the aggressive features of these tumors and particularly the cancer stem like compartment. Tumors with low levels of ESE3/EHF might be more prone to an aberrant response to IL-6 stimulation. Collectively, these data open new perspectives to specifically target this group of tumors with strategies reversing IL-6 activation. We report here that treatment with JAK-STAT inhibitors prevented self-renewal in vivo in prostate epithelial cells with ESE3/EHF knockdown. These data are clinically relevant and suggest the possibility of selective targeting of the CSC like compartment in ESE3low tumors.
Although the use and benefit of JAK-STAT inhibitors has been amply reported, our data introduce a novel concept that targeting IL-6-JAK-STAT3 signaling could be more effective in tumors in which both ESE3/EHF and IL-6 are altered. Further studies are necessary to demonstrate this concept. IL-6 has been shown to have context-dependent pro- and anti-inflammatory properties and this might influence greatly its oncogenic capabilities in different contexts [29]. Interestingly, recent data from our laboratory indicate that there are additional molecular mechanisms linking to IL-6/STAT3 activation in the context of ESE3/EHF loss in addition to the one described here. Collectively, our data support the notion that activation of the IL-6/STAT3 and inflammatory-like signaling leads to deleterious effects in normal prostate epithelial cells and tumors, leading to expansion of the cancer stem-like cell compartment and self-renewal properties. Understanding these mechanisms will support the possibility of targeting these aggressive tumors within the scope of development of precision medicine approaches for the prostate cancer management and treatment.
MATERIALS AND METHODS
Cell lines, transfection and selection of cell clones
Immortalized human prostate epithelial cells (PrECs) [15] and RPWE-1 with stable knockdown of ESE3/EHF by shRNAs were established as previously described [16]. LNCaP, DU145 and PC3 were obtained from ATCC and maintained in RPMI-1640 (Gibco) supplemented with 10% fetal bovine serum. DU145 cells expressing ESE3/EHF were generated after transfection with ESE3/EHF expression vector and selection with G418 [15]. Where indicated, PrECs, RWPE-1, ESE3KD-PrECs and ESE3KD-RWPE-1 cells were stimulated with IL-6 50 ng/mL for 2 or 4 h or DMSO as control.
RNA extraction, quantitative real-time RT-PCR and siRNAs
RNA was extracted by Direct-zol RNA Mini-prep kit (Zymo Research). Quantitative real-time RT-PCR (qRT-PCR) was carried out using 20 ng of RNA as template for SYBR Green Fast One Step kit (Qiagen). qRT-PCR primers are reported in Supplementary Table S1. For transient gene knockdown cells were transfected with siRNAs directed to IL-6 (siRNA Silencer select, Ambion) or control (siGL3) siRNA [17] using jetPRIME (Polyplus).
Soft agar and in vitro prostatosphere forming and self-renewal assay
Soft-agar assays were performed as previously described [32]. The prostatosphere assay was previously described [17]. The sphere forming efficiency (SFE) was determined as percentage of prostatosphere relative to the number of cells plated at the start of the experiment. Each experiment was carried out in triplicate and repeated at least three times.
Reporter constructs and luciferase assays
To analyze IL-6 activity we used IL-6 responsive luciferase reporter pGL3-basic-IL-6 provided by W. Farrar [33]. Luciferase reporter assays were performed as previously described [17]. Results were normalized to Renilla luciferase and expressed as Relative Luciferase Activity (RLA). Each experiment was performed in triplicate and repeated at least three times.
Animals and tumor xenografts
Mice were purchased from the Harlan Laboratories. Mice were maintained under pathogen-free conditions with food and water provided ad libitum and their general health status was monitored daily. All protocols involving animals were conducted in conformity with the institutional guidelines for animal experimentation and in compliance with national and international policies. Study protocol was approved by the Swiss Veterinary Authority. For in vivo self-renewal experiments prostatosphere-derived ESE3KD-PrECs cells (2 × 105 cells/site) were inoculated with Matrigel in the flank of NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice (n = 4/ group). Tumor size was monitored twice a week with caliper. Primary tumor xenografts derived from ESE3KD-PrECs prostatospheres were dissociated into single cell suspensions and implanted subcutaneously with Matrigel (2 × 105 cells/site) in NSG mice for two more generations (n = 4/group). For systemic treatment with JAK-STAT inhibitor (NVP), mice were injected with a dose of 100 mg/kg of NVP-BSK805 by oral gavage every two days for 4 weeks [34, 35].
Immunoblot and immunocytochemistry
Immunoblots were carried out using anti-IL-6 Rabbit Polyclonal antibody (NeoBioLab),1:500 in I-block and tubulin as control as previously described (Albino et al., 2016). For immunocytochemistry (ICC), harvested cells were washed in PBS by centrifugation and then the concentration adjusted to 5 × 106 cells/ml in PBS. Cells were attached to slides using Cytospin Cytocentifuge (Thermo Scientific) at 800 rpm for 4 minutes. Cells were fixed and permeabilized with Acetone:Methanol, 1:1. After blocking with 5% BSA cells were incubated with anti-STAT3 total (Cell Signaling #9139) 1:400, and anti-STAT3 TYR705 (Cell Signaling #9145) 1:400, antibodies. Cell nuclei were counterstained with hematoxylin solution and finally, the sections were dehydrated and mounted in a suitable organic mounting medium.
Flow cytometry
All steps for flow cytometry were performed in PBS supplemented with 0.5% BSA, and 2 mM EDTA. STAT3-TYR705 was purchased from Bioconcept (Anti-human Phospho-Stat3 /D3A7) alexa 647, cat. #4324) and used for analysis in accordance with the manufacturer’s instructions. Cell sorting was performed with a FACSAria III sorter (BD Biosciences).
Chromatin immunoprecipitation
Computational search for ETS binding sites on selected gene promoters was performed using Motifviz (biowulf.bu.edu/MotifViz). Chromatin immunoprecipitation was performed with anti-ESE3 (Clone 5A5.5, Lab Vision, Fremont, CA USA); anti AcH3 (Upstate, Millipore); anti H3K9 2met (Upstate, Millipore); anti H3K27 3met (Upstate, Millipore) and IgG control antibody. Samples were analyzed as previously described [16] by qRT-PCR. Primer sets are reported in Supplementary Table S1.
Correlation analysis, gene signature, functional annotation and gene set enrichment analysis (GSEA)
The EHRO, Taylor and Grasso [20, 21, 36] human prostate cancer datasets were retrieved from GEO. Only raw intensity data for prostate cancer samples were considered. Data were processed in R using the Bioconductor package “oligo” for Affymetrix arrays: sets were separately RMA (Robust Multi-Array Average) normalized (with background correction) and quantile normalized at the probe level. Log2, normalized expression values for ESE3/EHF and IL-6 genes were extracted and their correlation was tested by Pearson’s product moment correlation coefficient. Genes were centered and scaled. Scatter plots of ESE3/EHF vs IL-6 have been drawn and lines of best fit calculated. Correlation analysis was performed in indicated human prostate cancer datasets. Pearson test was used, which estimates a correlation value “r” and a significance p-val (r > 0 < 1, direct correlation; r < 0 > –1, inverse correlation).
To extract the gene signature, expression of genes EHF and IL-6 was dichotomized based on the following threshold (for EHF 25th percentile and for IL-6 75th percentile). Samples with low EHF and high IL-6 (n = 51, Ehro et al., 2013) were selected and compared to all the others (n = 494, Ehro et al., 2013) by t-test and results were retained if FDR (False Discovery Rate) < 0.05. The resulted lists of genes were then divided into activated and repressed genes respectively. Transcription factor analysis was carried using Enrich. For ETS occupancy we used published ChIP-Seq data for EHF in CALU3 cells [37] and ERG in VCaP / LNCaP [38]. The ChIP-Seq data was annotated using gencode-v19 human genome database. Functional annotation of the genes significantly deregulated in IL-6high/ESE3low was obtained using metacore tools. GSEA software was used to identify groups of functionally related genes. Gene sets with an FDR < 0.05 were considered significant. The gene ranking metric in the weighted enrichment score was SNR (signal-to-noise ratio), and P values were calculated using 1000 permutations of the genes.
ACKNOWLEDGMENTS AND FUNDING
This work was supported by grants from the Swiss Cancer League (KLS-2648-08-2010, KLS-3243-08-2010 and KLS 3243-08-2013), Swiss National Science Foundation (SNF 310030-146560-2013), Swiss Bridge Award, Ticino Foundation for Cancer Research, Virginia Boeger Foundation and Fidinam Foundation. We thank Enrica Mira Cato and Sandra Pinton for technical assistance.
CONFLICTS OF INTEREST
The authors declare no competing financial interests.
REFERENCES
1. Gronberg H. Prostate cancer epidemiology. Lancet. 2003; 361:859–864.
2. Jemal A, Siegel R, Xu J, Ward E. Cancer statistics. CA Cancer J Clin. 2010; 60:277–300.
3. Visvader JE, Lindeman GJ. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer. 2008; 8:755–768.
4. Ailles LE, Weissman IL. Cancer stem cells in solid tumors. Curr Opin Biotechnol. 2007; 18:460–466.
5. Maitland NJ, Collins AT. Prostate cancer stem cells: a new target for therapy. J Clin Oncol. 2008; 26:2862–2870.
6. Visvader JE. Cells of origin in cancer. Nature. 2011; 469:314–322.
7. Clevers H. The cancer stem cell: premises, promises and challenges. Nat Med. 2011; 17:313–319.
8. Sharrocks AD. The ETS-domain transcription factor family. Nat Rev Mol Cell Biol. 2001; 2:827–837.
9. Seth A, Watson DK. ETS transcription factors and their emerging roles in human cancer. Eur J Cancer. 2005; 41:2462–2478.
10. Tomlins SA, Rhodes DR, Perner S, Dhanasekaran SM, Mehra R, Sun XW, Varambally S, Cao X, Tchinda J, Kuefer R, Lee C, Montie JE, Shah RB, et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. 2005; 310:644–648.
11. Tomlins SA, Laxman B, Dhanasekaran SM, Helgeson BE, Cao X, Morris DS, Menon A, Jing X, Cao Q, Han B, Yu J, Wang L, Montie JE, et al. Distinct classes of chromosomal rearrangements create oncogenic ETS gene fusions in prostate cancer. Nature. 2007; 448:595–599.
12. Tomlins SA, Mehra R, Rhodes DR, Smith LR, Roulston D, Helgeson BE, Cao X, Wei JT, Rubin MA, Shah RB, Chinnaiyan AM. TMPRSS2:ETV4 gene fusions define a third molecular subtype of prostate cancer. Cancer Res. 2006; 66:3396–3400.
13. Kumar-Sinha C, Tomlins SA, Chinnaiyan AM. Recurrent gene fusions in prostate cancer. Nat Rev Cancer. 2008; 8:497–511.
14. Consortium F, the RP, Clst, Forrest AR, Kawaji H, Rehli M, Baillie JK, de Hoon MJ, Haberle V, Lassmann T, Kulakovskiy IV, Lizio M, Itoh M, et al. A promoter-level mammalian expression atlas. Nature. 2014; 507:462–470.
15. Cangemi R, Mensah A, Albertini V, Jain A, Mello-Grand M, Chiorino G, Catapano CV, Carbone GM. Reduced expression and tumor suppressor function of the ETS transcription factor ESE-3 in prostate cancer. Oncogene. 2008; 27:2877–2885.
16. Kunderfranco P, Mello-Grand M, Cangemi R, Pellini S, Mensah A, Albertini V, Malek A, Chiorino G, Catapano CV, Carbone GM. ETS transcription factors control transcription of EZH2 and epigenetic silencing of the tumor suppressor gene Nkx3.1 in prostate cancer. PLoS One. 2010; 5:e10547.
17. Albino D, Longoni N, Curti L, Mello-Grand M, Pinton S, Civenni G, Thalmann G, D'Ambrosio G, Sarti M, Sessa F, Chiorino G, Catapano CV, Carbone GM. ESE3/EHF controls epithelial cell differentiation and its loss leads to prostate tumors with mesenchymal and stem-like features. Cancer Res. 2012; 72:2889–2900.
18. Albino D, Civenni G, Dallavalle C, Roos M, Jahns H, Curti L, Rossi S, Pinton S, D'Ambrosio G, Sessa F, Hall J, Catapano CV, Carbone GM. Activation of the Lin28/let7 axis by loss of ESE3/EHF promotes a tumorigenic and stem-like phenotype in prostate cancer. Cancer Res. 2016.
19. Don-Doncow N, Marginean F, Coleman I, Nelson PS, Ehrnstrom R, Krzyzanowska A, Morrissey C, Hellsten R, Bjartell A. Expression of STAT3 in Prostate Cancer Metastases. Eur Urol. 2016.
20. Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, Arora VK, Kaushik P, Cerami E, Reva B, Antipin Y, Mitsiades N, Landers T, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010; 18:11–22.
21. Erho N, Crisan A, Vergara IA, Mitra AP, Ghadessi M, Buerki C, Bergstralh EJ, Kollmeyer T, Fink S, Haddad Z, Zimmermann B, Sierocinski T, Ballman KV, et al. Discovery and validation of a prostate cancer genomic classifier that predicts early metastasis following radical prostatectomy. PLoS One. 2013; 8:e66855.
22. Nguyen DP, Li J, Tewari AK. Inflammation and prostate cancer: the role of interleukin 6 (IL-6). BJU Int. 2014; 113:986–992.
23. Sasser AK, Sullivan NJ, Studebaker AW, Hendey LF, Axel AE, Hall BM. Interleukin-6 is a potent growth factor for ER-alpha-positive human breast cancer. FASEB J. 2007; 21:3763–3770.
24. Wegiel B, Bjartell A, Culig Z, Persson JL. Interleukin-6 activates PI3K/Akt pathway and regulates cyclin A1 to promote prostate cancer cell survival. Int J Cancer. 2008; 122:1521–1529.
25. Wegiel B, Bjartell A, Tuomela J, Dizeyi N, Tinzl M, Helczynski L, Nilsson E, Otterbein LE, Harkonen P, Persson JL. Multiple cellular mechanisms related to cyclin A1 in prostate cancer invasion and metastasis. J Natl Cancer Inst. 2008; 100:1022–1036.
26. Iliopoulos D, Hirsch HA, Wang G, Struhl K. Inducible formation of breast cancer stem cells and their dynamic equilibrium with non-stem cancer cells via IL6 secretion. Proc Natl Acad Sci USA. 2011; 108:1397–1402.
27. Iliopoulos D, Hirsch HA, Struhl K. An epigenetic switch involving NF-kappaB, Lin28, Let-7 MicroRNA, and IL6 links inflammation to cell transformation. Cell. 2009; 139:693–706.
28. Anestakis D, Petanidis S, Kalyvas S, Nday CM, Tsave O, Kioseoglou E, Salifoglou A. Mechanisms and applications of interleukins in cancer immunotherapy. Int J Mol Sci. 2015; 16:1691–1710.
29. Hunter CA, Jones SA. IL-6 as a keystone cytokine in health and disease. Nat Immunol. 2015; 16:448–457.
30. Longoni N, Sarti M, Albino D, Civenni G, Malek A, Ortelli E, Pinton S, Mello-Grand M, Ostano P, D'Ambrosio G, Sessa F, Garcia-Escudero R, Thalmann GN, et al. ETS transcription factor ESE1/ELF3 orchestrates a positive feedback loop that constitutively activates NF-kappaB and drives prostate cancer progression. Cancer Res. 2013; 73:4533–4547.
31. Culig Z. Proinflammatory cytokine interleukin-6 in prostate carcinogenesis. Am J Clin Exp Urol. 2014; 2:231–238.
32. Napoli S, Pastori C, Magistri M, Carbone GM, Catapano CV. Promoter-specific transcriptional interference and c-myc gene silencing by siRNAs in human cells. Embo J. 2009; 28:1708–1719.
33. Xiao W, Hodge DR, Wang L, Yang X, Zhang X, Farrar WL. Co-operative functions between nuclear factors NFkappaB and CCAT/enhancer-binding protein-beta (C/EBP-beta) regulate the IL-6 promoter in autocrine human prostate cancer cells. Prostate. 2004; 61:354–370.
34. Baffert F, Regnier CH, De Pover A, Pissot-Soldermann C, Tavares GA, Blasco F, Brueggen J, Chene P, Drueckes P, Erdmann D, Furet P, Gerspacher M, Lang M, et al. Potent and selective inhibition of polycythemia by the quinoxaline JAK2 inhibitor NVP-BSK805. Mol Cancer Ther. 2010; 9:1945–1955.
35. Marotta LL, Almendro V, Marusyk A, Shipitsin M, Schemme J, Walker SR, Bloushtain-Qimron N, Kim JJ, Choudhury SA, Maruyama R, Wu Z, Gonen M, Mulvey LA, et al. The JAK2/STAT3 signaling pathway is required for growth of CD44(+)CD24(−) stem cell-like breast cancer cells in human tumors. J Clin Invest. 2011; 121:2723–2735.
36. Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, Quist MJ, Jing X, Lonigro RJ, Brenner JC, Asangani IA, Ateeq B, Chun SY, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012; 487:239–243.
37. Fossum SL, Mutolo MJ, Yang R, Dang H, O'Neal WK, Knowles MR, Leir SH, Harris A. Ets homologous factor regulates pathways controlling response to injury in airway epithelial cells. Nucleic Acids Res. 2014; 42:13588–13598.
38. Yu J, Mani RS, Cao Q, Brenner CJ, Cao X, Wang X, Wu L, Li J, Hu M, Gong Y, Cheng H, Laxman B, Vellaichamy A, et al. An integrated network of androgen receptor, polycomb, and TMPRSS2-ERG gene fusions in prostate cancer progression. Cancer Cell. 2010; 17:443–454.