
INTRODUCTION
Pancreatic ductal adenocarcinoma (PDAC) is a devastating diagnosis for patients and their families [1]. In Japan, the incidence and mortality have shown an increasing trend over the past decades [2] and incidence and mortality rates are approximately twice of those in the United States [3]. To date, no efficient and affordable screening tests for PDAC are available [4].
A generally accepted definition of familial pancreatic cancer (FPC) is presence of at least a pair of affected first-degree relatives (FDRs: sibling–sibling or parent-child) in the family [5]. It is estimated that 5-10% of patients with PDAC is familial in the United States [6] and individuals with a family history of PDAC have been shown to have a greatly elevated risk of developing PDAC themselves [5]. A number of genes, including BRCA1, BRCA2 [7], PALB2 [8], and ATM [9], have been associated with-FPC in Western countries. The prevalence of deleterious mutations in these genes varies significantly in different populations (e.g., Ashkenazi Jews have high rates of germline BRCA2 mutations) [10].
Although there have been three studies reporting that a significant fraction of patients with PDAC have a family history of PDAC (10 out of 200, 5.0%; 40 out of 577, 6.9%; 28 out of 688, 4.1%) in the Japanese population [11–13], the prevalence of deleterious FPC susceptibility gene mutations in the Asian population is poorly described.
In the present study, we retrospectively reviewed 1,197 PDAC patients diagnosed at one of two hospitals to evaluate incidence of FPC and we performed targeted-sequencing of germline variants for previously reported FPC susceptibility genes in the Western countries in 54 patients with available DNA, who had a family history of PDAC, and 13 patients who had a personal history of breast or ovarian cancer.
RESULTS
Clinicopathologic features in PDAC patients with a family history of PDAC
A total of 955 patients were identified as having been diagnosed with PDAC between 2002 and 2013 in the National Cancer Center Hospital. Of these, 48 were excluded because of insufficient information, especially on their family histories, and eventually, 907 patients were enrolled, 68 (7.5%) of whom fulfilled the criterion of FPC in which at least a pair of FDRs had been diagnosed with PDAC. In addition, 290 patients were enrolled in the Tokyo Women’s Medical University Hospital. Among them, 20 (6.9%) met criteria for FPC. Clinicopathologic features compared between FPC patients and PDAC patients without a family history of PDAC in FDRs are summarized in Table 1. There were no significant differences between groups in terms of gender, age, smoking history, tumor location, UICC stage, family history of any cancer except PDAC in FDRs, personal medical history of other cancer, diabetes mellitus and chronic pancreatitis in both hospitals.
Table 1: Comparison of clinicopathologic features between PDAC patients with and without a family history of PDAC in FDRs
Demographics |
National Cancer Center Hospital |
Tokyo Women’s Medical University Hospital |
||||
|---|---|---|---|---|---|---|
A family history of PDAC in FDRs |
A family history of PDAC in FDRs |
|||||
Yes (n = 68) |
No (n = 839) |
P -value |
Yes (n = 20) |
No (n = 270) |
P -value |
|
Gender |
0.0741 |
0.3327 |
||||
Male |
32 (47.1%) |
491 (58.5%) |
9 (47.4%) |
163 (60.8%) |
||
Female |
36 (52.9%) |
348 (41.5%) |
10 (52.6%) |
105 (39.2%) |
||
Unknown |
0 |
0 |
1 |
2 |
||
Age |
||||||
Median, yrs |
64 |
65 |
69 |
68 |
||
Range, yrs |
35-83 |
33-90 |
47-82 |
26-95 |
||
Smoking history |
0.4472 |
0.3229 |
||||
Yes |
30 (44.8%) |
414 (50.0%) |
10 (55.6%) |
97 (41.6%) |
||
No |
37 (55.2%) |
414 (50.0%) |
8 (44.4%) |
137 (58.5%) |
||
Unknown |
1 |
11 |
2 |
36 |
||
Tumor location |
0.0585 |
0.7480 |
||||
Head |
24 (35.3%) |
416 (49.9%) |
9 (45.0%) |
138 (53.9%) |
||
Body/tail |
42 (61.8%) |
405 (48.6%) |
9 (45.0%) |
118 (46.1%) |
||
Whole |
2 (2.9%) |
13 (1.6%) |
0 |
0 |
||
Unknown |
0 |
5 |
2 |
14 |
||
UICC stage |
0.3712 |
Unevaluable |
||||
0/IA |
2 (2.9%) |
23 (2.8%) |
2 (10.0%) |
7 (2.8%) |
||
IB |
5 (7.4%) |
38 (4.5%) |
1 (5%) |
1 (0.4%) |
||
IIA |
4 (5.9%) |
124 (14.8%) |
0 |
20 (7.4%) |
||
IIB |
9 (13.2%) |
94 (11.2%) |
0 |
5 (7.9%) |
||
III |
15 (22.1%) |
199 (23.8%) |
6 (30.0%) |
90 (35.4%) |
||
IV |
33 (48.5%) |
358 (42.8%) |
9 (45.0%) |
131 (51.6%) |
||
Unknown |
0 |
3 |
0 |
16 |
||
Family history of any cancer in FDRs |
0.0762 |
0.4840 |
||||
Yes |
33 (52.3%) |
473 (64.2%) |
10 (50.0%) |
110 (40.9%) |
||
No |
30 (47.6%) |
264 (35.8%) |
10 (50.0%) |
159 (59.1%) |
||
Unknown |
5 |
102 |
0 |
1 |
||
Past medical history of other cancer |
0.3272 |
0.1052 |
||||
Yes |
7 (21.9%) |
145 (30.7%) |
6 (30.0%) |
40 (14.9%) |
||
No |
25 (78.1%) |
327 (69.3%) |
14 (70.0%) |
229 (85.1%) |
||
Unknown |
36 |
367 |
0 |
1 |
||
Diabetes mellitus |
0.4030 |
0.6238 |
||||
Yes |
20 (51.3%) |
214 (43.5%) |
5 (25.0%) |
86 (31.9%) |
||
No |
19 (48.7%) |
278 (56.5%) |
15 (75.0%) |
184 (68.1%) |
||
Unknown |
29 |
347 |
0 |
0 |
||
Chronic pancreatitis |
1.0000 |
1.0000 |
||||
Yes |
0 |
6 (2.9%) |
0 |
3 (1.1%) |
||
No |
11 (100%) |
199 (97.1%) |
20 (100%) |
267 (98.9%) |
||
Unknown |
57 |
634 |
0 |
0 |
||
FDRs, first-degree relatives; PDAC, pancreatic ductal adenocarcioma; UICC, the Union for International Cancer Control.
Analysis of germline variants
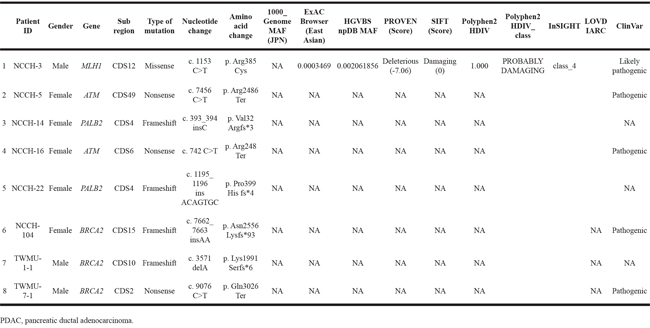
Among the 88 FPC patients we identified, germline DNA samples were available from 54 patients. We performed targeted deep sequencing using a massively parallel sequencer for 21 genes known to be associated with hereditary predispositions for pancreatic, breast, and ovarian cancers, namely, ATM, BARD1, BRCA1, BRCA2, BRIP1, CDH1, CHEK2, MLH1, MRE11, MSH2, MSH6, MUTYH, NBN, PALB2, PMS1, PMS2, PTEN, RAD50, RAD51C, STK11, and TP53. The unique sequence depth was 714.1× on average (range, 502.8× – 1126.1×). The FPC patients had 18 variations on average (range 9-28) in the 21 targeted genes. Eight (14.5%) of the 54 FPC patients carried deleterious variants, three in BRCA2, two in PALB2, two in ATM, and the remaining one in MLH1, as summarized in Table 2. These deleterious variants were verified successfully with Sanger sequencing. One of the mutations found in BRCA2 gene, c.9076C>T/p.Gln3026Ter, has previously been identified in Japanese families with a history of breast or ovarian cancer [14, 15]. Details for clinicopathologic features in FPC patients with the deleterious mutations are shown in Table 3. All of tumors having developed in patients with these deleterious mutations were histopathologically conventional ductal adenocarcinomas. The youngest age of onset for PDAC in the 8 patients was 45 years old. The patient (ID, NCCH-16) who carried a nonsense mutation in ATM had a personal history of breast cancer and ureter cancer. We also classified six mutations into variants of unknown significance (VUS) (Supplementary Table S1).
Table 2: Deleterious mutations in 21 genes associated with hereditary predisposition for pancreatic, breast and ovarian cancers in PDAC patients with a family history of PDAC
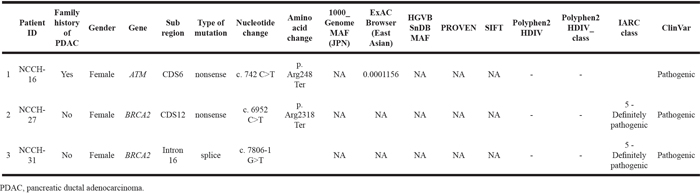
Table 3: Clinicopathologic data for each familial pancreatic cancer patient with a deleterious germline mutation
Patient ID |
Age |
Gender |
Gene with pathogenic mutation |
UICC-stage |
Smoking history (Brinkman index) |
Tumor location |
Family history of other cancer in FDRs |
Past medical history of other cancer |
Diabetes mellitus |
Chronic pancreatitis |
|
|---|---|---|---|---|---|---|---|---|---|---|---|
1 |
NCCH-3 |
62 |
Male |
MLH1 |
IV |
+ (630) |
Body/tail |
- |
- |
- |
- |
2 |
NCCH-5 |
55 |
Female |
ATM |
IV |
- |
Body/tail |
Father (Gastric cancer, Colon cancer, Prostate cancer) |
- |
- |
- |
3 |
NCCH-14 |
45 |
Female |
PALB2 |
IB |
- |
Body/tail |
- |
- |
- |
- |
4 |
NCCH-16 |
67 |
Female |
ATM |
IIB |
+ (510) |
Head |
- |
Breast cancer, Ureter cancer |
- |
- |
5 |
NCCH-22 |
58 |
Female |
PALB2 |
IIB |
- |
Body/tail |
Father (Brain tumor) |
- |
- |
- |
6 |
NCCH-104 |
52 |
Female |
BRCA2 |
IB |
- |
Body/tail |
- |
- |
- |
- |
7 |
TWMU-1-1 |
69 |
Male |
BRCA2 |
III |
- |
Body/tail |
- |
- |
- |
- |
8 |
TWMU-7-1 |
64 |
Male |
BRCA2 |
IB |
- |
Head |
- |
- |
+ |
- |
PDAC patients with a personal history of breast or ovarian cancer
PDAC patients with a history of breast and/or ovarian cancer may have a genetic predisposition common to FPC. Among the 907 PDAC patients in the National Cancer Center Hospital, 36 patients (4.0%) had a personal history of breast and/or ovarian cancer. Among 290 PDAC patients in the Tokyo Women’s Medical University Hospital, 10 (3.4%) had a personal history of breast cancer. Among the 36 who had a personal history of breast and/or ovarian cancer in the National Cancer Center Hospital, germline DNA was available from 13 individuals. Among these 13 patients, 4 fulfilled the FPC criteria and their germline DNAs had been already examined. Then, we additionally examined mutations in the 21 genes in the remaining 9 patients without a family history of PDAC, therefore eventually, the germline mutation was investigated in 63 patients in total in this study. Three (23%) of the 13 PDAC patients with a personal history of breast or ovarian cancer had deleterious mutations, two BRCA2 mutations and one ATM mutation, the latter of which was described in the previous section (NCCH-16) (Table 4). One of mutations found in BRCA2 gene, c.6952C>T/p.Arg2318Ter, has previously been identified in Japanese having a strong family history of breast cancer [15, 16]. Although four of these 13 patients had a family history of PDAC, only one (NCCH-16) of three patients with deleterious mutations had such a family history.
Table 4: Pathogenic mutations in 21 genes associated with hereditary predisposition for pancreatic, breast and ovarian cancers in PDAC patients with a personal history of breast and/or ovarian cancer
DISCUSSION
A better understanding of clinicopathologic and genomic features of FPC in each population should afford new opportunities for therapeutic intervention and clinical management of PDAC patients with a family history of PDAC. However, such features remain largely unexplored in Asian countries. Our present study provides compelling evidence that a subset of PDAC in the Japanese population is familial and attributable to germline mutations in known familial pancreatic cancer genes.
The prevalence of FPC found in the present study of a relatively large Japanese cohort from two hospitals is similar to that reported in the Western world [6]. Cho and colleague reported that 8 (7.2%) of 110 patients with PDAC met criteria for FPC in Korea [17]. Although the number of patients in the Korean study is small, the frequency of FPC is similar to our study. The age of onset of PDAC in some studies is reported to be slightly younger in FPC cases than in sporadic cases in the Western countries [18], though there was no significant difference in age in our Japanese population.
The heterogeneity of mutations uncovered in our Japanese cohort indicates the value of using a multiple-gene panel when evaluating patients with a family history of PDAC. Deleterious germline mutations in FPC cases were found in 14.5% of the Japanese cohort of FPC patients. Interestingly, these mutations were found to be heterogeneous but to primarily affect genes involved in DNA repair pathway, namely, BRCA2, PALB2, ATM, and MLH1 [19]. BRCA2 germline mutations are the most common in most Western cohorts of FPC [20, 21]. In the present study, 3.7% (2/54) of FPC patients had PALB2 deleterious mutations. Jones et al. [8] identified PALB2 mutations in 3 out of 96 American FPC families, which suggests that 3-4% of familial pancreatic cancer kindreds in Japan as well as USA likely to be attributable to germline PALB2 mutations. Studies in other populations show varying mutation frequencies, ranging from absent in Dutch (0 out of 31) to 3.7% (3 out of 81) in Germans [22, 23].
Identification of germline mutations in BRCA2, PALB2 and ATM may have valuable for implications of treatment. Cancers in which BRCA2, PALB2, or ATM have been biallelically inactivated are usually susceptible to poly(ADP-ribose) polymerase (PARP) inhibitors or platinum-based agents that facilitates double strand DNA breaks. This susceptibility could lead to an improved response and enhanced patient survival [24–26]. Consistent with this notion, Fogelman et al. [27] have recently shown that patients with a strong family history are more sensitive to platinum-based chemotherapy. Interestingly, PARP inhibition is currently being evaluated as a maintenance strategy after response to first-line platinum-based chemotherapy in patients with BRCA-related PDAC (https://clinicaltrials.gov/ct2/show/NCT02184195). In addition, our Japanese PDAC patients with a personal history of breast/ovarian cancer, although number is small, frequently carried deleterious mutations in these genes. This latter finding suggests that genetic testing is also valuable for treatment in such populations.
Following limitations could be considered in our study. First, our approach to classifying variants was conservative; several rare non-synonymous variants that were potentially deleterious were classified as VUS as noted in Supplementary Table S1, which could lead to underestimation of prevalence of deleterious mutation carriers. Second, the candidate target gene analysis approach employed in this study was intrinsically highly selective and, therefore, could lead to limited genetic results. Recently, Roberts et al. [28] conducted germline whole genome sequencing of 638 FPC patients and demonstrated that inherited PDAC-susceptible genes are highly heterogeneous, which has uncovered many genes, e.g., BUB1B, FANCC, and FANCG, in addition to the known classical causative FPC genes analyzed in this study. A Japanese large whole-exome sequencing project is ongoing to identify any characteristic or novel genes causative for FPC in Japan. Our approach would also miss some germline deletions. Finally, while we successfully identified germline alterations, we did not have tumor samples available to look for biallelic inactivation of these genes, and thus we were not able to definitively establish the functional loss of these genes.
The Japanese Familial Pancreatic Cancer Registry (JFPCR) was established in 2015 [29], 20 years after the American registry (the National Familial Pancreatic Tumor Registry, NFPTR) [30]. The significance of FPC remains to be undefined in a large extent in Asian counties. Establishing a screening program for early diagnosis is crucial to improving the prognosis of this intractable cancer. However, screening of general populations is not feasible because of the relatively low incidence of PDAC. Therefore, identification and evaluation of a risk of PDAC in FPC kindred may lead to establishment of highly efficient screening and improving of prognosis of PDAC in Asian populations.
In conclusion, 7.3% (88/1197) of PDAC patients in our Japanese cohort fulfilled the criterion of FPC; there was at least a pair of FDRs with PDAC in the kindred. We identified deleterious heterozygous germline mutations in well-established familial cancer-associated genes, BRCA2, PALB2, ATM and MLH1, in 14.5% (8/54) of our FPC patients. Our findings indicate that a subset of Japanese FPC patients may be associated with deleterious mutations of classical FPC genes as evidenced in Western populations.
MATERIALS AND METHODS
Ethics
The experimental protocols were approved by the institutional review board at the National Cancer Center (2013-292) and Tokyo Women’s Medical University (213C). Written informed consent was obtained from all patients. The methods were carried out in accordance with the approved guidelines.
Patients and tissue samples
We reviewed the National Cancer Center Hospital database and the Tokyo Women’s Medical University Hospital database of patients with pathologically proven PDAC including its variants, namely, adenosquamous carcinoma, mucinous noncystic carcinoma, and undifferentiated carcinoma, between 2002 and 2013 and 2006 and 2011, respectively. Three patients treated in Kyorin University Hospital were included in cases of Tokyo Women’s Medical University Hospital as consulting cases. On examination of medical interview sheets and medical records, cases with insufficient data were excluded.
We identified patients who fulfilled the criterion of FPC in which at least a pair of FDRs with PDAC and compared the following clinicopathologic features between them and PDAC patients without a family history of PDAC: gender, age, tumor location, clinical stage at diagnosis, risk factors (smoking history, diabetes mellitus and chronic pancreatitis), personal medical history of any cancer, and family histories of any cancer in FDRs (Table 1). Clinical stage was categorized following the UICC staging system (seventh edition).
Massively parallel sequencing of target genes
Germline DNA was extracted from peripheral blood leukocytes. A custom capture kit was designed using NimbleDesign (NimbleGen, Madison, WI) targeting exons and splice sites of 21 genes known to be associated with hereditary predispositions for pancreatic, breast and ovarian cancers, namely, ATM, BARD1, BRCA1, BRCA2, BRIP1, CDH1, CHEK2, MLH1, MRE11, MSH2, MSH6, MUTYH, NBN, PALB2, PMS1, PMS2, PTEN, RAD50, RAD51C, STK11, and TP53. Libraries were created using the SeqCap EZ Library (NimbleGen) and KAPA Library Preparation Kits (Kapa Biosysytems, Wilmington, MA) according to the manufacturers’ protocols. Massively parallel sequencing was performed on Illumina HiSeq2500 platforms (Illumina, San Diego, CA). Bases were called with the default setting using Illumina BCLFAST2 (Illumina). Paired-end reads were aligned to the human reference genome (GRCh37) using the Burrows-Wheeler Aligner [31]. A Genome Analysis Toolkit (GATK) was used to detect single-nucleotide substitutions and small insertions and deletions, using best practices from the GATK Website (available at: https://www.broadinstitute.org/gatk/) [32]. To maximize sensitivity to detect variants, no variant quality filters were applied. In-house script was applied to annotate variants.
Variant characterization
Variants in 21 genes were considered for analysis if they were (1) called nonreference by GATK; (2) predicted to affect the protein sequence or the splice site (i.e., ±5 base pairs); and (3) had an allele frequency of less than 1% in the 1000 Genome project [33, 34], dbSNP [35], ExAC Browser (available at Exome Aggregation Consortium, http://exac.broadinstitute.org), or the Japanese genetic variation (available at the Human Genetic Variation Browser, http://www.genome.med.kyoto-u.ac.jp/SnpDB/).
Variants were classified according to ClinVar [36]. In addition, for MLH1, MSH2, MSH6, MUTYH, PMS1, and PMS2, classification was according to the InSiGHT consortium (available at: http://insight-group.org/variants/database/) [37]. For BRCA1 and BRCA2, variants were classified using the database generated by Vallée et al. [38], accessed through the LOVD Website (available at: http://brca.iarc.fr/LOVD). These groups have classified large numbers of variants in BRCA1, BRCA2, MLH1, MSH2, MSH6, and PMS2, according to the International Agency for Research on Cancer system based on available information from the literature. Classes 1 and 2 are considered benign, class 3 is considered VUS, and classes 4 and 5 are considered pathogenic [39].
Rare non-synonymous variants that were not in these databases were classified based on the predicted effect on the protein product. Nonsense variants and variants changing the canonical splice-sites (i.e., ±2 base pairs), and frameshift insertions and deletions were considered pathogenic unless they occurred in the last exon. PROVEN (available at: http://provean.jcvi.org/index.php) [40], SIFT (available at: http://sift.jcvi.org) [41] and PolyPhen-2 (available at: http://genetics.bwh.harvard.edu/pph2/) [42] were used for identification of functional missense mutations along with a literature review. Taken together with this information, rare non-synonymous variants were classified as either deleterious, benign, or VUS.
Sanger sequencing
Variants were validated by Sanger sequencing. Polymerase chain reaction amplification was carried out using 20 ng of DNA with intronic primers flanking targeted exons as previously reported [43]. Polymerase chain reaction products were sequenced by use of a M13F primer (5’-GTAAAACGACGGCCAGT-3’) or a M13R primer (5’-CAGGAAACAGCTATGACC-3’) incorporated into the forward and reverse primers of each primer pair, respectively. Sequencing data were analyzed with Sequencher 5.0.1 software (Gene Codes, Ann Arbor, MI).
Statistics
Differences in variables between PDAC patients with and without a family history of PDAC were analyzed using χ2 test. P < 0.05 was considered statistically significant. All statistical analyses were performed using JMP ver.11 (SAS Institute, Cary, NC).
ACKNOWLEDGMENTS
We wish thank all patients and their families who contributed to this study. We also wish to express our appreciation to Ms. Hiroko Hosoi and Ms. Risa Usui (National Cancer Center Hospital) for clinical contributions.
CONFLICTS OF INTEREST
R.H.H. receives royalty payments from Myriad Genetics for the PALB2 invention in a relationship managed by Johns Hopkins University.
GRANT SUPPORT
This work was supported by the following grants: JSPS KAKENHI (Grant Number 24390090 to T.F.); the Project for Development of Innovative Research on Cancer Therapeutics, the Japan Agency for Medical Research and Development (P-DIRECT to S.Y., M.S., C.M. and T.F.); the National Cancer Center Research and Development Fund (25-A-1 and 28-A-1 to S.Y. and C.M.); the Takeda Science Foundation (to S.Y.); and the National Institutes of Health grants (CA62924 to R.H.H.). The National Cancer Center Biobank is supported by the National Cancer Center Research and Development Fund, Japan.
REFERENCES
1. Yachida S, Jones S, Bozic I, Antal T, Leary R, Fu B, Kamiyama M, Hruban RH, Eshleman JR, Nowak MA, Velculescu VE, Kinzler KW, Vogelstein B, Iacobuzio-Donahue CA. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature. 2010; 467: 1114-1117.
2. Katanoda K, Matsuda T, Matsuda A, Shibata A, Nishino Y, Fujita M, Soda M, Ioka A, Sobue T, Nishimoto H. An updated report of the trends in cancer incidence and mortality in Japan. Jpn J Clin Oncol. 2013; 43: 492-507.
3. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015; 65: 5-29.
4. Oshima M, Okano K, Muraki S, Haba R, Maeba T, Suzuki Y, Yachida S. Immunohistochemically detected expression of 3 major genes (CDKN2A/p16, TP53, and SMAD4/DPC4) strongly predicts survival in patients with resectable pancreatic cancer. Ann Surg. 2013; 258: 336-346.
5. Klein AP, Brune KA, Petersen GM, Goggins M, Tersmette AC, Offerhaus GJ, Griffin C, Cameron JL, Yeo CJ, Kern S, Hruban RH. Prospective risk of pancreatic cancer in familial pancreatic cancer kindreds. Cancer Res. 2004; 64: 2634-2638.
6. Klein AP, Hruban RH, Brune KA, Petersen GM, Goggins M. Familial pancreatic cancer. Cancer J. 2001; 7: 266-273.
7. Lal G, Liu G, Schmocker B, Kaurah P, Ozcelik H, Narod SA, Redston M, Gallinger S. Inherited predisposition to pancreatic adenocarcinoma: role of family history and germ-line p16, BRCA1, and BRCA2 mutations. Cancer Res. 2000; 60: 409-416.
8. Jones S, Hruban RH, Kamiyama M, Borges M, Zhang X, Parsons DW, Lin JC, Palmisano E, Brune K, Jaffee EM, Iacobuzio-Donahue CA, Maitra A, Parmigiani G, Kern SE, Velculescu VE, Kinzler KW, et al. Exomic sequencing identifies PALB2 as a pancreatic cancer susceptibility gene. Science. 2009; 324: 217.
9. Roberts NJ, Jiao Y, Yu J, Kopelovich L, Petersen GM, Bondy ML, Gallinger S, Schwartz AG, Syngal S, Cote ML, Axilbund J, Schulick R, Ali SZ, Eshleman JR, Velculescu VE, Goggins M, et al. ATM mutations in patients with hereditary pancreatic cancer. Cancer Discov. 2012; 2: 41-46.
10. Holter S, Borgida A, Dodd A, Grant R, Semotiuk K, Hedley D, Dhani N, Narod S, Akbari M, Moore M, Gallinger S. Germline BRCA Mutations in a Large Clinic-Based Cohort of Patients With Pancreatic Adenocarcinoma. J Clin Oncol. 2015; 33: 3124-3129.
11. Mizuno S, Nakai Y, Isayama H, Kawahata S, Saito T, Takagi K, Watanabe T, Uchino R, Hamada T, Miyabayashi K, Kogure H, Sasaki T, Yamamoto N, Sasahira N, Hirano K, Tsujino T, et al. Smoking, family history of cancer, and diabetes mellitus are associated with the age of onset of pancreatic cancer in Japanese patients. Pancreas. 2014; 43: 1014-1017.
12. Matsubayashi H, Maeda A, Kanemoto H, Uesaka K, Yamazaki K, Hironaka S, Miyagi Y, Ikehara H, Ono H, Klein A, Goggins M. Risk factors of familial pancreatic cancer in Japan: current smoking and recent onset of diabetes. Pancreas. 2011; 40: 974-978.
13. Inoue M, Tajima K, Takezaki T, Hamajima N, Hirose K, Ito H, Tominaga S. Epidemiology of pancreatic cancer in Japan: a nested case-control study from the Hospital-based Epidemiologic Research Program at Aichi Cancer Center (HERPACC). Int J Epidemiol. 2003; 32:257-262.
14. Sekine M, Nagata H, Tsuji S, Hirai Y, Fujimoto S, Hatae M, Kobayashi I, Fujii T, Nagata I, Ushijima K, Obata K, Suzuki M, Yoshinaga M, Umesaki N, Satoh S, Enomoto T, et al. Mutational analysis of BRCA1 and BRCA2 and clinicopathologic analysis of ovarian cancer in 82 ovarian cancer families: two common founder mutations of BRCA1 in Japanese population. Clin Cancer Res. 2001; 7: 3144-3150.
15. Sugano K, Nakamura S, Ando J, Takayama S, Kamata H, Sekiguchi I, Ubukata M, Kodama T, Arai M, Kasumi F, Hirai Y, Ikeda T, Jinno H, Kitajima M, Aoki D, Hirasawa A, et al. Cross-sectional analysis of germline BRCA1 and BRCA2 mutations in Japanese patients suspected to have hereditary breast/ovarian cancer. Cancer Sci. 2008; 99: 1967-1976.
16. Nakamura S, Takahashi M, Tozaki M, Nakayama T, Nomizu T, Miki Y, Murakami Y, Aoki D, Iwase T, Nishimura S, Yamauchi H, Ohsumi S, Baba S, Shimizu T. Prevalence and differentiation of hereditary breast and ovarian cancers in Japan. Breast Cancer. 2015; 22: 462-468.
17. Cho JH, Bang S, Park SW, Chung JB, Song SY. BRCA2 mutations as a universal risk factor for pancreatic cancer has a limited role in Korean ethnic group. Pancreas. 2008; 36: 337-340.
18. McFaul CD, Greenhalf W, Earl J, Howes N, Neoptolemos JP, Kress R, Sina-Frey M, Rieder H, Hahn S, Bartsch DK, European Registry of Hereditary P, Familial Pancreatic C, German National Case Collection for Familial Pancreatic C. Anticipation in familial pancreatic cancer. Gut. 2006; 55: 252-258.
19. Rahman N, Scott RH. Cancer genes associated with phenotypes in monoallelic and biallelic mutation carriers: new lessons from old players. Hum Mol Genet. 2007; 16: R60-66.
20. Hahn SA, Greenhalf B, Ellis I, Sina-Frey M, Rieder H, Korte B, Gerdes B, Kress R, Ziegler A, Raeburn JA, Campra D, Grutzmann R, Rehder H, Rothmund M, Schmiegel W, Neoptolemos JP, et al. BRCA2 germline mutations in familial pancreatic carcinoma. J Natl Cancer Inst. 2003; 95: 214-221.
21. Ghiorzo P, Pensotti V, Fornarini G, Sciallero S, Battistuzzi L, Belli F, Bonelli L, Borgonovo G, Bruno W, Gozza A, Gargiulo S, Mastracci L, Nasti S, Palmieri G, Papadia F, Pastorino L, et al. Contribution of germline mutations in the BRCA and PALB2 genes to pancreatic cancer in Italy. Fam Cancer. 2012; 11: 41-47.
22. Harinck F, Kluijt I, van Mil SE, Waisfisz Q, van Os TA, Aalfs CM, Wagner A, Olderode-Berends M, Sijmons RH, Kuipers EJ, Poley JW, Fockens P, Bruno MJ. Routine testing for PALB2 mutations in familial pancreatic cancer families and breast cancer families with pancreatic cancer is not indicated. Eur J Hum Genet. 2012; 20: 577-579.
23. Slater EP, Langer P, Niemczyk E, Strauch K, Butler J, Habbe N, Neoptolemos JP, Greenhalf W, Bartsch DK. PALB2 mutations in European familial pancreatic cancer families. Clin Genet. 2010; 78: 490-494.
24. Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, Martin NM, Jackson SP, Smith GC, Ashworth A. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005; 434: 917-921.
25. Ashworth A. A synthetic lethal therapeutic approach: poly(ADP) ribose polymerase inhibitors for the treatment of cancers deficient in DNA double-strand break repair. J Clin Oncol. 2008; 26: 3785-3790.
26. Mateo J, Carreira S, Sandhu S, Miranda S, Mossop H, Perez-Lopez R, Nava Rodrigues D, Robinson D, Omlin A, Tunariu N, Boysen G, Porta N, Flohr P, Gillman A, Figueiredo I, Paulding C, et al. DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. N Engl J Med. 2015; 373: 1697-1708.
27. Fogelman D, Sugar EA, Oliver G, Shah N, Klein A, Alewine C, Wang H, Javle M, Shroff R, Wolff RA, Abbruzzese JL, Laheru D, Diaz LA, Jr. Family history as a marker of platinum sensitivity in pancreatic adenocarcinoma. Cancer Chemother Pharmacol. 2015; 76: 489-498.
28. Roberts NJ, Norris AL, Petersen GM, Bondy ML, Brand R, Gallinger S, Kurtz RC, Olson SH, Rustgi AK, Schwartz AG, Stoffel E, Syngal S, Zogopoulos G, Ali SZ, Axilbund J, Chaffee KG, et al. Whole Genome Sequencing Defines the Genetic Heterogeneity of Familial Pancreatic Cancer. Cancer Discov. 2016; 6: 166-175.
29. Wada K, Takaori K, Traverso LW, Hruban RH, Furukawa T, Brentnall TA, Hatori T, Sano K, Takada T, Majima Y, Shimosegawa T. Clinical importance of Familial Pancreatic Cancer Registry in Japan: a report from kick-off meeting at International Symposium on Pancreas Cancer 2012. J Hepatobiliary Pancreat Sci. 2013; 20: 557-566.
30. Hruban RH, Petersen GM, Goggins M, Tersmette AC, Offerhaus GJ, Falatko F, Yeo CJ, Kern SE. Familial pancreatic cancer. Ann Oncol. 1999; 10: 69-73.
31. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009; 25: 1754-1760.
32. McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010; 20: 1297-1303.
33. Genomes Project C, Abecasis GR, Altshuler D, Auton A, Brooks LD, Durbin RM, Gibbs RA, Hurles ME, McVean GA. A map of human genome variation from population-scale sequencing. Nature. 2010; 467: 1061-1073.
34. Genomes Project C, Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE, Kang HM, Marth GT, McVean GA. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012; 491: 56-65.
35. Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, Sirotkin K. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001; 29: 308-311.
36. Landrum MJ, Lee JM, Riley GR, Jang W, Rubinstein WS, Church DM, Maglott DR. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014; 42: D980-985.
37. Thompson BA, Spurdle AB, Plazzer JP, Greenblatt MS, Akagi K, Al-Mulla F, Bapat B, Bernstein I, Capella G, den Dunnen JT, du Sart D, Fabre A, Farrell MP, Farrington SM, Frayling IM, Frebourg T, et al. Application of a 5-tiered scheme for standardized classification of 2,360 unique mismatch repair gene variants in the InSiGHT locus-specific database. Nat Genet. 2014; 46: 107-115.
38. Vallee MP, Francy TC, Judkins MK, Babikyan D, Lesueur F, Gammon A, Goldgar DE, Couch FJ, Tavtigian SV. Classification of missense substitutions in the BRCA genes: a database dedicated to Ex-UVs. Hum Mutat. 2012; 33: 22-28.
39. Grant RC, Selander I, Connor AA, Selvarajah S, Borgida A, Briollais L, Petersen GM, Lerner-Ellis J, Holter S, Gallinger S. Prevalence of germline mutations in cancer predisposition genes in patients with pancreatic cancer. Gastroenterology. 2015; 148: 556-564.
40. Choi Y, Sims GE, Murphy S, Miller JR, Chan AP. Predicting the functional effect of amino acid substitutions and indels. PLoS One. 2012; 7: e46688.
41. Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009; 4: 1073-1081.
42. Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010; 7: 248-249.
43. Sjoblom T, Jones S, Wood LD, Parsons DW, Lin J, Barber TD, Mandelker D, Leary RJ, Ptak J, Silliman N, Szabo S, Buckhaults P, Farrell C, Meeh P, Markowitz SD, Willis J, et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006; 314: 268-274.