
INTRODUCTION
Gastric adenocarcinoma remains the third most common cause of cancer-related mortality worldwide [1]. Helicobacter pylori infection is involved in the carcinogenesis process and has been classified as a class 1 carcinogen by the WHO [2]. The CagA protein produced by certain H. pylori strains is the main pathogenic factor implicated in gastric adenocarcinoma. CagA, once injected into the host cell, disrupts the cell junctions, in particular at the level of the E-cadherin-based adherens junctions [3], and induces an epithelial to mesenchymal transition (EMT) [4] which leads to the emergence of cells with cancer stem cell (CSC) properties [5]. EMT appears to be at the origin of the adenocarcinoma initiation [6, 7]. According to Lauren’s classification, 2 types of gastric carcinoma (GC) have been defined, the intestinal type and the diffuse type [8]. Recent reports on whole-genome sequencing and comprehensive molecular profiling identified new driver mutations in GC defining 4 main subtypes based on their molecular signature. Among the diffuse type GCs, in addition to well-known invalidating mutations of the E-cadherin cdh1 gene, invalidating mutations of the rhoA gene were also found, in nearly 30% and 15% of the cases, respectively [9, 10]. IQ-domain GTPase-activating proteins (IQGAPs) are an evolutionary conserved family of multi-domain proteins that regulate distinct cellular processes including cell adhesion, cell migration, response to extracellular signals and cytokinesis [11, 12]. There are 3 IQGAP proteins and among them IQGAP1, the first to be described, is the only one ubiquitously expressed. IQGAP1 is located on chromosome 15q26, which corresponds to a hotspot for gene amplification, in particular in diffuse type GCs [13]. IQGAP1 is a scaffold protein interacting with numerous partners which modulates the actin cytoskeleton via Rac1 and Cdc42, cell/cell adhesions via E-cadherin, plasticity and proliferation via the Wnt/ß-catenin proteins, β-catenin and APC, and MEK and Erk [11]. Thus, it appears that IQGAP1, as well as E-cadherin, could play a role in gastric carcinogenesis. Different studies linked IQGAP1 expression [13-17] and localization [18] to neoplasia. The role of IQGAP1 has been studied in different models. Li et al. showed that aged mice lacking IQGAP1 develop gastric hyperplasia, suggesting a role for IQGAP1 in maintaining epithelial integrity during ageing [19]. Furthermore, Cai et al., using a murine model, showed that a Helicobacter sp. infection leads to a progressive shift of IQGAP1 from cytoplasmic expression to cell surface expression [20].
The above observations led to the consideration that IQGAP1 may be implicated in GC initiation and progression in the presence of H. pylori. Consequently our aim was to determine whether IQGAP1 inhibition favours EMT and acquisition of CSC properties in an in vitro model (gastric epithelial cell lines) and whether IQGAP1 inhibition accentuates the H. pylori carcinogenesis. Furthermore, a study of IQGAP1 deletion in mice was performed in order to better determine the potential role of IQGAP1 in vivo in the development of neoplastic lesions after H. pylori infection.
RESULTS
Effect of IQGAP1 deletion on the development of lesions in the gastric mucosa of mice infected by H. pylori
In order to determine the role of IQGAP1 in the development of lesions in the gastric mucosa in response to Helicobacter infection, infection experiments were carried out on female 129/Bl/6 mice deleted for one allele of iqgap1 [19] and on their WT littermates. Mice were infected with either H. felis (an animal pathogen which is a strong inducer of gastric inflammation and pre-neoplastic lesions in mice), H. pylori SS1 which harbours a non-functional cag pathogenicity island (PAI), and H. pylori HPARE which harbours a functional cagPAI and CagA. The mice were sacrificed after 6 or 12 months for histopathological analyses as previously described [21].
Invalidation of one or two iqgap1 alleles was described to lead to the same phenotype. In order to confirm this point, groups of iqgap1-/- mice, iqgap1+/- mice and WT littermates, were infected or not (control) with only H. felis to determine the consequences of invalidating one or both iqgap1 alleles on the development of gastric inflammation and pre-neoplastic lesions 6 months post-infection (PI). We decided to work with heterozygous and not with homozygous mice in order to mimic as closely as possible the situation in patients.
No significant lesions were observed in uninfected mice at any time point, neither in WT nor in iqgap1+/- mice or iqgap1-/- mice (data not shown). After 6 and 12 months of infection of iqgap1+/- mice, both H. pylori strains and H. felis induced significant inflammation in the gastric mucosa and sub-mucosa associated with an increase in mucosal height (Figure 1), an atrophy with the replacement of parietal and chief cells by mucous-producing cells defining a mucinous metaplasia (Figure 1). Dysplastic lesions appeared 6 months PI. Their number and their severity increased overtime, reaching high-grade gastrointestinal intraepithelial neoplasia (GIN) at 12 months PI (Figure 1F-1G). At 6 months PI, no difference was observed between the WT and the iqgap1+/- mice (data not shown) except for GIN which developed in response to H. felis only in iqgap1+/- and to the same extent in iqgap1-/- mice, but not in WT mice (~40% of H. felis-infected iqgap1+/- mice and of H. felis-infected iqgap1+/- mice vs. 0% of H. felis WT mice; 8<n<10 in each group) (Table 1).
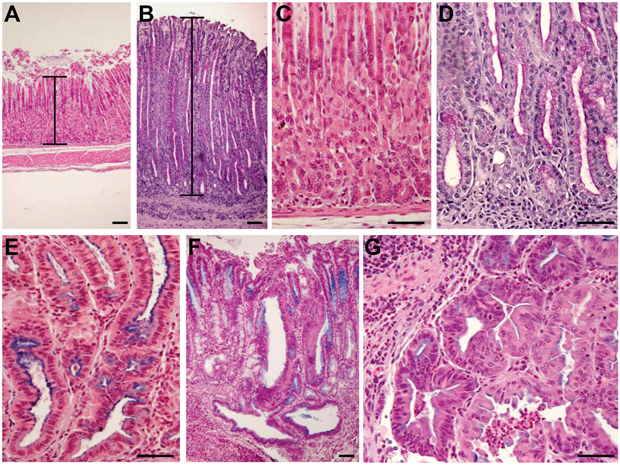
Figure 1: Development of histopathological lesions of the gastric mucosa after Helicobacter pylori infection in iqgap1+/- mice. Representative images of histopathologic features of the gastric mucosa of uninfected (A, C) or H. pylori SS1-infected (B, D, E, F, G) iqgap1+/- mice after 12 months. (A, C) Normal gastric mucosa. (B) H pylori-induced hyperplasia compared to the normal thickness of the mucosa of an uninfected control mouse (A). (D) Mucinous metaplasia with mucins producing cells replacing parietal and chief cells observed in normal gastric mucosa of a control mouse (C). (E) Pseudo-intestinal metaplasia with cells resembling enterocytes and expressing mucins mainly at the apical surface. (F-G) Dysplasia with gastrointestinal intraepithelial neoplasia (GIN) penetrating the muscularis mucosa. (G) GIN with an area of less cohesive dysplastic cells. Images are representative of the lesions obtained with Helicobacter felis and H. pylori SS1 and HPARE strains in WT and iqgap1+/- mice. Scale bar = 50 μm.
Table 1: Occurrence of gastrointestinal neoplasia (GIN) lesions in iqgap1 -/-, +/- and +/+ mice after Helicobacter infection with 3 different strains.
|
|
Number of mice with GIN / total number of mice |
Number of GIN lesions |
||||
|---|---|---|---|---|---|---|---|
WT |
+/- |
-/- |
WT |
+/- |
-/- |
||
M6 |
Control |
0/9 |
0/10 |
0/8 |
|
|
|
|
H. felis |
0/10 |
4/10 |
4/9 |
0 |
4 |
4 |
|
H. pylori SS1 |
0/7 |
0/9 |
|
0 |
0 |
|
|
H. pylori HPARE |
0/10 |
0/14 |
|
0 |
0 |
|
M12 |
Control |
0/9 |
0/10 |
|
0 |
0 |
|
|
H. felis |
12/17 |
17/18 |
|
29 |
52 |
|
|
H. pylori SS1 |
1/18 |
5/18 |
|
1 |
5 |
|
|
H. pylori HPARE |
0/20 |
3/20 |
|
0 |
3 |
|
For all mice studied GIN lesions have been evaluated on 2 separated tissue section per mouse. For each group of mice (7<n<20 per experimental condition), the number of mice with GIN compared to the total number of mice in the corresponding group is indicated. The total number of GIN lesions per group corresponds to the sum of all GIN lesions recorded for all mice of the same group (some mice harbouring several GIN lesions).
At 12 months PI, H. felis induced more hyperplasia and dysplasia in iqgap1+/- mice compared to the WT mice (Figure 2). The non-functional cagPAI H. pylori strain SS1 only induced more hyperplasia in iqgap1+/- mice compared to the WT mice (Figure 2). The most important effects were observed in response to the H. pylori strain HPARE harbouring a functional cagPAI, with iqgap1+/- infected mice developing significantly greater inflammation, hyperplasia, atrophy, metaplasia and dysplasia (Figure 2). All of the analyzed mice were consistently positive for H. pylori 12 months PI as confirmed by direct culture of the bacteria and/or PCR amplification of stomach samples (data not shown). Twelve months PI with H. felis, almost all of the iqgap1+/- mice (94.5%) developed GIN lesions compared to 70.5% of their WT littermates, and the total number of GIN lesions observed in iqgap1+/- mice was almost twice that of WT mice (52 vs. 29) (Table 1). Furthermore, iqgap1+/- mice infected with H. pylori developed 8 times more GIN lesions than their WT littermates (21% vs. 2.6% of mice infected with H. pylori SS1 and HPARE).
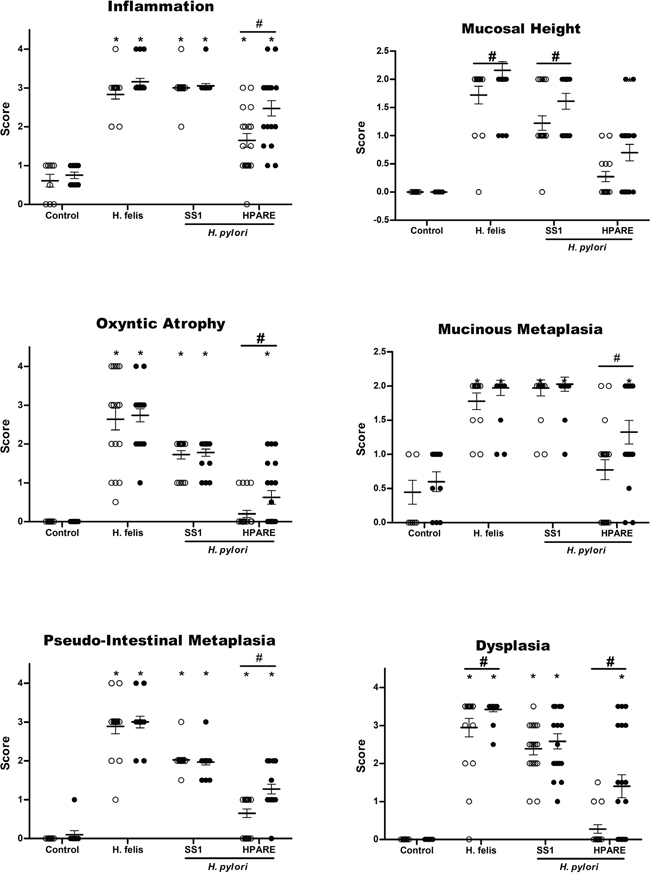
Figure 2: Evaluation of gastric histopathological lesions 12 months post-infection with Helicobacter. Scores for gastric inflammation, mucosal height, oxyntic atrophy, mucinous metaplasia, pseudo-intestinal metaplasia, and dysplasia determined on WT (light grey) and iqgap1+/- (black) mice after 12 months of infection with Helicobacter pylori SS1, HPARE or Helicobacter felis (17 < n < 20) or uninfected controls (9 < n < 10) are shown for comparison. Data represent the mean ± SD. *, p < 0.05 compared to uninfected controls; #, p < 0.05 compared to WT mice.
Expression and localization of IQGAP1 in response to H. pylori infection in vivo and in gastric cancer
The expression of IQGAP1 and E-cadherin was evaluated by immunohistochemistry (IHC) on the gastric mucosa of the corpus region of the stomach of WT mice 12 months PI with Helicobacter. In uninfected mice, IQGAP1 expression was detectable mainly in gastric epithelial cells compared to stromal cells and infiltrated leucocytes, and was located mainly at cell-cell junctions on the surface of parietal and chief cells, with a more intense and diffuse cytoplasmic pattern of expression in chief cells (Figure 3A). In GIN induced after H. felis or H. pylori infection, IQGAP1 remained localized at cell-cell junctions but was also detected more diffusely in the cytoplasm. E-cadherin was detected at cell-cell junctions of gastric epithelial cells in uninfected mice; its expression was lower in chief cells than in parietal or surface epithelial cells (Figure 3A). In GIN, E-cadherin localization was more diffuse but always present at cell-cell junctions.
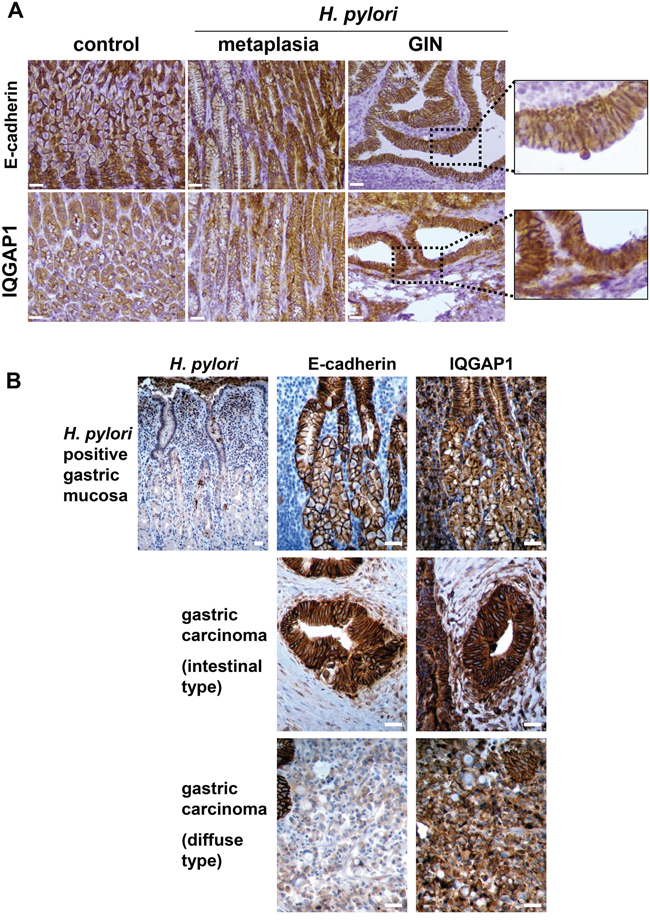
Figure 3: Expression of IQGAP1 and E-cadherin in gastric mucosa of mice and humans infected by Helicobacter pylori and in GIN and gastric carcinoma. Representative images of immunohistochemistry detection (in brown) of H. pylori, IQGAP1, and E-cadherin on gastric PET-sections. (A) Gastric tissue sections from uninfected (control) or H. pylori SS1-infected wild type (WT) mice (12 months) having developed metaplasia of the mucinous type (left side of the metaplasia images) and/or pseudo-intestinal type (right side of the metaplasia images) and GIN. (B) Representative gastric tissue sections from only gastric cancer patients with H. pylori associated chronic gastritis (n = 6) (top row), with moderately differentiated intestinal-type adenocarcinoma (n = 6) (second row) and with diffuse type adenocarcinoma comprised of E-cadherin-negative isolated signet ring cells (n = 3) (third row). Scale bars, 25 μm.
The expression of these proteins was evaluated by IHC on tissue sections of fundic gastric mucosa from H. pylori-infected patients and on gastric adenocarcinoma samples (Figure 3B). IQGAP1 and E-cadherin were expressed predominantly at the epithelial cell-cell junctions in gastritis. IQGAP1 was also present in the cytoplasm but to a lesser extent. E-cadherin was poorly expressed in diffuse type GC as already reported [22], whereas in the intestinal type, E-cadherin was localized at the cell-cell junctions and to a lesser extent into the cytoplasm. IQGAP1 localization differed in GC according to the histology. In the intestinal type, IQGAP1 was expressed mainly in the cytoplasm and reduced at cell/cell junctions, whereas in the diffuse type IQGAP1 was mainly membrane related (Figure 3A).
Effect of IQGAP1 knock-down on cancer stem cell-like properties in vitro
H. pylori induces an EMT in vitro which leads to the emergence of cells expressing CD44 and possessing CSC-like properties [4-6]. As IQGAP1 is a component of adherens junctions and as these structures are destabilized following infection, the impact of IQGAP1 knock-down on H. pylori-promoted-EMT and CSC properties was evaluated. Therefore, gastric epithelial cells were transfected with siRNA targeting IQGAP1 (siIQGAP1) or negative control siRNA (siCtrl) and were infected or not with H. pylori (Figure 4). Upon IQGAP1 silencing, the percentage of cells exhibiting a “hummingbird” phenotype significantly increased from 5 to 25% in AGS and MKN-45 cells, from 5 to 10% in MKN-74 cells (Figure 4A-4B). The “hummingbird” phenotype was even more prevalent in cells infected with H. pylori and transfected with siIQGAP1 reaching 50%, 30% and 17% for AGS, MKN-45 and MKN-74 cells, respectively.
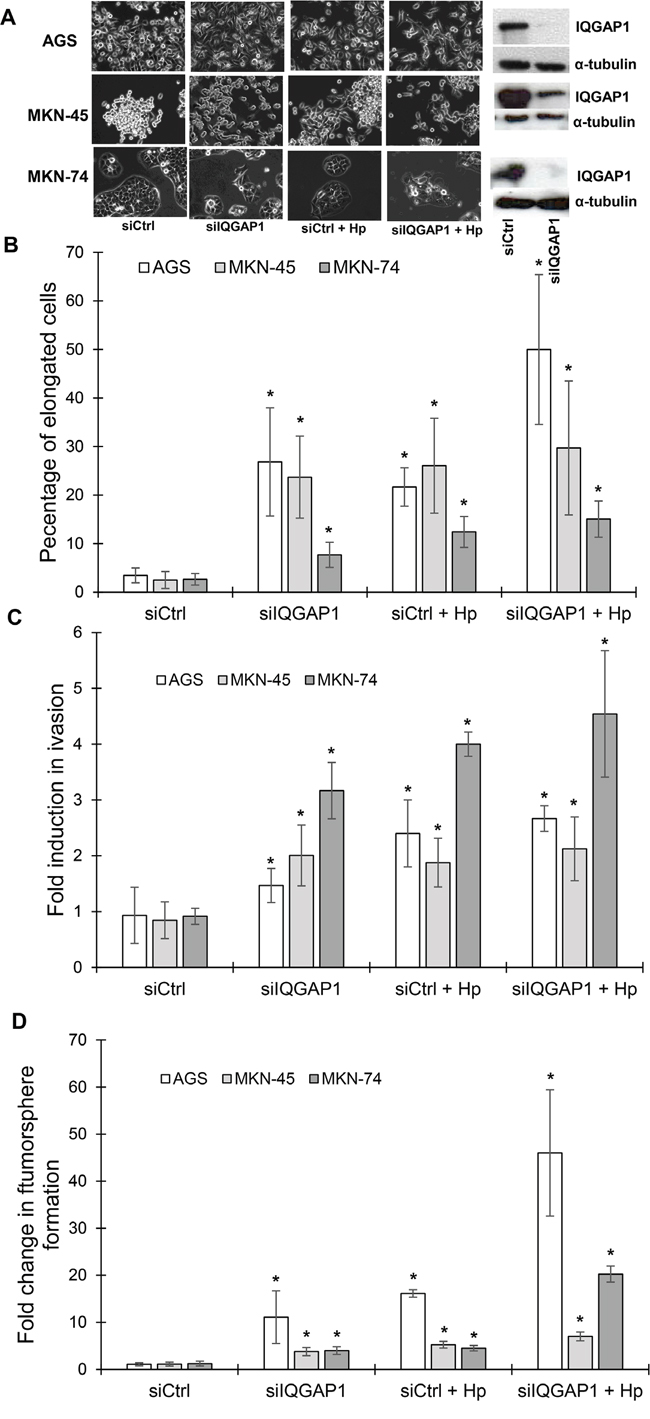
Figure 4: IQGAP1 inhibition mimicks Helicobacter pylori induced effects on gastric epithelial cell morphology, invasion capacities and tumorsphere formation. AGS, MKN-45 and MKN-74 cells were transfected in a double round of transfection with the negative control (siCtrl) or IQGAP1 (siIQGAP1) siRNA, and infected or not with H. pylori 7.13 for 18 to 24 h. (A) Representative phase-contrast images of transfected cells after infection or not with H. pylori 7.13; and representative images of western blotting experiments showing the inhibition of expression of IQGAP1 by siIQGAP1 compared to siCtrl transfected cells. α-tubulin detection was used as a loading control of equal amounts of proteins. Scale bar: 10 μm. (B) Quantification of the percentage of cells harboring an elongated phenotype. (C) Quantification of cellular invasion after 18 h of infection in the Transwell invasion assay. * p<0.05 vs. uninfected siCtrl cells. (D) Quantification of spheroids obtained under non-adherent culture conditions after 5 days of infection in the tumorsphere assay.
Some CSC-like properties of infected and/or transfected cells were studied. All of the cells transfected with siIQGAP1 were significantly more invasive (Transwell assay) than siCtrl transfected cells, indicating that IQGAP1 inhibition promotes invasion (Figure 4C). The most important effect was observed in MKN-74 cell line: transfected cells were 3 times more invasive and transfected and infected cells were 4 times more invasive than control cells. MKN-74 cells correspond to cells having intact cell-cell junctions in contrast to AGS and MKN-45 cells which have altered cell-cell junction due to the cdh1 mutation. The same results were obtained concerning the capacity of the cells to form tumorspheres (Figure 4D). Indeed, the cells with the strongest ability to form tumorspheres were those infected and transfected with siIQGAP1, implying that the inhibition of IQGAP1 promotes tumorsphere formation in response to H. pylori infection, especially in AGS and MKN-74 cells where the association of the transfection and the infection led to 40 and 20 times more tumorspheres, respectively.
Effects of H. pylori on the localization and expression of IQGAP1, CD44 and EMT-mediating transcription factors in gastric epithelial cells in vitro
To study the localization of IQGAP1, CD44 and mesenchymal markers, MKN-74 gastric epithelial cells (forming cell-cell junctions) and AGS gastric epithelial cells (cdh1 mutated, reduced cell/cell junctions) were infected with the cagPAI positive H. pylori strain 7.13 for 24 h (Figures 5 and 6). In the 2 cell lines, H. pylori infection induced an increase in CD44, in the EMT markers Snail1, Zeb1 and Vimentin and in IQGAP1 at the mRNA and protein levels (Figures 5 and 6). In MKN-74 cells, IQGAP1 localization changed with H. pylori infection: IQGAP1 was translocated from the cell-cell junctions to the cytoplasm and to membrane ruffles (Figure 5A). It is also interesting to note that inhibiting IQGAP1 and infecting the cells with H. pylori led to the formation of compact cell clusters with a spheroid appearance at the cell periphery and expressing high quantities of CD44 and Snail1. Furthermore, the immunofluorescence staining performed on MKN-74 confirmed that siIQGAP1 transfection favors a mesenchymal phenotype whereas the elongated cells highly express Zeb1 and to a lesser extent Snail1. The same trends were obtained with AGS cells in which IQGAP1 was mainly expressed in the cytoplasm and to a lesser extent at the cell-cell junctions and was translocated after infection to the plasma membrane (Figure 6). These results were partly confirmed in vivo in the mouse model, iqgap1 +/- and -/- mice showing a higher expression of Snail and Zeb1 compared to WT mice (Supplementary Figure S1). Infection with H. felis significantly increased the expression of CD44 and Zeb1 in iqgap1 +/- mice compared to uninfected WT mice, and their expression tended to be higher in H. felis infected iqgap1+/- mice than in H. felis infected WT mice (Supplementary Table S1).
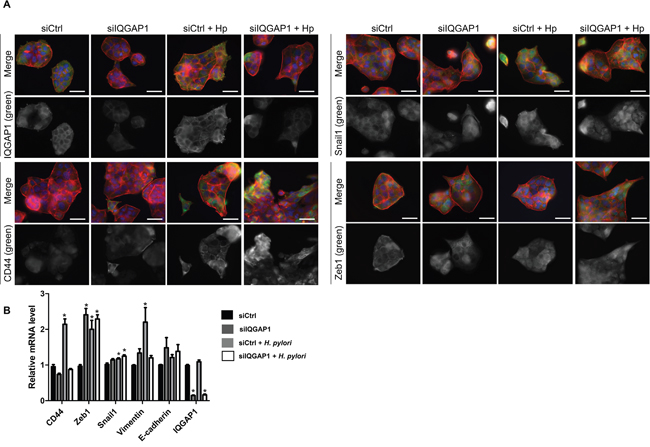
Figure 5: IQGAP1 inhibition accentuates Helicobacter pylori effect on EMT and CD44 expression in MKN-74 cells. MKN74 cells were cocultured with H. pylori 7.13 for 24 h. (A) Representative images of immunofluorescent staining of IQGAP1, CD44, Snail1 and Zeb1 alone (in green) or combined with F-actin (in red, phalloidin staining) and nuclei (in blue, DAPI staining). Scale bars, 15 μm. (B) Relative expression levels of the mRNA encoding IQGAP1, CD44, Zeb1, Snail1, Vimentin, E-cadherin in uninfected cells, siIQGAP1-transfected cells, infected cells and infected and siIQGAP1-transfected cells by RT-qPCR (relative to HPRT1 housekeeping gene mRNA) (n=6).
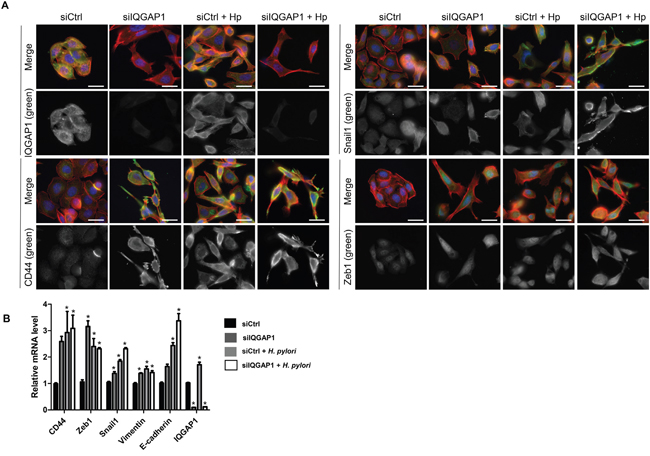
Figure 6: IQGAP1 inhibition accentuates Helicobacter pylori effect on EMT and CD44 expression in AGS cells. AGS cells were cocultured with H. pylori 7.13 for 24 h. (A) Representative images of immunofluorescent staining of IQGAP1, CD44, Snail1 and Zeb1 alone (in green) or combined with F-actin (in red, phalloidin staining) and nuclei (in blue, DAPI staining). Scale bars, 10 μm. (B) Relative expression levels of the mRNA encoding IQGAP1, CD44, Zeb1, Snail1, Vimentin, E-cadherin in uninfected cells, siIQGAP1-transfected cells, infected cells and infected and siIQGAP1-transfected cells by RT-qPCR (relative to HPRT1 housekeeping gene mRNA) (n=6).
DISCUSSION
We previously reported that the EMT-like changes induced during H. pylori infection are associated with the emergence of CD44+ cells possessing CSC-like properties [5, 6]. Here, inhibition of IQGAP1 induced a similar EMT-like process due to the adherens junction destabilization and this effect was more pronounced with H. pylori infection.
Interestingly, we show that reducing IQGAP1 by using siRNA, led to CD44 upregulation, as well as the acquisition of invasive properties and formation of tumorspheres in vitro, 2 hallmarks of CSC-like cells. In addition, our in vitro data revealed for the first time that the association of IQGAP1 inhibition and H. pylori infection favors EMT and the acquisition of CSC properties. This is in agreement with different reports which considered IQGAP1 as a potential target of H. pylori in disrupting epithelial polarity [23]. Furthermore, the infection leads to an increase in IQGAP1 expression and also to a delocalization of IQGAP1 from cell-cell junctions and cytoplasm to the plasma membrane confirmed, using different models, the results described by Conlin et al. [24]. IQGAP1 knock-down led to a mesenchymal phenotype acquisition in both AGS and MKN-74 cells where IQGAP1 is not located at the same site. Indeed, in spite of the different IQGAP1 localization, observed effects are similar indicating that the effects are independent of the IQGAP1 localization.
In this study, we showed for the first time that deletion of one IQGAP1 allele in mice significantly promotes the development of high grade dysplasia in response to infection with pro-carcinogenic H. pylori strains. This mechanism appears to be due to a promotion and acceleration of the process of lesion development, as GIN was induced by H. felis after 6 months of infection in iqgap1+/- mice, whereas it appeared after more than 12 months in WT mice [21, 25]. Surprisingly, we were not able to reproduce the results published by Li et al., who observed spontaneous hyperplasia of the gastric mucosa in heterozygous and homozygous mice invalidated for iqgap1 compared to WT littermates [19]. There were no apparent differences concerning these two independent studies, as both were performed on a 129/BL6 genetic background, with the exception of the environmental housing conditions which may differ significantly from one laboratory to another (our colony was confirmed to be negative for all pathogenic organisms including Helicobacter spp. before experimental infection). In the present study, aged iqgap1+/- mice showed no more basal inflammation or alterations of the gastric mucosa than their WT littermates, and infection with the cagA-positive HPARE H. pylori strain induced more inflammation and pre-neoplastic lesions in iqgap1+/- mice than in WT littermates after 12 months. Finally, iqgap1+/- mice developed 8 times more GIN in response to H. pylori infection than their WT littermates. This result suggests that the deletion of only one IQGAP1 allele could alter cell-cell junctions and favor the pro-inflammatory and pro-carcinogenic effects of cagA-positive H. pylori strains, and consequently the development of pre-neoplastic lesions in ageing animals. In humans, the carcinogenic potential of H. pylori is mainly associated with CagA, which can be considered to be an oncoprotein as its expression in transgenic mice is sufficient to induce carcinoma without any other stimuli [25]. These results support the hypothesis that the IQGAP1 protein is essential to maintaining the integrity of the gastric epithelium against the effects of CagA injected into host cells by carcinogenic H. pylori strains. Indeed, H. pylori and particularly the oncoprotein CagA, are known to induce a disturbance of the signalling pathways regulating the integrity of the adherens junctions and the equilibrium between cellular proliferation and the differentiation state [26-28]. Here we report that cagA-positive H. pylori strains induce a destabilization of cell/cell junctions with a delocalization of IQGAP1 and its partner E-cadherin. Little is known about the molecular events leading to the development of diffuse type gastric adenocarcinoma, as no pre-malignant precursor lesions or markers have been described. Diffuse type GCs are associated with mutations in the cdh1 gene encoding E-cadherin in about 30% of cases. This was confirmed experimentally in one study carried out on transgenic mice in which the invalidation of one cdh1 allele led to the development of diffuse type gastric adenocarcinoma in response to N-methyl-N-nitrosourea [22]. For the remaining 70% of the cases, other gene deregulations or mutations have been proposed. In two recent reports from the Consortium of the Human Genome Atlas and from Wang et al., a molecular classification of gastric adenocarcinoma in 4 main subclasses was proposed based on their molecular profile of mutations and genetic stability [9, 10]. These two studies do not report IQGAP1 mutations among the most frequent ones described, but in 2005 Morris et al. reported some specific IQGAP1 mutations in a few cases of diffuse type GC [29], showing that IQGAP1 mutations are not commonly found in GC. However, our results are in agreement with those of Takemoto et al. concerning the localization of IQGAP1. They stated that IQGAP1 was frequently observed diffusely in the cytoplasm in intestinal type tumors whereas it was expressed at the cell membrane in diffuse type tumors [18]. In our study, the same trend was observed for neoplastic lesions in the mouse model of H. pylori infection and in patients with GC.
Our results prove that IQGAP1 is critical for the maintenance of normal cell physiology limiting carcinogenesis especially during H. pylori infection.
MATERIALS AND METHODS
Mice
Experiments on mice were carried out in Level 2 animal facilities at the University of Bordeaux in agreement with the local Ethics Committee (approval number 50120141-A) and in conformity with the French Committee of Genetic Engineering (approval number 4608). The iqgap1-null 129/BL6 were kindly provided by A. Bernards (Massachusetts General Hospital Center for Cancer Research, Charlestown, MA, USA) [19]. Heterozygous iqgap1+/- 129/BL6 males were bred with wild type (WT) C57Bl/6 females, and heterozygous iqgap1+/- females and WT littermates were generated. Groups of WT and iqgap1+/- mice (7 < n < 17 for each group) were force-fed with H. pylori SS1 H. pylori HPARE, Helicobacter felis (both kindly provided by A. Labigne, Pasteur Institute, Paris, France) or phosphate buffered saline (PBS) for control mice, at 6 weeks of age with 200 μl of 10.8 CFU/ml of Helicobacter sp. suspension every other day for a total of three doses [21]. A group of iqgap1-/- mice was challenged with Helicobacter felis (n = 9) or PBS (n = 8) for 6 months. After 6 or 12 months of infection, mice were euthanized by cervical dislocation; stomachs were collected aseptically, opened along the greater curvature, washed in PBS and sectioned longitudinally into 2 pieces along the smaller curvature from the squamocolumnar junction through the pylorus. The first part of the stomach was fixed for 24 h in 3.7% neutral-buffered formalin (Sigma, Saint-Quentin Fallavier, France), followed by standard histological processing and paraffin embedding. The second part was homogenized in 200 μL of brucella broth to confirm H. pylori colonization by bacterial culture and PCR as previously described [21].
Information on protocols used for bacterial culture, tissue processing, histopathological scoring, statistical analysis, cell culture experiments, and qRT-PCR is provided in the Supplementary Materials and Methods. AGS, MKN-45 and MKN-74 cell lines were used for the in vitro part. AGS cells are known to be predisposed to the EMT process since they have a cdh1 mutation. Thus, they are a good model to study the H. pylori transforming effect. AGS cells were used to study the role of IQGAP1 independently of its junctional role.
ACKNOWLEDGMENTS
This project was supported by the French ‘Association pour la Recherche contre le Cancer’ (grant number 8412), ‘Institut National du Cancer’ (grant number 07/3D1616/IABC-23-12/NC-NG), ‘Conseil Regional d’Aquitaine’ (grant number 20071301017 and 20081302203) and the SIRIC BRIO (Site de Recherche Intégrée sur le Cancer -Bordeaux Recherche Intégrée Oncologie) [Grant: INCa-DGOS-Inserm 6046]. We thank André Bernards (Massachusetts General Hospital Center for Cancer Research, Charlestown, MA, USA) for giving access to the iqgap1 knockout mice and for helpful discussions; Patrick Auguste (INSERM U1029, University of Bordeaux, Bordeaux, France) for IQGAP1 siRNAs and antibodies; Denis Collet (Digestive Surgery of Haut-Lévèque Hospital, University Hospital of Bordeaux, France) and Serge Evrard and Isabelle Soubeyran (Bergonié Institute, Bordeaux, France) and Geneviève Belleannée (Pathology Department, Haut Leveque Hospital, University Hospital Center, Pessac, france) for providing human gastric tissue samples from consenting patients; David Cappellen (EA2406 University of Bordeaux) and Violaine Moreau (INSERM U1053, University of Bordeaux, Bordeaux, France) for helpful discussions; DBS is supported by the National Institutes of Health Intramural Research Program and Laura Biguet for technical support.
CONFLICTS OF INTEREST
All the authors of this paper declare that they have no conflict of interest.
REFERENCES
1. Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. International journal of cancer 2014; 136:E359-86.
2. Schistosomes, liver flukes and Helicobacter pylori. IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Lyon, 7-14 June 1994. IARC Monogr Eval Carcinog Risks Hum 1994; 61:1-241.
3. Amieva MR, Vogelmann R, Covacci A, Tompkins LS, Nelson WJ, Falkow S. Disruption of the epithelial apical-junctional complex by Helicobacter pylori CagA. Science 2003; 300:1430-4.
4. Baud J, Varon C, Chabas S, Chambonnier L, Darfeuille F, Staedel C. Helicobacter pylori initiates a mesenchymal transition through ZEB1 in gastric epithelial cells. PloS one 2013; 8:e60315.
5. Bessède E, Staedel C, Acuna Amador LA, Nguyen PH, Chambonnier L, Hatakeyama M, Belleannee G, Mégraud F, Varon C. Helicobacter pylori generates cells with cancer stem cell properties via epithelial-mesenchymal transition-like changes. Oncogene 2013; 33:4123-31.
6. Bessède E, Dubus P, Mégraud F, Varon C. Helicobacter pylori infection and stem cells at the origin of gastric cancer. Oncogene. 2015 May 14; 34:2547-55.
7. Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, Campbell LL, Polyak K, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008; 133:704-15.
8. Lauren P. The two histological main types of gastric carcinoma: diffuse and so-called intestinal-type carcinoma. an attempt at a histo-clinical classification. Acta Pathol et Microbiol Scand 1965; 64:31-49.
9. Network TCGAR. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014; 513:202-9.
10. Wang K, Yuen ST, Xu J, Lee SP, Yan HH, Shi ST, Siu HC, Deng S, Chu KM, Law S, Chan KH, Chan AS, et al. Whole-genome sequencing and comprehensive molecular profiling identify new driver mutations in gastric cancer. Nature Gen 2014; 46:573-82.
11. Brown MD, Sacks DB. IQGAP1 in cellular signaling: bridging the GAP. Trends Cell Biol 2006; 16:242-9.
12. Noritake J, Watanabe T, Sato K, Wang S, Kaibuchi K. IQGAP1: a key regulator of adhesion and migration. J Cell Sci 2005; 118:2085-92.
13. Sugimoto N, Imoto I, Fukuda Y, Kurihara N, Kuroda S, Tanigami A, Kaibuchi K, Kamiyama R, Inazawa J. IQGAP1, a negative regulator of cell-cell adhesion, is upregulated by gene amplification at 15q26 in gastric cancer cell lines HSC39 and 40A. J Human Gen 2001; 46:21-5.
14. Briggs MW, Sacks DB. IQGAP proteins are integral components of cytoskeletal regulation. EMBO Rep 2003; 4:571-4.
15. Jadeski L, Mataraza JM, Jeong HW, Li Z, Sacks DB. IQGAP1 stimulates proliferation and enhances tumorigenesis of human breast epithelial cells. J Biol Chem 2008; 283:1008-17.
16. Nabeshima K, Shimao Y, Inoue T, Koono M. Immunohistochemical analysis of IQGAP1 expression in human colorectal carcinomas: its overexpression in carcinomas and association with invasion fronts. Cancer Lett 2002; 176:101-9.
17. Walch A, Seidl S, Hermannstadter C, Rauser S, Deplazes J, Langer R, von Weyhern CH, Sarbia M, Busch R, Feith M, Gillen S, Hofler H, et al. Combined analysis of Rac1, IQGAP1, Tiam1 and E-cadherin expression in gastric cancer. Mod Pathol 2008; 21:544-52.
18. Takemoto H, Doki Y, Shiozaki H, Imamura H, Utsunomiya T, Miyata H, Yano M, Inoue M, Fujiwara Y, Monden M. Localization of IQGAP1 is inversely correlated with intercellular adhesion mediated by e-cadherin in gastric cancers. Int JCancer 2001; 91:783-8.
19. Li S, Wang Q, Chakladar A, Bronson RT, Bernards A. Gastric hyperplasia in mice lacking the putative Cdc42 effector IQGAP1. Mol Cell Biol 2000; 20:697-701.
20. Cai X, Carlson J, Stoicov C, Li H, Wang TC, Houghton J. Helicobacter felis eradication restores normal architecture and inhibits gastric cancer progression in C57BL/6 mice. Gastroenterology 2005; 128:1937-52.
21. Varon C, Dubus P, Mazurier F, Asencio C, Chambonnier L, Ferrand J, Giese A, Senant-Dugot N, Carlotti M, Mégraud F. Helicobacter pylori infection recruits bone marrow-derived cells that participate in gastric preneoplasia in mice. Gastroenterology 2012; 142:281-91.
22. Humar B, Blair V, Charlton A, More H, Martin I, Guilford P. E-cadherin deficiency initiates gastric signet-ring cell carcinoma in mice and man. Cancer Res 2009; 69:2050-6.
23. Osman MA, Bloom GS, Tagoe EA. Helicobacter pylori-induced alteration of epithelial cell signaling and polarity: a possible mechanism of gastric carcinoma etiology and disparity. Cytoskeleton 2013; 70:349-59.
24. Conlin VS, Curtis SB, Zhao Y, Moore ED, Smith VC, Meloche RM, Finlay BB, Buchan AM. Helicobacter pylori infection targets adherens junction regulatory proteins and results in increased rates of migration in human gastric epithelial cells. Infect Imm 2004; 72:5181-92.
25. Ohnishi N, Yuasa H, Tanaka S, Sawa H, Miura M, Matsui A, Higashi H, Musashi M, Iwabuchi K, Suzuki M, Yamada G, Azuma T, et al. Transgenic expression of Helicobacter pylori CagA induces gastrointestinal and hematopoietic neoplasms in mouse. Proc Nat Acad Scie U SA 2008; 105:1003-8.
26. Franco AT, Johnston E, Krishna U, Yamaoka Y, Israel DA, Nagy TA, Wroblewski LE, Piazuelo MB, Correa P, Peek RM, Jr. Regulation of gastric carcinogenesis by Helicobacter pylori virulence factors. Cancer Res 2008; 68:379-87.
27. Murata-Kamiya N, Kurashima Y, Teishikata Y, Yamahashi Y, Saito Y, Higashi H, Aburatani H, Akiyama T, Peek RM, Jr., Azuma T, Hatakeyama M. Helicobacter pylori CagA interacts with E-cadherin and deregulates the beta-catenin signal that promotes intestinal transdifferentiation in gastric epithelial cells. Oncogene 2007; 26:4617-26.
28. Saito Y, Murata-Kamiya N, Hirayama T, Ohba Y, Hatakeyama M. Conversion of Helicobacter pylori CagA from senescence inducer to oncogenic driver through polarity-dependent regulation of p21. J Ex Med 2010; 207:2157-74.
29. Morris LE, Bloom GS, Frierson HF, Jr., Powell SM. Nucleotide variants within the IQGAP1 gene in diffuse-type gastric cancers. Genes, chromosomes & cancer 2005; 42:280-6.